 Yi-Ting Lu
Yi-Ting Lu Peng Fan*†
Peng Fan*† Xu Meng
Xu Meng- Department of Cardiology, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
Hypertension is an important risk factor in many conditions and creates a heavy burden of disease and mortality globally. Polygenic hypertension is the most common form; however, it is increasingly recognized that monogenic hypertension is not rare, especially in patients with electrolyte disorders. Single genetic alterations are associated with plasma volume expansion and catecholamines/sympathetic excess with simultaneously increased potassium excretion in the urine and potassium intracellular shift. Early-onset refractory hypertension and profound hypokalemia are characteristics of monogenic hypertension. However, accumulated evidence shows the existence of phenotypic heterogeneity in monogenic hypertension meaning that, even for mild symptoms, clinicians cannot easily exclude the possibility of monogenic hypertension. Genetic, epigenetic and non-genetic factors are all possible mechanisms influencing phenotypic diversity. Genetic sequencing is a precise and efficient method that can broaden the mutant gene spectrum of the disease and is very helpful for understanding the pathophysiology of monogenic hypertension. Genetic sequencing, along with biochemical tests and imaging modalities, is essential for the early diagnosis and targeted management of monogenic hypertension to avoid long-term catastrophic complications.
Introduction
Hypertension is one of the leading causes of death worldwide, affecting more than 1.1 billion people (1), and is a major risk factor for cardiovascular disease (CVD) and stroke (2). On the basis of its genetic contribution, hypertension can be classified into two types: polygenic hypertension and monogenic hypertension. Polygenic hypertension is affected by complex genetic variants and also lifestyle and environmental factors, such as smoking status, alcohol consumption, dietary salt intake, obesity, and physical motion (3, 4). Polygenic hypertension without identifiable causes is the most common form. In contrast, monogenic hypertension is an inherited hypertension disease caused by single genetic variants that follow Mendelian inheritance.
Monogenic hypertension is almost always associated with electrolyte disturbances, with hypokalemia commonly seen. Based on salt sensitivity and the level of aldosterone, monogenic hypertension combined with hypokalemia could be classified into three categories, salt insensitive hypertension, salt-sensitive hypertension with low/high aldosterone levels (Figure 1). Hypokalemia is generally attributed to increased potassium excretion or intracellular metastasis, while decreased potassium intake is relatively uncommon (5). Low potassium levels can induce multiple cardiac blocks or arrhythmias, sometimes requiring emergency medical care (6). Refractory hypertension is also frequent in patients with monogenic hypertension, defined as the failure to control blood pressure when given five or more various classes of antihypertensive agents (7). Long-term uncontrolled hypertension may develop complex complications, including severe targeted organ damage and even cardiovascular death events. Early recognition and individualized treatment are of paramount importance for the above situations.
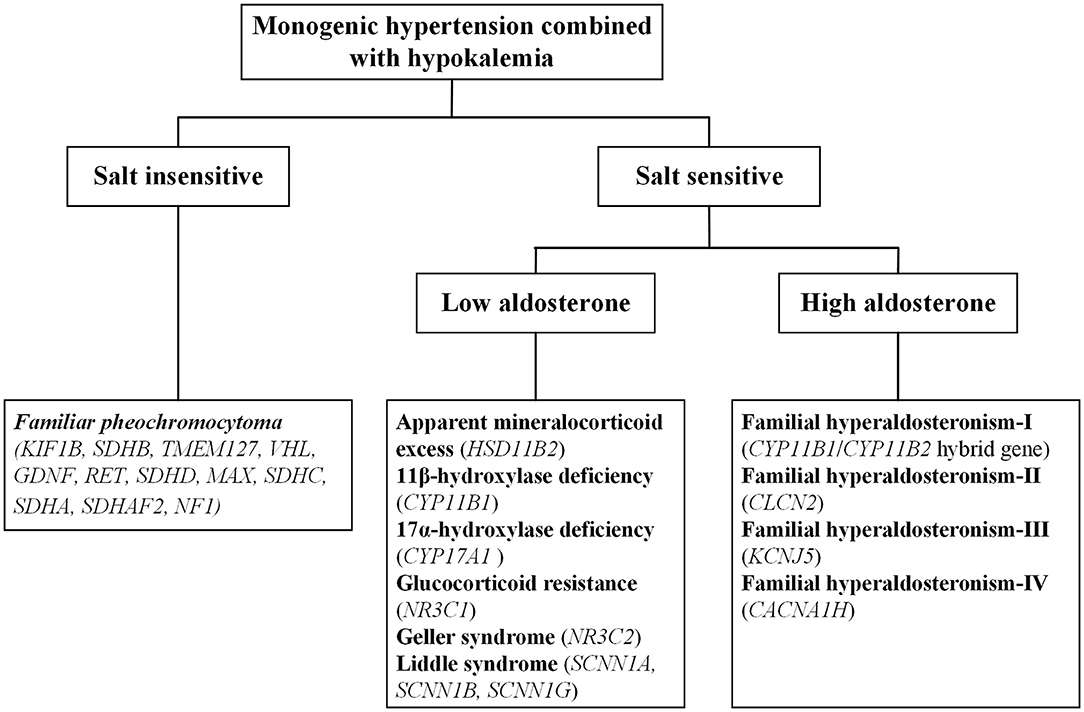
Figure 1. The overall categorization of monogenic hypertension combined with hypokalemia.
There are three basic mechanisms leading to the condition of expansive volume and hypokalemia: mineralocorticoid excess, salt retention and sympathetic activation. On this basis, various forms of monogenic hypokalemic hypertension commonly manifest as early-onset refractory hypertension, profound hypokalemia and metabolic alkalosis; except for pheochromocytoma (PCC), all forms of monogenic hypertension also have low levels of renin. However, it is vital to note that not all monogenic hypertension patients present with typical phenotypes. In recent years, advanced gene testing techniques have identified a series of atypical cases with monogenic hypertension that may previously have been overlooked or misdiagnosed as primary hypertension (8–11).
In this review, we summarize different kinds of monogenic hypertension combined with hypokalemia. We describe their mechanisms, clinical manifestations, and treatment strategies. Additionally, possible reasons for the phenotypic diversity of monogenic hypertension will be discussed.
Genetics of Monogenic Hypertension
Familial hyperaldosteronism (FH) is a rare autosomal dominant disorder of hypertension. It encompasses a group of conditions characterized by early-onset severe hypertension, hypokalemia, metabolic alkalosis, and a high plasma aldosterone/renin ratio (ARR>20). According to the underlying genetic defect, FH can be divided into four types, from I to IV. Diagnostically, it is hard to distinguish various subtypes of FH from sporadic primary aldosteronism (PA) just by clinical and biochemical observations, raising up the importance of genetic testing.
FH-I was firstly described by Sutherland et al. (12), representing about <1% of PA patients (13). For glucocorticoids have a significant positive effect on the disease, it was also named glucocorticoid remediable aldosteronism (GRA). In 1992, Lifton et al. (14) discovered the root cause of FH-I. Asymmetrical cross-over between the 11β-hydroxylase gene (CYP11B1) and the aldosterone synthase gene (CYP11B2) results in a chimeric gene, which possesses an adrenocorticotropic hormone (ACTH)-responsive promotor region at the 5' end of CYP11B1 and the aldosterone synthase coding region at the 3' end of CYP11B2. The gold standard diagnosis is to confirm the chimeric gene by genetic sequencing. PA subjects combined with a PA-positive family history, early stroke, or early onset of the disease are recommended to take a genetic diagnosis for FH-I (15). Affected individuals are associated with a high incidence of the thoracoabdominal aneurysm and cerebrovascular complications (such as cerebral aneurysm, hemorrhagic stroke) (16, 17). In terms of treatment, low dose glucocorticoid is advised to inhibit the concentration of ACTH. Moreover, the key therapeutic target is normal smooth blood pressure, not the normalization of all biochemical parameters, which may lead to unnecessary side effects.
