 Cheng Cheng
Cheng Cheng Lizhi Chen†
Lizhi Chen†- Department of Pediatrics, The First Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
The WT1 variant is confirmed to be pathogenic for Denys–Drash syndrome (DDS), a rare disorder characterized by early-onset nephrotic syndrome and renal failure, pseudo-hermaphroditism, and a high risk of Wilms' tumor. Several cases of DDS presenting with atypical hemolytic uremic syndrome (aHUS) have been reported. Here we report the case of a 2-year-old child who was diagnosed with WT1 missense variant, associated with DDS and initial presentation of aHUS. Complement factor H autoantibodies were negative. Complement regulatory system-related gene variants were not found, but a de novo heterozygous c.754G>A missense variant in exon 9 of WT1 gene was detected, resulting in a p. Asp252Asn substitution, by next-generation sequencing. The patient was a female morphologically but proved to be a genetic male because of karyotype 46, XY with normally developed female external genitalia. Bilateral nephrectomy and renal transplantation were performed 1 year later, and there was no recurrence of aHUS at 10 months after transplantation.
Introduction
Denys–Drash syndrome (DDS) is a rare genetic disorder characterized by early-onset nephrotic syndrome that rapidly progresses to renal failure during the first few years of life, pseudo-hermaphroditism, and a high risk of developing Wilm's tumor (1). Wilms' tumor 1 (WT1) gene variants have been shown to be pathogenic for DDS. The WT1 gene encodes proteins that regulate progenitor cells and their differentiation, especially those of the gonads, uteri, and kidneys (2, 3). The initial manifestations of DDS can be vague and indistinguishable. Here we report the case of a 2-year-old girl who initially presented with atypical hemolytic uremic syndrome (aHUS) and was eventually diagnosed with DDS with WT1 gene heterozygous variant. HUS is a triad of microangiopathic hemolytic anemia, thrombocytopenia, and AKI. aHUS is known to be caused by genetic alterations in the complement alternative pathway or formation of complement factor H (CFH) autoantibodies, leading to complement system dysregulation (4, 5).
Case Description
