 Pei Qian
Pei Qian Ying Bao
Ying Bao Hui-mei Huang
Hui-mei Huang Lei Suo
Lei Suo- Department of Nephrology, Xi’an Children's Hospital, Xi'an, China
Background: X-linked Alport syndrome (XLAS) is caused by pathogenic variants in COL4A5 and is characterized by progressive kidney disease, hearing loss, and ocular abnormalities.The aim of this study was to identify gene mutations in a Chinese family with XLAS, confirm a diagnosis, and provide an accurate genetic counseling.
Methods: The proband was a 5-year-old male with microscopic hematuria and a family history of renal disease in 5 relatives.His relatives had microhematuria with or without proteinuria. His maternal uncle developed renal failure at the age of 35 years. He was evaluated by renal biopsy,whole-exome sequencing (WES) and whole-genome sequencing (WGS) for Alport syndrome. RT-PCR and cDNA Sanger sequencing were performed on RNA extracted from the skin of the proband. Then, a splicing reporter minigene assay was used to examine the effect of the variation on the splicing of the primary transcript in transfected cells.
Results: Pathological examination of the kidney of the proband revealed diffuse thinning of the glomerular basement membrane, and immunofluorescence analysis indicated normal expression of the α5 chain in the basement membrane. No phenotype-associated candidate variant was detected in the proband via WES. A novel deep intronic COL4A5 variant (c.385–716G > A), which is segregated with disease in this family, was identified using WGS. In-vitro minigene assay and in-vivo RT-PCR analysis demonstrated that the variant could produce both normal and abnormal transcripts. The abnormal transcripts showed that the variant activated a cryptic splice site, introducing a 147 bp pseudoexon into the mRNA sequence and consequently generating a premature termination codon (p.G129Afs*38) and leading to frameshifting and truncation of the α5 (collagen IV) protein.
Conclusion: This is the first report of the novel c.385–716G > A splicing mutation in the COL4A5 gene, which illustrates the importance of performing WGS to find additional mutations in WES-negative patients with highly suspected forms of genetic diseases. The same results obtained from the in-vitro and in-vivo splicing experiments confirm the consistency between the minigene assay and RT-PCR analysis. In addition, this study highlights the importance of functional analysis in diagnosis and genetic counseling in AS.
Introduction
Alport syndrome (AS) is a rare genetic disease of the glomerular basement membrane (GBM) caused by mutations in the COL4A3, COL4A4, and COL4A5 genes, with a prevalence ranging from 1 in 5,000 to 1 in 53,000 individuals in different populations (1). Microhematuria is the most common symptom, and some patients will gradually develop proteinuria; most cases of AS will progress to end-stage renal disease (ESRD) by 30 years of age (2). The pathogenic variant of the COL4A5 gene (NM: 000495.5), which encodes α5 chain (collagen IV), causes X-linked AS that accounts for approximately 85% of the cases of AS (3). Some experts have recently asserted that patients with hematuria, thin basement membrane nephropathy (TBMN), and heterozygous mutations in COL4A3 or COL4A4 should be classified as cases of autosomal Alport syndrome (4). XLAS will not be the most frequent form of Alport syndrome if named in that manner. As in many heritable diseases, there is vast intra- and inter-familial variability and allelic heterogeneity in the phenotypic expression of XLAS, such as in disease severity, onset age, and progression (5). In male XLAS patients, the genotype-phenotype correlation is evident; whereas patients with the nonsense variants show the most severe, early-onset ESRD phenotype, those with a splice variant and missense variant show moderate and mild phenotypes, respectively (6). A previous study reported that heterozygous female patients usually have a milder and more variable clinical course compared with male patients with XLAS (7). However, a retrospective analysis of female Japanese patients with XLAS revealed that phenotypes in these patients may be severe and that 15% of patients reached ESRD by the age of 40 years (8). So, genetic testing is valuable for conformation of clinical diagnosis, providing prognostic advice and genetic counseling.
In recent years, Next-generation sequencing (NGS) has greatly improved the rate of diagnosis in XLAS. Despite these technological advances, at least one-fifth of XLAS patients still have no clearly defined molecular diagnosis (9), and this deficiency hampers a deeper understanding of the genotype–phenotype relationship in patients with XLAS. Standard NGS data analysis focuses on coding exons and splice sites and can miss the mutations in deep introns and promoters. However, Whole-genome sequencing (WGS) not only comprehensively covers exons but also detects deep intronic and intergenic variations, but interpretation of non-coding variants via WGS is challenging. It has been shown that splice variants account for approximately 15% of all cases of XLAS (6). It was found significant differences in renal survival that are based on the abnormal splicing patterns of the COL4A5 primary transcript (6). However, the functional outcomes caused by many splice-region variants have not been investigated in AS patients. Further functional studies on the variations in COL4A5 that may affect splicing are therefore necessary to identify the aberrant splicing patterns of the COL4A5 primary transcript, define the relationship between the genotype and clinical phenotype, and predict the renal prognosis in AS. In this report, we identified a novel deep intronic variant of the COL4A5 gene via WGS that could not have been detected by WES alone. Via a combination of these in-vivo, in-silico, and in-vitro analyses, the variant was found to impair the splicing process. After functional analysis, we concluded that this deep intronic variant in COL4A5 is responsible for this family.
