 Saghar Yousefnia1
Saghar Yousefnia1 Farzad Seyed Forootan2,3*Shiva Seyed Forootan4
Farzad Seyed Forootan2,3*Shiva Seyed Forootan4 Mohammad Hossein Nasr Esfahani2
Mohammad Hossein Nasr Esfahani2 Ali Osmay Gure5*
Ali Osmay Gure5* Kamran Ghaedi1,2*
Kamran Ghaedi1,2*- 1Department of Cell and Molecular Biology and Microbiology, Faculty of Biological Science and Technology, University of Isfahan, Isfahan, Iran
- 2Department of Cellular Biotechnology at Cell Science Research Center, Royan Institute of Biotechnology, ACECR, Isfahan, Iran
- 3Legal Medicine Research Center, Legal Medicine Organization, Tehran, Iran
- 4Department of Molecular and Clinical Pharmacology, MRC Centre for Drug Safety Science, Institute of Translational Medicine, University of Liverpool, Liverpool, United Kingdom
- 5Department of Molecular Biology and Genetics, Faculty of Science, Bilkent University, Ankara, Turkey
Breast cancer stem cells (BCSCs) are the minor population of breast cancer (BC) cells that exhibit several phenotypes such as migration, invasion, self-renewal, and chemotherapy as well as radiotherapy resistance. Recently, BCSCs have been more considerable due to their capacity for recurrence of tumors after treatment. Recognition of signaling pathways and molecular mechanisms involved in stemness phenotypes of BCSCs could be effective for discovering novel treatment strategies to target BCSCs. This review introduces BCSC markers, their roles in stemness phenotypes, and the dysregulated signaling pathways involved in BCSCs such as mitogen-activated protein (MAP) kinase, PI3K/Akt/nuclear factor kappa B (NFκB), TGF-β, hedgehog (Hh), Notch, Wnt/β-catenin, and Hippo pathway. In addition, this review presents recently discovered molecular mechanisms implicated in chemotherapy and radiotherapy resistance, migration, metastasis, and angiogenesis of BCSCs. Finally, we reviewed the role of microRNAs (miRNAs) in BCSCs as well as several other therapeutic strategies such as herbal medicine, biological agents, anti-inflammatory drugs, monoclonal antibodies, nanoparticles, and microRNAs, which have been more considerable in the last decades.
Introduction
The most common cancer among women all over the world is breast cancer (BC). The prevalence of BC is increasing; according to the recent report (2020), 279,100 new cases and 42,690 deaths of BC have been estimated in the United States (https://doi.org/10.3322/caac.21590). Approximately 25% of cancers in women and 15% of cancer deaths are caused by BC. About 0.8–1% of all BCs are due to male breast carcinoma, although the incidence of BC in men has increased recently. It has been estimated that the prevalence of BC in the world will reach 2.3 million by 2030 (1, 2). The most important factors for promoting BC are high expression level of estrogen, estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2). On the other hand, mutation in tumor suppressor genes such as BRCA1 and BRCA2 is the most important reason for familial BC (3). There are two types (ductal and lobular) and multiple subtypes [luminal A and B, basal-like triple negative breast cancer (TNBC), HER2+, and claudin-low] of BC that are classified by molecular and histological phenotypes. Each type and subtype of BC are categorized to non-invasive and invasive (2, 4–7). A population of cancer cells in BC tissues makes the treatment of BC obscure. Cancer stem cells (CSCs) are a type of BC cells with some stemness phenotypes such as self-renewal, differentiation, metastasis, migration, and therapeutic resistance that make tumors more progressive and aggressive (8–10). Breast tumor consists of 2% breast cancer stem cells (BCSCs), in which their resistance to chemotherapy and radiotherapy can cause treatment failure and disease recurrence (8). There is a controversy among researchers about the origin of BCSCs, in which normal stem cells, progenitor cells, or differentiated cells can be considered as an origin of BCSCs (11). Deregulation of signaling pathways including mitogen-activated protein (MAP) kinase, PI3K/Akt/nuclear factor kappa B (NFκB), TGF-β, hedgehog (Hh), Notch, Wnt/β-catenin, and Hippo pathway in normal stem cells, progenitor cells, or differentiated cells may transform them to CSCs due to genetic and epigenetic changes (12–14). In addition, the expression of the specific molecules in these signaling pathways can be deregulated in BCSCs as well as microRNAs (miRNAs). It has been reported that there are several miRNAs that have been upregulated in BCSCs (Table 1). Downregulation of these miRNAs has been suggested as a novel treatment strategy of BC (Table 2). Alternative strategies are being used to target BCSCs directly. In this review, we have summarized the malignant characterization of BCSCs such as drug and radiotherapy resistance, metastasis and angiogenesis, biomarkers, and mechanisms involved in drug and radiotherapy resistance. We have assessed signaling pathways that are responsible for maintaining stemness phenotypes as well as miRNAs in BCSC. The recognition of signaling pathways and molecular mechanisms involved in stemness phenotypes of BCSCs would be effective for discovering novel treatment strategies to target BCSCs. Finally, we have reviewed several therapeutic strategies that have been investigated in the last decades.
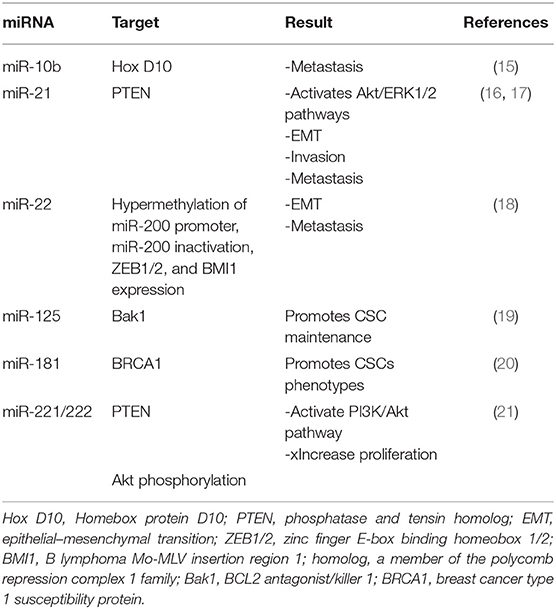
Table 1. MicroRNAs (miRNAs) upregulated in breast cancer stem cells (BCSCs).
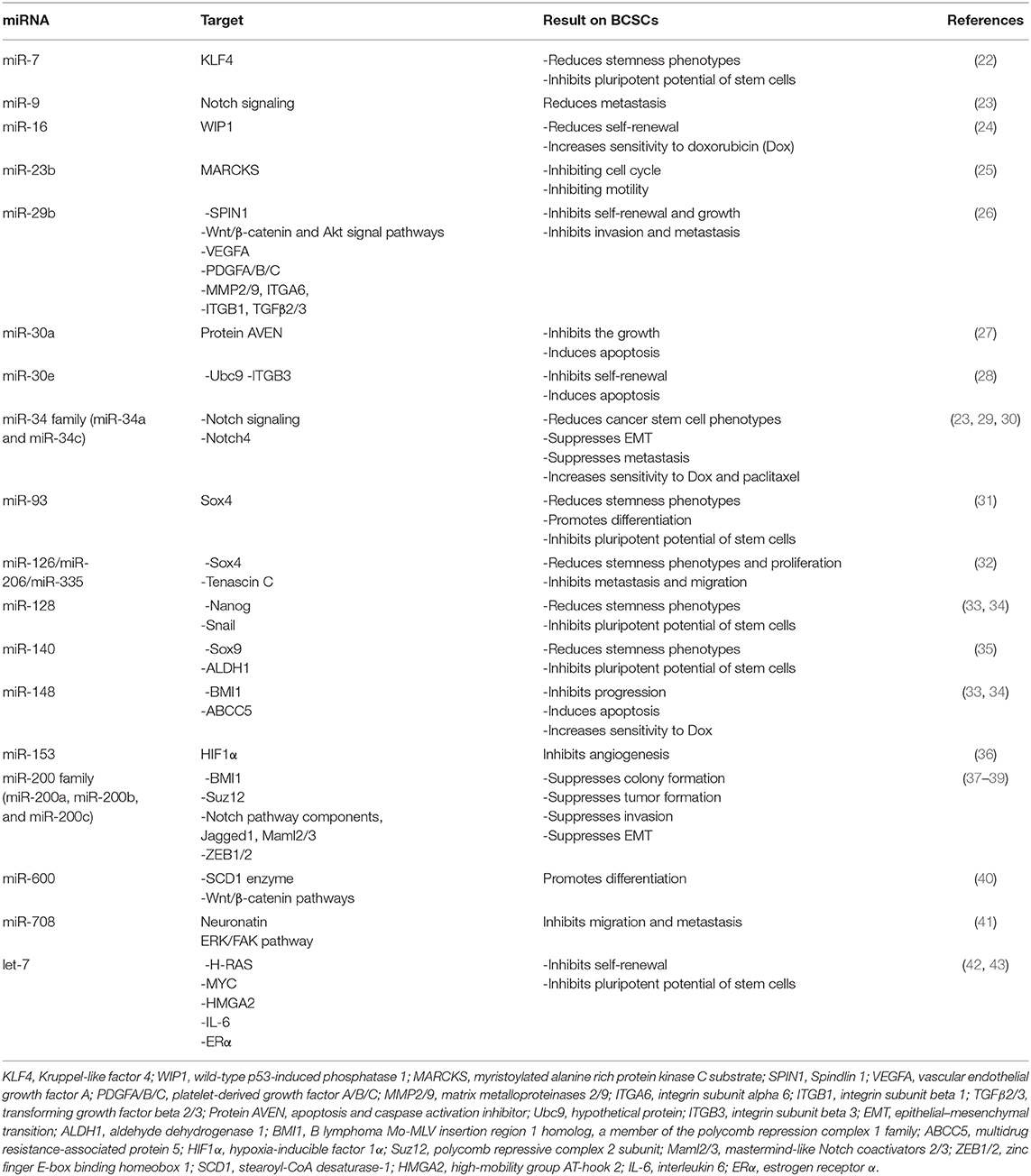
Table 2. MicroRNAs (miRNAs) targeting breast cancer stem cells (BCSCs).
BCSC Markers
BCSCs express variety of specific markers such as CD44+/CD24−, CD326 (EpCAM), epithelial specific antigen (ESA), and aldehyde dehydrogenase (ALDH) activity in different types of BC. Unlike CD24, known as a differentiated BC cells marker, CD44 has been identified as a stemness marker on the surface of BCSCs. CD44 is bonded to hyaluronic acid (HA) and extracellular matrix proteins such as osteopontin (OPN) and matrix metalloprotease (MMP) (44). CD44 with receptor tyrosine kinase (RTK) may control cell adhesion, migration, and transmission of proliferation signals in BCSCs (45). Several signaling pathways such as Rho GTPases, Ras-MAPK, and PI3K/Akt involved in the regulation of cell adhesion, migration, invasion, and epithelial–mesenchymal transition (EMT) are mediated by CD44 (46). However, CD44+ cannot be very helpful as a definite marker for isolation and detection of BCSCs in all types of BC (5, 47, 48). Luminal and basal/mesenchymal BCs have CD44−/CD24+ and CD44+/CD24− phenotypes, respectively, whereas basal/epithelial BC is positive for both markers (49). Frequency of BCSCs with CD44+/CD24− is significantly more in TNBCs (44). In addition, PR and ER messenger RNA (mRNA) levels in cells with the markers of CD44+/CD24−/ALDH+ significantly are less in comparison with cells with phenotype of CD44−/CD24+ (11). ALDH activity is a better predictive marker in parallel with higher tumorigenic activity in vivo in comparison with CD44/CD24 markers (50, 51). ALDH enzyme is responsible for intracellular aldehyde oxidation and has a critical role in differentiation of stem cells (52). To detect ALDH activity using Aldeflour assay kit, ALDH converts BODIPY-aminoacetaldehyde substrate to BODIPY-aminoacetate, a fluorescent product detectable by flow cytometry (51). The other important marker is ESA or CD326. ESA is a protein marker that is expressed on the surface of BCSCs essential for cell adhesion, proliferation, migration, and invasion of BC cells through Wnt signaling pathway (53). A regulated intramembrane proteolysis by ADAM metallopeptidase domain 17 (ADAM17) and Presenilin-2 (PSEN2) involves breakage of EpCAM intracellular domain(EpICD). EpICD binds to a half LIM domains 2 (FHL2) and β-catenin and forms a nuclear protein complex, which expresses genes involved in stemness physiological features (54). The other markers mostly used for isolation and identification of BCSCs in all types of BCs are CD133, CD166, Lgr5, CD47, and ABCG2 (55). A recent study indicated that transglutaminase (TG2) is expressed highly in CSCs and is involved in the expression of CSC markers, proliferation, drug resistance, migration, invasion, and EMT of CSCs. This protein is dependent to Ca2+ and GTP localized in cytosol, nucleus, cell membrane, and extracellular environment and can be converted to both open (Ca2+-bonded cross-linking form) and closed (GTP-bonded signaling form) configurations. Closed configuration has a vital role in BC progression and CSC survival through activation of NFκβ, Akt, and focal adhesion kinase (FAK) signaling (56).