A 2-year-old girl had diffusely distributed rashes followed by abdominal pain, non-bloody loose stools (once a day, which resolved in 2 days), anasarca, and anuria for a month. The patient was admitted to a local hospital on October 18, 2018 with anemia (hemoglobin, 87 g/L), thrombocytopenia (platelet, PLT 82 × 109/L), stage 3 acute kidney injury (AKI) according to the guidelines of Kidney Disease: Improving Global Outcomes (KDIGO) (serum creatinine 961 μmol/L; creatinine clearance, 4.6 ml/min/1.73m2; and blood urea nitrogen 77.2 mmol/L) (6). Initial urinalysis showed urine protein 3+; 24-h proteinuria was 0.16 g (24-h urine amount: 40 ml), and other blood biochemistry analyses demonstrated hypoalbuminemia (albumin, ALB 30.5 g/L) and hyperlipidemia (total cholesterol, TC 6.42 mmol/L). There was no abdominal pain, loose stools, or vomiting. Continuous renal replacement therapy (CRRT) was initiated the first time. The child was transferred to our hospital 7 days later. Physical examination revealed hypertension (blood pressure was initially normal), pallor, generalized edema, and hepatomegaly. Routine blood tests showed moderate anemia with increased reticulocyte percentage (5.39%), while PLT returned to baseline levels. Urinalysis showed 1+ hematuria and 3+ proteinuria but improved hypoalbuminemia (ALB 31 g/L) and hyperlipidemia (TC, 5.8 mmol/L). Other blood chemistry analyses were consistent with KDIGO stage 3 AKI, and lactate dehydrogenase was elevated (386 U/L). Helmet erythrocytes were observed in peripheral blood smears. Coombs test and paroxysmal nocturnal hemoglobinuria screening tests were negative. Platelet activation tests and complement C3 levels were normal. Stool culture was negative for Escherichia coli. aHUS was, thus, diagnosed. Therefore, the patient underwent alternate-day CRRT and six sequences of therapeutic plasma exchange. Hemolysis still progressed, and renal function did not recover after treatment (Figure 1). Eculizumab was inaccessible since it had just been approved in the Chinese market, and renal biopsy was not performed due to coagulation issues. However, further measures were taken to determine the cause of aHUS. CFH autoantibodies were negative. Next-generation sequencing assay for the nephrology panel (MyGenostics) was performed using peripheral blood samples from the patient and her parents. Genes that are known to be associated with aHUS, including complement regulatory protein-coding genes and other related genes (C3, C4, C5, CFB, CFH, CFI CFHR1, CFHR3, CFHR4, CFHR5, THBD, PLG, and DGKE), were included. However, no variants were detected. Unexpectedly, a de novo heterozygous c.754G>A missense variant in exon 9 of the WT1 gene (NM_001198551.1: c.754G>A) was