FH-II, first reported by Stowasser et al. (18), might be the most common form of FH. In 2018, Scholl et al. (19) demonstrated a gain of function mutation of the gene CLCN2 in several affected subjects, which encodes voltage-gated chloride channel, ClC-2, that is expressed in adrenal glomerulosa. The mutation makes the glomerular cell membrane depolarize easily and activate voltage-gated calcium channels, thereby upregulating the expression of CYP11B2 which encodes aldosterone synthase, the sole enzyme for aldosterone biosynthesis (20). Recently, a specific hypertensive mouse model with the missense mutation homologous to the CLCN2 mutation related to the most frequent FH-II was reported by Schewe et al. (21) that further confirmed the role of ClC-2 in the regulation of aldosterone generation. Compared with FH-I, patients with FH-II are also associated with aldosterone-producing adenoma (APA) or bilateral adrenal hyperplasia (BAH). According to previous diagnostic criteria, when two or more family members suffer from PA, FH-II can be diagnosed after excluding the possibility of other FH forms (22). With the discovery of the new pathogenic gene variant, genetic testing may be a standard method for diagnosing FH-II. Different from FH-I, FH-II is unresponsive to glucocorticoids, but unilateral adrenalectomy combined with mineralocorticoid antagonists can relieve symptoms.
FH-III accounts for about 0.3% of PA (23) and is presumably caused by mutation of KCNJ5 (24), which encodes a G protein-activated inward rectifying potassium channel, GIRK4. The mutation affects GIRK4 selectivity, leading to loss of potassium selectivity in adrenal cortical cells, enhancing sodium conductance, and an increased influx of sodium and depolarization of the membrane, finally resulting in elevated expression of CYP11B2 (25). So far, six different germline mutations in KCNJ5 gene have been identified, and the severity of clinical manifestation is variable (26). Severe hyperaldosteronism symptoms and BAH are typical features of FH-III patients. Adrenal computed tomography (CT) and adrenal venous sampling are necessary to indicate the disease, and genetic testing should be suggested for patients with PA. Bilateral adrenalectomy and mineralocorticoid antagonists usually dramatically alleviate symptoms.
FH-IV is a rare form of FH with an unclear prevalence worldwide that is caused by germline CACNA1H mutations (27). CACNA1H is abundantly expressed in the adrenal zona glomerulosa and encodes the α subunit of a T-type calcium channel (Cav3.2). Abnormal activation of Cav3.2 increases calcium influx, leading to abnormal aldosterone synthesis (28). Clinically, the symptoms of FH-IV are similar to other forms of FH, with low specificity. Gene testing is advised for young (≤10 years old) with hypertension and PA (29). There is currently no specific treatment for FH-IV. Based on the severity of a patient's condition, clinicians generally administer mineralocorticoid antagonists or perform adrenalectomy (27).
Apparent Mineralocorticoid Excess (AME) is inherited as an autosomal recessive trait caused by inactivating HSD11B2 variants. The gene is located at cytogenetic locus 16q and is responsible for the synthesis of 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) (30). 11β-HSD2 is a member of the short-chain dehydrogenase/reductase family and is richly expressed in the salivary gland, skin, colon, placenta, thymus, vascular vessel, kidney and brain (31, 32). In renal epithelial tissue, 11β-HSD2 is principally co-localized with the mineralocorticoid receptors (MRs) in aldosterone-sensitive distal nephron (ASDN) (33). The MRs share a similar affinity for aldosterone and cortisol without selective characteristics and the circulating concentration of cortisol is 100–1000 times higher than aldosterone (33). Physiologically, the enzyme 11β-HSD2 catalyzes the metabolic conversion of cortisol to cortisone and corticosterone to 18-OH-deoxycorticosterone, respectively and thereby prevents the MRs from cortisol over-saturation (Figure 2). However, because of the mutated gene, the metabolic impairment of cortisol results in cortisol-mediated overstimulation of the MRs. It is worth noting that the clinical phenotype of AME is variable, and based on varying degrees of enzyme deficiency, AME can be divided into two types: a severe phenotype (AME-I) and a mild phenotype (AME-II). Patients with homozygous mutations display low birth weight, developmental retardation, unrelenting salt-sensitive hypertension in childhood. Nevertheless, those patients with heterozygous mutations usually show a mild or moderate phenotype, such as late-onset slight hypertension or staying normotensive, barely with electrolyte disturbances (34, 35). Previously, a high ratio of urinary cortisol to cortisone (F/E) was considered a basis for AME diagnosis. Recent studies (36, 37) show that serum F/E and urinary exosomes miRNA levels are more sensitive for the prediction of AME-II. In addition, it is necessary to distinguish the disease from those with acquired suppression of the activity of HSD11B2 by the ingestion of excessive liquorice or carbenoxolone (38). Treatments including MR antagonist spironolactone, dexamethasone can effectively alleviate AME.
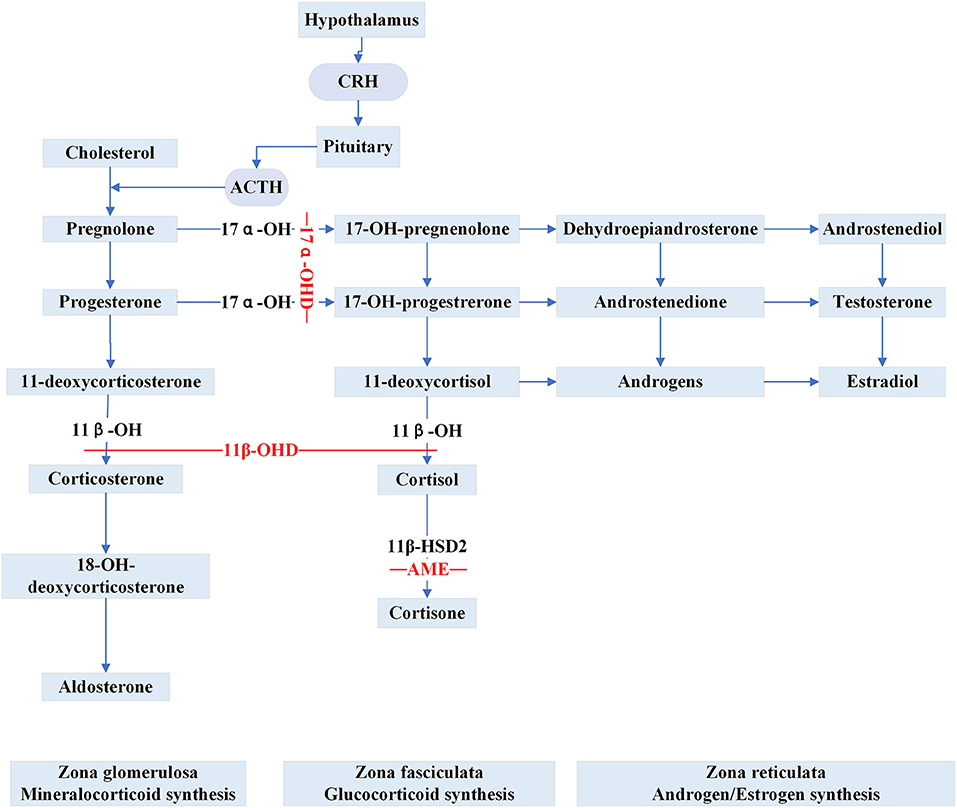
Figure 2. Steroid synthesis pathway and reduced activity of peripheral steroid hormone-metabolizing enzymes that catalyze the synthesis or inactivation of the corresponding hormones (11β-OHD, 17α-OHD, AME). 11β-OH, 11β-hydroxylase enzyme; 11β-OHD, 11β-hydroxylase deficiency; 17α-OH, 17α-hydroxylase enzyme; 17α-OHD, 17α-hydroxylase deficiency; 11β-HSD2, 11β-hydroxysteroid dehydrogenase type 2; AME, apparent mineralocorticoid excess.