Materials and methods
Clinical data
The study subjects included members of a Chinese family who is a 3-generation pedigree (Figure 1A and Table 1). The proband (III-2) was a 5-year-old boy., who was found to have microhematuria, but no proteinuria. No abnormalities were found in routine blood, serum chemical, or immunological tests. The results of pure-tone audiometry and eye examination were normal. Abdominal ultrasound examination, vascular color ultrasound, and electrocardiogram of the left renal vein showed no obvious abnormalities. His mother (II-2) presented with microhematuria and proteinuria with normal renal function, her quantity of proteinuria was 10∼15 mg/kg/24 h. His sister (III-1), older female cousin (III-3), and grandmother (I-1) all presented with microhematuria with normal renal function. His maternal uncle (II-3) presented with microhematuria and proteinuria and developed stage 5 chronic kidney disease by the age of 35 years, his quantity of proteinuria was 35∼40 mg/kg/24 h. Except for the proband's uncle, all the others had normal kidney function and blood pressure. In addition, the maternal uncle (II-3) and mother (II-2) of the proband had high-frequency sensorineural hearing impairment. Because the proband had long lasting microscopic hematuria and a family history of renal disease in 5 relatives, a kidney biopsy was performed on the proband due to the suspicion of AS.
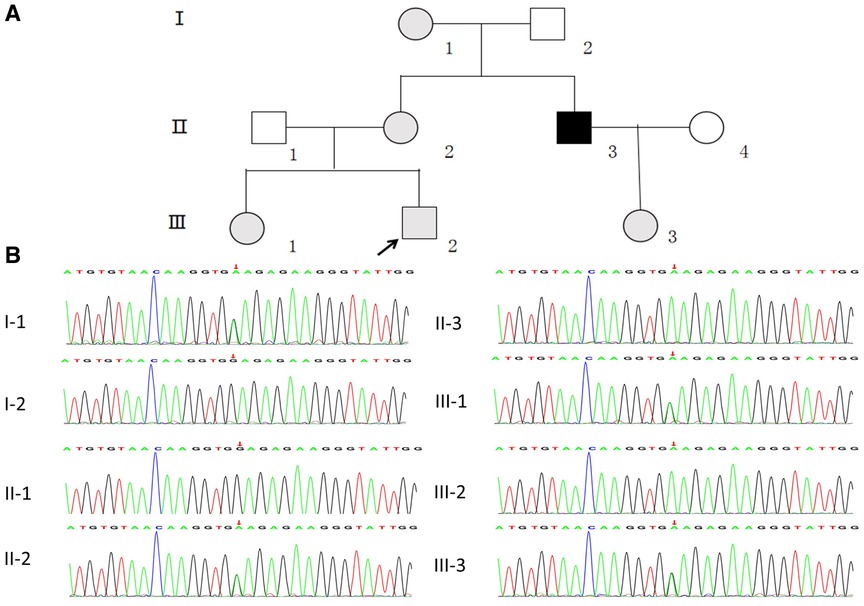
Figure 1. (A) pedigree analysis of the family. The gray, black, and white symbols represent the individuals with hematuria or proteinuria, renal failure, and healthy renal physiology, respectively. The proband is denoted by the black arrow. (B) Sequencing results showing the COL4A5 mutations in the proband and his family members.
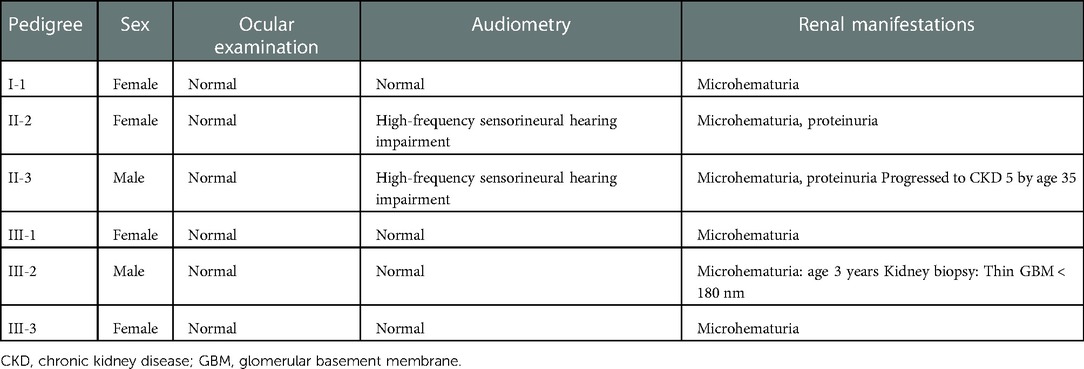
Table 1. Clinical features of affected individuals in the family.