detected, resulting in a p. Asp252Asn substitution (NP_001185480.1: p. Asp252Asn), and this was further verified by Sanger sequencing (Figure 2). Variant analysis according to the American College of Medical Genetics and Genomics standards and guidelines revealed PS1 (sequence variation had previously been reported to be pathogenic for DDS in the Human Gene Mutation Database) + PS2 (de novo and no family history) + PM2 (sequence variation absent from controls), which was regarded as pathogenic for DDS (RefSNP number: rs28941778). The karyotype of this patient was suspected to be 46, XY. As described earlier, DDS is a condition characterized by the triad of nephropathy, genitalia abnormality, and a high risk of Wilm's tumor (1). The patient was a female but later proved to be a genetic male owing to the 46, XY karyotype; however, normally developed female external genitalia was observed. Abdominal computed tomography did not reveal cryptorchidism or abnormal renal mass. There was no improvement in renal function following therapy; hence, the patient received regular peritoneal dialysis and underwent renal transplantation 1 year later; however, prior to that, bilateral nephrectomy was performed for the prevention of Wilm's tumor. Gross specimen examination demonstrated many dysplastic glomeruli and partial sclerotic glomeruli, protein casts, and calcium depositions in the renal tubes, with vacuolar degeneration in the epithelium of the proximal tubes, thickened arteriolar walls and narrowed lumens, and interstitial inflammation infiltration. There was no recurrence of aHUS 10 months after transplantation.
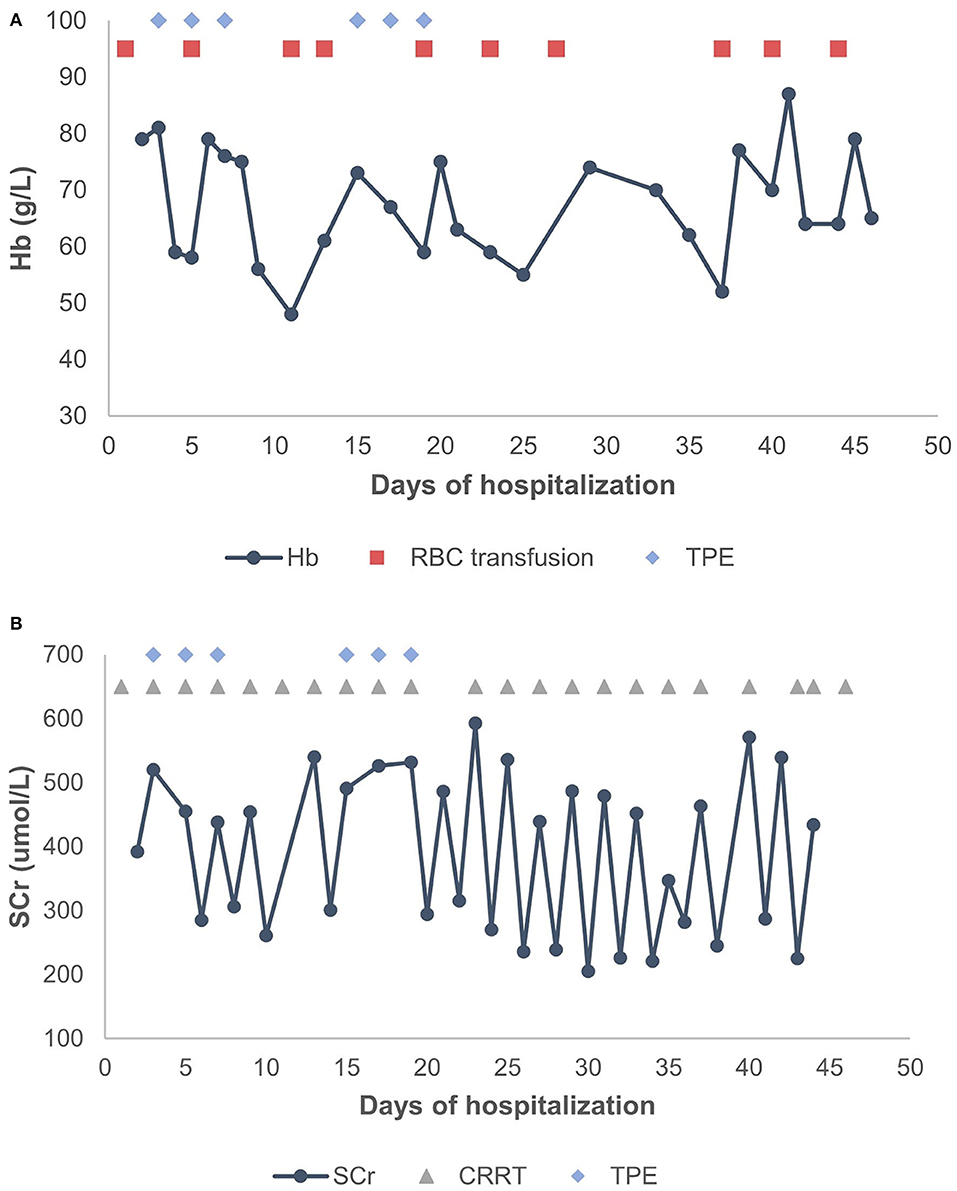
Figure 1. (A) Changes in Hb levels after treatment. (B) Changes in SCr levels after treatment. TPE, therapeutic plasma exchange; CRRT, continuous renal replacement therapy; RBC, red blood cell; Hb, hemoglobin; SCr, serum creatinine.
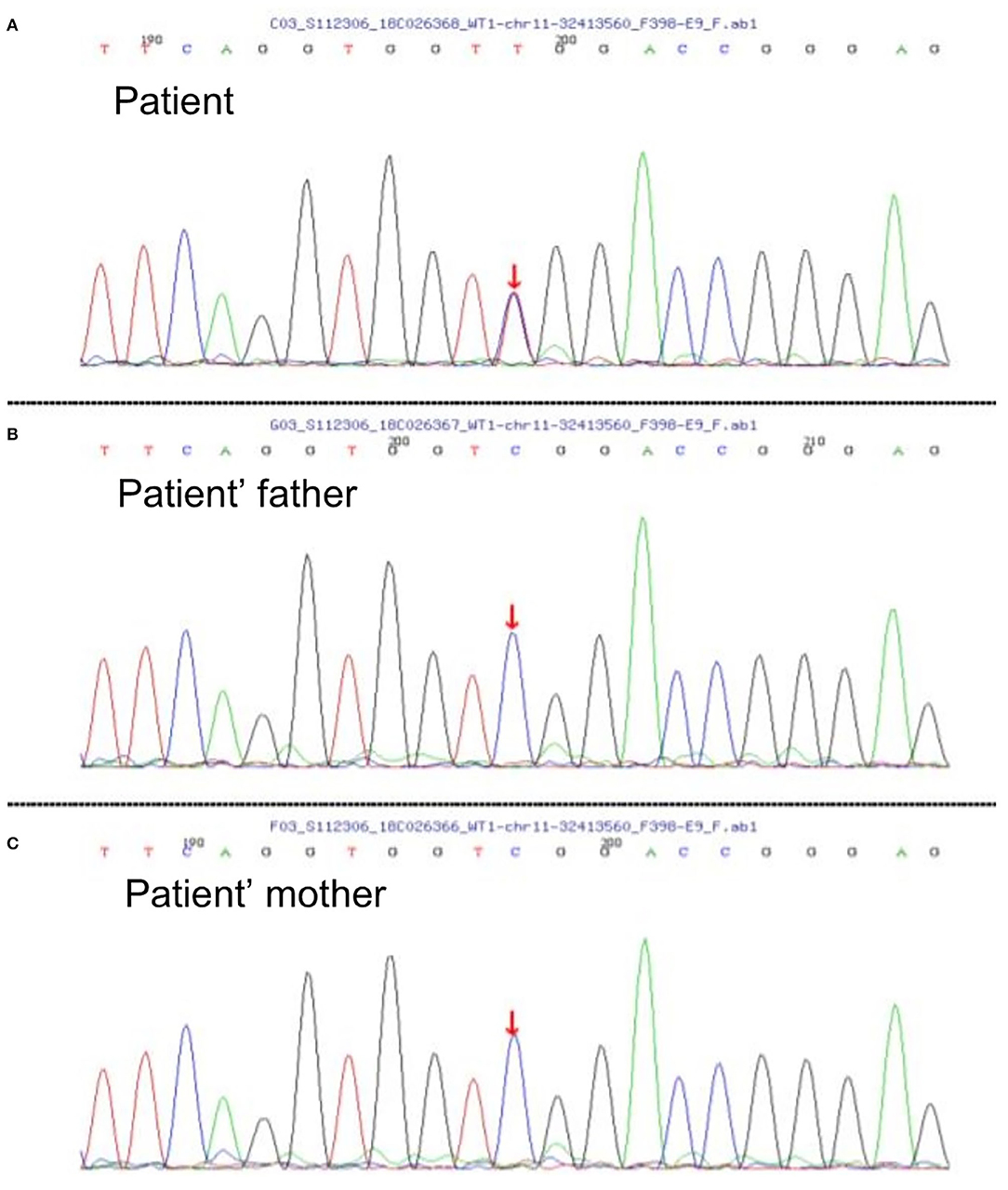
Figure 2. (A) Diagram of the patient's de novo heterozygous c.754G>A missense variant in exon 9 of WT1 gene; nucleotide 754 in the coding region was altered from guanine to adenine, resulting in a change of amino acid 252 from aspartic to asparagine. This missense variant has been reported to be pathogenic to Denys–Drash syndrome (DDS) according to the Human Gene Mutation Database, and it was evaluated to be pathogenic to DDS according to the American College of Medical Genetics and Genomics standards and guidelines. (B,C) Diagrams of WT1 genes of the patient's parents showing no variation. Since reverse reads were used by Sanger sequencing, the bases shown in the peak diagram are the reverse complementary sequence C>T of the altered bases.