Congenital adrenal hyperplasia (CAH) is a class of steroidogenic disorders transmitted in an autosomal recessive fashion. It is caused by variants in genes encoding enzymes involved in adrenal steroid synthesis. 11β-hydroxylase deficiency (11β-OHD) and 17α-hydroxylase deficiency (17α-OHD) are two forms of CAH typically showing early-onset hypertension combined with hypokalemia caused by the accumulation of ACTH and mineralocorticoids. Figure 2 shows detailed pathways for the synthesis of steroids.
11β-OHD is the second major subtype of CAH, accounting for 0.2–8% of all CAH cases while 21-hydroxysteroid deficiency is the most common subtype of CAH (39, 40). Mutation in CYP11B1, encoding steroid 11β-hydroxylase (11β-OH), results in a series of pathophysiological changes, and the mutation has a significant racial specificity (41). Deficiency of 11β-OH leads to impaired conversion of 11-deoxycortisol and 11-deoxycorticosterone to cortisol and corticosterone, respectively, resulting in an elevated ACTH level and superfluous adrenal androgens. Typically, apart from mineralocorticoid excess, patients may display hyperandrogenemia, which leads to masculinization of genitalia in females, pseudoprecocious puberty in males, and premature bone maturation in both. Curiously, the degree of excessive MR activation is not strongly correlated with the extent of virilization. Some severely virilized women have normal blood pressure, while mild individuals may experience resistant hypertension or even fatal cardiovascular accidents (42). Because of the variability of clinical manifestations, the possibility of 11β-OHD should be considered when encountering hypertensive patients with hyperandrogenemia and hyperadrenocorticism (41). The raised levels of 11-deoxycortisol, 11-deoxycorticosterone, and androgen are robust clues for the biochemical diagnosis of suspected 11β-OHD patients. Appropriate glucocorticoid supplementation is helpful in improving hypertension, hypokalemia and end-organ damage (43). Some patients with refractory hypertension or severe hyperandrogenism that is unresponsive to glucocorticoid therapy are recommended to undergo bilateral adrenalectomy. If necessary, surgical correction for patients with genital deformities is feasible according to the patients' will. Moreover, aromatase inhibitor therapy may be useful to improve the height prognosis of 11β-OHD patients, especially for those with very late bone ages (44).
17α-OHD is a syndrome caused by the mutation in CYP17A1. The function of 17α-hydroxylase enzyme (17α-OH) is to catalyze the conversion of pregnenolone/progesterone to 17-OH-prenenolone/17-OH-progesterone, respectively. Defective 17α-OH causes the insufficient secretion of cortisol, gonadal steroids, adrenal androgens, and because of a feedback increase in ACTH, the production of excess mineralocorticoids (45). Classical characteristics are sexual infantilism (secondary sexual sign hypoplasia, primary amenorrhea), and delayed puberty. Recently, Wu et al. (46) reported reduced bone density and adrenal masses in affected cases, but the underlying pathogenesis is not yet clear. Physical and biochemical tests are useful for detecting disease, while genetic sequencing is a standard way to confirm a diagnosis. Routine treatment of 17α-OHD includes MR antagonists, supplement of sex steroids and glucocorticoid; if the defective karyotype is 46, XY, prophylactic gonadectomy is also executed to prevent the genesis of gonadal tumors (47, 48). Clinicians should also pay close attention to the bone mineral density of patients with prolonged glucocorticoid therapy to prevent the occurrence of adverse events such as fractures (49).
Glucocorticoid resistance (GCCR) is a hereditary monogenic disorder transmitted in an autosomal dominant pattern, caused by variants in NR3C1, which encodes the glucocorticoid receptor (GR). The defective mutated gene leads to glucocorticoid resistance, which may be due to impaired GR binding affinity, reduced GR number, or impaired GR signal transduction function (50–52). The inactivation of GR results in a feedback activation of the hypothalamic-pituitary-adrenal (HPA) axis with the augmentation of ACTH, steroids, such as cortisol and androgens. Consequently, residual cortisol and mineralocorticoid precursor substances (deoxycortisol and corticosterone) are associated with resistant hypertension and hypokalemia. The typical features are characterized by a significant increase in plasma cortisol and ACTH levels, virilization in females and pseudoprecocious in males but without evidence of Cushing syndrome (53). Also, fatigue and anxiety are common characteristics of GCCR (54). High serum cortisol and urinary free cortisol are sensitive biomarkers, and their levels do not decrease after adrenal suppression by dexamethasone leading to the suspicion of GCCR. Low dose dexamethasone can be considered to suppress the secretion of ACTH, subsequently reducing the over-production of mineralocorticoids and androgens. To antagonize the androgen effect, clinicians sometimes administer aldosterone antagonists (53). When facing complex complications like cerebral infarction caused by severe long-term hypertension, high doses of dexamethasone are suitable (55).
Geller syndrome, also known as pregnancy-exacerbated hypertension, is a rare autosomal dominant disorder resulting from a gain-of-function mutation in the MR gene NR3C2, and is characterized by its constitutive activation (56). Changes in the selectivity and affinity of mutant MR for its ligand lead to their abnormal activation not only by aldosterone but also some steroids cortisone and progesterone (57). The disease typically presents at an early age and shows excess mineralocorticoid symptoms. The level of progesterone is significantly upregulated during pregnancy; therefore, pregnant women with the mutation display more severe symptoms (58). Definitive diagnosis can be achieved by genetic sequencing. The MR antagonist, spironolactone, is absolutely contraindicated because it is an agonist for mutant MR. For pregnant women with exacerbated hypertension, termination of pregnancy is advised to improve symptoms efficiently.
PCC refers to tumors originating from adrenal chromaffin cells with an estimated incidence of 3–8 cases per million people per year (59), and the ones arising from extra-adrenal chromaffin tissues are called paraganglioma (PGL). Germline mutations exist in more than 40% of patients with PCC or PGL, and it is mainly inherited in an autosomal dominant manner (60). Hereditary PCC can occur solitarily or as local manifestations of some genetic tumor syndromes, such as multiple endocrine neoplasia, neurofibromatosis, von Hippel-Lindau (61). Patients carrying susceptible genes are with the great possibility of developing PCC/PGL at a young age (62). The identification of susceptibility genes has been a research hotspot recently, which facilitates the diagnosis of patients in asymptomatic period (63). To date, the following tumor susceptibility genes have been reported: KIF1B, SDHB, TMEM127, VHL, GDNF, RET, SDHD, MAX, SDHC, SDHA, SDHAF2, NF1, with further novel mutations continually being discovered (64, 65). Continuous or paroxysmal hypertension, headaches, palpitations, diaphoresis, pallor are typical symptoms because of high concentrations of catecholamines. Genetic testing should be considered in all patients with PCC/PGL, particularly for those with a positive family history, or single and multifocal metastasis (64, 66, 67). Plasma free metanephrines (MNs) or all-day urinary free MNs are sensitive markers for PCC while spot urinary free MNs/creatinine is a powerful biomarker for PGL (68, 69). Imaging modalities such as CT, magnetic resonance imaging (MRI), positron emission tomography/CT (PET/CT) are beneficial for tumor localization. After a definite diagnosis, functional PCC is often removed surgically with α-adrenergic antagonists taken during the perioperative period. Annual biochemical follow-up of patients after surgery should last at least 10 years, longer for high-risk patients (64).