NGS and direct sanger sequencing
We collected 4 ml of peripheral blood samples from the proband and his family members for WES and WGS. Sanger sequencing was used to confirm the candidate variant. Genomic DNA samples were fragmented to approximately 200–300 bp by using ultrasound, and DNA libraries were prepared using the established Illumina paired-end protocol. The xGen Exome Research Panel v1.0 (Integrated DNA Technologies) was used as the whole-exon capture chip in the WES, which was performed in an Illumina NovaSeq 6,000 Series Sequencer (PE150). Whole-genome libraries were prepared according to the standardized on-machine sequencing operation procedures of the DNBSEQ-T7 platform. The raw sequencing data were aligned against the reference human genome (hg19) by using the BWA software. Repeated reads were excluded, and then statistical analysis was performed on the remaining reads. The GATK software was used to analyze SNPs and indels. According to the sequencing and mutation quality, the detected SNPs and indels were filtered and screened for high-quality and reliable mutations. For the detected high-quality variants, self-developed variant-annotation software was used to perform association annotation at major databases, such as OMIM, HGMD, ClinVar, and frequency databases, including dbSNP, 1,000 Genomes, ExAC, and ESP. Via protein structure prediction software, such as Provean, SIFT, Polyphen2-HVAR, Polyphen2-HDIV, M-Cap, Revel, Mutationtster, and MaxEntScan cleavage site prediction software, the hazards were analyzed, and the possible harmful effects on the protein structure were screened out variation. Variant loci were graded for pathogenicity according to the American College of Medical Genetics and Genomics (ACMG) guidelines (10). Copy number variation (CNV) detection was performed using self-developed CNV software. The genes included in the CNV interval were analyzed; related data, such as those from Decipher, ClinVar, OMIM, DGV, and ClinGen, were associated; and the reported disease genes were annotated. According to the annotation information and frequency database, the hazard level of CNV was comprehensively assessed. Sanger sequencing was performed on the samples of all available family members based on the results from NGS.
In silico splicing assay
We used publicly available in silico analysis tools to predict the effects of the c.385–716G > A variant on the splicing of COL4A5 exon 6. These tools were Human Splicing Finder [http://www.umd.be/HSF3/HSF.html], BDGP: Splice Site Prediction by Neural Network [http://www.fruitfly.org/seq_tools/splice.html], and NetGene2 Server [http://www.cbs.dtu.dk/services/NetGene2/].
Hybrid minigene assay
We used a minigene plasmid based on the pMini-CopGFP vector (Beijing Hitrobio Biotechnology Co., Ltd., Beijing, China). Briefly, we cloned DNA fragments into pMini-CopGFP by using GenScript (Piscataway, NJ), and the BamHI and XhoI restriction sites around the target variations in the COL4A5 gene (both wild-type [WT] and mutant-type [MT] with exons and introns) according to the manufacturer's instructions (primers available upon request).The hybrid minigenes were confirmed by sequencing and then transfected into 293 T cells by using Lipofectamine™ 2,000 (Thermo Fisher Scientifc, Waltham, MA, United States). The cells were harvested 24–48 h later, and total RNA was extracted using the RNeasy Plus Mini Kit (QIAGEN, Hilden, Germany). Subsequently, 1.5 µg of the total RNA was used to generate cDNA by using Multiscribe RT Polymerase (AB systems) according to the manufacturer's instructions. pMini-CopGFP-specific primers (available upon request) were used to amplify cDNA to determine the splicing patterns. The PCR products were analyzed using electrophoresis on a 1.5% agarose gel, followed by direct sequencing. The amplicon included COL4A5 exons 6–8 in the case of normal splicing normal.
Reverse transcription (rt)-PCR analysis
We obtained a skin sample from the proband. Total RNA was extracted from the skin sample and a blood sample derived from a healthy control, and then subjected to RT-PCR for COL4A5 primary transcript. Total RNA was isolated using TRIzol Reagent (Invitrogen, Carlsbad, CA). Then, 1 μg of total RNA was reverse-transcribed to generate cDNA by using PrimeScript RT Reagent Kit with gDNA Eraser (TaKaRa Biotechnology (Dalian) Co., Ltd., Dalian, China). Duplex PCR was performed (primers available upon request) to amplify a 985 bp fragment of the COL4A5 cDNA encompassing exons 1–17. Then, the PCR products were directly sequenced.
Results
Histopathology of renal biopsy
Renal histopathological features of proband are displayed in Figure 2. No glomerular immunoglobulin or complement component was observed via immunofluorescence staining. The expression patterns of the positive control (collagen IV α1 chain), collagen IV α3 chain and collagen IV α5 chain were normal. Light microscopy showed slight proliferation of mesangial cells and stroma, but no crescents were observed. The vacuolar degeneration of the capillary endothelial cells was observed via electron microscopy (EM). The parietal layer of the renal follicle was not significantly thickened, and the basement membrane was diffusively thinned to a thickness of < 180 nm. Comprehensive light microscopy, immunofluorescence analyses, and EM showed that the glomerular lesions were mild, and the morphology indicated thin basement-membrane nephropathy.