Discussion
DDS is a rare genetic disorder characterized by early-onset nephrotic syndrome that rapidly progresses to renal failure during the first few years of life, pseudo-hermaphroditism, and a high risk of developing Wilm's tumor (1). It has been proven that constitutional variants in the zinc finger (ZF) motif of the WT1 gene are associated with DDS (2). The WT1 gene is located at chromosome 11p13 and contains 10 coding exons. It encodes a transcription factor that contains four Cys2-His2 ZF DNA-binding domains at the C-terminus and a proline/glutamine-rich regulatory domain at the N-terminus (7). There are two alternative splicing resulting in four distinct WT1 isoforms: 17 amino acids encoded by exon 5 are inserted into the proline-terminus, producing WT1 +17AA/-17AA isoforms, and three amino acids lysine-threonine-serine (KTS) encoded by exon 9 are inserted between ZF3 and ZF4, producing WT1 +KTS/-KTS isoforms (8). The WT1 gene is essential for the development of both kidneys and the genital system, and it is expressed in renal podocytes throughout life (9). Most of the WT1 variations in DDS are heterozygous germ-line variations that occur in exon 8 or 9, encoding protein ZF2 or ZF3, with a frequent variant of p. Arg394Trp substitution, leading to either DNA binding capacity alteration or isoform imbalance (2, 10).
The case of DDS that we report here had a de novo heterozygous c.754G>A; p. Asp252Asn missense variant in exon 9 of WT1. Unexpectedly, the patient initially presented with symptoms of aHUS until the diagnosis of DDS was made based on early-onset nephropathy, male pseudo-hermaphroditism, and WT1 sequence variation. Enterocolitis caused by Shiga toxin-producing Escherichia coli accounts for most of the HUS cases in children (3). In contrast, the atypical form of HUS is often caused by genetic alterations in the complement alternative pathway or by CFH autoantibody formation, leading to complement system overactivation (4). Non-Shiga toxin-induced diarrhea prior to aHUS has been reported in some pediatric cases (11, 12). Our case did not have any underlying diseases associated with HUS. However, she neither harbored a variant related to the complement regulatory system nor synthesized CFH antibody.