Liddle syndrome (LS) is an autosomal dominant disorder caused by gain-of-function mutations in the genes that encode the epithelial sodium channel (ENaC) subunits. Germline mutations in SCNN1A, SCNN1B, SCNN1G, encoding α, β, and γ subunits, respectively, lead to sodium reabsorption and blood volume expansion by affecting specific characteristics of the ENaC (70). In comparison, mutations in SCNN1B and SCNN1G are the most frequent forms of LS (8). In 2017, Salih et al. (71) reported a Caucasian family with LS and identified a new heterozygous missense mutation in SCNN1A through exome sequencing. Definitive diagnosis mainly depends on genetic screening. Early diagnosis and therapy are necessary for LS can cause sudden death, stroke, and end-stage renal failure (8). However, the prevalence of LS in the hypertensive population is still unclear. Two studies have shown the prevalence of LS among Chinese young hypertensive patients to be 1.52% and 0.91% (72, 73). Nevertheless, the discovery of new pathogenic genes and variable penetrance indicates that the prevalence of LS should be much higher than currently estimated. LS patients show remarkable response to the ENaC blocker, amiloride, and the potassium-sparing diuretic, triamterene. A salt-limited diet is also recommended on the basis of medication (74). The long-term prognosis of LS is not clear and requires further investigation.
Pathophysiology of Monogenic Hypertension
Plasma volume expansion and catecholamines/sympathetic excess are two distinct mechanisms that contribute to hypertension, the former being the more common cause. ENaC channels are abundantly distributed in the apical portion of epithelial cells of the connecting duct and the distal convoluted tubule. ENaC controls the rate-limiting step of sodium reabsorption from the lumen into the epithelial cells in the final pathway of volume expansion. There is a highly conserved proline-rich region named the PY motif in all carboxyl-terminal of three subunits. The ubiquitin ligase, NEDD4-2, can bind to the PY motif and initiate the ubiquitination of ENaC and endocytotic degradation (75). Mutations in ENaC subunit genes can directly increase the density and the open probability of channels resulting in sodium retention (LS).
In addition, an important factor influencing the expression of ENaC channels is excessive mineralocorticoid effects, which can be divided into three pathways: (1) abnormal affinity of steroid hormone receptors resulting from MR and GR genes variants (Geller Syndrome and GCCR); (2) reduced activity of peripheral steroid hormone-metabolizing enzymes that catalyze the synthesis or inactivation of corresponding hormones (AME, 11β-OHD, 17α-OHD); (3) excessive aldosterone synthesis (FH-I, II, III, IV). The over-bound of mineralocorticoid/MR complex up-regulates the expression of serum and glucocorticoid-inducible kinase 1 (SGK1), thus regulating the activity of ENaC in various ways. First, SGK1 directly phosphorylates NEDD4-2 by binding to its amino acid residues and reduces the ubiquitination of ENaC channels. Second, SGK1 can promote the combination of NEDD4-2 and 14-3-3 protein, leading to the conformational change of NEDD4-2, which diminishes the NEDD4-2/ ENaC interaction (76). Moreover, SGK1 could enhance the electrophysiological function of ENaC through directly phosphorylating or influencing the transcription process of its α subunit (77).
Catecholamines/sympathetic excess is another mechanism of monogenic hypertension that occurs in PCC/PGL. Various clusters' genetic mutations which involve activation of hypoxia-angiogenic pathways or RAS and kinase signaling pathways lead to the development of catecholamine-producing neuroendocrine neoplasms (64). Activation of the sympathetic nervous system mediates the contraction of the renal vasculature, hence stimulating the secondary secretion of renin and aldosterone which, in turn, increases the reabsorption of sodium and water (78).
Pathophysiology of Hypokalemia
Increased potassium excretion in the urine and intracellular shift are two common causes of hypokalemia, a typical electrolyte disturbance of monogenic hypertension. The kidney plays a vital role in maintaining potassium balance in the body. Under normal physiological conditions, 90% of excreted potassium is passed in the urine with only 10% via the digestive tract. When 24-h urinary potassium is >15 mEq, inappropriate renal potassium loss is indicated (15). Differential transmembrane concentrations of potassium and potential gradients drive tubule potassium secretion via the reabsorption of sodium through ENaC located in the distal convoluted tubule. The activity of ENaC can be elevated through mineralocorticoid effects by increasing the number of channels and the channel opening time, which is responsible for partial forms of monogenic hypertension combined with hypokalemia.
Hyperactive ENaC and excess mineralocorticoid effect are associated with the elevated secretion of potassium. Physiologically, sodium ions in the lumen enter the cell through ENaC along the electrochemical gradient and are pumped across the basolateral membrane through the Na+/K+-ATPase, which drives the transport of potassium into the cell, followed by the secretion into urine through different potassium channels (79). Due to hyperactive ENaC, the increase of sodium reabsorption results in a luminal negative potential gradient, stimulating the elevated potassium secretion to maintain electrochemical equilibrium. Similarly, excess mineralocorticoid effects magnify the activity of ENaC, followed by too much potassium loss. The renal outer medullary K+ channel located in the distal tubule is one of the key transport portions of potassium (80). Maxi-K channel also plays a vital role in secreting potassium, whose function could be activated by increased luminal flow rate and aldosterone (81, 82).
The vast majority of potassium (98%) is present in intracellular fluid. The maintenance of potassium distribution between intracellular and extracellular fluids, referred to as the internal potassium balance, is also an intrinsic mechanism for potassium homeostasis. Insulin and catecholamines are the two most important hormones that affect the internal potassium balance by moving potassium into cells (82). The high levels of catecholamines released by PCC can increase the activity of Na+/K+-ATPase by blocking the α receptors or activating β2 receptors, thus facilitating the migration of potassium into cells. In a nutshell, the extent of sodium reabsorption in the ASDN, the flow rate of fluid in the distal convoluted tubule, acid-base disorders, the state of α/ β2 receptors, and the concentration of aldosterone and arginine vasopressin are all key factors in determining the degree of potassium secretion.
Phenotypic Heterogeneity
The clinical phenotypes and genotypes of monogenic hypertension show considerable heterogeneity, which may hamper the diagnosis of the disease. With the same genetic mutation, patients may display various degrees of clinical manifestation, ranging from milder symptoms encompassing normotension or normokalemic to severe, life-threatening conditions (20, 46, 70, 83, 84). Genetic interaction, epigenetic modifier and non-genetic factors, such as age, environmental and nutritional factors, are closely associated with the variable phenotype of monogenic hypertension.
Genotype-Phenotype Correlation
Firstly, some mutations which result in partial activity defects of relevant enzymes are possibly responsible for some mild phenotypes, which may be misdiagnosed easily (38, 85). For instance, patients with the mutations in exon 3 of gene CYP11B1 tend to present as a non-classic phenotype, similar to polycystic ovary syndrome (41). Second, the overlap of germline and somatic mutations may be one of the causes of phenotypic variability. Lin et al. (86) first reported two GRA affected siblings, who also had a somatic KCNJ5 mutation, showing the rare phenotype of adrenal adenoma, hypertension and lower blood potassium level compared with most GRA patients. In addition, mutations in different regions of ClC-2 may affect channel activity to varying degrees, partly explaining the phenotypic heterogeneity of FH-II (87).
Epigenetic Modification
Epigenetic modification plays a critical role in explaining the phenotypic heterogeneity of monogenic hypertension beyond genetic defects. It refers to a reversible change in gene function but without altering the DNA sequence in the nucleus. DNA methylation is one of the first modification pathways to be discovered. High methylation levels of the HSD11B2 promoter are associated with increased blood pressure and is one of the factors influencing the onset of hypertension (88). Multiple lines of evidence show that the higher the degree of HSD11B2 promoter methylation, the lower the activity of 11β-HSD2, which possibly explains the inconsistent phenotype of hypertension among AME patients (9, 89). Furthermore, the role of non-coding microRNAs and histone modification in regulating blood pressure has been reviewed by Burrello et al. (90), but the mechanisms of these process that affect the phenotype of monogenic hypertension remain to be identified.