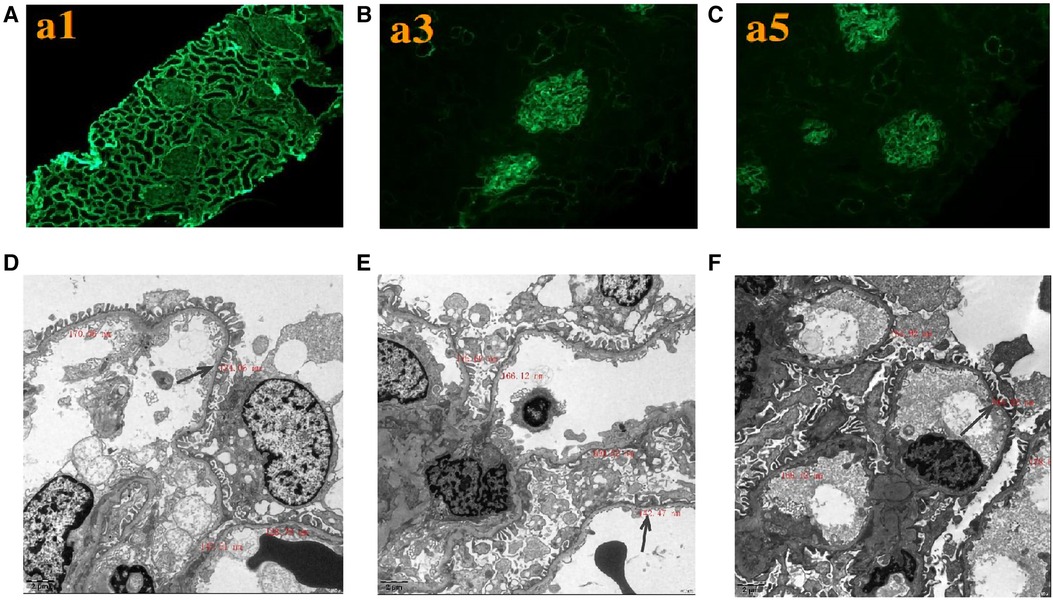
Figure 2. Renal expression patterns of α1, α3, and α5 and renal ultrastructure of the proband. Immunofluorescence staining of the kidney for (A) α1, (B) α3, and (C) α5. × 200. Electron microscopy images of his kidney show that the thickness of the basement membrane ranges around 124.06 nm (D), 142.47 nm (E), and 164.92 nm (F). EM × 50,000.
Mutation analysis
WES was performed first, but no candidate variant related to the phenotype was found. Thus, WGS was performed next since it has higher coverage than WES. The WGS test results revealed a novel hemizygous variant in intron 6 of the proband (chrX: 107813927, Ghg19, NM_000495.5: c.385–716G > A). He was found to inherit this variant from his mother. Pedigree validation via Sanger sequencing revealed that all the affected female patients (I-1, II-2, III-1, III-2, and III-3) and the affected male patient (the maternal uncle of the proband, II-3) in this pedigree were heterozygous for this variant (c.385–716G > A), whereas the asymptomatic family members did not carry the variant (Figure 1B). Additionally, the phenotypes and genotypes of the proband and the 5 other patients in the family were co-isolated. The discovered variant (c.385–716G > A) is a deep intronic variant (> 100 bp from the splice site), absent from the control databases or from the Single Nucleotide Polymorphism Database (dbSNP) and ClinVar database. Additionally, it has not been reported to be associated with any disease.
In silico splicing analysis
No significant effect of this variant on splicing was detected by Human Splicing Finder and NetGene. The BDGP online software suggested that the variant produced a new splice donor site located in c.385–716G > A.
In vitro splicing analysis
As the discovered variant is located in intron 6 of the COL4A5 gene, we suspected that it might affect the splicing of the primary COL4A5 transcript. We amplified the COL4A5 DNA fragments spanning exons 6–8 and then cloned them into the minigene pMini-CopGFP vector (Figure 3A). Afterward, 293 T cells were transfected with the WT and MT splice-region variants for functional assessment (see Materials and Methods). Agarose gel electrophoresis (Figure 3B) showed that, after the intron sequence was spliced, the cells transfected with the WT vector yielded the expected 241-bp amplicon, containing COL4A5 exons 6–8, flanked by the exons of the vector. In contrast, the cells transfected with the MT vector produced two amplicons, one with the normal size of 241 bp, and the other with the size of 388 bp amplicon, with the additional 147 bp fragment added from exon 6 of COL4A5. The variant disrupts the splicing acceptor site of intron 6, preventing the removal of intron 6 from the primary transcript and thus resulting in the extra 147 bp. The change in the splicing results in the truncation of exon 6 from C.385–764 to C.385–618 (Figure 3C), and this new mRNA sequence contains a premature termination codon (p. G129Afs*38, Figure 3D).