It has been reported that the use of radiation to destroy cancer cells after surgery may convert differentiated cancer cells to CSCs through the expression of CSC markers such as Oct4/Sox2/KLF4. Therefore, in some cancer cases, radiation is not recommended, as it can involve recurrence and metastasis (57). Hypoxia, generated in the depths of the tumor due to lack of oxygen and blood vessels, may regulate the expression of genes involved in CSCs. It may increase the number of CSCs through the conversion of differentiated cancer cells to CSCs (4).
Signaling Pathways Regulate BCSCs
It has been noted that a number of signaling pathways including MAP kinase, PI3K/Akt/NFκB, TGF-β, hedgehog (Hh), Notch, Wnt/β-catenin, and Hippo signaling have been implicated in stemness maintenance and regulation of self-renewal, metastasis, and therapeutic resistance into CSCs (12, 14, 56–61). Deregulation of these pathways in normal stem cells may transform them to CSCs. CSCs markers could show a vital role in the regulation of signaling pathways. There is a relationship between CD24 and Sonic hedgehog (SHH), as knocking down CD24 in breast cancer cells have demonstrated increased proliferation, invasion, and tumorigenicity through higher expression of SHH, GLI1, and MMP2. CD24 suppresses the malignant phenotype of BCSCs by downregulating SHH expression through STAT1 inhibition (12) (Figure 1). However, cells with high expression of CD44 show higher expression level of β-catenin and Notch1 and Ki67 (62). CD44-PKC-Nanog signaling axis is involved in BCSCs. Binding of CD44 with protein kinase Cε (PKCε) is mediated by hyaluronan and regulates human breast tumor cells and BCSC functions. Activated PKCε increases phosphorylation of Nanog, a stem cell marker. Phosphorylated Nanog is translocated into the nucleus and increases miR-21 expression and decreases tumor suppressor program cell death protein 4 (PDCD4) expression. Along with this process, inhibitors of apoptotic proteins (IAPs) and MD11 are upregulated and then leads to antiapoptosis and chemotherapy resistance in BCSCs (63). The other pathway that is mediated by CD44–hyaluronan is HA/CD44-induced c-Jun signaling. Activated c-Jun is translocated into the nucleus and stimulates miR-21 expression, antiapoptosis protein Bcl-2, and IAPs (64). In addition, HA–CD44 interaction promotes c-Src kinase activation and Twist phosphorylation. Phosphorylated Twist is translocated into the nucleus and activates miR-10a expression through binding to Twist binding site(s). This process leads to a decrease in tumor suppressor HOXD10, upregulation of RhoA/RhoC, and stimulation of RhoGTPase-Rho kinase (ROK). ROK activation leads to cytoskeleton activation and breast cancer cell invasion (65) (Figure 1).
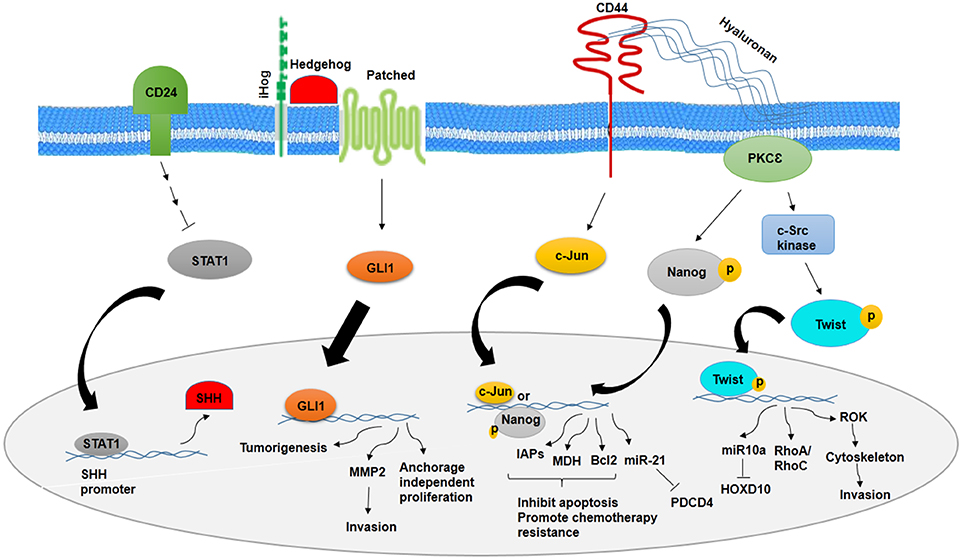
Figure 1. Role of CD24 and CD44 in inhibition and promotion of BCSCs, respectively. CD24 inhibits stemness phenotypes of BCSCs through inhibition of STAT1 and SHH. However, CD44 promotes chemotherapy resistance and invasion of BCSCs through CD44–PKCε-Nanog signaling axis and inducing c-Jun signaling.
In addition, EMT is a process that converts cells with epithelial to mesenchymal phenotypes through decrease in cell–cell adhesion and increase in cell metastasis (40). There are many mediators such as growth factors (epidermal growth factor and TGF-β) that have been involved in several signaling pathways including TGF-β, PI3K/Akt, Wnt, and MAPK and regulate EMT (66). Induction of EMT in normal and cancer cells can generate CSCs with high expression of respective markers, mammosphere formation, colony formation, as well as invasion and metastasis (67). One of the most important levels in the regulation of EMT is the regulation of EMT-inducing transcription factors such as ZEB, Snail, Slug, and Twist. These transcription factors induce EMT through activation of the specific molecular programs in different signaling pathways, which mediate repression of epithelial markers including E-cadherin and induce mesenchymal markers such as vimentin (68, 69). It has been reported that high expression level of ZEB1 has been associated with the activity of several specific molecules, FOXC2, NFκB, SOX2, BCL6, and HIF1α involved in EMT and malignancy of BCSCs (68). Overall, EMT transcription factors contributed to the regulation of signaling pathways in CSCs (66).
MAPK Signaling Pathway
Several studies have shown MAPK signaling activity in aggressive BC and BCSCs. One of the pathways activates MAPK signaling in BCSCs, which is mediated by epidermal growth factor receptor (EGFR) and HER2. In this way, activated ERK stimulates MAPK interacting kinase (MNK) signaling, and then, MNK activates XIAP, its downstream protein. XIAP is an apoptotic inhibitor protein that is upregulated in inflammatory and aggressive BC and links NFκβ signaling activity to MAPK signaling (14). On the other hand, IAP upregulates Snai2 expression, an EMT factor that has an association with increased stem cell properties. Inhibition of IAP protein decreases MAPK activity through downregulation of EGFR and Snai2 expression (70) (Figure 2).
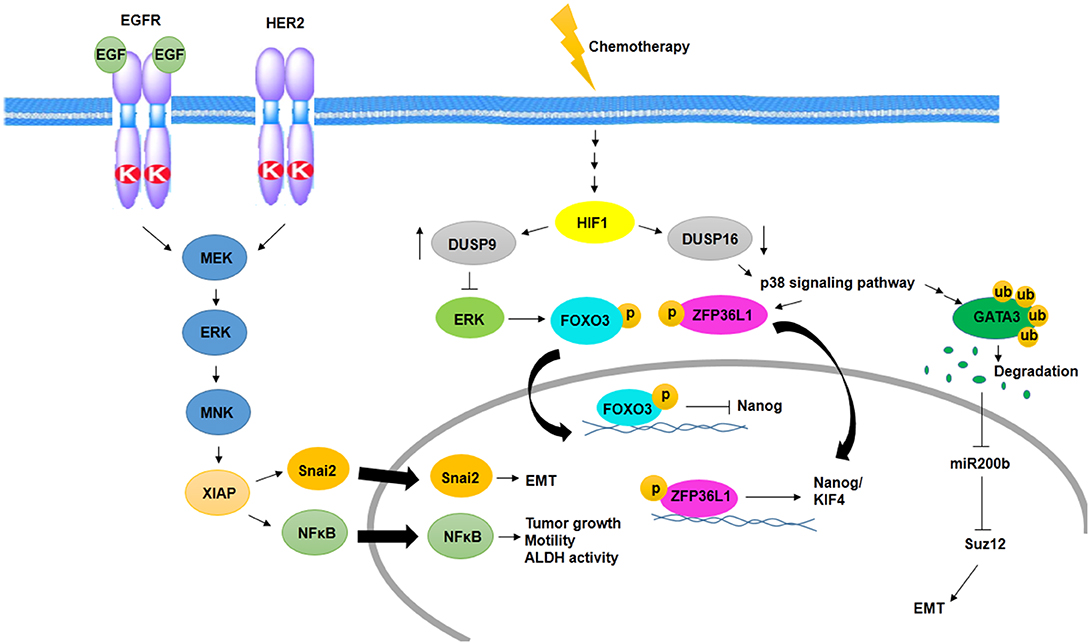
Figure 2. Activation of MAPK signaling pathway in BCSCs. Cooperation between EGFR and HER2 promotes EMT, tumor growth, motility, and ALDH activity through activation of MNK and XIAP in a MAPK-dependent manner. In addition, chemotherapy promotes EMT and induces MAPK signaling pathway and expression of Nanog/KIF4 in a HIF1-dependent manner.
One of the other MAPK activating signaling pathways in BCSC resistance to chemotherapy is mediated by hypoxia-inducible factor-1 (HIF1)-dependent manner. Chemotherapy in an HIF1-dependent manner regulates the expression of dual specificity phosphatase (DUSP9) and DUSP16. Reciprocally, increased expression of DUSP9 inhibits ERK and decreased expression of DUSP16, which activates p38 signaling pathway. ERK inhibition leads to increased expression of Nanog through decreased phosphorylation of FoxO3, and p38 activation stabilizes transcripts of stemness genes such as Nanog and KIF4 via ZFP36L1 phosphorylation, which has been known as a Nanog and KIF4 mRNA binding protein (71) (Figure 2).
p38δ MAPK is involved in cancer cell progression, metastasis, and regulation of BCSC phenotypes as well as EMT. p38δ MAPK-mediated EMT is promoted by the inhibition of miR200b, the downstream of p38γ MAPK. p38γ MAPK inhibits miR200b through induction of GATA3 ubiquitylation and its degradation. Then, expression of a polycomb group protein, Suz12, which is a target of miR200b, promotes EMT (72) (Figure 2).