Non-Genetic Factors
Age along with environmental and nutritional factors, including obesity, diabetes, daily sodium and potassium intake, may also have an effect on the disease phenotype (11, 91). Tapolyai et al. (92) reported the phenomenon of elderly patients demonstrating Liddle's-like syndrome but with the negative family history. Several elderly patients presenting with Liddle syndrome phenotype also have been discovered and identified without genetic mutation, rasing the hypothesis that there is a correlation between age or medication and ENaC channel activity (93, 94). Several studies (95, 96) have confirmed that the level of cortisol and the ratio of F/E decrease with age and suggested the activity of 11β-HSD2 gradually reduces with age, which may relate to variable clinical phenotype of AME. The the exact mechanism between sodium, potassium intake levels and the diverse phenotype of monogenic hypertension deserves further study.
Conclusions
Table 1 summarizes the basic characteristics of different monogenic forms of hypertension combined with hypokalemia. Although the epidemiology of monogenic hypertension is not completely clear and requires further study, we suggest that monogenic hypertension is not an uncommon cause of secondary hypertension, especially for those with simultaneous electrolyte disturbances. We do require useful strategies to identify phenotypic heterogeneity to avoid severe complications of monogenic hypertension. Genetic testing, which is precise and efficient, is beneficial for the early identification of patients, for targeted therapy and better management of affected subjects, particularly for those with atypical phenotypes, thus improving the prognosis of the disease. Overall, there is great optimism that the morbidity and mortality of monogenic hypertension will decrease further with the advancement of genetically driven individualized treatment.
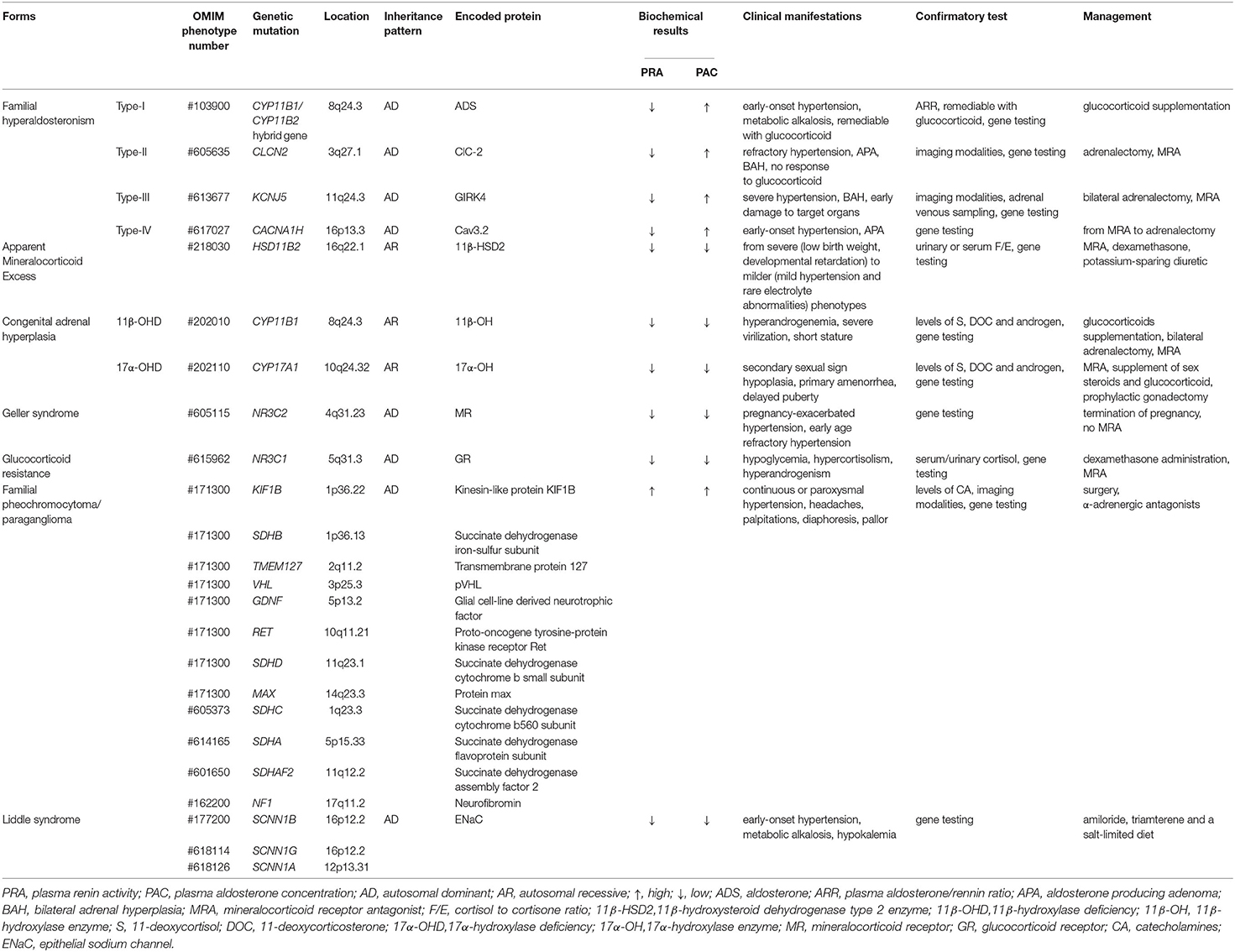
Table 1. Basic characteristics of different monogenic forms of hypertension with hypokalemia.
Author Contributions
All authors are responsible for the literature review, drafting and revision of the manuscript, and approved the final version of the manuscript.
Funding
This work was supported by the Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019XK320057), CAMS Innovation Fund for Medical Sciences (2016-I2M-1-002), and the National Key Research and Development Program of China (2016YFC1300100).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Islam MS. Hypertension: from basic research to clinical practice. Adv Exp Med Biol. (2017) 956:1–2. doi: 10.1007/5584_2017_30
2. James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. (2014) 311:507–20. doi: 10.1001/jama.2013.284427
3. Mente A, O'Donnell MJ, Rangarajan S, McQueen MJ, Poirier P, Wielgosz A, et al. Association of urinary sodium and potassium excretion with blood pressure. N Engl J Med. (2014) 371:601–11. doi: 10.1056/NEJMoa1311989
4. Shimosawa T, Fukunaga A, Inoue Y, Chandraratne N, Yamaguchi M, Kuwahara K, et al. Healthy lifestyle index and its association with hypertension among community adults in Sri Lanka: a cross-sectional study. PLoS ONE. (2020) 15:e0226773. doi: 10.1371/journal.pone.0226773
5. Kardalas E, Paschou SA, Anagnostis P, Muscogiuri G, Siasos G, Vryonidou A. Hypokalemia: a clinical update. Endocr Connect. (2018) 7:R135–46. doi: 10.1530/EC-18-0109
6. Martinez-Morillo E, Alvarez FV. Management of potassium results in haemolysed plasma samples at the emergency department laboratory. Clin Chem Lab Med. (2019) 57:e271–3. doi: 10.1515/cclm-2019-0393
7. Calhoun DA, Booth JN III, Oparil S, Irvin MR, Shimbo D, Lackland DT, et al. Refractory hypertension: determination of prevalence, risk factors, and comorbidities in a large, population-based cohort. Hypertension. (2014) 63:451–8. doi: 10.1161/HYPERTENSIONAHA.113.02026
8. Kozina AA, Trofimova TA, Okuneva EG, Baryshnikova NV, Obuhova VA, Krasnenko AY, et al. Liddle syndrome due to a novel mutation in the gamma subunit of the epithelial sodium channel (ENaC) in family from Russia: a case report. BMC Nephrol. (2019) 20:389. doi: 10.1186/s12882-019-1579-4
9. Pizzolo F, Friso S, Morandini F, Antoniazzi F, Zaltron C, Udali S, et al. Apparent mineralocorticoid excess by a novel mutation and epigenetic modulation by HSD11B2 promoter methylation. J Clin Endocrinol Metab. (2015) 100:E1234–41. doi: 10.1210/jc.2015-1760
10. Buyukkaragoz B, Yilmaz AC, Karcaaltincaba D, Ozdemir O, Ludwig M. Liddle syndrome in a Turkish family with heterogeneous phenotypes. Pediatr Int. (2016) 58:801–4. doi: 10.1111/ped.12985
11. Baudrand R, Vaidya A. The low-renin hypertension phenotype: genetics and the role of the mineralocorticoid receptor. Int J Mol Sci. (2018) 19:546. doi: 10.3390/ijms19020546
12. Sutherland DJ, Ruse JL, Laidlaw JC. Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. Can Med Assoc J. (1966) 95:1109–19.