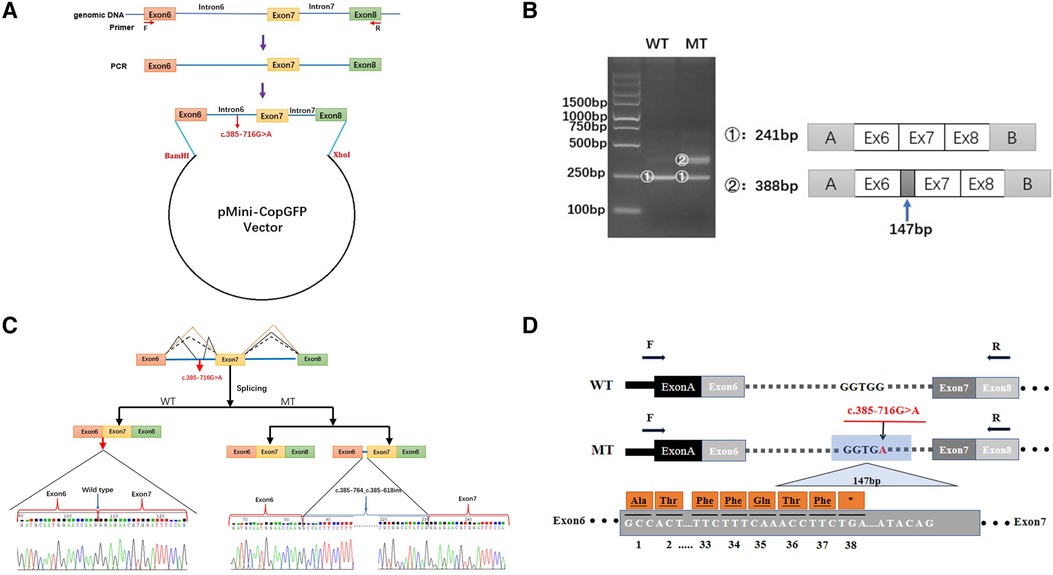
Figure 3. Minigene-assay transcript analysis. (A) COL4A5 exons 6–8, spanning the intron variants, were cloned into the pMini-CopGFP vector. (B) Electrophoresis of the reverse transcription-polymerase chain reaction products of the minigene transcripts. The wild type (WT) exhibited a single, full-length band (241 bp), and the mutant type (MT) exhibited two bands (the WT band and 147 bp insert band). The drawing on the right shows the predicted WT and MT amplicon sizes. (C) Schematic diagram showing the variation and its consequences. The WT situation is indicated by the orange line. The variation produces two transcripts, produced via WT and MT splicing (indicated by black dashes and lines, respectively). The change of c.385-716G > A introduces a new splice site, resulting in a cryptic exon activation between exons 6 and 7 and creating a transcript with a 147 bp insertion. (D) The structure of the WT COL4A5 transcript fragment spanning exons 6–8 and that of the MT version, generated by the 385-716G > A variation. A 147 bp sequence around the variation is introduced to the mRNA to form a pseudo-exon between exons 6 and 7. A TGA termination codon appeared at 114 bp in the 147 bp fragment, resulting in translational termination of the frameshifted polypeptide after 37 amino acids are translated (modified and referenced from Verdura E (23)).
In vivo validation
To confirm the result from the minigene assay, we assessed the proband for the predicted splicing aberration. Gel electrophoresis (Figure 4A) showed the two predicted bands (WT and MT) from the skin sample of the proband, proving that the dermal cells of the proband produced the WT and MT COL4A5 mRNA variants. The doublet was purified from the gel and subjected to Sanger sequencing for confirmation. The results (Figure 4B) confirmed that the variant (c.385–716G > A) produces both the WT and MT transcripts. The MT transcript produces an abnormal mRNA that includes a 147 bp fragment from intron 6, a cryptic exon, and a premature termination codon (p. G129Afs*38). Because there is only a small size difference between the WT and MT transcripts, the electrophoretic bands form a doublet and are difficult to distinguish from each other. The results from our in vitro and in vivo splicing assays are consistent.
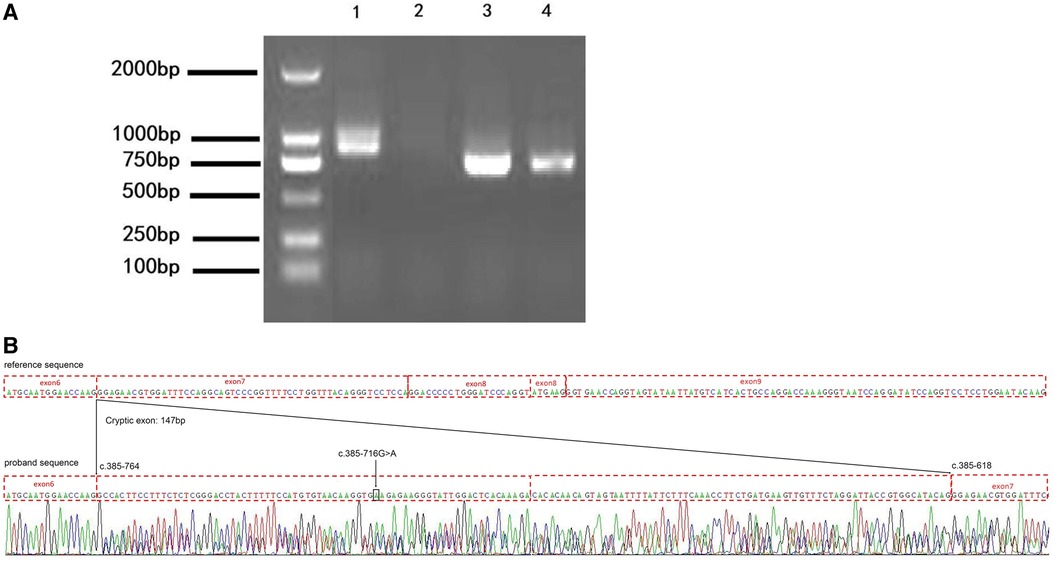
Figure 4. The variation (c.385-716G > A) perturbs the splicing of the COL4A5 transcript. (A) Agarose gel electrophoresis results of the reverse transcription–polymerase chain reaction (RT-PCR) analysis of the patient (1 and 3, skin) and healthy control (2 and 4, blood) for the presence of the target (1 and 2) and reference (3 and 4) mRNAs (985 bp and 843 bp amplicon size, respectively). (B) Results of the Sanger sequencing of the RT-PCR products. c.385-716 G > A, black arrow and box position. The mutation introduces a 147 bp cryptic exon between exons 6 and 7 (c.385-764 and c.385-618 border exons 6 and 7, respectively).