This is the first case of DDS presenting with HUS reported in China; four similar cases have been reported previously in other countries (Table 1). Manivel et al. first noted a patient with a 46, XY karyotype who was born with ambiguous genitalia and had developed HUS at 26 months of age (13). Renal biopsy was consistent with the diagnosis of HUS, while bilateral nephrectomy demonstrated diffuse mesangial sclerosis (DMS) at 32 months of age. A few decades later, Sherbotie et al. described two patients with DDS who had also developed features of aHUS (14). The first case was a 13-month-old boy who developed aHUS 1 month after nephrotic syndrome and urinary tract infection with Citrobacter and Enterococcus. The other patient was a 16-month-old boy who manifested aHUS and DDS at the same time. Normal serum C3 levels were revealed, and WT1 exon 9 missense variants were identified in both 46, XY males with undescended testes and grade III hypospadias. Renal failure was irreversible, and transplantations were eventually carried out in both cases. The renal pathologies revealed diffuse sclerosis. Alge et al. recently reported an 8-month-old female infant who had aHUS as the initial manifestation of DDS (15). Whole exome sequencing identified a pathogenic de novo heterozygous variant in the WT1 gene with predisposing variants in the CFH gene and an unknown variant in the DGKE gene. Acquired causes of aHUS were ruled out. Karyotyping confirmed that the patient was 46, XX with normal external genitalia, but without ovaries. In contrast to the previously reported cases, our patient was a 46, XY child with female physical appearance and external genitalia. In addition, our case had normal C3 levels, as in the case reported by Sherbotie et al. which is not uncommon in aHUS (11). WT1 sequence variants associated with DDS were detected in our patient as well as in the cases reported by Sherbotie et al. and Alge et al. Causative variants related to aHUS and autoantibodies against CFH were screened in our case and the case of Alge et al. but no direct pathogenic result was obtained.
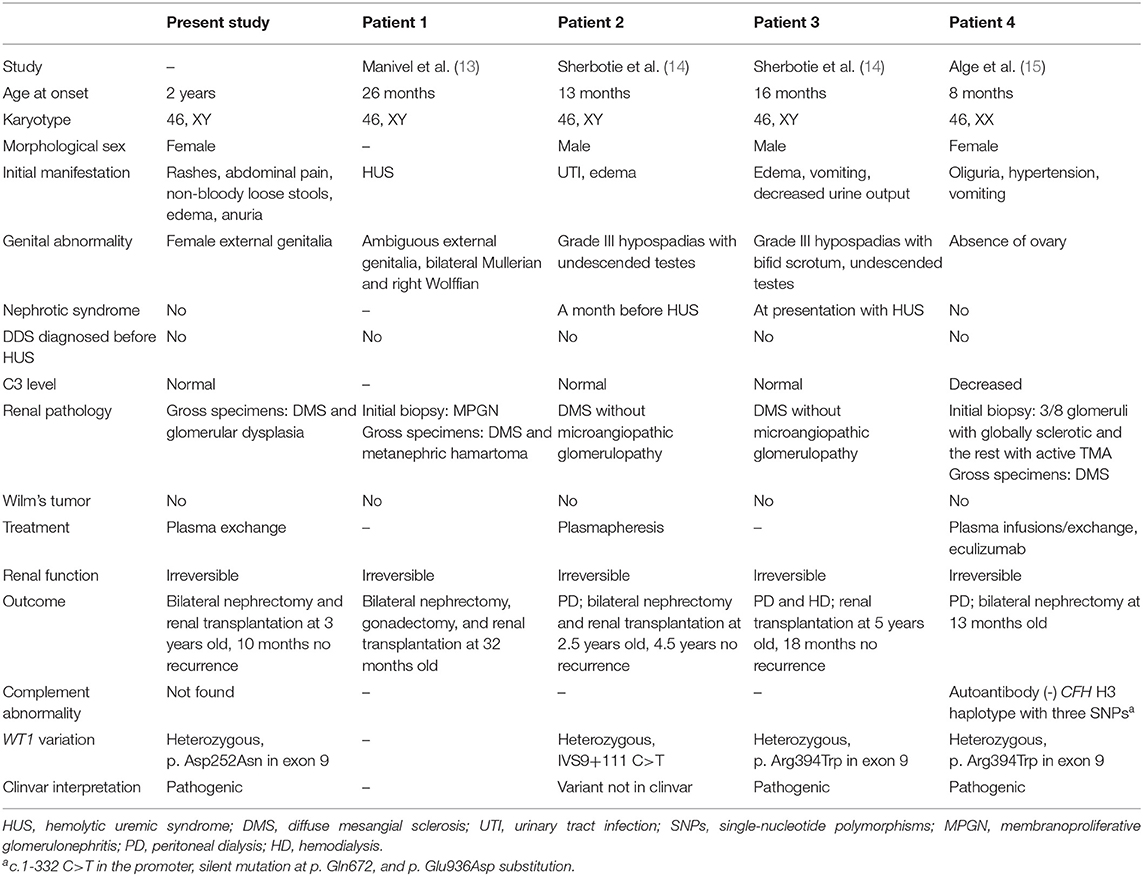
Table 1. Clinical finding in cases of WT1 variants associated with Denys–Drash syndrome (DDS) presenting as HUS.