13. Hirsch JS, Hong S. The demystification of secondary hypertension: diagnostic strategies and treatment algorithms. Curr Treat Options Cardiovasc Med. (2019) 21:90. doi: 10.1007/s11936-019-0790-8
14. Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, et al. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature. (1992) 355:262–5. doi: 10.1038/355262a0
15. Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2016) 101:1889–916. doi: 10.1210/jc.2015-4061
16. Shahrrava A, Moinuddin S, Boddu P, Shah R. A case of glucocorticoid remediable aldosteronism and thoracoabdominal aneurysms. Case Rep Endocrinol. (2016) 2016:2017571. doi: 10.1155/2016/2017571
17. Al Romhain B, Young AM, Battacharya JJ, Suttner N. Intracranial aneurysm in a patient with glucocorticoid-remediable aldosteronism. Br J Neurosurg. (2015) 29:715–7. doi: 10.3109/02688697.2015.1023775
18. Stowasser M, Gordon RD, Tunny TJ, Klemm SA, Finn WL, Krek AL. Familial hyperaldosteronism type II: five families with a new variety of primary aldosteronism. Clin Exp Pharmacol Physiol. (1992) 19:319–22. doi: 10.1111/j.1440-1681.1992.tb00462.x
19. Scholl UI, Stolting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet. (2018) 50:349–54. doi: 10.1038/s41588-018-0048-5
20. Stowasser M, Wolley M, Wu A, Gordon RD, Schewe J, Stolting G, et al. Pathogenesis of familial hyperaldosteronism type II: new concepts involving anion channels. Curr Hypertens Rep. (2019) 21:31. doi: 10.1007/s11906-019-0934-y
21. Schewe J, Seidel E, Forslund S, Marko L, Peters J, Muller DN, et al. Elevated aldosterone and blood pressure in a mouse model of familial hyperaldosteronism with ClC-2 mutation. Nat Commun. (2019) 10:5155. doi: 10.1038/s41467-019-13033-4
22. Pons Fernandez N, Moreno F, Morata J, Moriano A, Leon S, De Mingo C, et al. Familial hyperaldosteronism type III a novel case and review of literature. Rev Endocr Metab Disord. (2019) 20:27–36. doi: 10.1007/s11154-018-9481-0
23. Mulatero P, Tauber P, Zennaro MC, Monticone S, Lang K, Beuschlein F, et al. KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension. (2012) 59:235–40. doi: 10.1161/HYPERTENSIONAHA.111.183996
24. Choi M, Scholl UI, Yue P, Bjorklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. (2011) 331:768–72. doi: 10.1126/science.1198785
25. Monticone S, Hattangady NG, Nishimoto K, Mantero F, Rubin B, Cicala MV, et al. Effect of KCNJ5 mutations on gene expression in aldosterone-producing adenomas and adrenocortical cells. J Clin Endocrinol Metab. (2012) 97:E1567–72. doi: 10.1210/jc.2011-3132
26. Monticone S, Tetti M, Burrello J, Buffolo F, De Giovanni R, Veglio F, et al. Familial hyperaldosteronism type III. J Human Hypertens. (2017) 31:776. doi: 10.1038/jhh.2017.34
27. Scholl UI, Stolting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife. (2015) 4:e06315. doi: 10.7554/eLife.06315
28. Daniil G, Fernandes-Rosa FL, Chemin J, Blesneac I, Beltrand J, Polak M, et al. CACNA1H mutations are associated with different forms of primary aldosteronism. EBioMed. (2016) 13:225–36. doi: 10.1016/j.ebiom.2016.10.002
29. Boulkroun S, Fernandes-Rosa FL, Zennaro MC. Old and new genes in primary aldosteronism. Best Pract Res Clin Endocrinol Metab. (2020) 34:101375. doi: 10.1016/j.beem.2020.101375
30. Mune T, Rogerson FM, Nikkila H, Agarwal AK, White PC. Human hypertension caused by mutations in the kidney isozyme of 11 beta-hydroxysteroid dehydrogenase. Nat Genet. (1995) 10:394–9. doi: 10.1038/ng0895-394
31. Woods C, Tomlinson JW. The dehydrogenase hypothesis. Adv Exp Med Biol. (2015) 872:353–80. doi: 10.1007/978-1-4939-2895-8_16
32. Almanzar G, Mayerl C, Seitz JC, Hofner K, Brunner A, Wild V, et al. Expression of 11beta-hydroxysteroid-dehydrogenase type 2 in human thymus. Steroids. (2016) 110:35–40. doi: 10.1016/j.steroids.2016.03.019
33. Zennaro MC, Boulkroun S, Fernandes-Rosa F. Inherited forms of mineralocorticoid hypertension. Best Pract Res Clin Endocrinol Metab. (2015) 29:633–45. doi: 10.1016/j.beem.2015.04.010
34. Alzahrani AS, Aljuhani N, Qasem E, Almohanna M, Hamoudh E, AlOmair A, et al. Apparent mineralocorticoid excess caused by a novel mutation in 11-beta hydroxysteroid dehydrogenase type 2 enzyme: its genetics and response to therapy. Endocr Pract. (2014) 20:e151–6. doi: 10.4158/EP14094.CR
35. Kucuk N, Yavas Abali Z, Abali S, Canpolat N, Yesil G, Turan S, et al. A rare cause of hypertension in childhood: Answers. Pediatr Nephrol. (2020) 35:79–82. doi: 10.1007/s00467-019-04329-0
36. Tapia-Castillo A, Baudrand R, Vaidya A, Campino C, Allende F, Valdivia C, et al. Clinical, biochemical, and genetic characteristics of “nonclassic” apparent mineralocorticoid excess syndrome. J Clin Endocrinol Metab. (2019) 104:595–603. doi: 10.1210/jc.2018-01197
37. Tapia-Castillo A, Guanzon D, Palma C, Lai A, Barros E, Allende F, et al. Downregulation of exosomal miR-192-5p and miR-204-5p in subjects with nonclassic apparent mineralocorticoid excess. J Transl Med. (2019) 17:392. doi: 10.1186/s12967-019-02143-8
38. Funder JW. Apparent mineralocorticoid excess. J Steroid Biochem Mol Biol. (2017) 165:151–3. doi: 10.1016/j.jsbmb.2016.03.010
39. Torok D. Congenital adrenal hyperplasia. Exp Suppl. (2019) 111:245–60. doi: 10.1007/978-3-030-25905-1_12
40. Bulsari K, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 11beta-hydroxylase deficiency. Endocrine. (2017) 55:19–36. doi: 10.1007/s12020-016-1189-x
41. Wang D, Wang J, Tong T, Yang Q. Non-classical 11beta-hydroxylase deficiency caused by compound heterozygous mutations: a case study and literature review. J Ovarian Res. (2018) 11:82. doi: 10.1186/s13048-018-0450-8
42. Rösler A, Leiberman E, Sack J, Landau H, Benderly A, Moses SW, et al. Clinical variability of congenital adrenal hyperplasia due to 11β-hydroxylase deficiency. Hormones. (1982) 16:133–41. doi: 10.1159/000179494
43. Goyal A, Boro H, Khadgawat R. Male gender identity and reversible hypokalemic hypertension in a 46,XX child with 11-beta-hydroxylase deficiency congenital adrenal hyperplasia. Cureus. (2019) 11:e5248. doi: 10.7759/cureus.5248
44. Atay Z, Turan S, Bugdayci O, Guran T, Bereket A. Restoration of height after 11 years of letrozole treatment in 11beta-hydroxylase deficiency. Hormone Res Paediatrics. (2019) 92:203–8. doi: 10.1159/000501456
45. Kronenberg S, Melmed S, Polonsky KS. Adrenal Cortex and Endocrine Hypertension. In: Melmed S, Polonsky KS, Larsen PR, Kronenberg S, editors. Williams Textbook of Endocrinology. Elsevier Health Sciences (2015) p. 490–589.
46. Wu CM, Fan SS, Qian YY, Zhou YY, Jin J, Dai ZJ, et al. 17 alpha-hydroxylase/17, 20-lyase deficiency: clinical and molecular characterization of eight Chinese patients. Endocr Pract. (2017) 23:576–82. doi: 10.4158/EP161610.OR
47. Auchus RJ. Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J Steroid Biochem Mol Biol. (2017) 165:71–8. doi: 10.1016/j.jsbmb.2016.02.002
48. Kardelen AD, Toksoy G, Bas F, Yavas Abali Z, Gencay G, Poyrazoglu S, et al. A Rare cause of congenital adrenal hyperplasia: clinical and genetic findings and follow-up characteristics of six patients with 17-hydroxylase deficiency including two novel mutations. J Clin Res Pediatr Endocrinol. (2018) 10:206–15. doi: 10.4274/jcrpe.0032
49. Riehl G, Reisch N, Roehle R, Claahsen van der Grinten H, Falhammar H, Quinkler M. Bone mineral density and fractures in congenital adrenal hyperplasia: findings from the dsd-LIFE study. Clin Endocrinol (Oxf). (2020) 92:284–94. doi: 10.1111/cen.14149
50. Charmandari E, Kino T, Souvatzoglou E, Vottero A, Bhattacharyya N, Chrousos GP. Natural glucocorticoid receptor mutants causing generalized glucocorticoid resistance: molecular genotype, genetic transmission, and clinical phenotype. J Clin Endocrinol Metab. (2004) 89:1939–49. doi: 10.1210/jc.2003-030450
51. Pujols L, Mullol J, Roca-Ferrer J, Torrego A, Xaubet A, Cidlowski JA, et al. Expression of glucocorticoid receptor alpha- and beta-isoforms in human cells and tissues. Am J Physiol Cell Physiol. (2002) 283:C1324–31. doi: 10.1152/ajpcell.00363.2001
52. Charmandari E, Kino T, Ichijo T, Chrousos GP. Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab. (2008) 93:1563–72. doi: 10.1210/jc.2008-0040
53. van Rossum EF, Lamberts SW. Glucocorticoid resistance syndrome: a diagnostic and therapeutic approach. Best Pract Res Clin Endocrinol Metab. (2006) 20:611–26. doi: 10.1016/j.beem.2006.09.005
54. Donner KM, Hiltunen TP, Janne OA, Sane T, Kontula K. Generalized glucocorticoid resistance caused by a novel two-nucleotide deletion in the hormone-binding domain of the glucocorticoid receptor gene NR3C1. Eur J Endocrinol. (2013) 168:K9–18. doi: 10.1530/EJE-12-0532
55. Tatsi C, Xekouki P, Nioti O, Bachrach B, Belyavskaya E, Lyssikatos C, et al. A novel mutation in the glucocorticoid receptor gene as a cause of severe glucocorticoid resistance complicated by hypertensive encephalopathy. J Hypertens. (2019) 37:1475–81. doi: 10.1097/HJH.0000000000002048
56. Melcescu E, Koch CA. Syndromes of mineralocorticoid excess. In: Koch CA, Chrousos GP, editors. Endocrine Hypertension: Underlying Mechanisms and Therapy. Totowa, NJ: Humana Press (2013) p. 33–50.
57. Kang S-H, Lee H-A, Lee E, Kim M, Kim I. Histone deacetylase inhibition, but not a mineralocorticoid receptor antagonist spironolactone, attenuates atypical transcription by an activating mutant MR (MRS810L). Clin Exp Pharmacol Physiol. (2016) 43:995–1003. doi: 10.1111/1440-1681.12614
58. Geller DS, Farhi A, Pinkerton N, Fradley M, Moritz M, Spitzer A, et al. Activating mineralocorticoid receptor mutation in hypertension exacerbated by pregnancy. Science. (2000) 289:119–23. doi: 10.1126/science.289.5476.119
59. Ariton M, Juan CS, AvRuskin TW. Pheochromocytoma: clinical observations from a Brooklyn tertiary hospital. Endocr Pract. (2000) 6:249–52. doi: 10.4158/EP.6.3.249
60. Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. (2015) 11:101–11. doi: 10.1038/nrendo.2014.188
61. Tsirlin A, Oo Y, Sharma R, Kansara A, Gliwa A, Banerji MA. Pheochromocytoma: a review. Maturitas. (2014) 77:229–38. doi: 10.1016/j.maturitas.2013.12.009
62. Nolting S, Ullrich M, Pietzsch J, Ziegler CG, Eisenhofer G, Grossman A, et al. Current management of pheochromocytoma/paraganglioma: a guide for the practicing clinician in the era of precision medicine. Cancers (Basel). (2019) 11:1505. doi: 10.3390/cancers11101505
63. Bausch B, Schiavi F, Ni Y, Welander J, Patocs A, Ngeow J, et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. (2017) 3:1204–12. doi: 10.1001/jamaoncol.2017.0223
64. Lenders JWM, Eisenhofer G. Update on modern management of pheochromocytoma and paraganglioma. Endocrinol Metab (Seoul). (2017) 32:152–61. doi: 10.3803/EnM.2017.32.2.152
65. Sbardella E, Cranston T, Isidori AM, Shine B, Pal A, Jafar-Mohammadi B, et al. Routine genetic screening with a multi-gene panel in patients with pheochromocytomas. Endocrine. (2018) 59:175–82. doi: 10.1007/s12020-017-1310-9
66. Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2014) 99:1915–42. doi: 10.1210/jc.2014-1498
67. Plouin PF, Amar L, Dekkers OM, Fassnacht M, Gimenez-Roqueplo AP, Lenders JW, et al. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol. (2016) 174:G1–10. doi: 10.1530/EJE-16-0033
68. Sbardella E, Grossman AB. Pheochromocytoma: an approach to diagnosis. Best Pract Res Clin Endocrinol Metab. (2019) 34:101346. doi: 10.1016/j.beem.2019.101346
69. Zuo M, Zhen Q, Zhang X, Zou W, Yang X, Tian G, et al. High specificity of spot urinary free metanephrines in diagnosis and prognosis of pheochromocytomas and paragangliomas by HPLC with electrochemical detection. Clin Chimica Acta. (2018) 478:82–9. doi: 10.1016/j.cca.2017.12.026
70. Tetti M, Monticone S, Burrello J, Matarazzo P, Veglio F, Pasini B, et al. Liddle syndrome: review of the literature and description of a new case. Int J Mol Sci. (2018) 19:812. doi: 10.3390/ijms19030812
71. Salih M, Gautschi I, van Bemmelen MX, Di Benedetto M, Brooks AS, Lugtenberg D, et al. A missense mutation in the extracellular domain of alphaenac causes liddle syndrome. JASN. (2017) 28:3291–9. doi: 10.1681/ASN.2016111163