Discussion
In this study, we reported a heterozygous deep intronic variant of the COL4A5 gene (c.385–716G > A) through WGS, and this variant could not have been detected using WES alone. The variant initially was classified as “a variant of uncertain significance” according to the ACMG guidelines (PM2 + PP1-Moderate). However, based on its co-isolation with the family phenotype and our in vitro and in vivo results on its impact on the splicing of the COL4A5 transcript, the mutation was finally classified as “pathogenic” (PM2 + PS3 + PP1-Strong). We concluded that this mutation is responsible for the disease of the proband, who was accordingly diagnosed with AS. All the affected family members carried this variant, and it was not detected in the father or maternal grandfather of the proband, who were unaffected. The maternal uncle of the proband developed ESRD at the age of 35 years, but the 80-year-old grandmother and 40-year-old mother of the proband had normal renal function when this research article was prepared. The male members of the family who carried the variant tended to have more severe phenotypes than the female members with the variant, indicating a typical X-linked inheritance pattern, and the severity of the disease varied among the pedigree individuals.
A growing number of diseases are attributed to abnormal splicing (11). Such variants reported by previous studies on splicing mutations are mostly located at evolutionarily conserved sites within exon–intron boundaries. However, few reports have focused on the effect of intron variation. In this study, we found that a deep intron variation in COL4A5 can also lead to abnormal splicing. Some deep intronic variants of COL4A5 were found to exhibit pathogenicity by generating cryptic exons (12). These findings highlight the importance of identifying splice variants of COL4A5, rare functional intronic variants in particular. However, a consensus approach for detecting deep intronic variants has not been established (13). Although intron variations can be detected via WGS, it is difficult to distinguish intron variations that cause splicing errors from harmless polymorphisms. Currently, the best approach to identify pathogenic splicing mutations is to use a combination of these in vivo, in silico, and in vitro analyses. Although various in silico approaches have been developed to assess the effects of sequence variations, the most reliable approach to evaluating splicing pathogenicity is through in vivo analyses (14), which are based on analysis of the relevant transcripts directly in infected tissues. However, cDNA analysis of COL4A5 using renal mRNA is not a common approach. It is often difficult to amplify COL4A5 transcripts extracted from peripheral leukocytes, and analysis of transcripts via targeting genomic DNA variants is also unsatisfactory. Currently, when in vivo splicing studies are not available, the most effective method for functional analysis of intron variations is based on the hybrid minigene assay (13, 15). In this study, our in silico assessment predicted that the variation might affect the splicing, but the results from different algorithms were inconsistent. Subsequently, our in-vitro hybrid minigene assay and RT-PCR analysis of the skin cells from the patient confirmed that the variation perturbs the splicing; the variant vector could simultaneously express the WT and MT transcripts. The MT transcript introduced a 147 bp fragment from intron 6, a cryptic exon, and a premature termination codon (p. G129Afs * 38). This truncating mutation may result in a variable deposition of COL4A5-expressed proteins in the tissue. The degree of consistency between the minigene analysis and patient RNA analysis has not been fully established to date. Via transcript analysis of the patient skin, this study confirmed that the discovered variant perturbed the splicing, and both this in-vivo transcript analysis and in-vitro minigene assay yielded the same results.
The particular form of competitive splicing pattern that we detected in this study has been reported in other studies, with previous findings confirming the presence of both wild-type and abnormal transcripts leading to the truncated COL4A5 variant in the kidney of a male patient with a splicing variant (16). In this study, none of the affected patients had abnormal eye or hearing abnormalities, except for the mother and maternal uncle of the proband. This observation may reflect the multiorgan-specific splicing pattern of COL4A5 (16). In addition, the amount of the WT COL4A5 transcript expressed may also vary among different cell types. The inner ear cells of some patients may not be able to produce COL4A5 transcripts normally or may not splice the primary transcript correctly, resulting in the hearing loss observed in some members of this family. The clinical phenotypes of the female patients in this study were generally mild, these patients showed both normally and abnormally spliced transcripts, the frequency of normal COL4A5 transcripts in the affected women may partly explain the differences in disease severity and phenotypic differences among the members of this family. In XLAS, men show more severe phenotypes than women due to the X-linked inheritance pattern. Men with XLAS can present with proteinuria and hematuria in early childhood and develop ESRD at a median age of 25–35 years (17). Regarding splice site mutations, Jais et al. have reported a 70% probability of ESRD development by the age of 30 (18). The mean onset age of ESRD in patients with splice-site variants is 28 (11). The median ages of ESRD in patients with a truncating splicing mutation and in those with a non-truncating splicing mutation have been reported to be 20 and 29 years old, respectively, indicating that non-truncating splicing variants cause more modest phenotypes than truncating ones (19). The fact that the renal outcomes of male XLAS patients with abnormal splicing differ depending on whether the mutations cause truncated or non-truncated proteins also suggests that for an accurate prognostic prediction, it is important to define transcript patterns when detecting splice-site mutations or abnormal splicing mutations. The mutation found in this study causes truncation of α5 (collagen IV). A previous study reported that the incomplete mutation of the splice site may produce both normal and abnormal kidney mRNA, leading to milder phenotypes (20). In this study, the maternal uncle of the proband developed renal failure by the age of 35 years, who exhibited a milder phenotype presumably because of producing a mixture of both WT and MT mRNAs. The normal transcript may have prevented the progression to the typically severe phenotype of XLAS. It has previously been reported that men with XLAS with positive a5 (IV) staining in the kidney often carry non-truncating mutations (mainly missense mutations) and show a milder clinical phenotype (21). Studies on patients with splicing site variation at the genomic level have found that positive α5 staining (collagen IV) in patient tissues indicates non-truncating transcriptional variation (19). In the present study, the proband showed positive a5 (IV collagen) staining in the kidney, but the transcript analysis revealed a truncating mutation, different from the non-truncating variation reported previously, which also calls into question the belief that renal immunohistochemical analysis for a5 (IV) predicts the genotype.
Although there is no specific treatment for AS, its management is aimed at delaying progression to renal failure using nephroprotective drugs (e.g., renin-angiotensin inhibitors). In this study, none of them received any treatment except the proband's uncle, who received peritoneal dialysis and hypertension treatment before diagnosis. It has been found that splicing regulation can induce a non-truncated transcription and produce a protein that partially compensates for the loss of the full-length protein, thereby improving the symptoms of the corresponding disease (22). Therapeutic approaches using splicing modulation that can alter truncating transcription to non-truncating transcription have been found effective in the treatment of XLAS (19). Understanding the differences between truncating non-sense mutations and non-truncating mutations is essential to assess renal prognosis, perform genetic counseling, and develop treatment strategies. Our study provides a foundation for the future gene therapy of this pathogenic variant.
Conclusion
A novel pathogenic COL4A5 deep intronic variant was identified in a Chinese family with AS through WGS, and its splicing effect was verified using the minigene assay and in vivo analysis. This study illustrates the importance of performing WGS to find additional mutations and confirm the consistency between the minigene assay and RT-PCR analysis. Differences in the proportions of the normal and abnormal transcripts in affected individuals carrying splice region variants may be responsible for the broad expressivity within this family, which provides insights into genotype-phenotype correlations.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: [INSERT REPOSITORY AND ACCESSION] Clinvar database (ClinVar Wizard Submission: SUB11982751).
Ethics statement
The studies involving human participants were reviewed and approved by All the procedures were reviewed and approved by the Clinical Research Ethics Committee of the Ethics Committee of Xi 'an Children's Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
PQ reviewed the literature and contributed to manuscript writing. YB was responsible for revision of the manuscript and funding acquisition. H-mH and LS curated the data and were involved in the methodology and validation. YH, Z-jL, and MZ contributed to the acquisition and analysis of clinical data. All authors contributed to the article and approved the submitted version.