Among the existing cases, two underwent renal biopsies at the time of HUS diagnosis, and thrombotic microangiopathy (TMA) was biopsy-proven. At 5 to 6 months later, DMS was demonstrated by bilateral nephrectomy when transplantation was performed, suggesting that HUS occurred prior to DDS. Additionally, DMS was also evident in two other cases a few years after disease onset. Unfortunately, our case was diagnosed based on clinical manifestations and genetic sequencing since the patient was unable to tolerate renal biopsy at the beginning. Gross pathology after nephrectomy revealed glomerular dysplasia and sclerosis.
WT1-encoded proteins are crucial in regulating cell growth and maintaining the normal function of renal podocytes, the cells that comprise the outer layer of the glomerular filtration barrier. Sequence variants that influence ZFs and disturb the splicing of +KTS/-KTS isoforms result in dedifferentiation, abnormal proliferation, or morphological alteration in mutant podocytes (16, 17). Although the mechanisms are still not well-understood, studies have proposed that reduced WT1 expression, platelet-derived growth factor-α (PDGF-α), and transforming growth factor-β1 (TGF-β1) overexpression and upregulated Pax-2 expression play roles in this regard (17). Consequently, abnormal podocytes disrupt the integrity of the glomerular filtration barrier, leading to remarkable proteinuria in DDS, while nephrotic-range proteinuria promotes the release of various procoagulants, creating an environment with a high risk of TMA (18). A recent study also found that podocyte injuries are commonly seen in renal TMA (19). Vascular endothelial growth factor (VEGF) secreted by podocytes is important for the survival of endothelial cells, podocytes, and mesangial cells. Its concentration is maintained by the appropriate size selectivity of the podocyte slit diaphragm; however, this selectivity may be affected by severe proteinuria (20). A study in adult mice has shown that a reduction in the podocyte-derived VEGF level is sufficient to induce profound TMA (21), while children with different nephropathies accompanied by nephrotic-range proteinuria have been reported to have aHUS as a complication (22). In addition, podocyte-derived VEGF participates in complement activity regulation, whereas dysregulation of complement pathways predisposes to TMA (23). Thus, emerging findings suggest that decreased VEGF concentration due to podocyte dysfunction contributes to the development of renal TMA (21, 22). Among the five above-mentioned cases of DDS with aHUS, four harbored causative variants in exon 9 of the WT1 gene. The two cases described by Sherbotie et al. were diagnosed with nephrotic syndrome, while our patient and the case reported by Alge et al. had urine protein 3+ and 4+ by dipstick analysis, respectively. We thus hypothesize that podocytopathy in WT1 variants may be one of the pathogenic factors contributing to the occurrence of aHUS.