PI3K/Akt/NFκB Signaling Pathway
It has been reported that the PI3K/Akt/NFκB axis has been involved in the characterization and drug resistance of BCSCs. Suppressed PI3K/Akt in CSCs overcomes the multidrug resistance (MDR) phenotype through induction of cell apoptosis. The role of PI3K/Akt in BCSCs may be mediated by HER2, which makes tumors more aggressive through unknown mechanism (58). HER2 dysregulation through overexpression of HER2 and its ligand, loss of particular phosphatase, altered dimerization, and decreased receptor turnover lead to an increase in signals and cancer cell proliferation (1). The relevance of HER2 with ALDH1 and phospho-Akt has been shown (1).
On the other hand, inflammation may show a critical role in the relapse of BC through the NFκB–IL-6 pathway, which is involved in BCSC characterization and recurrence of tumor progression. NFκB regulates interleukin-6 (IL-6) and helps in the maintenance of BCSC and conversion of non-CSC to CSC. In addition, some alterations in PI3K/mTOR signaling has been detected in triple negative BC. PI3K/mTOR inhibition through PI3K/mTOR inhibitor or TORC1 inhibitor increases mammosphere formation, a CSC enrichment model, and expression of CSC markers as well as fibroblast growth factor 1 (FGF1) and Notch1 expression in CSCs through increased mitochondrial metabolism (73). Hence, this pathway could be used as a target to make CSCs sensitive to inhibitors of TORC1, which eradicates CSCs. BCSC phenotypes can be mediated by T-box transcription factor 3 (Tbx3) through FGF/Tbx3 signaling pathway. Tbx3 is a member of ancient proteins that is associated with a variety of cancers such as gastric, pancreatic, liver, bladder, and ovary as well as BC. Overexpression of T-box is critical for the maintenance of CSC phenotypes such as self-renewal, drug resistance, and metastasis (74).
It has been reported that steroid hormones are an important player for induction and progression of BC. Despite involvement of hormones and hormone receptors in BC, most studies have reported that BCSCs are hormone receptor negative. It is suggested that ER+ to ER− transition is mediated by genetic and epigenetic changes. Proliferation and expansion of ER− CSCs could be driven by ER+ BC cells. The secretion of FGF in ER+ BC is through ER signaling, by induction of EGF receptor, Notch, and Wnt signaling through FGF paracrine manner in ER− CSCs (75). In addition, progesterone induces proliferation of BCSCs indirectly through NFκB signaling. NFκB signaling activated in PR+ cells mediates binding of Receptor activator of NFκB (RANK) with its ligand, RANKL, as a paracrine signal transduction in PR-negative CSCs (75).
TGF-β Signaling Pathway
The other signaling pathway implicated in BCSCs is TGF-β signaling pathway. TGF-β signaling in BCSCs may be mediated through cytokine receptor signaling indirectly by stimulating expression of family with sequence similarity 3-member C (FAM3C)/interleukin-like EMT inducer (ILEI), an oncogenic protein. Interaction of ILEI with LIFR, its receptor, promotes cytokine receptor signaling through STAT3 activation to drive EMT and metastasis (59). Besides, mTOR signaling, which stabilizes stemness and drug resistance of BCSCs, is mediated by TGF-β. Stable TGF-β exposure promotes proliferation and drug resistance of BCSCs through increased mTOR signaling (76). TGF-β signaling can be regulated by long non-coding RNA (lncRNA), linc-ROR. Linc-ROR is a prognostic and oncogenic factor that is critical in cancer stem cell maintenance. Overexpression of linc-ROR upregulates TGF-β signaling, and then, TGF-β signaling promotes proliferation and invasion of BCSCs (77). Activated Smad and non-Smad through TGF-β signaling pathway leads to the induction hyaluronan synthase 2 (Has2) and Has2 antisense (HAS2-AS) transcripts, high mobility group AT-Hook 2 (Hmga2), and EMT inducing transcription factors such as Snail, Snai1, Hmga2, and fibronectin 1 (78). In addition, induction of Hmga2, Has2-AS, and Has2 are accompanied by activation of Akt and Erk1/2 MAPK signaling. These factors have been contributed to cancer stem cell maintenance, migration, and EMT of BCSCs (78).
Wnt/β-Catenin and Notch Signaling Pathway
Wnt and Notch signaling pathways are other signaling pathways involved in cancer stem cell maintenance (60, 61). Wnt and Notch signaling pathways may be activated by HIF-2α overexpression, which is mediated by hypoxia. HIF-2α overexpression promotes stem cell phenotype conversion, drug resistance of BCSCs, and overexpression of BCSC markers (79). Among several factors that have been involved in Wnt signaling, overexpression of glycogen synthase kinase 3 beta (GSK3β) correlates with poor patient survival. Overexpression of GSK3β induces EMT and cancer stem cell properties in BCSCs (80). The contribution of Wnt signaling in stemness phenotypes of CSCs is mediated by programmed death 1 (PD-1), which is overexpressed in BCSCs. In addition, Wnt activators and Wnt inhibitors up- and down-regulate PD-1, respectively. High expression of PD-1 is associated with stem cell features (81). Recently, it has been reported that kinesin family member 11 (KIF11), a motor protein essential in mitosis, contributed to the regulation of Wnt/β-catenin in BCSCs. It increases stemness phenotypes of BCSCs through activation of Wnt/β-catenin (82). The other protein that regulates Wnt/β-catenin in BCSCs is Nectin-4. Nectin-4, a junction protein, is well-known as a BCSC marker and is implicated in metastasis of BC and BCSCs. Overexpression of Nectin-4 upregulates Wnt/β-catenin signaling pathway, which is implicated in EMT, metastasis, and proliferation of BCSCs (83). Wnt/β-catenin signaling regulates EMT by induction of ZEB1 expression through binding β-catenin to ZEB1 promoter. In addition, ZEB1 promoter has two STAT3 binding sites, suggesting strong correlation between ZEB1 and STAT3 (68). In addition, cytokeratin 5, which is enriched in BCSCs, alters the function of β-catenin in two ways. Knockdown of cytokeratin 5 interrupts the transcription activity of β-catenin, even in response to Wnt stimuli. On the other hand, cytokeratin 5 induced by Wnt stimuli promotes deficiency of β-catenin and the other junction protein of cell membrane such as E-cadherin. It suggests dual function of cytokeratin 5 in relation to β-catenin in the cell membrane of BCSCs (84).
It has been shown that there is an association between estrogen signaling and Notch signaling in ERα+ BC cells, which is mediated by Delta-like protein 1 (DLL1), Notch ligand. Upregulation of Notch signaling by overexpression of DLL1 promotes proliferation, migration, angiogenesis, and cancer stem cell phenotypes in ERα+ BC cells. This function for DLL1 is specific in ERα+ BC cells. Thus, it suggests that estrogen and estrogen signaling stabilizes DLL1 through inhibition of ubiquitylation and degradation of DLL1 (85).
In BCSCs, activated Notch signaling by jagged1 leads to Akt phosphorylation and IκB kinase α (IKKα), NFκB, and Notch1 activation. Recruitment of IKKα, NFκB, and Notch1 to antiapoptotic gene cIAP-2 promoter increases expression of cIAP-2 (86) (Figure 3). Along with Notch1, activated Notch3 has contributed to the maintenance of BCSCs properties. It is mediated by overexpression of cartilage oligomeric matrix protein (COMP) in BCSCs. COMP increases interaction of Notch3 and jagged1, which leads to overactivation of Notch3 signaling pathway (87).
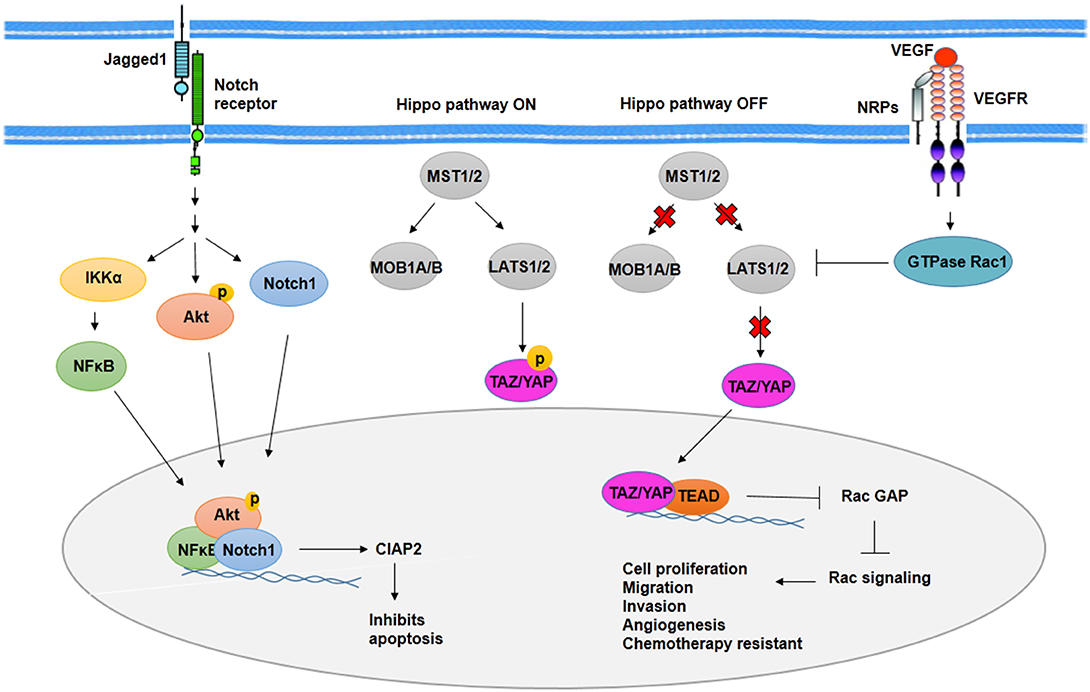
Figure 3. Notch signaling activity and Hippo pathway in BCSCs. As transcription factors, Notch1, phosphorylated Akt, and NFκB induce expression of CIAP2 and inhibit apoptosis through Notch signaling activity in BCSCs. On the other hand, Hippo pathway promotes cell proliferation, migration, invasion, angiogenesis, and chemotherapy resistance in BCSCs through translocation of YAP/YAZ into the nucleus and activation of Rac signaling. In addition, VEGFR/NRPs promotes stemness phenotypes of BCSCs, indirectly through inhibition of Hippo pathway.
Hedgehog Signaling Pathway
One of the signaling pathways upregulated in aggressive subtype of breast cancer is the hedgehog signaling pathway (61). There is an association between overexpression of hedgehog signaling pathway regulators such as SHH, DHH, IHH, PTCH1, SMO, and GLI1 with proliferation, migration, and aggressiveness of BC (88). It has been observed that SHH/GLI1+ samples express high levels of vimentin and Snail. It suggests that the hedgehog signaling pathway and SHH/GLI1 axis are involved in EMT, migration, and invasion of BCSCs (89).
Hippo Signaling Pathway
Among signaling pathways involved in CSCs, the Hippo pathway plays an important role in organogenesis and regeneration. Molecules that regulate Hippo signaling included regulatory kinases such as MST1, MST2, LATS1, LATS2, SAV1, MOB kinase 1A, MOB kinase 1B, and transcriptional coactivators such as TAZ and YAP. Alteration in Hippo signaling through deregulation of these molecules leads to increased cell proliferation and tissue overgrowth in mammary. Phosphorylated TAZ and YAP mediated by activation of kinase cascade suppresses the inclusion of TAZ and YAP into the nucleus as a result of the overexpression of TAZ and YAP and deregulation of kinases. The inclusion of TAZ and YAP into the nucleus leads to transcription of TAZ and YAP target genes involved in cell proliferation, migration, and invasion as well as resistance to chemotherapy (Figure 3). Overexpression of TAZ/YAP has been reported in BCSCs to lead to increased activity of MDR protein and cooperation between TAZ and ECM (90).