Acknowledgments
We are grateful to the patient and his family, as well as the help extended by the physicians. We wish to thank the staffs of Chigene® Translational Medicine Research Center Co., Ltd. for providing assistance with NGS and the analysis of genetic diseases.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hertz JM, Thomassen M, Storey H, Flinter F. Clinical utility gene card for: alport syndrome - update 2014. Eur J Hum Genet. (2015) 23:e1–e4. doi: 10.1038/ejhg.2014.254
2. Kashtan CE, Gross O. Clinical practice recommendations for the diagnosis and management of alport syndrome in children, adolescents, and young adults-an update for 2020. Pediatr Nephrol. (2021) 36:711–9. doi: 10.1007/s00467-020-04819-6
3. Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter F. Expert guidelines for the management of alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol. (2013) 24:364–75. doi: 10.1681/ASN.2012020148
4. Kashtan CE, Ding J, Garosi G, Heidet L, Massella L, Nakanishi K, et al. Alport syndrome: a unified classification of genetic disorders of collagen IV alpha345: a position paper of the alport syndrome classification working group. Kidney Int. (2018) 93:1045–51. doi: 10.1016/j.kint.2017.12.018
5. Tsiakkis D, Pieri M, Koupepidou P, Demosthenous P, Panayidou K, Deltas C. Genotype-phenotype correlation in X-linked alport syndrome patients carrying missense mutations in the collagenous domain of COL4A5. Clin Genet. (2012) 82:297–9. doi: 10.1111/j.1399-0004.2012.01849.x
6. Yamamura T, Horinouchi T, Aoto Y, Lennon R, Nozu K. The contribution of COL4A5 splicing variants to the pathogenesis of X-linked alport syndrome. Front Med (Lausanne). (2022) 9:841391. doi: 10.3389/fmed.2022.841391
7. King K, Flinter FA, Nihalani V, Green PM. Unusual deep intronic mutations in the COL4A5 gene cause X linked alport syndrome. Hum Genet. (2002) 111:548–54. doi: 10.1007/s00439-002-0830-3
8. Yamamura T, Nozu K, Fu XJ, Nozu Y, Ye MJ, Shono A, et al. Natural history and genotype-phenotype correlation in female X-linked alport syndrome. Kidney Int Rep. (2017) 2:850–5. doi: 10.1016/j.ekir.2017.04.011
9. Moriniere V, Dahan K, Hilbert P, Lison M, Lebbah S, Topa A, et al. Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J Am Soc Nephrol. (2014) 25:2740–51. doi: 10.1681/ASN.2013080912
10. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
11. Bekheirnia MR, Reed B, Gregory MC, McFann K, Shamshirsaz AA, Masoumi A, et al. Genotype-phenotype correlation in X-linked alport syndrome. J Am Soc Nephrol. (2010) 21:876–83. doi: 10.1681/ASN.2009070784
12. Dhir A, Buratti E. Alternative splicing: role of pseudoexons in human disease and potential therapeutic strategies. Febs J. (2010) 277:841–55. doi: 10.1111/j.1742-4658.2009.07520.x
13. Horinouchi T, Nozu K, Yamamura T, Minamikawa S, Nagano C, Sakakibara N, et al. Determination of the pathogenicity of known COL4A5 intronic variants by in vitro splicing assay. Sci Rep. (2019) 9:12696. doi: 10.1038/s41598-019-48990-9
14. Baralle D, Lucassen A, Buratti E. Missed threads. The impact of pre-mRNA splicing defects on clinical practice. Embo Rep. (2009) 10:810–6. doi: 10.1038/embor.2009.170
15. Malone AF, Funk SD, Alhamad T, Miner JH. Functional assessment of a novel COL4A5 splice region variant and immunostaining of plucked hair follicles as an alternative method of diagnosis in X-linked alport syndrome. Pediatr Nephrol. (2017) 32:997–1003. doi: 10.1007/s00467-016-3565-4
16. Nozu K, Vorechovsky I, Kaito H, Fu XJ, Nakanishi K, Hashimura Y, et al. X-linked alport syndrome caused by splicing mutations in COL4A5. Clin J Am Soc Nephrol. (2014) 9:1958–64. doi: 10.2215/CJN.04140414
17. Yamamura T, Horinouchi T, Nagano C, Omori T, Sakakibara N, Aoto Y, et al. Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked alport syndrome. Kidney Int. (2020) 98:1605–14. doi: 10.1016/j.kint.2020.06.038
18. Jais JP, Knebelmann B, Giatras I, Marchi M, Rizzoni G, Renieri A, et al. X-linked alport syndrome: natural history in 195 families and genotype- phenotype correlations in males. J Am Soc Nephrol. (2000) 11:649–57. doi: 10.1681/ASN.V114649
19. Horinouchi T, Nozu K, Yamamura T, Minamikawa S, Omori T, Nakanishi K, et al. Detection of splicing abnormalities and genotype-phenotype correlation in X-linked alport syndrome. J Am Soc Nephrol. (2018) 29:2244–54. doi: 10.1681/ASN.2018030228
20. Nozu K, Nakanishi K, Abe Y, Udagawa T, Okada S, Okamoto T, et al. A review of clinical characteristics and genetic backgrounds in alport syndrome. Clin Exp Nephrol. (2019) 23:158–68. doi: 10.1007/s10157-018-1629-4
21. Said SM, Fidler ME, Valeri AM, McCann B, Fiedler W, Cornell LD, et al. Negative staining for COL4A5 correlates with worse prognosis and more severe ultrastructural alterations in males with alport syndrome. Kidney Int Rep. (2017) 2:44–52. doi: 10.1016/j.ekir.2016.09.056
22. Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. (2016) 44:6549–63. doi: 10.1093/nar/gkw533
Keywords: Alport syndrome, COL4A5, splice site mutation, whole-genome sequencing, RT-PCR, minigene
Citation: Qian P, Bao Y, Huang H, Suo L, Han Y, Li Z and Zhang M (2023) A deep intronic splice variant of the
Received: 1 August 2022; Accepted: 26 December 2022;
Published: 13 January 2023.
Edited by:
Michael L. Moritz, University of Pittsburgh, United StatesReviewed by:
Kandai Nozu, Kobe University, JapanAgne Cerkauskaite Kerpauskiene, Vilnius University, Lithuania
© 2023 Qian, Bao, Huang, Suo, Han, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Bao YmFveWJqQDEyNi5jb20=
Specialty Section: This article was submitted to Pediatric Nephrology, a section of the journal Frontiers in Pediatrics