The limitations of this case report include the following: [1] renal pathology was not obtained at diagnosis, but it is of importance in understanding disease progression and distinguishing the initiating etiology and [2] multiplex ligation-dependent probe amplification analysis of CFHR1/CFHR3 deletion and CFH/CFHR1 hybrid gene, two important predisposing variants of aHUS that could not be detected by standard sequencing (24, 25), was not performed. The etiology and pathogenesis of DDS with WT1 variation presenting as aHUS is still unknown. The case reported here adds value to existing cases as it provides evidence that the association between DDS and aHUS is probably not coincidental. Genetic sequencing is useful in determining the underlying disease or comorbidity when both genetic and acquired causes of aHUS are ruled out. Whether DDS is a predisposing factor for aHUS or whether these conditions comprise another clinical syndrome requires further study.
Patient Perspective
The following patient perspective was provided by the patient's parents.
“It was very sudden that our child became ill and deteriorated unexpectedly quickly. It had been nearly a month since the disease started, but we still decided to take a chance for therapeutic plasma exchange, although renal function failed to recover. Later, the WES and karyotyping results were astonishing. We transitioned from hemodialysis to peritoneal dialysis for long-term renal replacement while we were preparing and waiting for transplantation. Renal transplantation was successful, and our child is currently receiving regular anti-rejection treatments. We hope that our case will add new findings and contribute to the learning of these rare diseases.”
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author/s.
Ethics Statement
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Patient Consent
Informed consent was obtained from the patient's parents for the publication of this case report.
Author Contributions
XJ conceptualized, designed the study, reviewed, and revised the manuscript. LC, CC, SW, and ZL collected, analyzed, interpreted clinical, imaging, genetic data, and were responsible for manuscript writing. All the authors contributed to the manuscript and approved the final version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge that this case report first appeared on the 18th Congress of the International Pediatric Nephrology Association as a poster presentation. We would like to thank the patient and her family, for allowing this case to be published for the development of the field of clinical medicine, and all the physicians involved in the case. We would also like to thank Editage (www.editage.com) for English language editing.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2020.605889/full#supplementary-material
References
1. Denys P, Malvaux P, Van Den Berghe H, Tanghe W, Proesmans W. Association d'un syndrome anatomo-pathologique de pseudohermaphrodisme masculin, d'une tumeur de Wilms, d'une nephropathie parenchymateuse et d'un mosaicisme XX/XY [Association of an anatomo-pathological syndrome of male pseudohermaphroditism, Wilms' tumor, parenchymatous nephropathy and XX/XY mosaicism]. Arch Fr Pediatr. (1967) 24:729–39.
2. Pelletier J, Bruening W, Kashtan CE, Mauer SM, Manivel JC, Striegel JE, et al. Germline mutations in the Wilms' tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell. (1991) 67:437–47. doi: 10.1016/0092-8674(91)90194-4
3. Lipska BS, Ranchin B, Iatropoulos P, Gellermann J, Melk A, Ozaltin F, et al. Genotype-phenotype associations in WT1 glomerulopathy. Kidney Int. (2014) 85:1169–78. doi: 10.1038/ki.2013.519
4. Cody EM, Dixon BP. Hemolytic uremic syndrome. Pediatr Clin North Am. (2019) 66:235–46. doi: 10.1016/j.pcl.2018.09.011
5. Dixon BP, Gruppo RA. Atypical hemolytic uremic syndrome. Pediatr Clin North Am. (2018) 65:509–25. doi: 10.1016/j.pcl.2018.02.003
6. Barry R, James MT. Guidelines for classification of acute kidney diseases and disorders. Nephron. (2015) 131:221–6. doi: 10.1159/000441425
7. Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell. (1990) 60:509–20. doi: 10.1016/0092-8674(90)90601-A