72. Wang LP, Yang KQ, Jiang XJ, Wu HY, Zhang HM, Zou YB, et al. Prevalence of liddle syndrome among young hypertension patients of undetermined cause in a Chinese population. J Clin Hypertens. (2015) 17:902–7. doi: 10.1111/jch.12598
73. Liu K, Qin F, Sun X, Zhang Y, Wang J, Wu Y, et al. Analysis of the genes involved in Mendelian forms of low-renin hypertension in Chinese early-onset hypertensive patients. J Hypertens. (2018) 36:502–9. doi: 10.1097/HJH.0000000000001556
74. Pagani L, Diekmann Y, Sazzini M, De Fanti S, Rondinelli M, Farnetti E, et al. Three reportedly unrelated families with liddle syndrome inherited from a common ancestor. Hypertension. (2018) 71:273–9. doi: 10.1161/HYPERTENSIONAHA.117.10491
75. Malik B, Price SR, Mitch WE, Yue Q, Eaton DC. Regulation of epithelial sodium channels by the ubiquitin-proteasome proteolytic pathway. Am J Physiol Renal Physiol. (2006) 290:F1285–94. doi: 10.1152/ajprenal.00432.2005
76. Baines D. Kinases as targets for ENaC regulation. Curr Mol Pharmacol. (2013) 6:50–64. doi: 10.2174/18744672112059990028
77. Boyd C, Náray-Fejes-Tóth A. Gene regulation of ENaC subunits by serum- and glucocorticoid-inducible kinase-1. Am J Physiol Renal Physiol. (2005) 288:F505–12. doi: 10.1152/ajprenal.00242.2004
78. Sata Y, Head GA, Denton K, May CN, Schlaich MP. Role of the sympathetic nervous system and its modulation in renal hypertension. Front Med. (2018) 5:82. doi: 10.3389/fmed.2018.00082
79. Kamel KS, Schreiber M, Halperin ML. Renal potassium physiology: integration of the renal response to dietary potassium depletion. Kidney Int. (2018) 93:41–53. doi: 10.1016/j.kint.2017.08.018
80. Pearce D, Soundararajan R, Trimpert C, Kashlan OB, Deen PM, Kohan DE. Collecting duct principal cell transport processes and their regulation. CJASN. (2015) 10:135–46. doi: 10.2215/CJN.05760513
81. Valinsky WC, Touyz RM, Shrier A. Aldosterone, SGK1, and ion channels in the kidney. Clin Sci. (2018) 132:173–83. doi: 10.1042/CS20171525
82. Palmer BF, Clegg DJ. Physiology and pathophysiology of potassium homeostasis: core curriculum 2019. Am J Kidney Dis. (2019) 74:682–95. doi: 10.1053/j.ajkd.2019.03.427
83. Bogdanovic R, Kuburovic V, Stajic N, Mughal SS, Hilger A, Ninic S, et al. Liddle syndrome in a Serbian family and literature review of underlying mutations. Eur J Pediatrics. (2012) 171:471–8. doi: 10.1007/s00431-011-1581-8
84. Molnar A, Patocs A, Liko I, Nyiro G, Racz K, Toth M, et al. An unexpected, mild phenotype of glucocorticoid resistance associated with glucocorticoid receptor gene mutation case report and review of the literature. BMC Med Genet. (2018) 19:37. doi: 10.1186/s12881-018-0552-6
85. Sousa Paredes SC, Marques O, Alves M. Partial deficiency of 17alpha-hydroxylase: a rare cause of congenital adrenal hyperplasia. BMJ Case Rep. (2019) 12:e230778. doi: 10.1136/bcr-2019-230778
86. Lin YF, Peng KY, Chang CH, Hu YH, Wu VC, Chueh JS, et al. Adrenalectomy completely cured hypertension in patients with familial hyperaldosteronism type I who had somatic KCNJ5 mutation. J Clin Endocrinol Metab. (2019) 104:5462–6. doi: 10.1210/jc.2019-00689
87. De Sousa K, Abdellatif AB, El Zein RM, Zennaro MC. Molecular mechanisms in primary aldosteronism. J Mol Endocrinol. (2019) 242:R67–79. doi: 10.1530/JOE-19-0193
88. Friso S, Pizzolo F, Choi SW, Guarini P, Castagna A, Ravagnani V, et al. Epigenetic control of 11 beta-hydroxysteroid dehydrogenase 2 gene promoter is related to human hypertension. Atherosclerosis. (2008) 199:323–7. doi: 10.1016/j.atherosclerosis.2007.11.029
89. Carvajal CA, Tapia-Castillo A, Vecchiola A, Baudrand R, Fardella CE. Classic and nonclassic apparent mineralocorticoid excess syndrome. J Clin Endocrinol Metab. (2020) 105:dgz315. doi: 10.1210/clinem/dgz315
90. Burrello J, Monticone S, Buffolo F, Tetti M, Veglio F, Williams TA, et al. Is there a role for genomics in the management of hypertension? Int J Mol Sci. (2017) 18:1131. doi: 10.3390/ijms18061131
91. Hirohama D, Fujita T. Evaluation of the pathophysiological mechanisms of salt-sensitive hypertension. Hypertens Res. (2019) 42:1848–57. doi: 10.1038/s41440-019-0332-5
92. Tapolyai M, Uysal A, Dossabhoy NR, Zsom L, Szarvas T, Lengvarszky Z, et al. High prevalence of liddle syndrome phenotype among hypertensive US Veterans in Northwest Louisiana. J Clin Hypertens. (2010) 12:856–60. doi: 10.1111/j.1751-7176.2010.00359.x
93. Kashif Nadeem M, Ling C. Liddle's-like syndrome in the elderly. J Clin Hypertens. (2012) 14:728. doi: 10.1111/j.1751-7176.2012.00683.x
94. Pepersack T, Allegre S, Jeunemaitre X, Leeman M, Praet JP. Liddle syndrome phenotype in an octogenarian. J Clin Hypertens. (2015) 17:59–60. doi: 10.1111/jch.12450
95. Campino C, Martinez-Aguayo A, Baudrand R, Carvajal CA, Aglony M, Garcia H, et al. Age-related changes in 11beta-hydroxysteroid dehydrogenase type 2 activity in normotensive subjects. Am J Hypertens. (2013) 26:481–7. doi: 10.1093/ajh/hps080
Keywords: monogenic hypertension, hypokalemia, phenotypic variability, pathophysiology, genetic sequencing
Citation: Lu Y-T, Fan P, Zhang D, Zhang Y, Meng X, Zhang Q-Y, Zhao L, Yang K-Q and Zhou X-L (2021) Overview of Monogenic Forms of Hypertension Combined With Hypokalemia. Front. Pediatr. 8:543309. doi: 10.3389/fped.2020.543309
Received: 16 March 2020; Accepted: 29 December 2020;
Published: 25 January 2021.
Edited by:
Oswin Grollmuss, Université Paris-Sud, FranceReviewed by:
Daniela Anne Braun, University Hospital Münster, GermanyXin Tian, Shaanxi Provincial Hospital of Traditional Chinese Medicine, China
Copyright © 2021 Lu, Fan, Zhang, Zhang, Meng, Zhang, Zhao, Yang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xian-Liang Zhou, emhvdXhpYW5saWFuZzAzMjZAaG90bWFpbC5jb20=; Peng Fan, ZmFucGVuZzEyNnB1bWNAMTI2LmNvbQ==
†These authors have contributed equally to this work