Toll-Like Receptor Activity
Toll-like receptors (TLRs) are a class of proteins implicated in inflammatory signaling and tumorigenesis (91). TLRs are overexpressed in a variety of cancers such as esophageal, gastric, as well as BC (91). Among TLRs, TLR2, and TLR3 are overexpressed in BCSCs. TLR2 and its ligand, high-mobility-group box 1 (HMGB1), display a key role in BCSC phenotypes such as self-renewal and metastatic and tumorigenic activity through inducing TGF-β and IL-6 secretion, Smad3 and STAT3 activation, and IκBα phosphorylation (92). In addition, activation of TLR3 promotes BC cells to BCSC phenotypes. Activation of TLR3 leads to coactivation of β-catenin and NFκB signaling pathways essential for increased BCSC phenotypes (91).
Chemotherapy and Radiotherapy Resistance in BCSCs
BCSCs are the main cause of chemotherapy and radiotherapy resistance of BC. These cells may up- or down-regulated essential genes and then initiate signaling pathways, which make tumors more aggressive than before and resistant to therapy. Several mechanisms and molecules have been proposed in therapeutic resistance of BCSCs. There are two main mechanisms: pump and non-pump resistance. Non-pump resistance is related to stimulation of BCL2 protein, an antiapoptotic protein, whereas pump resistance is related to overexpression of ABCG2. ABCG2 can flush out the chemotherapy drugs, which dilutes the concentration of intracellular drug (93). It has been reported that Survivin, an inhibitor of apoptosis, is overexpressed in BC cells as well as BCSCs. Survivin overexpression is related to reduced apoptosis and increased drug resistance. Downregulation of Survivin with anticancer agents such as prodigiosin may make BCSCs more sensitive to chemotherapeutic agents (94). Furthermore, ALDH is contributed to BCSC therapy resistance through direct and indirect manners. In direct manner, ALDH removes oxygen radicals that are produced by chemo- and radiotherapy, but in indirect situation, it produces nicotinamide adenine dinucleotide phosphate (NADP), an antioxidant agent (95). In addition, one of the specific mechanisms of BCSCs that protects them against reactive oxygen species (ROS) production by radiotherapy is using an ROS scavenging factor. ROS scavenging is mediated by the upregulation of genes such as catalase (CAT), superoxide dismutase (SOD), and glutathione peroxidase (GPx) in BCSCs (95).
Various studies have reported other important molecules involved in chemotherapy and radiotherapy resistance of BCSCs. Caveolin-1 is involved in chemoresistance of BC, its expression which is accompanied by coexpression of β-catenin and ABCG2. Caveolin-1 is upregulated after BC chemotherapy in BCSCs. Caveolin-1 promotes β-catenin/ABCG2 signaling via GSK3β suppression and Akt activation (13). In addition, protein kinase D1 (PRKD1) has been identified as a critical modulator of stemness phenotypes and therapy resistance of BCSCs within GSK3/β-catenin signaling. PRKD1 inhibitor and miR-34a, which target PRKD1 through binding to the PRKD1 3′-untranslated region (UTR), suppress tumor growth and proliferation as well as drug resistance and induce apoptosis (96). Cells with stem-like phenotypes such as having ALDH activity and drug resistance exhibit high mitochondrial mass (97). It proposes that chemo-resistance ability of BCSCs is driven by high function of mitochondria (97).
Recent studies suggested that the level of cholesterol may limit the efficiency of radiotherapy in cancer patients. There is a relationship between plasma levels of lipoproteins and radio-resistance in inflammatory BC and BCSCs. Transporters of cholesterol (lipoproteins) such as high-density lipoprotein (HDL), low-density lipoprotein (LDL), and very-low-density lipoprotein (VLDL) may regulate radiotherapy sensitivity. Decreasing cellular cholesterol through treatment with cholesterol-lowering agents such as HDL decreases radiotherapy resistance, whereas VLDL decreases radiotherapy sensitivity (98).
EGFR and HER2, two growth factor receptors that have been overexpressed in BCSCs, have an important role in radiotherapy resistance of BCSCs. EGFR and HER2 inhibitors such as GW572016 may make BCSCs sensitive to radiotherapy as well as inhibit proliferation through inhibition of receptor phosphorylation (99). The therapeutic resistance that is modulated by EMT has been detected in various types of cancer. Activation of genes related to EMT such as Snail and Slug accompanied with activation of Notch signaling pathway suppressed the expression of genes related to p53-mediated apoptosis (100). One of the other prosurvival factor that is involved in EMT and is induced through radiation in HER2+ BCSCs is NFκB (100). NFκB induces expression of HER2. HER2, as a receptor, activates STAT3, which upregulates stemness markers. It suggests that there is a relationship between EMT, cell stemness, and radioresistance of BCSCs (100). Non-CSCs show more level of apoptosis during radiotherapy, whereas CSCs confront with radiotherapy-induced damages through activation of repair mechanisms (101). Activation of repair mechanisms due to radiotherapy-induced damages is mediated by zinc finger E-box binding homeobox 1 (ZEB1). ZEB1, as a transcription factor, not only has been implicated in embryonic development through EMT induction but also may promote therapy resistance as well as metastasis and invasion in BCSCs. The role of ZEB1 in radiotherapy resistance is modulated by ATM and ATM and RAD3-related kinase (ATR) kinases. Genetic deficiencies and DNA variations that are caused by radiotherapy will be repaired through an ATM–ZEB1–CHK1 signaling axis activation. Activated ATM and ATR kinases through radiotherapy-induced damages phosphorylate ZEB1. ZEB1 interacts with deubiquitinase, ubiquitin-specific peptidase 7 (USP7) and promotes the deubiquitination ability of USP7 and then stabilizes CHK1 (102) (Figure 4). Thus, targeting ZEB1 by inhibitors such as miR-205 and miR-200c as radiosensitizer may be used as strategies for confronting with radio-resistance of BCSCs (102). In addition, ZEB1 is contributed to non-CSC conversion to CSCs in basal type of BC by inducing TGF-β, a microenvironment stimuli of basal BC (102).
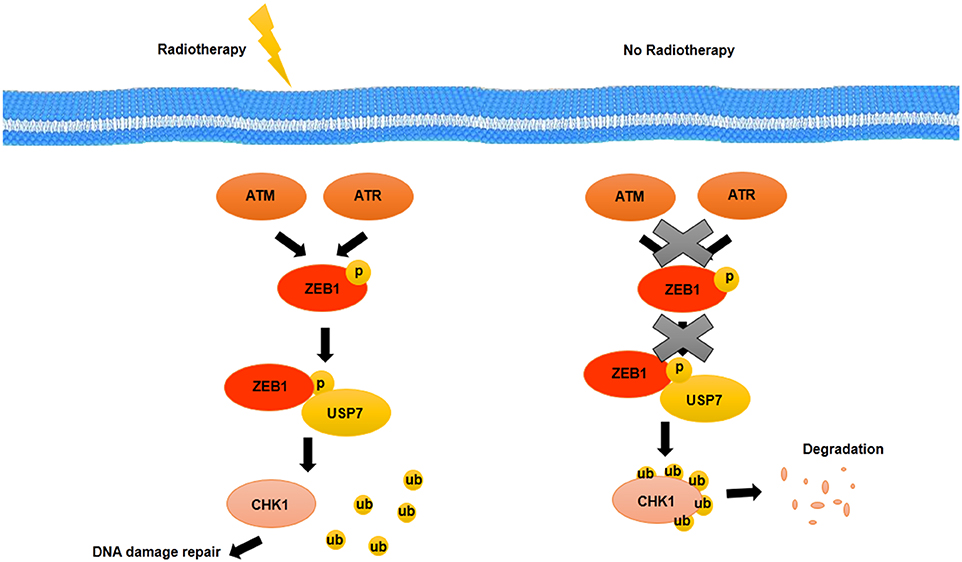
Figure 4. Activation of repair mechanisms due to radiotherapy induced-damages in BCSCs. DNA damage induced by radiotherapy is repaired through an ATM–ZEB1–CHK1 signaling axis. Activated ATM and ATR kinases phosphorylate ZEB1. ZEB1 interacts with USP7, a deubiquitylating enzyme, and promotes the deubiquitylation ability of USP7 that stabilizes CHK1.
Metastasis in BCSCs
Metastasis has been described by two potential ability of BCSCs, migration, and invasion. As mentioned above, not only EMT is an important process in chemotherapy and radiotherapy resistance but also it is implicated in metastasis of BCSCs. Snail, a transcriptional repressor, is one of the most important inducer of EMT through repression of E-cadherin expression. In addition, the expression of snail maintains malignancy phenotypes of BCSCs such as metastasis. Therefore, it could be suggested that snail is a potential target to prevent metastasis of BCSCs (40). Along with snail, ZEB1, an EMT inducer, promotes metastasis and tumor progression in BCSCs through repression of E-cadherin expression as well as epithelial polarity factors such as HUGL2, Crumbs3, and Pals1-associated tight junction (PATJ) expression. This leads to adhesion reduction and tumor cell dissociation and finally increased invasion (102). Additionally, deubiquitinating enzymes (DUBs), especially USP37 and USP2, are involved in EMT, metastasis, as well as chemotherapy resistance of BC and BCSCs. USP37 and USP2 are overexpressed in metastatic BC and BCSCs. USP37 mediates hedgehog signaling pathway through stabilizing components such as Smo and GLI-1. Finally, hedgehog signaling drives to stemness, invasion, and EMT of BCSCs (103). In addition, USP2 activates Bmi1 and EMT by stabilizing Twist through removing ubiquitylation mediated by beta-transducin repeats-containing proteins (β-TrCP), a cellular E3 ubiquitin ligase (104).
Extracellular matrix protein 1 (ECM1) is an important prognosis marker for the invasion of breast cancer as well as migration and drug resistance. In addition, ECM1 controls the expression of genes involved in EMT and BCSC maintenance through induction of Mucin1 (MUC1). The cytoplasmic tail of MUC1 has a physical association with β-catenin that stabilizes and increases posttranslational expression of β-catenin. Therefore, altered expression of β-catenin changes the expression of genes related to BCSC phenotypes such as invasion and EMT progression (105).
CD36, a member of the scavenger receptor family with binding to its receptor, thrombospondin-1 (TSP-1) plays an important role in metastasis of BCSCs through several pathways. CD36 binds to TSP-1 and TGF-β1 and promotes proliferation and migration of BCSCs. On the other hand, it can interact with NFκB and STAT3, which express genes related to carcinogenesis. In addition, CD36 has a main role in BCSC metastasis through uptaking fatty acids and regulating 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2) enzyme and β-oxidation of fatty acids (74). Furthermore, induction of NFκB signaling pathway as a result of phosphodiesterase 3A (PDE3A)-suppressed cAMP/PKA promotes expression of OCT4 as well as translocation of CCDC88A, which boots invasion and metastasis of BCSCs (106).
The role of CD44 in cancer progression, invasion, metastasis, and angiogenesis of BCSCs is remarkable. There are several ligands such as hyaluronic acid, osteopontin, and matrix metalloprotease that bond to CD44. Binding ligands to CD44 induces cleavage of CD44 at extracellular and transmembrane domain sites by membrane-type 1 matrix metalloprotease (MT1-MMP) and presenilin-1/g secretase, respectively. Then, the intracellular domain fragment of CD44 (CD44-ICD) displaces into the nucleus and activates expression of genes related to migration, invasion, metastasis, and angiogenesis of BCSCs (107).
Angiogenesis in BCSCs
Cell survival mediated by hypoxic condition after radiotherapy is contributed to the angiogenesis of CSCs (101). In both hypoxic and normoxic conditions, CSCs show high expression of vascular endothelial growth factor (VEGF). In addition, HIF-1, which is stabilized in hypoxic condition, regulates expression of VEGF (101). VEGF signaling has been contributed to proliferation, drug resistance, and cancer stem cell phenotypes as well as angiogenesis (108). Activated VEGF signaling reveals to be responsible for activation of GTPase Rac1, which then inhibits Hippo kinase LATS. The inhibition of Hippo kinase LATS leads to the activation of Hippo effector TAZ and YAP. TAZ and YAP, in a complex with transcription factor TEAD, repress expression of Rac GTPase-activating protein (Rac GAP) and then activate Rac signaling pathway, which promotes proliferation, angiogenesis, and the other cancer stem cell phenotypes (108) (Figure 3). In addition, VEGF-A/neuropilin-1 (NRP-1) axis is contributed to stemness properties such as angiogenesis through activating Wnt/β-catenin pathway.
The role of CD44 has been confirmed in angiogenesis as well as metastasis. CD44 binds to its ligand, hyaluronic acid, as well as VEGF through its binding domains and increases angiogenesis (107).
miRNAs Upregulated in BCSCs
miRNAs are introduced as one of the non-coding RNAs classes that suppress gene expression through inhibition of translation. Several studies suggest that several miRNAs regulate stemness phenotypes of BCSCs through targeting genes involved in signaling pathways. Table 1 presents miRNAs that are upregulated in BCSCs and are involved in BCSC maintenance and phenotypes.
BCSCs and Therapy Strategies
Substantial shreds of evidence support that resistance of BCSCs to chemotherapy and radiotherapy have been associated with metastasis and relapse of BC after treatment (109). Therefore, discovering potential strategies and targeting BCSCs could be effective to overcome the relapse of BC. Recently, various strategies such as herbal medicine, biological agents, anti-inflammatory drugs and monoclonal antibodies, nanoparticles, and microRNAs have been studied for targeting BCSCs. Signaling pathways that are dysregulated in BCSCs are the best candidate for developing novel strategies for targeting BCSCs. In addition to signaling pathway inhibitors such as Notch and γ-secretase inhibitor (MK-0752), hedgehog inhibitor (Vismodegib, GDC-0449), Wnt signaling pathway inhibitor (PKF118-310 and pyrvinium pamoate), PI3K/Akt/mTOR pathway inhibitor (Everolimus, RAD001), CXCR2 inhibitor (Reparixin), and EGFR/HER2 inhibitors (Lapatinib and Herceptin), which target signaling pathways directly (110), offer several breast cancer treatment strategies via targeting BCSCs.
Herbal Medicine
Recently, several studies have implicated natural components and herbal medicine in BC treatment by targeting BCSCs (111–113). These components have several advantages such as fewer side effects in comparison with non-natural drugs. Recently, it has been shown that Viola odorata extract, a herbal medicine, decreases stemness potency and cell viability of BC-cell-derived mammospheres as well as induction of apoptosis (114). It has been reported that a natural component of brown algae, phloroglucinol (PG), suppresses BCSC proliferation through reduction of Oct4, Sox2, β-catenin, and CD44 expression. In addition, it inhibits KRAS, RAF1/ERK, and PI3K/Akt signaling pathways in BCSCs (109). Capsaicin, an active ingredient of chili peppers, has shown anticancer and antiproliferation activity on BCSCs through inhibition of translocation Notch intracellular membrane domain (NICD) into the nucleus and inhibition of Notch signaling (115). In addition, Psoralidin (Pso), a small herbal molecule effectively inhibits Notch1 signaling, which leads to reduction in growth, migration, and invasion of BCSCs through downregulation of vimentin and β-catenin and upregulation of E-cadherin expression (116).
Nobiletin, a flavonoid derived from citrus peel, has anticancerous activity by binding to CD36 and suppressing CD36/STAT3/κ-κB signaling axis, which inhibits angiogenesis, migration, and metastasis of BCSCs (74). Apigenin, the other flavonoid detected in many herbal medicine, suppresses stemness properties of BC cells by inhibiting YAP/TAZ activity through disrupting YAP/TAZ-TEADs complex (79).
5,6,7-Trihydroxy-2-phenyl-4H-1-benzopyran-4-one (Baicalein), an active component of Scutellaria baicalensis Georgi, targets BCSCs and suppresses stemness potential of BCSCs by induction of apoptosis through inducing ROS and changing mitochondrial membrane potentials as well as downregulating Wnt/β-catenin pathway (117).
Biological Agents
Implicating biological agents against BCSCs have been observed in several researches. FK506-binding protein-like (FKBPL) and AD-01 (one of the its derivatives) are novel biological agents that have an effective role in inhibiting tumor growth initiation, angiogenesis, and metastasis of BC cells as well as BCSCs through targeting CD44 pathway and downregulating DLL4, a Notch ligand, and Notch4 (118, 119). Disabling mitochondria in BCSCs could be introduced as an effective way of eradicating BCSCs. Mitocans are a group of components that target mitochondria and induce apoptosis in BCSCs. They could be suggested as anticancer agents. Mito Vitamin E succinate (MitoVES) is a component of mitocans that induces apoptosis in BCSCs through targeting mitochondria complex II (120). Targeting PI3K/Akt signaling pathway, as an important pathway in MDR of BCSCs, has been suggested as an antineoplastic strategy to overcome the MDR phenotype of BCSCs. PI3K inhibitors such as BKM120 inhibit PI3K/Akt signaling, which leads to activation of apoptotic caspases such as caspase-3/7 and caspase-9 (58). In addition, a novel chemical component, 1-ferrocenyl-3-(4-methylsulfonylphenyl)propen-1-one (FMSP), targets several signaling pathways such as Wnt/β-catenin and Notch through downregulating Wnt1, Notch1, β-catenin, SOX2, CXCR4, and ALDH1A1 as well as apoptosis induction through ROS production (121). In addition, 5-aza-2′-deoxycytidine (DAC), an epigenetic drug, induces differentiation, S phase cell cycle arrest through induced expression of p53, p21, p16, p15, BRCA1, and BRCA2, and reduced expression of ABCG2 through demethylation mechanism (122).
Anti-inflammatory Agents and Monoclonal Antibodies
Developing anti-inflammatory drugs and monoclonal antibodies could be useful for preventing proliferation, metastasis, and drug resistance of BCSCs. Aspirin (ASA) prodrug is one of the anti-inflammatory drugs that has shown antineoplastic activity without toxicity through targeting NFκB–IL6 pathway and cyclooxygenase (COX)/prostaglandin in BCSCs (123, 124). In addition, ASA blocks angiogenic factors that are secreted by CSCs and contributed to tumor angiogenesis. It blocks paracrine–autocrine signaling between endothelial cells and CSCs (125).
Previously, it has been shown that EGFR is a critical player in tumor initiation, metastasis, and drug resistance of BC and BCSCs. Therefore, implication of EGFR monoclonal antibody such as Cetuximab effectively could be suggested as a useful strategy to reduce BCSC population (126). Bevacizumab, an anti-VEGF antibody targets VEGF and suppresses angiogenesis of BCSCs (127). Recently, BCSC markers such as CD44 and EpCAM have been used for developing therapeutically monoclonal antibodies. For example, MT110, anti-EpCAM and P245, and anti-CD44 are potential therapeutic monoclonal antibodies that have exhibited anticancer activity against BCSCs (128).
Nanoparticles
Recently, nanoparticles have been an interesting approach for targeting cancer cells especially CSCs. Specific phenotypes of CSCs can be used for designing nanoparticles. Among nanomaterials, mesoporous silica particles have demonstrated efficient drug delivery in cancer therapy. It has been designed based on glycolytic phenotype and Notch activity of CSCs. Uptaking mesoporous silica nanoparticles accompanied with γ-secretase inhibitor by BCSCs targets Notch signaling pathway and eliminates stemness properties of BCSCs.
MicroRNAs
miRNAs are the most attractive topics in BCSCs. They have been implicated in targeting BCSCs through several signaling pathways. Table 2 presents tumor-suppressing miRNAs that are downregulated in BCSCs and are implicated for targeting BCSCs as a novel strategy with their suggested functional pathways.
Conclusion
BCSCs are one of the main reasons for tumor development and recurrence of BC after treatment. These cells have some specific markers: CD44+/CD24−, ALDH activity, and ESA or CD326 (EpCAM), CD133, CD166, Lgr5, CD47, and ABCG2 in different types of BCs. BC cells' conversion to BCSCs due to genetic and epigenetic changes leads to deregulation of several signal transduction pathways such as MAP kinase, PI3K/Akt/NFκB, TGF-β, Wnt/β-catenin, Notch, hedgehog, and Hippo signaling. Deregulation of these signaling pathways and other factors leads to several stemness phenotypes such as chemotherapy and radiotherapy resistance, EMT, invasion, migration, and angiogenesis. Additionally, the role of miRNAs in stemness features of BCSCs is remarkable. A variety of miRNAs upregulated in BCSCs are contributed to stemness phenotypes of BCSCs. Recently, implicating novel treatment strategies such as herbal medicine, biological agents, anti-inflammatory drugs, monoclonal antibodies, nanoparticles, and microRNAs that target different signaling pathways and other deregulated genes directly or indirectly could be effective for targeting BCSCs.
Author Contributions
SY and SS: conception, providing the data and design, manuscript writing. FS, MN, AG, and KG: conception and final approval of manuscript.
Funding
This project was funded by a grant-in-aid of research from Iran National Science Foundation (Award no. 93021399) to KG, as the Principal Investigator.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We sincerely thank our colleagues at Royan Institute for Biotechnology and University of Isfahan for their valuable discussions.
Abbreviations
ADAM17, ADAM metallopeptidase domain 17; ALDH, aldehyde dehydrogenase; ASA, aspirin; ATM, ataxia telangiectasia mutated; ATR, ATM- and RAD3-related; DAC, 5-aza-2′-deoxycytidine; β-TrCP, beta-transducin repeat-containing proteins; BC, breast cancer; BCSCs, breast cancer stem cells; CSCs, cancer stem cells; COMP, cartilage oligomeric matrix protein; CAT, catalase; COX, cyclooxygenase; DLL, delta like protein; DUBs, deubiquitinating enzymes; DUSP9/DUSP16, dual specificity phosphatase; EpICD, EpCAM intracellular domain; EGFR, epidermal growth factor receptor; EMT, epithelial–mesenchymal transition; ESA, epithelial specific antigen; ER, estrogen receptor; ECM1, extracellular matrix protein 1; FAM3C, family with sequence similarity 3 member C; FMSP, 1-ferrocenyl-3-(4-methylsulfonylphenyl)propen-1-one; PDE3A, phosphodiesterase 3A; FGF1, fibroblast growth factor 1; FKBPL, FK506-binding protein like; FAK, focal adhesion kinase; FHL2, four and a half LIM domains 2; GPx, glutathione peroxidase; GSK3β, glycogen synthase kinase 3 beta; Hh, hedgehog; HDL, high-density lipoproteins; Hmga2, high mobility group AT-Hook 2; HMGB1, high-mobility-group box 1; HER2, human epidermal growth factor receptor 2; Has2, hyaluronan synthase 2; HAS2-AS, hyaluronan synthase 2-antisense; HA, hyaluronic acid; HMGCS2, 3-hydroxy-3-methylglutaryl-CoA synthase 2; HIF1, hypoxia-inducible factor-1; IKKα, IκB kinase α; IAPs, inhibitors of apoptosis proteins; IL-6, interleukin-6; ILEI, interleukin-like EMT inducer; CD44-ICD, intracellular domain fragment of CD44; KIF11, kinesin family member 11; LncRNA, long non-coding RNA; LDL, low-density lipoproteins; MNK, MAPK interacting kinase; MMP, matrix metalloprotease; MT1-MMP, membrane type 1-matrix metalloprotease; MitoVES, Mito Vitamin E succinate; MUC1, mucin1; MDR, multidrug resistance; NRPs, neuropilins; NADP, nicotinamide adenine dinucleotide phosphate; NICD, notch intracellular membrane domain; OPN, osteopontin; PATJ, Pals1-associated tight junction; PG, phloroglucinol; PSEN2, presenilin-2; PR, progesterone receptor; PDCD4, program cell death protein 4; PD-1, programmed death 1; PKCε, protein kinase Cε; PRKD1, protein kinase D1; Pso, psoralidin; Rac GAP, Rac GTPase-activating protein; RANKL, RANK ligand; ROS, reactive oxygen species; RANK, receptor activator of NFκB; RTK, receptor tyrosine kinase; ROK, rho kinase; SHH, Sonic hedgehog; SOD, superoxide dismutase; Tbx3, T-box transcription factor 3; TSP-1, thrombospondin-1; TLRs, Toll-like receptors; TGF-β, Transforming growth factor beta; TG2, transglutaminase; TNBC, triple negative breast cancer; USP, ubiquitin specific peptidase; VEGF, vascular endothelial growth factor; VLDL, very-low-density lipoproteins; ZEB1, zinc finger E-box binding homeobox 1.
References
1. Alanazi IO, Khan Z. Understanding EGFR signaling in breast cancer and breast cancer stem cells: overexpression and therapeutic implications. Asian Pac J Cancer Prev. (2016) 17:445–53. doi: 10.7314/APJCP.2016.17.2.445
2. Nounou MI, ElAmrawy F, Ahmed N, Abdelraouf K, Goda S, Syed-Sha-Qhattal H. Breast cancer: conventional diagnosis treatment modalities recent patents technologies. Breast Cancer (Auckl). (2015) 9:17–34. doi: 10.4137/BCBCR.S29420
3. Kaminska M, Ciszewski T, Łopacka-Szatan K, Miotła P, Starosławska E. Breast cancer risk factors. Prz Menopauzalny. (2015) 14:196–202. doi: 10.5114/pm.2015.54346
4. Semenza GL. Regulation of the breast cancer stem cell phenotype by hypoxia-inducible factors. Clin Sci. (2015) 129:1037–45. doi: 10.1042/CS20150451
5. Yousefnia S, Ghaedi K, Seyed Forootan F, Nasr Esfahani MH. Characterization of the stemness potency of mammospheres isolated from the breast cancer cell lines. Tumour Biol. (2019) 41:1010428319869101. doi: 10.1177/1010428319869101
6. Perou CM, Sørlie T, Eisen MB, Van De Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. (2000) 406:747–52. doi: 10.1038/35021093
7. Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. (2001) 98:10869–74. doi: 10.1073/pnas.191367098
8. Lin CY, Barry-Holson KQ, Allison KH. Breast cancer stem cells: are we ready to go from bench to bedside? Histopathology. (2016) 68:119–37. doi: 10.1111/his.12868
9. Louie E, Nik S, Chen JS, Schmidt M, Song B, Pacson C, et al. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and reoxygenation. Breast Cancer Res. (2010) 12:R94. doi: 10.1186/bcr2773
10. Michor F, Polyak K. The origins and implications of intratumor heterogeneity. Cancer Prev Res (Phila). (2010) 3:1361–4. doi: 10.1158/1940-6207.CAPR-10-0234
11. Sin WC, Lim CL. Breast cancer stem cells-from origins to targeted therapy. Stem Cell Investig. (2017) 4:96. doi: 10.21037/sci.2017.11.03
12. Suyama K, Onishi H, Imaizumi A, Shinkai K, Umebayashi M, Kubo M, et al. CD24 suppresses malignant phenotype by downregulation of SHH transcription through STAT1 inhibition in breast cancer cells. Cancer Lett. (2016) 374:44–53. doi: 10.1016/j.canlet.2015.12.013
13. Wang Z, Wang N, Li W, Liu P, Chen Q, Situ H, et al. Caveolin-1 mediates chemoresistance in breast cancer stem cells via β-catenin/ABCG2 signaling pathway. Carcinogenesis. (2014) 35:2346–56. doi: 10.1093/carcin/bgu155
14. Evans MK, Brown MC, Geradts J, Bao X, Robinson TJ, Jolly MK, et al. XIAP regulation by MNK links MAPK and NFκB signaling to determine an aggressive breast cancer phenotype. Cancer Res. (2018) 78:1726–38. doi: 10.1158/0008-5472.CAN-17-1667
15. Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. (2007) 449:682. doi: 10.1038/nature06174
16. Han M, Liu M, Wang Y, Chen X, Xu J, Sun Y, et al. Antagonism of miR-21 reverses epithelial-mesenchymal transition cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN. PLoS ONE. (2012) 7:e39520. doi: 10.1371/journal.pone.0039520
17. Han M, Liu M, Wang Y, Mo Z, Bi X, Liu Z, et al. Re-expression of miR-21 contributes to migration and invasion by inducing epithelial-mesenchymal transition consistent with cancer stem cell characteristics in MCF-7 cells. Molecular and Cellular Biochem. (2012) 363:427–36. doi: 10.1007/s11010-011-1195-5
18. Song SJ, Poliseno L, Song MS, Ala U, Webster K, Ng C, et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell. (2013) 154:311–24. doi: 10.1016/j.cell.2013.06.026
19. Liu Z, Liu H, Desai S, Schmitt DC, Zhou M, Khong HT, et al. miR-125b functions as a key mediator for snail-induced stem cell propagation and chemoresistance. J Biol Chem. (2013) 288:4334–45. doi: 10.1074/jbc.M112.419168
20. Liu L, Zhou W, Cheng C-T, Ren X, Somlo G, Fong MY, et al. TGFβ induces “BRCAness” and sensitivity to PARP inhibition in breast cancer by regulating DNA-repair genes. Molecular Cancer Res. (2014) 12:1597–609. doi: 10.1158/1541-7786.MCR-14-0201
21. Li B, Lu Y, Wang H, Han X, Mao J, Li J, et al. miR-221/222 enhance the tumorigenicity of human breast cancer stem cells via modulation of PTEN/Akt pathway. Biomed Pharmacotherapy. (2016) 79:93–101. doi: 10.1016/j.biopha.2016.01.045
22. Okuda H, Xing F, Pandey PR, Sharma S, Watabe M, Pai SK, et al. miR-7 suppresses brain metastasis of breast cancer stem-like cells by modulating KLF4. Cancer Res. (2013) 73:1434–44. doi: 10.1158/0008-5472.CAN-12-2037
23. Mohammadi-Yeganeh S, Mansouri A, Paryan M. Targeting of miR9/NOTCH1 interaction reduces metastatic behavior in triple-negative breast cancer. Chemical Biol Drug Design. (2015) 86:1185–91. doi: 10.1111/cbdd.12584
24. Zhang X, Wan G, Mlotshwa S, Vance V, Berger FG, Chen H, et al. Oncogenic Wip1 phosphatase is inhibited by miR-16 in the DNA damage signaling pathway. Cancer Res. (2010) 70:7176–86. doi: 10.1158/0008-5472.CAN-10-0697
25. Ono M, Kosaka N, Tominaga N, Yoshioka Y, Takeshita F, Takahashi R-U, et al. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. (2014) 7:ra63–ra63. doi: 10.1126/scisignal.2005231
26. Wolfson B, Eades G, Zhou Q. Roles of microRNA-140 in stem cell-associated early stage breast cancer. World J Stem Cells. (2014) 6:591. doi: 10.4252/wjsc.v6.i5.591
27. Ouzounova M, Vuong T, Ancey P-B, Ferrand M, Durand G, Kelm FL-C, et al. MicroRNA miR-30 family regulates non-attachment growth of breast cancer cells. BMC Genomics. (2013) 14:139. doi: 10.1186/1471-2164-14-139
28. Yu F, Deng H, Yao H, Liu Q, Su F, Song E. Mir-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene. (2010) 29:4194. doi: 10.1038/onc.2010.167
29. Park EY, Chang E, Lee EJ, Lee H-W, Kang H-G, Chun K-H, et al. Targeting of miR34a-NOTCH1 axis reduced breast cancer stemness and chemoresistance. Cancer Res. (2014) 74:7573–82. doi: 10.1158/0008-5472.CAN-14-1140
30. Yu F, Jiao Y, Zhu Y, Wang Y, Zhu J, Cui X, et al. MicroRNA 34c gene down-regulation via DNA methylation promotes self-renewal and epithelial-mesenchymal transition in breast tumor-initiating cells. J Biol. Chem. (2012) 287:465–73. doi: 10.1074/jbc.M111.280768
31. Liu S, Patel SH, Ginestier C, Ibarra I, Martin-Trevino R, Bai S, et al. MicroRNA93 regulates proliferation and differentiation of normal and malignant breast stem cells. PLoS Genetics. (2012) 8:e1002751. doi: 10.1371/journal.pgen.1002751
32. Tavazoie SF, Alarcón C, Oskarsson T, Padua D, Wang Q, Bos PD, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. (2008) 451:147. doi: 10.1038/nature06487
33. Qian P, Banerjee A, Wu Z-S, Zhang X, Wang H, Pandey V, et al. Loss of SNAIL regulated miR-128-2 on chromosome 3p22. 3 targets multiple stem cell factors to promote transformation of mammary epithelial cells. Cancer Res. (2012) 72:6036–50. doi: 10.1158/0008-5472.CAN-12-1507
34. Zhu Y, Yu F, Jiao Y, Feng J, Tang W, Yao H, et al. Reduced miR-128 in breast tumorinitiating cells induces chemotherapeutic resistance via Bmi-1 and ABCC5. Clin Cancer Res. (2011) 17:7105–15. doi: 10.1158/1078-0432.CCR-11-0071
35. Li Q, Yao Y, Eades G, Liu Z, Zhang Y, Zhou Q. Downregulation of miR-140 promotes cancer stem cell formation in basal-like early stage breast cancer. Oncogene. (2014) 33:2589. doi: 10.1038/onc.2013.226
36. Liang H, Xiao J, Zhou Z, Wu J, Ge F, Li Z, et al. Hypoxia induces miR-153 through the IRE1a-XBP1 pathway to fine tune the HIF1a/VEGFA axis in breast cancer angiogenesis. Oncogene. (2018) 37:1961. doi: 10.1038/s41388-017-0089-8
37. Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. (2009) 138:592–603. doi: 10.1016/j.cell.2009.07.011
38. Lim Y.-Y., Wright JA, Attema JL, Gregory PA, Bert AG, Smith E, et al. Epigenetic modulation of the miR-200 family is associated with transition to a breast cancer stem-cell-like state. J Cell Sci. (2013) 126:2256–66. doi: 10.1242/jcs.122275
39. Song C, Liu L-Z, Pei X-Q, Liu X, Yang L, Ye F, et al. miR-200c inhibits breast cancer proliferation by targeting KRAS. Oncotarget. (2015) 6:34968. doi: 10.18632/oncotarget.5198
40. El Helou R, Pinna G, Cabaud O, Wicinski J, Bhajun R, Guyon L, et al. miR-600 acts as a bimodal switch that regulates breast cancer stem cell fate through WNT signaling. Cell Rep. (2017) 18:2256–68. doi: 10.1016/j.celrep.2017.02.016
41. Ryu S, McDonnell K, Choi H, Gao D, Hahn M, Joshi N, et al. Suppression of miRNA-708 by polycomb group promotes metastases by calcium-induced cell migration. Cancer Cell. (2013) 23:63–76. doi: 10.1016/j.ccr.2012.11.019
42. Cai W-Y, Wei T-Z, Luo Q-C, Wu Q-W, Liu Q-F, Yang M, et al. The Wntβ-catenin pathway represses let-7 microRNA expression through transactivation of Lin28 to augment breast cancer stem cell expansion. J Cell Sci. (2013) 126:2877–89. doi: 10.1242/jcs.123810
43. Piskounova E, Polytarchou C, Thornton JE, LaPierre RJ, Pothoulakis C, Hagan JP, et al. Lin28A and Lin28B inhibit let-7 microRNA biogenesis by distinct mechanisms. Cell. (2011) 147:1066–79 doi: 10.1016/j.cell.2011.10.039
44. Idowu MO, Kmieciak M, Dumur C, Burton RS, Grimes MM, Powers CN, et al. CD44+/CD24–/low cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Hum Pathol. (2012) 43:364–73. doi: 10.1016/j.humpath.2011.05.005
45. Zöller M. CD44. can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. (2011) 11:254–67. doi: 10.1038/nrc3023
46. Al-Othman N, Alhendi A, Ihbaisha M, Barahmeh M, Alqaraleh M, Al-Momany BZ. Role of CD44 in breast cancer. Breast Dis. (2019) 39:1–13. doi: 10.3233/BD-190409
47. Ricardo S, Vieira AF, Gerhard R, Leitão D, Pinto R, Cameselle-Teijeiro JF, et al. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Pathol. (2011) 64:937–46. doi: 10.1136/jcp.2011.090456
48. Liu Y, Nenutil R, Appleyard M, Murray K, Boylan M, Thompson A, et al. Lack of correlation of stem cell markers in breast cancer stem cells. Br J Cancer. (2014) 110:2063–71. doi: 10.1038/bjc.2014.105
49. Feifei N, Mingzhi Z, Yanyun Z, Huanle Z, Fang R, Mingzhu H, et al. MicroRNA expression analysis of mammospheres cultured from human breast cancers. Journal of cancer research and clinical oncology. (2012) 138:1937–44. doi: 10.1007/s00432-012-1272-5
50. Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, et al. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers. Clin Cancer Res. (2009) 15:4234–41. doi: 10.1158/1078-0432.CCR-08-1479
51. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. (2007) 1:555–67. doi: 10.1016/j.stem.2007.08.014
52. Storms RW, Trujillo AP, Springer JB, Shah L, Colvin OM, Ludeman SM, et al. Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity. Proc Natl Acad Sci. (1999) 96:9118–23. doi: 10.1073/pnas.96.16.9118
53. Baeuerle P, Gires O. EpCAM (CD326) finding its role in cancer. Br J Cancer. (2007) 96:417–23. doi: 10.1038/sj.bjc.6603494
54. Warneke V, Behrens H, Haag J, Krüger S, Simon E, Mathiak M, et al. Members of the EpCAM signalling pathway are expressed in gastric cancer tissue and are correlated with patient prognosis. Br J Cancer. (2013) 109:2217–27. doi: 10.1038/bjc.2013.536
55. Collina F, Di Bonito M, Li Bergolis V, De Laurentiis M, Vitagliano C, Cerrone M, et al. Prognostic value of cancer stem cells markers in triple-negative breast cancer. Biomed Res Int. (2015) 2015:158682. doi: 10.1155/2015/158682
56. Eckert RL, Fisher ML, Grun D, Adhikary G, Xu W, Kerr C. Transglutaminase is a tumor cell and cancer stem cell survival factor. Mol Carcinog. (2015) 54:947–58. doi: 10.1002/mc.22375
57. Wang Y, Li W, Patel SS, Cong J, Zhang N, Sabbatino F, et al. Blocking the formation of radiation-induced breast cancer stem cells. Oncotarget. (2014) 5:3743–55. doi: 10.18632/oncotarget.1992
58. Hu Y, Guo R, Wei J, Zhou Y, Ji W, Liu J, et al. Effects of PI3K inhibitor NVP-BKM120 on overcoming drug resistance and eliminating cancer stem cells in human breast cancer cells. Cell Death Dis. (2015) 6:e2020. doi: 10.1038/cddis.2015.363
59. Woosley AN, Dalton AC, Hussey GS, Howley BV, Mohanty BK, Grelet S, et al. TGFβ promotes breast cancer stem cell self-renewal through an ILEI/LIFR signaling axis. Oncogene. (2019) 38:3794–811. doi: 10.1038/s41388-019-0703-z
60. Jang GB, Kim JY, Cho SD, Park KS, Jung JY, Lee HY, et al. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci Rep. (2015) 5:12465. doi: 10.1038/srep12465
61. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. (2015) 12:445–64. doi: 10.1038/nrclinonc.2015.61
62. Cui J, Li P, Liu X, Hu H, Wei W. Abnormal expression of the Notch and Wnt/β-catenin signaling pathways in stem-like ALDHhiCD44+ cells correlates highly with Ki-67 expression in breast cancer. Oncol Lett. (2015) 9:1600–6. doi: 10.3892/ol.2015.2942
63. Bourguignon LY, Spevak CC, Wong G, Xia W, Gilad E. Hyaluronan-CD44 interaction with PKCε promotes oncogenic signaling by the stem cell marker, Nanog and the production of microRNA-21 leading to downregulation of the tumor suppressor protein, PDCD4, anti-apoptosis and chemotherapy resistance in breast tumor cells. J Biol Chem. (2009) 284:26533–46. doi: 10.1158/1538-7445.AM10-2039
64. Chen L, Bourguignon LY. Hyaluronan-CD44 interaction promotes c-Jun signaling and miRNA21 expression leading to Bcl-2 expression and chemoresistance in breast cancer cells. Mol Cancer. (2014) 13:52. doi: 10.1186/1476-4598-13-52
65. Bourguignon LY, Wong G, Earle C, Krueger K, Spevak CC. Hyaluronan-CD44 interaction promotes c-Src-mediated Twist signaling, microRNA-10b expression and RhoA/RhoC upregulation leading to Rho-Kinase-associated cytoskeleton activation and breast tumor cell invasion. J Biol Chem. (2010) 285:36721–35. doi: 10.1074/jbc.M110.162305
66. Stefania DD, Vergara D. The many-faced program of epithelial-mesenchymal transition: a system biology-based view. Front Oncol. (2017) 7:274. doi: 10.3389/fonc.2017.00274
67. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. (2008) 133:704–15. doi: 10.1016/j.cell.2008.03.027
68. Simeone P, Trerotola M, Franck J, Cardon T, Marchisio M, Fournier I, et al. The multiverse nature of epithelial to mesenchymal transition. Sem Cancer Biol. (2019) 58:1–10. doi: 10.1016/j.semcancer.2018.11.004
69. Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. (2018) 18:128–34. doi: 10.1038/nrc.2017.118
70. Majorini MT, Manenti G, Mano M, De Cecco L, Conti A, Pinciroli P, et al. cIAP1 regulates the EGFR/Snai2 axis in triple-negative breast cancer cells. Cell Death Differ. (2018) 25:2147–64. doi: 10.1038/s41418-018-0100-0
71. Lu H, Tran L, Park Y, Chen I, Lan J, Xie Y, et al. Reciprocal regulation of DUSP9 and DUSP16 expression by HIF1 controls ERK and p38 MAP kinase activity and mediates chemotherapy-induced breast cancer stem cell enrichment. Cancer Res. (2018) 78:4191–202. doi: 10.1158/0008-5472.CAN-18-0270
72. Xu M, Wang S, Wang Y, Wu H, Frank JA, Zhang Z, et al. Role of p38γ MAPK in regulation of EMT and cancer stem cells. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:3605–17. doi: 10.1016/j.bbadis.2018.08.024
73. Bhola NE, Jansen VM, Koch JP, Li H, Formisano L, Williams JA, et al. Treatment of triple-negative breast cancer with TORC1/2 inhibitors sustains a drug-resistant and notch-dependent cancer stem cell population. Cancer Res. (2016) 76:440–52. doi: 10.1158/0008-5472.CAN-15-1640-T
74. Sp N, Kang D, Kim D, Park J, Lee H, Kim H, et al. Nobiletin inhibits CD36-dependent tumor angiogenesis, migration, invasion, and sphere formation through the Cd36/Stat3/Nf-κb signaling axis. Nutrients. (2018) 10:772. doi: 10.3390/nu10060772
75. Nandy SB, Gangwani L, Nahleh Z, Subramani R, Arumugam A, de la Rosa JM, et al. Recurrence and metastasis of breast cancer is influenced by ovarian hormone's effect on breast cancer stem cells. Future Oncol. (2015) 11:983–95. doi: 10.2217/fon.14.301
76. Katsuno Y, Meyer DS, Zhang Z, Shokat KM, Akhurst RJ, Miyazono K, et al. Chronic TGF-β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci Signal. (2019) 12:eaau8544. doi: 10.1126/scisignal.aau8544
77. Hou L, Tu J, Cheng F, Yang H, Yu F, Wang M, et al. Long noncoding RNA ROR promotes breast cancer by regulating the TGF-β pathway. Cancer Cell Int. (2018) 18:142. doi: 10.1186/s12935-018-0638-4
78. Kolliopoulos C, Lin CY, Heldin CH, Moustakas A, Heldin P. Has2 natural antisense RNA and Hmga2 promote Has2 expression during TGFβ-induced EMT in breast cancer. Matrix Biol. (2018) 80:29–45. doi: 10.1016/j.matbio.2018.09.002
79. Yan Y, Liu F, Han L, Zhao L, Chen J, Olopade OI, et al. HIF-2α promotes conversion to a stem cell phenotype and induces chemoresistance in breast cancer cells by activating Wnt and Notch pathways. J Exp Clin Cancer Res. (2018) 37:256. doi: 10.1186/s13046-018-0925-x
80. Vijay GV, Zhao N, Den Hollander P, Toneff MJ, Joseph R, Pietila M, et al. GSK3β regulates epithelial-mesenchymal transition and cancer stem cell properties in triple-negative breast cancer. Breast Cancer Res. (2019) 21:37. doi: 10.1186/s13058-019-1125-0
81. Castagnoli L, Cancila V, Cordoba-Romero SL, Faraci S, Talarico G, Belmonte B, et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene. (2019) 38:4047–60. doi: 10.1038/s41388-019-0700-2
82. Pei YY, Li GC, Ran J, Wan XH, Wei FX, Wang L. Kinesin family member 11 enhances the self-renewal ability of breast cancer cells by participating in the Wnt/β-catenin pathway. J Breast Cancer. (2019) 22:522–32. doi: 10.4048/jbc.2019.22.e51
83. Siddharth S, Goutam K, Das S, Nayak A, Nayak D, Sethy C, et al. Nectin-4 is a breast cancer stem cell marker that induces WNT/β-catenin signaling via Pi3k/Akt axis. Int J Biochem Cell Biol. (2017) 89:85–94. doi: 10.1016/j.biocel.2017.06.007
84. McGinn O, Ward AV, Fettig LM, Riley D, Ivie J, Paul KV, et al. Cytokeratin 5 alters β-catenin dynamics in breast cancer cells. Oncogene. (2020) 39:2478–92. doi: 10.1038/s41388-020-1164-0
85. Kumar S, Srivastav RK, Wilkes DW, Ross T, Kim S, Kowalski J, et al. Estrogen-dependent DLL1-mediated Notch signaling promotes luminal breast cancer. Oncogene. (2019) 38:2092–107. doi: 10.1038/s41388-018-0562-z
86. Hossain F, Sorrentino C, Ucar DA, Peng Y, Matossian M, Wyczechowska D, et al. Notch signaling regulates mitochondrial metabolism and NF-kB activity in Triple-negative breast cancer cells via IKKa-dependent non-canonical pathways. Front Oncol. (2018) 8:575. doi: 10.3389/fonc.2018.00575
87. Papadakos KS, Bartoschek M, Rodriguez C, Gialeli C, Jin SB, Lendahl U, et al. Cartilage Oligomeric Matrix Protein initiates cancer stem cells through activation of Jagged1-Notch3 signaling. Matrix Biol. (2018) 81:107–21. doi: 10.1016/j.matbio.2018.11.007
88. Riaz SK, Khan JS, Shah STA, Wang F, Ye L, Jiang WG, et al. Involvement of hedgehog pathway in early onset, aggressive molecular subtypes and metastatic potential of breast cancer. Cell Commun Signal. (2018) 16:3. doi: 10.1186/s12964-017-0213-y
89. Riaz SK, Ke Y, Wang F, Kayani MA, Malik MFA. Influence of SHH/GLI1 axis on EMT mediated migration and invasion of breast cancer cells. Sci Rep. (2019) 9:6620. doi: 10.1038/s41598-019-43093-x
90. Maugeri-Saccà M, De Maria R. Hippo pathway and breast cancer stem cells. Crit Rev Oncol Hematol. (2016) 99:115–22. doi: 10.1016/j.critrevonc.2015.12.004
91. Jia D, Yang W, Li L, Liu H, Tan Y, Ooi S, et al. β-Catenin and NF-κB co-activation triggered by TLR3 stimulation facilitates stem cell-like phenotypes in breast cancer. Cell Death Differ. (2015) 22:298–310. doi: 10.1038/cdd.2014.145
92. Conti L, Lanzardo S, Arigoni M, Antonazzo R, Radaelli E, Cantarella D, et al. The noninflammatory role of high mobility group box 1/Toll-like receptor 2 axis in the self-renewal of mammary cancer stem cells. FASEB J. (2013) 27:4731–44. doi: 10.1096/fj.13-230201
93. Sun M, Yang C, Zheng J, Wang M, Chen M, Le DQS, et al. Enhanced efficacy of chemotherapy for breast cancer stem cells by simultaneous suppression of multidrug resistance and antiapoptotic cellular defense. Acta Biomater. (2015) 28:171–82. doi: 10.1016/j.actbio.2015.09.029
94. Yu CJ, Ou JH, Wang ML, Jialielihan N, Liu YH. Elevated survivin mediated multidrug resistance and reduced apoptosis in breast cancer stem cells. J BUON. (2015) 20:1287–94.
95. Bozorgi A, Khazaei M, Khazaei MR. New findings on breast cancer stem cells: a review. J Breast Cancer. (2015) 18:303–12. doi: 10.4048/jbc.2015.18.4.303
96. Kim do Y, Park EY, Chang E, Kang HG, Koo Y, Lee EJ, et al. A novel miR-34a target, protein kinase D1, stimulates cancer stemness and drug resistance through GSK3/β-catenin signaling in breast cancer. Oncotarget. (2016) 7:14791–802. doi: 10.18632/oncotarget.7443
97. Farnie G, Sotgia F, Lisanti MP. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget. (2015) 6:30472–86. doi: 10.18632/oncotarget.5401
98. Wolfe AR, Atkinson RL, Reddy JP, Debeb BG, Larson R, Li L, et al. High-density and very-low-density lipoprotein have opposing roles in regulating tumor-initiating cells and sensitivity to radiation in inflammatory breast cancer. Int J Radiat Oncol Biol Phys. (2015) 91:1072–80. doi: 10.1016/j.ijrobp.2014.12.039
99. Zhou H, Kim YS, Peletier A, McCall W, Earp HS, Sartor CI. Effects of the EGFR/HER2 kinase inhibitor GW572016 on EGFR-and HER2-overexpressing breast cancer cell line proliferation, radiosensitization, and resistance. Int J Radiat Oncol Biol Phys. (2004) 58:344–52. doi: 10.1016/j.ijrobp.2003.09.046
100. Nantajit D, Lin D, Li JJ. The network of epithelial-mesenchymal transition: potential new targets for tumor resistance. J Cancer Res Clin Oncol. (2015) 141:1697–713. doi: 10.1007/s00432-014-1840-y
101. Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol. (2008) 26:2839–45. doi: 10.1200/JCO.2007.15.1829
102. Zhang P, Sun Y, Ma L. ZEB1: at the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle. (2015) 14:481–7. doi: 10.1080/15384101.2015.1006048
103. Qin T, Li B, Feng X, Fan S, Liu L, Liu D, et al. Abnormally elevated USP37 expression in breast cancer stem cells regulates stemness, epithelial-mesenchymal transition and cisplatin sensitivity. J Exp Clin Cancer Res. (2018) 37:287. doi: 10.1186/s13046-018-0934-9
104. He J, Lee HJ, Saha S, Ruan D, Guo H, Chan CH. Inhibition of USP2 eliminates cancer stem cells and enhances TNBC responsiveness to chemotherapy. Cell Death Dis. (2019) 10:285. doi: 10.1038/s41419-019-1512-6
105. Lee K, Nam K, Oh S, Lim J, Kim R, Shim D, et al. ECM1 regulates tumor metastasis and CSC-like property through stabilization of β-catenin. Oncogene. (2015) 34:6055–65. doi: 10.1038/onc.2015.54
106. Hao N, Shen W, Du R, Jiang S, Zhu J, Chen Y, et al. Phosphodiesterase 3A represents a therapeutic target that drives stem cell-like property and metastasis in breast cancer. Mol Cancer Ther. (2019) 19:868–81. doi: 10.1158/1535-7163.MCT-18-1233
107. Senbanjo LT, Chellaiah MA. CD44: a multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front Cell Dev Biol. (2017) 5:18. doi: 10.3389/fcell.2017.00018
108. Elaimy AL, Guru S, Chang C, Ou J, Amante JJ, Zhu LJ, et al. VEGF-neuropilin-2 signaling promotes stem-like traits in breast cancer cells by TAZ-mediated repression of the Rac GAP β2-chimaerin. Sci Signal. (2018) 11:eaao6897. doi: 10.1126/scisignal.aao6897
109. Kim RK, Uddin N, Hyun JW, Kim C, Shu Y, Lee SJ. Novel anticancer activity of phloroglucinol against breast cancer stem-like cells. Toxicol Appl Pharmacol. (2015) 286:143–50. doi: 10.1016/j.taap.2015.03.026
110. Chiotaki R, Polioudaki H, A, Theodoropoulos P. Cancer stem cells in solid and liquid tissues of breast cancer patients: characterization and therapeutic perspectives. Curr Cancer Drug Targets. (2015) 15:256–69. doi: 10.2174/1568009615666150211102503
111. Abdal Dayem A, Choi HY, Yang GM, Kim K, Saha SK, Cho SG. The anti-cancer effect of polyphenols against breast cancer and cancer stem cells: molecular mechanisms. Nutrients. (2016) 8:E581. doi: 10.3390/nu8090581
112. Dandawate PR, Subramaniam D, Jensen RA, Anant S. Targeting cancer stem cells and signaling pathways by phytochemicals: novel approach for breast cancer therapy. Semin Cancer Biol. (2016) 40:192–208. doi: 10.1016/j.semcancer.2016.09.001
113. Sotillo WS, Villagomez R, Smiljanic S, Huang X, Malakpour A, Kempengren S, et al. Anti-cancer stem cell activity of a sesquiterpene lactone isolated from Ambrosia arborescens and of a synthetic derivative. PLoS One. (2017) 12:e0184304. doi: 10.1371/journal.pone.0184304
114. Yousefnia S, Naseri D, Forootan FS, Tabatabaeian M, Moattar F, Ghafghazi T, et al. Suppressive role of Viola odorata extract on malignant characters of mammosphere-derived breast cancer stem cells. Clin Transl Oncol. (2020) 22:1–16. doi: 10.1007/s12094-020-02307-9
115. Shim Y, Song JM. Quantum dot nanoprobe-based high-content monitoring of notch pathway inhibition of breast cancer stem cell by capsaicin. Mol Cell Probes. (2015) 29:376–81. doi: 10.1016/j.mcp.2015.09.004
116. Suman S, Das T, Damodaran C. Silencing NOTCH signaling causes growth arrest in both breast cancer stem cells and breast cancer cells. Br J ca ncer. (2013) 109:2587–96. doi: 10.1038/bjc.2013.642
117. Koh SY, Moon JY, Unno T, Cho SK. Baicalein suppresses stem cell-like characteristics in radio-and chemoresistant MDA-MB-231 human breast cancer cells through up-regulation of IFIT2. Nutrients. (2019) 11:624. doi: 10.3390/nu11030624
118. McClements L, Yakkundi A, Papaspyropoulos A, Harrison H, Ablett MP, Jithesh PV, et al. Targeting treatment-resistant breast cancer stem cells with FKBPL and its peptide derivative, AD-01, via the CD44 pathway. Clin Cancer Res. (2013) 19:3881–93. doi: 10.1158/1078-0432.CCR-13-0595
119. McClements L, Annett S, Yakkundi A, O'Rourke M, Valentine A, Moustafa N, et al. FKBPL and its peptide derivatives inhibit endocrine therapy resistant cancer stem cells and breast cancer metastasis by downregulating DLL4 and Notch4. BMC Cancer. (2019) 19:351. doi: 10.1186/s12885-019-5500-0
120. Yan B, Stantic M, Zobalova R, Bezawork-Geleta A, Stapelberg M, Stursa J, et al. Mitochondrially targeted vitamin E succinate efficiently kills breast tumour-initiating cells in a complex II-dependent manner. BMC Cancer. (2015) 15:401. doi: 10.1186/s12885-015-1394-7
121. Nourbakhsh M, Farzaneh S, Taghikhani A, Zarghi A, Noori S. The effect of a newly synthesized ferrocene derivative against MCF-7 breast cancer cells and spheroid stem cells through ROS production and inhibition of JAK2/STAT3 signaling pathway. Anticancer Agents Med Chem. (2020). doi: 10.2174/1871520620666200101151743
122. Phan NL, Trinh NV, Pham PV. Low concentrations of 5-aza-2′-deoxycytidine induce breast cancer stem cell differentiation by triggering tumor suppressor gene expression. Onco Targets Ther. (2016) 9:49–59. doi: 10.2147/OTT.S96291
123. Kastrati I, Litosh VA, Zhao S, Alvarez M, Thatcher GR, Frasor J. A novel aspirin prodrug inhibits NFκB activity and breast cancer stem cell properties. BMC Cancer. (2015) 15:845. doi: 10.1186/s12885-015-1868-7
124. Saha S, Mukherjee S, Khan P, Kajal K, Mazumdar M, Manna A, et al. Aspirin suppresses the acquisition of chemoresistance in breast cancer by disrupting an NFκB-IL6 signaling axis responsible for the generation of cancer stem cells. Cancer Res. (2016) 76:2000–12. doi: 10.1158/0008-5472.CAN-15-1360
125. Maity G, Chakraborty J, Ghosh A, Haque I, Banerjee S, Banerjee SK. Aspirin suppresses tumor cell-induced angiogenesis and their incongruity. J Cell Commun Signal. (2019) 13:491–502. doi: 10.1007/s12079-018-00499-y
126. Tanei T, Choi DS, Rodriguez AA, Liang DH, Dobrolecki L, Ghosh M, et al. Antitumor activity of Cetuximab in combination with Ixabepilone on triple negative breast cancer stem cells. Breast Cancer Res. (2016) 18:6. doi: 10.1186/s13058-015-0662-4
127. Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, et al. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci U S A. (2012) 109:2784–9. doi: 10.1073/pnas.1018866109
Keywords: angiogenesis, breast cancer stem cell, chemotherapy and radiotherapy resistance, invasion, metastasis
Citation: Yousefnia S, Seyed Forootan F, Seyed Forootan S, Nasr Esfahani MH, Gure AO and Ghaedi K (2020) Mechanistic Pathways of Malignancy in Breast Cancer Stem Cells. Front. Oncol. 10:452. doi: 10.3389/fonc.2020.00452
Received: 13 December 2019; Accepted: 13 March 2020;
Published: 30 April 2020.
Edited by:
Leonardo Freire-de-Lima, Federal University of Rio de Janeiro, BrazilReviewed by:
Mariane B. Melo, Massachusetts Institute of Technology, United StatesDaniele Vergara, University of Salento, Italy
Copyright © 2020 Yousefnia, Seyed Forootan, Seyed Forootan, Nasr Esfahani, Gure and Ghaedi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Farzad Seyed Forootan, ZnNmb3Jvb3RhbkBnbWFpbC5jb20=; ZnNmb3Jvb3RhbkByb3lhbmluc3RpdHV0ZS5vcmc=; Ali Osmay Gure, YWd1cmVAYmlsa2VudC5lZHUudHI=; Kamran Ghaedi, a2FtcmFuZ2hhZWRpQHNjaS51aS5hYy5pcg==; a2FtcmFuZ2hhZWRpQHlhaG9vLmNvbQ==; a2FtcmFuZ2hhZWRpQHJveWFuaW5zdGl0dXRlLm9yZw==