8. Haber DA, Sohn RL, Buckler AJ, Pelletier J, Call KM, Housman DE. Alternative splicing and genomic structure of the Wilms tumor gene WT1. Proc Natl Acad Sci USA. (1991) 88:9618–22. doi: 10.1073/pnas.88.21.9618
9. Pritchard-Jones K, Fleming S, Davidson D, Bickmore W, Porteous D, Gosden C, et al. The candidate Wilms' tumour gene is involved in genitourinary development. Nature. (1990) 346:194–7. doi: 10.1038/346194a0
10. Little M, Holmes G, Bickmore W, van Heyningen V, Hastie N, Wainwright B. DNA binding capacity of the WT1 protein is abolished by Denys-Drash syndrome WT1 point mutations. Hum Mol Genet. (1995) 4:351–8. doi: 10.1093/hmg/4.3.351
11. Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. (2010) 5:1844–59. doi: 10.2215/CJN.02210310
12. Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, Macher MA, Niaudet P, Guest G, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. (2007) 18:2392–400. doi: 10.1681/ASN.2006080811
13. Manivel JC, Sibley RK, Dehner LP. Complete and incomplete Drash syndrome: a clinicopathologic study of five cases of a dysontogenetic-neoplastic complex. Hum Pathol. (1987) 18:80–9. doi: 10.1016/S0046-8177(87)80199-5
14. Sherbotie JR, van Heyningen V, Axton R, Williamson K, Finn LS, Kaplan BS. Hemolytic uremic syndrome associated with Denys-Drash syndrome. Pediatr Nephrol. (2000) 14:1092–7. doi: 10.1007/s004670000389
15. Alge JL, Wenderfer SE, Hicks J, Bekheirnia MR, Schady DA, Kain JS, et al. Hemolytic uremic syndrome as the presenting manifestation of WT1 mutation and Denys-Drash syndrome: a case report. BMC Nephrol. (2017) 18:243. doi: 10.1186/s12882-017-0643-1
16. Yang AH, Chen JY, Chen BF. The dysregulated glomerular cell growth in Denys-Drash syndrome. Virchows Arch. (2004) 445:305–14. doi: 10.1007/s00428-004-1069-2
17. Morrison AA, Viney RL, Saleem MA, Ladomery MR. New insights into the function of the Wilms tumor suppressor gene WT1 in podocytes. Am J Physiol Renal Physiol. (2008) 295:F12–7. doi: 10.1152/ajprenal.00597.2007
18. Chen G, Liu H, Liu F. A glimpse of the glomerular milieu: from endothelial cell to thrombotic disease in nephrotic syndrome. Microvasc Res. (2013) 89:1–6. doi: 10.1016/j.mvr.2013.06.011
19. Hu YF, Tan Y, Yu XJ, Wang H, Wang SX, Yu F, et al. Podocyte Involvement in Renal Thrombotic Microangiopathy: A Clinicopathological Study. Am J Nephrol. (2020) 51:752–60. doi: 10.1159/000510141
20. Katavetin P, Katavetin P. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. (2008) 359:205–7. doi: 10.1056/NEJMc080770
21. Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, et al. VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med. (2008) 358:1129–36. doi: 10.1056/NEJMoa0707330
22. Noris M, Mele C, Remuzzi G. Podocyte dysfunction in atypical haemolytic uraemic syndrome. Nat Rev Nephrol. (2015) 11:245–52. doi: 10.1038/nrneph.2014.250
23. Keir LS, Firth R, Aponik L, Feitelberg D, Sakimoto S, Aguilar E, et al. VEGF regulates local inhibitory complement proteins in the eye and kidney. J Clin Invest. (2017) 127:199–214. doi: 10.1172/JCI86418
24. Valoti E, Alberti M, Tortajada A, Garcia-Fernandez J, Gastoldi S, Besso L, et al. A novel atypical hemolytic uremic syndrome-associated hybrid CFHR1/CFH gene encoding a fusion protein that antagonizes factor H-dependent complement regulation. J Am Soc Nephrol. (2015) 26:209–19. doi: 10.1681/ASN.2013121339
Keywords: denys-drash syndrome (DDS), pseudohermaphroditism, atypical hemolytic uremic syndrome (aHUS), case report [publication type], wilms tumor 1 gene (WT1)
Citation: Cheng C, Chen L, Wen S, Lin Z and Jiang X (2020) Case Report: Denys–Drash Syndrome With WT1 Causative Variant Presenting as Atypical Hemolytic Uremic Syndrome. Front. Pediatr. 8:605889. doi: 10.3389/fped.2020.605889
Received: 13 September 2020; Accepted: 03 November 2020;
Published: 18 December 2020.
Edited by:
Jakub Zieg, University Hospital in Motol, CzechiaReviewed by:
Thaitana Evilen da Silva, PreScouter, United StatesMartin Bezdíčka, Charles University, Czechia
Scott E. Wenderfer, Baylor College of Medicine, United States
Copyright © 2020 Cheng, Chen, Wen, Lin and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyun Jiang, anhpYW95QG1haWwuc3lzdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship