 Fengyang Wang
Fengyang Wang Huijuan Peng2
Huijuan Peng2 Guiyu Lou
Guiyu Lou- 1Henan Provincial Institute of Medical Genetics, Henan Provincial People’s Hospital, People’s Hospital of Zhengzhou University, Zhengzhou, China
- 2Department of Ultrasonography, Henan Provincial People’s Hospital, People’s Hospital of Zhengzhou University, Zhengzhou, China
Objective: Our study aimed to collect fetuses with recurrent 1q21.1 deletion or duplication syndrome for systematic clinical phenotype analysis to further delineate the intrauterine phenotype features of the two reciprocal syndromes.
Methods: Prenatal samples, including amniotic fluid and chorionic villus samples, were obtained by amniocentesis and chorionic villus sampling at our center, respectively. In total, 43 fetuses were diagnosed with recurrent 1q21.1 deletion or duplication syndrome via array comparative genomic hybridization (array CGH) or copy number variation sequencing (CNV-seq). Prenatal clinical data, pregnancy outcomes, and individual conditions after birth were collected.
Results: In total, 20 fetuses were diagnosed with 1q21.1 deletion syndrome, and 11 had abnormal ultrasound findings. The most common ultrasound features were renal anomalies, musculoskeletal abnormalities, and increased NT. Other less common ultrasound findings encompassed neurologic abnormalities, cardiovascular defects, absence of the gallbladder, intrauterine growth retardation, and cervical cystic hygroma. On the other hand, 23 fetuses had reciprocal 1q21.1 duplication syndrome, 11 of which had abnormal ultrasound findings, mainly nasal bone abnormalities, cardiovascular defects, increased NT, and neurologic abnormalities.
Conclusions: Our case study suggested that the prenatal clinical phenotypes of the recurrent 1q21.1 deletion syndrome and reciprocal duplication syndrome fetuses were highly diverse with incomplete penetrance. Additionally, our findings should expand the intrauterine phenotype associated with the recurrent 1q21.1 region by a series of prenatal ultrasonic anomalies in this work that were described for the first time, which might broaden knowledge of the genotype and phenotype correlation.
Introduction
The chromosomal distal 1q21.1 recurrent region is widely recognized for its reciprocal 1q21.1 deletion syndrome (OMIM612474) and 1q21.1 duplication syndrome (OMIM612475), which spans 817 kb of a unique DNA sequence (chr1:146577486–147394506, hg19) and includes at least 7 genes. The mechanism of deletion and duplication in the chromosomal 1q21.1 recurrent region is most likely due to the low copy repeat cluster that can mediate genomic rearrangements (1, 2). Based on the chromosomal breakpoints BP1 to BP4, the 1q21.1 band is divided into distinct proximal region (chr1:145386507–145748064, hg19) and distal region (chr1:146577486–147394506, hg19) that cluster CNVs into two classes. Class I deletions and duplications occur between BP3 and BP4 involving only the distal 1q21.1 region, while class II deletions and duplications occur between BP1/BP2 and BP4 containing both the proximal and distal region (1).
To date, the clinical phenotypic features of 1q21.1 deletion or duplication syndrome have been well-defined with incomplete penetrance and variable expressivity (3–5). Individuals with 1q21.1 deletion syndrome manifested a broad spectrum of nonspecific features, such as developmental delay, including gross motor, fine motor and speech delay; mild to moderate dysmorphic facial features, including eye anomalies, large ears with deficient helices, microretrognathia, bulbous nose, dental deformities and cleft lip/palate; congenital heart defects, including atrial septal defects, ventricular septal defects, pulmonary valve stenosis, patent ductus arteriosus, patent foramen ovale, ascending aorta dilatation and bicuspid valve; genitourinary abnormalities, especially renal anomalies; psychiatric and behavioral conditions, such as schizophrenia, ADHD, ataxia, disruptive behavior, and behavioral disorders; neurologic abnormalities, including seizures, microcephaly, intellectual disability, learning difficulties, and hypotonia; growth abnormalities, including short stature; skeletal abnormalities, including polydactyly and clinodactyly, syndactyly, scoliosis, joint laxity and nail dystrophy; and other clinical phenotype features, including failure to thrive, constipation and gastroesophageal reflux disease (1–3, 5–9).
On the other hand, 1q21.1 duplication syndrome shares clinical features similar to reciprocal deletion syndrome, including developmental delay (speech and gross motor delays, fine motor delay), dysmorphic facial features (eye abnormalities, prominent forehead, broad nasal bridge, long philtrum, low-set ears, cleft palate), cardiovascular anomalies (patent ductus arteriosus, patent foramen ovale, atrial septal defects, carotid artery stenosis, tricuspid regurgitation, enlarged right atrium, aortic arch hypoplasia), psychiatric and behavioral conditions (ADHD, autism spectrum disorder, abnormalities of gait/agility, mood disorder/social or behavioral anomalies), and neurologic abnormalities (seizures, macrocephaly, brain anomalies, hypotonia). Other clinical phenotypes include scoliosis gastric ulcers. Although the clinical phenotypes were similar, the deletions seemed to be more strongly associated with renal anomalies, skeletal abnormalities, and microcephaly. However, the duplications were more strongly associated with autism spectrum disorder and macrocephaly (1–4, 6, 7, 9–11).
Conversely, unlike the well-characterized postnatal clinical phenotype, limited information has been collected on the intrauterine phenotype features of 1q21.1 deletion and duplication syndrome. The prenatal diagnosis of 1q21.1 deletion and duplication syndrome remain challenging. To better understand the relationship between genotype and prenatal phenotype features, we examined the prenatal clinical phenotype of 20 fetuses with 1q21.1 deletion syndrome and 23 fetuses with reciprocal duplication syndrome. The results were compared with the fetuses reported in the literature to achieve a comprehensive overview of the intrauterine phenotypes of 1q21.1 recurrent region. To our knowledge, this is the largest prenatal study of recurrent 1q21.1 deletion and duplication syndrome involving systematic clinical analysis.
Methods
Clinical data
We retrospectively reviewed 43 fetuses with recurrent 1q21.1 deletion and duplication syndrome from May 2020–March 2024 who came to our center for genetic counseling. Maternal age at diagnosis ranged from 17–41 years, with a mean age of 30 years. The mean gestational age at diagnosis was 20 years, ranging from 11–30 weeks (Tables 1, 2). All the pregnant women were offered serological screening during the second trimester and routine ultrasound screening during the gestation period. The main indications for prenatal diagnosis include abnormal ultrasound findings, advanced maternal age, high risks of T21 and T18, and other reasons for pregnancy (Tables 1, 2). Follow-up data on pregnancy outcome, childbirth, birth weight, parental phenotype, age at verbal language and walking, growth, and development, intelligence, communication and social interaction were collected.
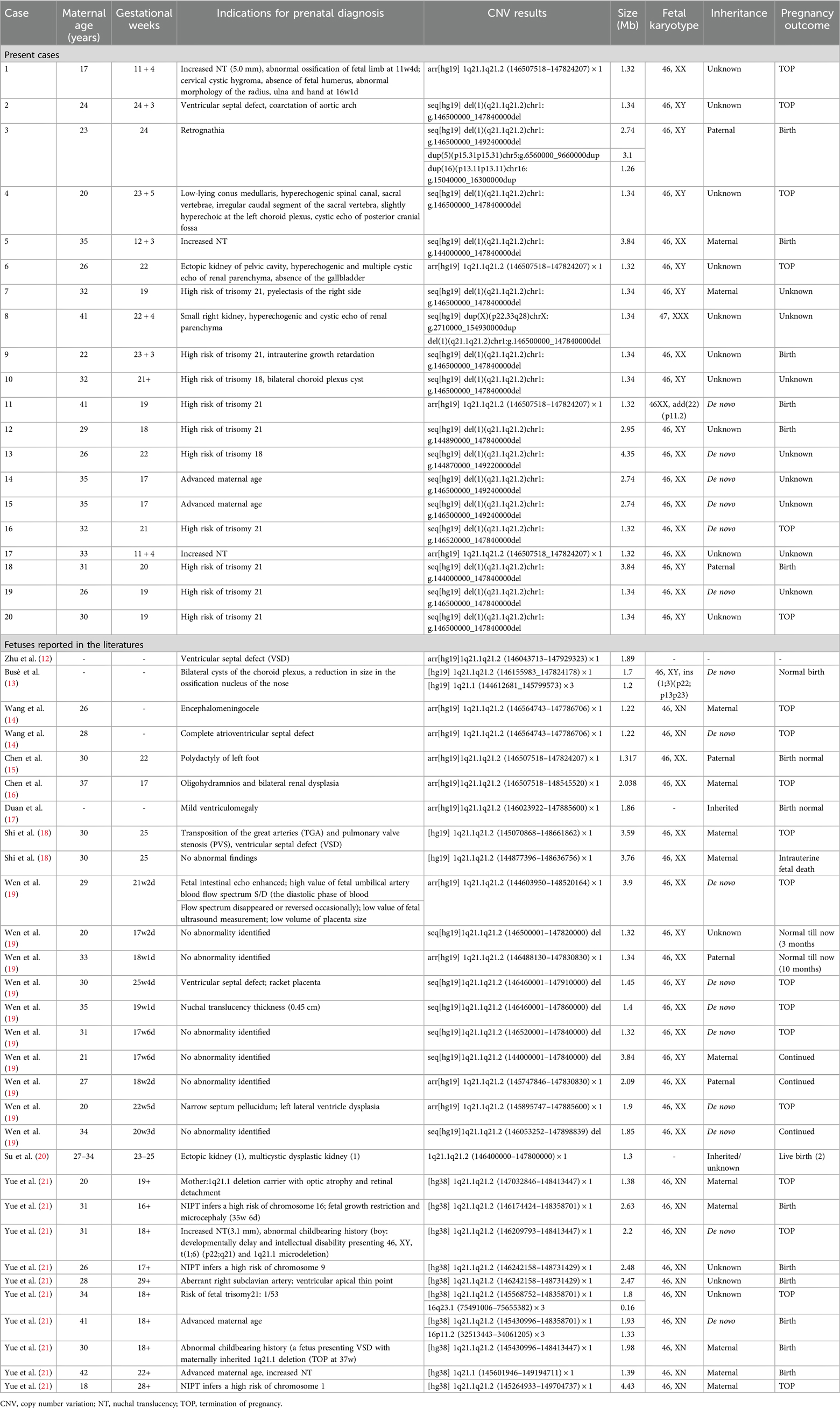
Table 1. Clinical data of all the reported fetuses with 1q21.1 microdeletion syndrome.
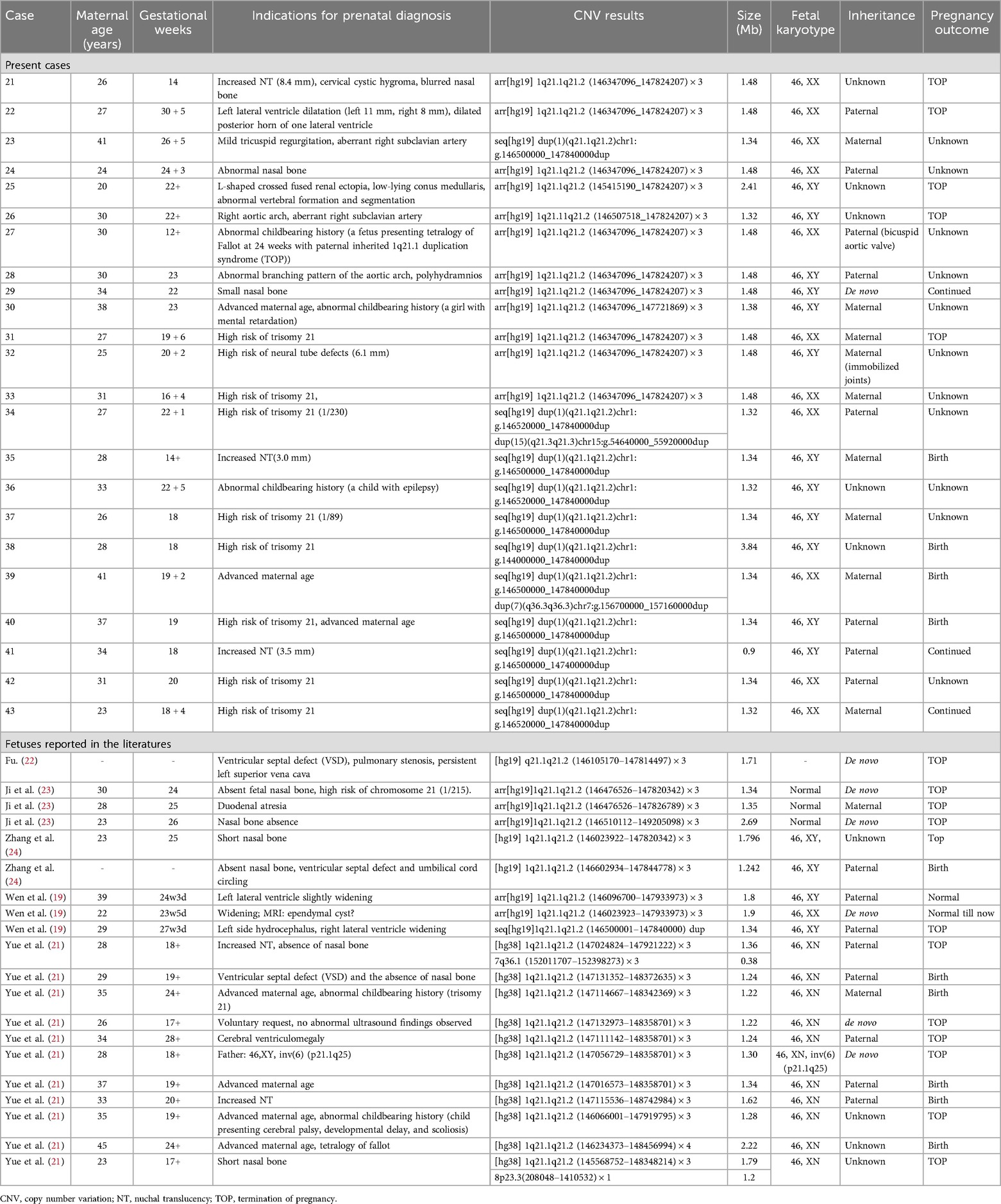
Table 2. Clinical data of all the reported fetuses with 1q21.1 microduplication syndrome.
Cytogenetic and molecular genetic analyses
Amniotic fluid, chorionic villus, or umbilical cord blood samples were obtained according to the gestational week. Peripheral blood samples from the parents were also collected from patients who required segregation analysis. G banding karyotype analysis was performed for all amniotic fluid and umbilical cord blood samples after cell culture. The genomic DNA of the clinical samples was extracted with a Qiagen genomic DNA extraction kit. DNA concentration and purity were then detected by a NanoDrop 2000 UV‒Vis spectrophotometer (Thermo). Array comparative genomic hybridization (array CGH) analysis was performed using 8 × 60 K Agilent Oligo arrays, while copy number variation sequencing (CNV-seq) was performed via the NextSeq CN500 platform of Berry Genomics. All the patients underwent short tandem repeat (STR) analysis with a GoldeneyeTM DNA identification system kit (Peoplespot) to test for maternal contamination.
Results
Intrauterine phenotypes of 1q21.1 deletion syndrome
In our study, twenty fetuses were diagnosed with 1q21.1 deletion syndrome (Table 1, Figure 1). Eleven fetuses (11/20, 55%) had abnormal ultrasound findings, while the remaining nine fetuses (9/20, 45%) were referred for prenatal diagnosis only due to a high risk of age (2/20, 10%) or serological screening (7/20, 35%). The prenatal ultrasound features mainly encompassed renal anomalies (3/20, 15%), musculoskeletal abnormalities (3/20, 15%), increased NT (3/20, 15%), neurologic abnormalities (2/20, 10%), and cardiovascular defects (1/20, 5%). Renal anomalies mainly included hyperechogenic and cystic echoes of the renal parenchyma (2/20, 10%), ectopic kidney (1/20, 5%), pyelectasis (1/20, 5%), and small kidney (1/20, 5%). Musculoskeletal abnormalities in our study included the absence of a fetal humerus and abnormal morphology of the radius, ulna, and hand in Case 1; retrognathia in Case 3; and an irregular caudal segment of the sacral vertebra in Case 4. The neurologic abnormalities included bilateral choroid plexus cysts in Case 10, a low-lying conus medullaris, hyperechoic choroid plexus and spinal canal and cystic echo of the posterior cranial fossa in Case 4. Cardiovascular defects included ventricular septal defects and coarctation of the aortic arch in Case 2. Other rare ultrasonic anomalies included the absence of the gallbladder in Case 6, intrauterine growth retardation in Case 9 and cervical cystic hygroma in Case 1.
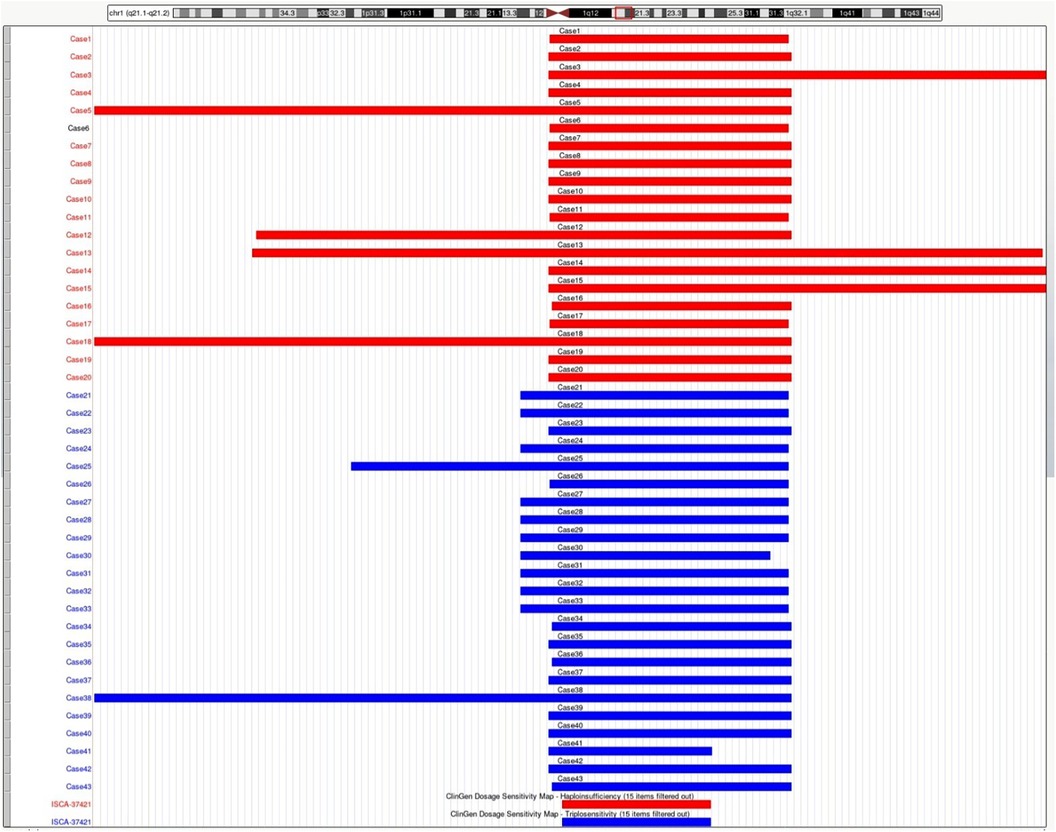
Figure 1. Schematic representation of the deleted or duplicated fragments in the present cases. Genomic parameters are from GRCh38/hg38.
Intrauterine phenotypes of 1q21.1 duplication syndrome
A total of twenty-three fetuses were diagnosed with 1q21.1 duplication syndrome (Table 2, Figure 1), among which ten presented with abnormal ultrasound findings (10/23, 43.5%). The most prevalent abnormalities identified by prenatal ultrasonography included nasal bone abnormalities (3/23, 13%), cardiovascular defects (3/23, 13%), increased NT (3/23, 13%), and neurologic abnormalities (2/23, 8.7%). Cardiovascular defects contained aberrant right subclavian artery in Case 23 and Case 26, mild tricuspid regurgitation in Case 23, a right aortic arch in Case 26, and an abnormal branching pattern of the aortic arch in Case 28. Neurologic abnormalities included left lateral ventricle dilatation, dilated posterior horn of one lateral ventricle in Case 22, and low-lying conus medullaris in Case 25. Other rare ultrasonic anomalies included abnormal vertebral formation and segmentation and L-shaped crossed fused renal ectopia in Case 25, cervical cystic hygroma in Case 21, and polyhydramnios in Case 28. The remaining thirteen fetuses showed no structural anomalies, and the prenatal diagnosis reasons included a high risk of serological screening (8/23, 34.8%), advanced maternal age (3/23, 13%), and abnormal childbearing history (2/23, 8.7%).
Cytogenetic and molecular analyses
All 43 fetuses underwent G-banded karyotype analysis, except for Case 1, 5, 21, and 35. Two abnormal karyotypes were found, namely, 47, XXX in Case 8 and 46, XX, add (24) (p11.2) in Case 11. All fetuses in our study had a common deletion or duplication in the 1q21.1 recurrent region, ranging from 0.9 Mb–4.35 Mb (Figure 1). CNV analysis also revealed an additional 3.10 Mb duplication of 5p15.31 and a 1.26 Mb duplication of 16p13.11p13.11 in Case 3 and a 1.28 Mb duplication of 15q21.3 in Case 34, all of which were of uncertain significance.
Parental analysis of the 1q21.1 region revealed that the deletions occurred de novo in four cases and were inherent in four cases, while duplications were inherent in 15 cases. In addition, the symptomatic father of Case 27 was described with a bicuspid aortic valve. The mother of Case 32 showed immobilized joints in both wrists, legs, and feet. The segregation was not completed due to unavailable parent samples among the remaining 20 cases.
Follow-up
The follow-up revealed that ten fetuses continued pregnancy, including six (Case 3, 5, 9, 11, 12, and 18) with 1q21.1 deletion syndrome and four (Case 35, 38, 39, and 40) with 1q21.1 duplication syndrome. The children were delivered at gestational weeks ranging from 35–40, and the mean birth weight was 3.4 kg (Table 3). Furthermore, deletions or duplications in six children were inherited from normal parents, while one was de novo. The oldest child is currently three years and four months old, while the youngest is nine months old. Case 11 and Case 12 have already attended kindergarten. All the children could speak and walk at the normal stage. No abnormal findings related to growth and development, intelligence, communication, or social interaction existed. However, Case 18 was diagnosed with megacolon at two months after birth and underwent surgery at four months. In addition, Case 11 and Case 39 seem more intelligent than the siblings or peer group based on the parents' statements.
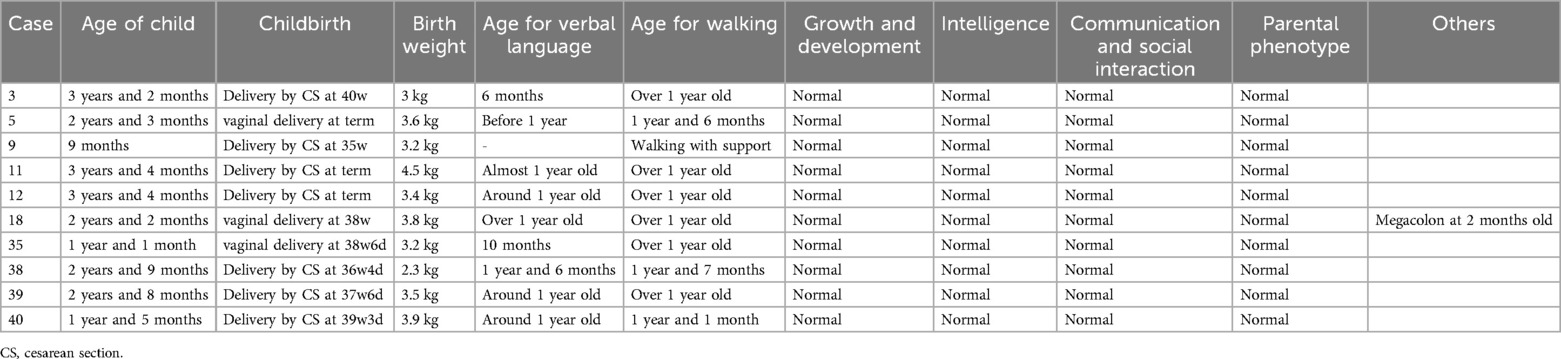
Table 3. Clinical data of 10 children that were born in present cases.
Six pregnancies (Case 1, 2, 4, 6, 16, and 20) with 1q21.1 deletion and five (Case 21, 22, 25, 26, and 31) with 1q21.1 duplication were selectively aborted. The remaining patients were lost to follow-up due to a telephone outage or call rejection.
Discussion
In our study, 20 fetuses with 1q21.1 deletion syndrome and 23 fetuses with 1q21.1 duplication syndrome were described with detailed prenatal manifestations. The prenatal phenotypes of both 1q21.1 deletion and duplication syndrome fetuses were variable in the family series, ranging from a normal phenotype to musculoskeletal system abnormalities, cardiovascular defects, abnormal nervous system, and renal anomalies. The CNVs of most cases in our study were clustered into Class I deletions and duplications with only the distal region of the 1q21.1 band, while CNVs of only five cases (Case 5, 12, 13, 18, 38) were divided into Class II deletions and duplications with both the proximal and distal region of the 1q21.1 band (Figure 1). Despite the larger deletion or duplication region in the five Class II CNVs, the prenatal phenotype of the fetuses only contained increased NT and high risk of trisomy 21 or 18 without other abnormal ultrasonic findings (Tables 1, 2). It indicated that the severity of prenatal clinical phenotype might not be proportional to the CNVs size.
Most structural malformations in our study were reported prenatally for the first time and were consistent with those reported in the postnatal literature. The ultrasound findings, including retrognathia and abnormal morphology of the hand, described in Case 3 and Case 1, respectively, were once observed in a 27-year-old female patient (5). Additionally, a spinal anomaly, such as an irregular caudal segment of the sacral vertebra of Case 4, was also postnatally described as scoliosis by Digilio et al. (6). Case 7 had pyelectasis, while Case 8 had a small right kidney, which was consistent with two patients with hydronephrosis (9) and three affected patients with renal asymmetry (8). The ultrasound images of Case 23 showed tricuspid regurgitation, which had been reported in an infant (7). In contrast, some clinical phenotypes have never been reported in the literature, including the absence of fetal humerus and abnormal morphology of the radius and ulna in Case 1 and the absence of the gallbladder in Case 6.
To date, most research on 1q21.1 deletion and duplication syndrome has focused on postnatal cases. Prenatal diagnosis and genetic counseling of 1q21.1 recurrent region syndrome remain difficult due to the lack of consistent and complete prenatal phenotype features. To the best of our knowledge, only 31 fetuses with 1q21.1 deletion syndrome have been reported in the literature (Table 1), while 20 fetuses with 1q21.1 duplication syndrome have been reported (Table 2). To make a clear diagnosis of 1q21.1 deletion and duplication syndrome in the prenatal clinical phenotype, we summarize the prenatal ultrasound phenotypes of all the fetuses reported in the literature that cover the whole 1q21.1 recurrent deletion and duplication region combined with our present cases.
Fetuses with 1q21.1 deletion syndrome in our cases and the literature (Table 1, 4) commonly manifested cardiovascular defects (7/51, 13.7%), neurologic abnormalities (7/51, 13.7%), renal anomalies (6/51, 11.8%), increased NT (6/51, 11.8%), musculoskeletal abnormalities (5/51, 9.8%) and other rare phenotypes, including intrauterine growth retardation (2/51, 3.9%), placental abnormalities (2/51, 3.9%), absence of the gallbladder (1/51, 1.9%), cervical cystic hygroma (1/51, 1.9%), oligohydramnios (1/51, 1.9%), and fetal intestinal echo enhanced (1/51, 1.9%). The most common prenatal phenotype of cardiovascular defects was VSD, which was observed in 7.8% (4/51), followed by AVSD (1/51, 1.9%), PVS (1/51, 1.9%), TGA (1/51, 1.9%), ARSA (1/51, 1.9%), coarctation of the aortic arch (1/51, 1.9%), high value of s/d (1/51, 1.9%) and ventricular apical thin point (1/51, 1.9%). The main neurologic abnormalities of fetuses with 1q11.23 deletion syndrome described thus far included bilateral choroid plexus cysts (2/51, 3.9%), low-lying conus medullaris (1/51, 1.9%), hyperechogenic spinal canal (1/51, 1.9%), hyperechoic choroid plexus (1/51, 1.9%), cystic echo of posterior cranial fossa (1/51, 1.9%), encephalomeningocele (1/51, 1.9%), ventriculomegaly (1/51, 1.9%), narrow septum pellucidum (1/51, 1.9%), lateral ventricle dysplasia (1/51, 1.9%) and microcephaly (1/51, 1.9%) (12–21).
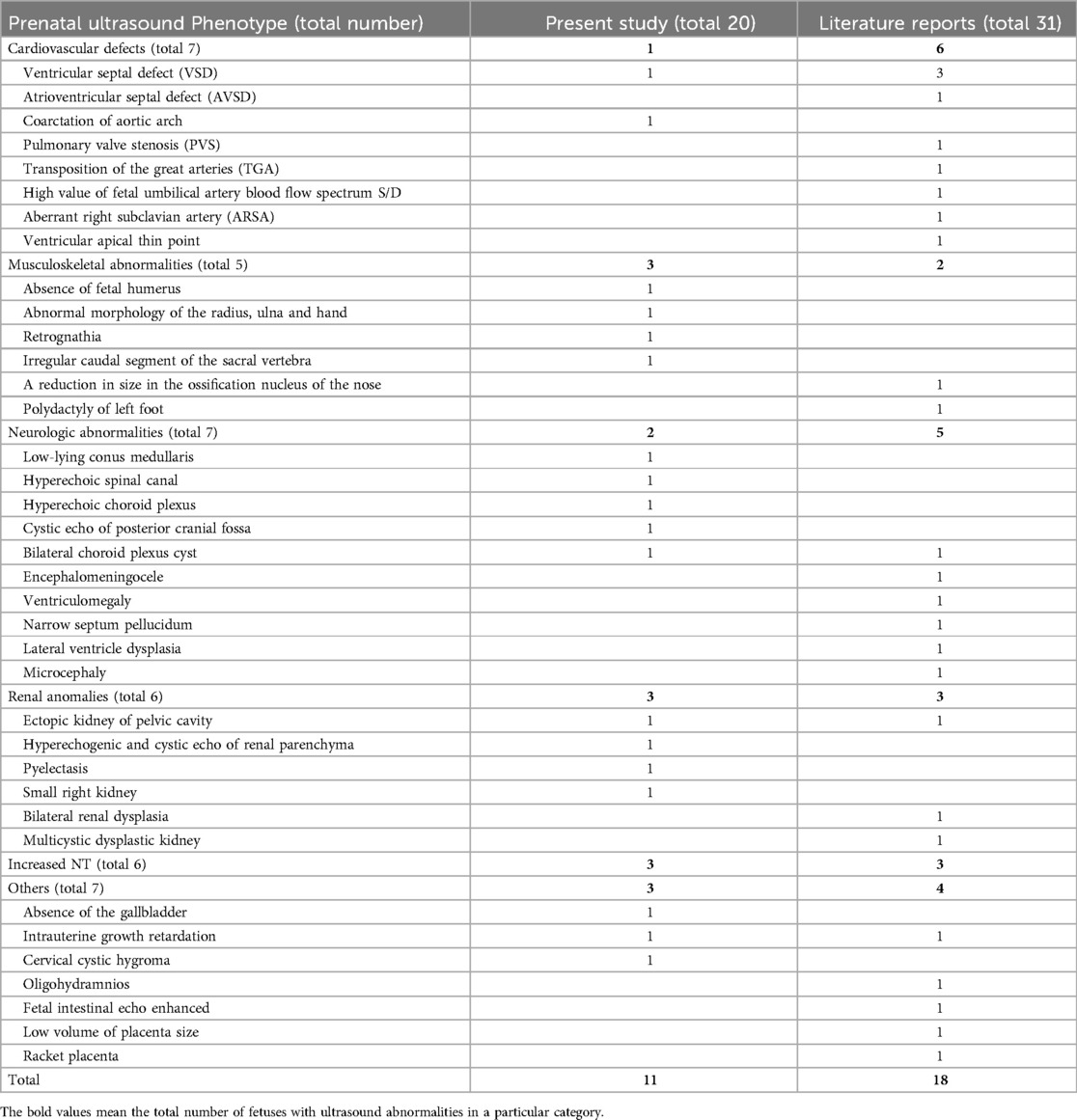
Table 4. Summarization of the prenatal ultrasound phenotype of all reported fetuses with 1q21.1 deletion syndrome.
Based on previous case reports and our current data (Table 2, 5), fetuses with 1q21.1 duplication syndrome were associated with a broad range of prenatal ultrasonic phenotypes, with nasal bone abnormalities (10/43, 23.3%), cardiovascular defects (7/43, 16.3%), neurologic abnormalities (6/43, 13.9%) and increased NT (5/43, 11.6%) being the most frequent. Other rare ultrasonic phenotypes included L-shaped crossed-fused renal ectopia (1/43, 2.3%), polyhydramnios (1/43, 2.3%), cervical cystic hygroma (1/43, 2.3%), duodenal atresia (1/43, 2.3%) and abnormal vertebral formation and segmentation (1/43, 2.3%). Cardiovascular defects mainly included VSD (3/43, 7%), ARSA (2/43, 4.7%), TR (1/43, 2.3%), RAA (1/43, 2.3%), PS (1/43, 2.3%), PLSVC (1/43, 2.3%) and TOF (1/43, 2.3%). The most common abnormality of the nervous system was lateral ventricle widening (3/43, 7%), followed by dilation of the posterior horn of the lateral ventricle (1/43, 2.3%), low-lying conus medullaris (1/43, 2.3%), ependymal cyst (1/43, 2.3%), hydrocephalus (1/43, 2.3%), and ventriculomegaly (1/43, 2.3%) (19, 21–24).
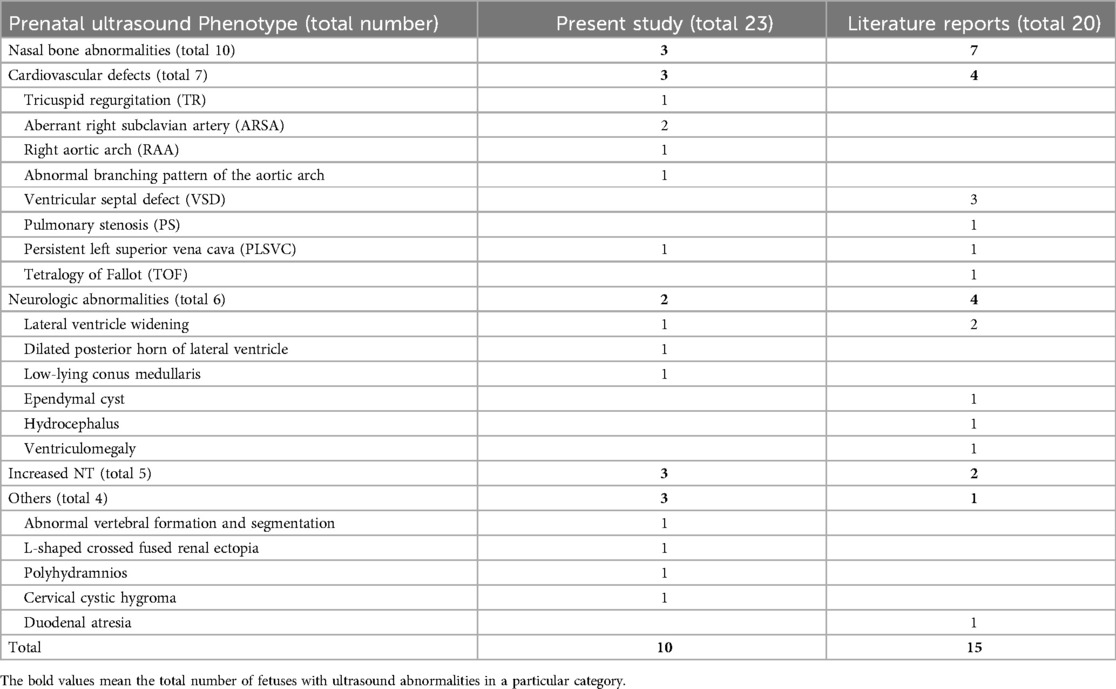
Table 5. Summarization of the prenatal ultrasound phenotype of all reported fetuses with 1q21.1 duplication syndrome.
According to Tables 4, 5, consistent with postnatal reports, abnormal nasal bone function was more prevalent in fetuses with 1q21.1 duplication syndrome than in those with 1q21.1 deletion syndrome. On the other hand, renal abnormalities and musculoskeletal abnormalities were more common in fetuses with 1q21.1 deletion syndrome. This result indicated that the duplication or deletion of individual genes in the 1q21.1 recurrent region may be responsible for the difference. In addition, penetrance in all the prenatal cases might be evaluated at 56.8% (29/51) for 1q21.1 deletion syndrome and 58.1% (25/43) for reciprocal 1q21.1 duplication syndrome, which are both higher than the 36.9% and 29.1% reported for deletion and duplication, respectively, by Rosenfeld et al. (25). However, the penetrance might be overestimated due to that all the cases came to our center exhibited abnormal ultrasound findings or a high risk of prenatal screen. More prenatal cases are necessary to characterize the intrauterine phenotype of 1q21.1 deletion or duplication syndrome.
In the follow-up stage, we found that all the ten fetuses that were delivered showed normal postnatal phenotype, including birth weight, age for verbal language and walking, growth and development, intelligence, communication and social interaction, which seemed to be contrary to the well-defined postnatal phenotypes of 1q21.1 deletion or duplication syndrome in the literatures. Further analysis revealed that six out of the ten fetuses (Case 11, 12, 18, 38, 39, 40) were referred to prenatal diagnosis only due to high risk of trisomy 21 or advanced maternal age, without any ultrasonic anomaly during the pregnancy. Although the remaining four cases exhibited ultrasonic anomaly, including two fetuses (Case 5, 35) with increased NT in the first trimester, one (case 9) with intrauterine growth retardation, however, only one (Case 3) exhibited structural anomaly -retrognathia. Parental analysis revealed that deletions or duplications in six cases (Case 3, 5, 18, 35, 39, 40) were inherited from normal parents, while one (Case 11) was de novo. These findings may provide important cue for genetic counselors and pregnant women when considering the pregnancy outcome of fetuses with 1q21.1 deletion or duplication syndrome, especially when there were parental inheritance and no abnormal ultrasonic findings prenatally.
One major limitation of our case series is that not all cases underwent parental analysis, which may attenuate the genotype and phenotype correlation, especially when considering the incomplete penetrance and variable expressivity. The other limitation is that the pregnancy outcomes of about half of the cases were unavailable, leading to inadequate analysis of postnatal outcomes.
Conclusion
In summary, the prenatal ultrasound findings of both 1q21.1 deletion and duplication syndrome were highly variable. Prenatal ultrasound findings, including cardiovascular defects, neurologic abnormalities, and increased NT were commonly associated with both 1q21.1 deletion and duplication syndrome. However, nasal bone abnormalities were the most common intrauterine phenotype of 1q21.1 duplication syndrome, while 1q21.1 deletion syndrome was more frequently associated with renal anomalies and musculoskeletal abnormalities. Our case series might expand the prenatal phenotype of 1q21.1 deletion and duplication syndrome by a series of new ultrasound features. Our findings provided cue insights into the genotype and prenatal phenotype correlation of 1q21.1 deletion and duplication syndrome through summarizing the prenatal phenotypes, which might benefit prenatal diagnosis and genetics counseling of the recurrent 1q21.1 region. Considering phenotypic heterogeneity and incomplete penetrance, further studies are required to elucidate the prenatal phenotype and genotype correlation of the recurrent 1q21.1 region.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Ethics Committee of Henan Provincial People's Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
FW: Data curation, Formal Analysis, Investigation, Writing – original draft. HP: Data curation, Writing – original draft. GL: Funding acquisition, Writing – review & editing. YR: Writing – review & editing. SL: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Henan Province Medical Science and Technology Public Relations Plan Province Department joint construction project (No. SBGJ2018062) and the China and Health Science and Technology Innovation Leading Talent Training Project of Henan Province (No. YXKC2021001).
Acknowledgments
We would like to acknowledge all the pregnant women who consented to participate in this research.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Brunetti-Pierri N, Berg JS, Scaglia F, Belmont J, Bacino CA, Sahoo T, et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. (2008) 40(12):1466–71. doi: 10.1038/ng.279
2. Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. (2008) 359(16):1685–99. doi: 10.1056/NEJMoa0805384
3. Bernier R, Steinman KJ, Reilly B, Wallace AS, Sherr EH, Pojman N, et al. Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet Med. (2016) 18(4):341–9. doi: 10.1038/gim.2015.78
4. Verhagen JM, de Leeuw N, Papatsonis DN, Grijseels EW, de Krijger RR, Wessels MW. Phenotypic variability associated with a large recurrent 1q21.1 microduplication in a three-generation family. Mol Syndromol. (2015) 6(2):71–6. doi: 10.1159/000431274
5. Upadhyai P, Amiri EF, Guleria VS, Bielas SL, Girisha KM, Shukla A. Recurrent 1q21.1 deletion syndrome: report on variable expression, nonpenetrance and review of literature. Clin Dysmorphol. (2020) 29(3):127–31. doi: 10.1097/MCD.0000000000000327
6. Digilio MC, Bernardini L, Consoli F, Lepri FR, Giuffrida MG, Baban A, et al. Congenital heart defects in recurrent reciprocal 1q21.1 deletion and duplication syndromes: rare association with pulmonary valve stenosis. Eur J Med Genet. (2013) 56(3):144–9. doi: 10.1016/j.ejmg.2012.12.004
7. Pang H, Yu X, Kim YM, Wang X, Jinkins JK, Yin J, et al. Disorders associated with diverse, recurrent deletions and duplications at 1q21.1. Front Genet. (2020) 11:577. doi: 10.3389/fgene.2020.00577
8. Edwards SD, Schulze KV, Rosenfeld JA, Westerfield LE, Gerard A, Yuan B, et al. Clinical characterization of individuals with the distal 1q21.1 microdeletion. Am J Med Genet A. (2021) 185(5):1388–98. doi: 10.1002/ajmg.a.62104
9. Bourgois A, Bizaoui V, Colson C, Vincent-Devulder A, Molin A, Gerard M, et al. Phenotypic and genotypic characterization of 1q21.1 copy number variants: a report of 34 new individuals and literature review. Am J Med Genet A. (2024) 194(3):e63457. doi: 10.1002/ajmg.a.63457
10. Gourari I, Schubert R, Prasad A. 1q21.1 duplication syndrome and epilepsy: case report and review. Neurol Genet. (2018) 4(1):e219. doi: 10.1212/NXG.0000000000000219
11. Huang TT, Xu HF, Wang SY, Lin WX, Tung YH, Khan KU, et al. Identification of 1q21.1 microduplication in a family: a case report. World J Clin Cases. (2023) 11(4):874–82. doi: 10.12998/wjcc.v11.i4.874
12. Zhu X, Li J, Ru T, Wang Y, Xu Y, Yang Y, et al. Identification of copy number variations associated with congenital heart disease by chromosomal microarray analysis and next-generation sequencing. Prenat Diagn. (2016) 36(4):321–7. doi: 10.1002/pd.4782
13. Buse M, Cuttaia HC, Palazzo D, Mazara MV, Lauricella SA, Malacarne M, et al. Expanding the phenotype of reciprocal 1q21.1 deletions and duplications: a case series. Ital J Pediatr. (2017) 43(1):61. doi: 10.1186/s13052-017-0380-x
14. Wang HD, Liu L, Wu D, Li T, Cui CY, Zhang LZ, et al. Clinical and molecular cytogenetic analyses of four families with 1q21.1 microdeletion or microduplication. J Gene Med. (2017) 19(4):e2948. doi: 10.1002/jgm.2948
15. Chen CP, Chang SY, Chen YN, Chern SR, Wu PS, Chen SW, et al. Prenatal diagnosis of a familial 1q21.1–q21.2 microdeletion in a fetus with polydactyly of left foot on prenatal ultrasound. Taiwan J Obstet Gynecol. (2018) 57(5):739–44. doi: 10.1016/j.tjog.2018.08.024
16. Chen CP, Huang JP, Chen YY, Chern SR, Wu PS, Chen SW, et al. Detection of a familial 1q21.1 microdeletion and concomitant CHD1l mutation in a fetus with oligohydramnios and bilateral renal dysplasia on prenatal ultrasound. Taiwan J Obstet Gynecol. (2019) 58(6):859–63. doi: 10.1016/j.tjog.2019.07.031
17. Duan HL, Zhu XY, Zhu YJ, Wu X, Zhao GF, Wang WJ, et al. The application of chromosomal microarray analysis to the prenatal diagnosis of isolated mild ventriculomegaly. Taiwan J Obstet Gynecol. (2019) 58(2):251–4. doi: 10.1016/j.tjog.2019.01.015
18. Shi SS, Lin SB, Cai DL, Wen WR, Li RM. Discordance of cardiovascular abnormalities in a monozygotic twin pair carrying a class II 1q21.1 microdeletion. Taiwan J Obstet Gynecol. (2020) 59(1):123–6. doi: 10.1016/j.tjog.2019.11.019
19. Wen X, Xing H, Qi K, Wang H, Li X, Zhu J, et al. Analysis of 17 prenatal cases with the chromosomal 1q21.1 copy number variation. Dis Markers. (2022) 2022:5487452. doi: 10.1155/2022/5487452
20. Su J, Qin Z, Fu H, Luo J, Huang Y, Huang P, et al. Association of prenatal renal ultrasound abnormalities with pathogenic copy number variants in a large Chinese cohort. Ultrasound Obstet Gynecol. (2022) 59(2):226–33. doi: 10.1002/uog.23702
21. Yue F, Yang X, Jiang Y, Li S, Liu R, Zhang H. Prenatal phenotypes and pregnancy outcomes of fetuses with recurrent 1q21.1 microdeletions and microduplications. Front Med. (2023) 10:1207891. doi: 10.3389/fmed.2023.1207891
22. Fu F, Deng Q, Lei TY, Li R, Jing XY, Yang X, et al. Clinical application of SNP array analysis in fetuses with ventricular septal defects and normal karyotypes. Arch Gynecol Obstet. (2017) 296(5):929–40. doi: 10.1007/s00404-017-4518-2
23. Ji X, Pan Q, Wang Y, Wu Y, Zhou J, Liu A, et al. Prenatal diagnosis of recurrent distal 1q21.1 duplication in three fetuses with ultrasound anomalies. Front Genet. (2018) 9:275. doi: 10.3389/fgene.2018.00275
24. Zhang H, Yue F, Zhang X, He J, Jiang Y, Liu R, et al. Prenatal detection of distal 1q21.1q21.2 microduplication with abnormal ultrasound findings: two cases report and literature review. Medicine. (2021) 100(1):e24227. doi: 10.1097/MD.0000000000024227
Keywords: 1q21.1 deletion syndrome, 1q21.1 duplication syndrome, prenatal ultrasound phenotype, array CGH, CNV-seq
Citation: Wang F, Peng H, Lou G, Ren Y and Liao S (2025) Prenatal ultrasound phenotype of fetuses with recurrent 1q21.1 deletion and duplication syndrome. Front. Pediatr. 12:1504122. doi: 10.3389/fped.2024.1504122
Received: 30 September 2024; Accepted: 27 December 2024;
Published: 7 January 2025.
Edited by:
Jordi Pérez-Tur, Spanish National Research Council (CSIC), SpainReviewed by:
Gabriela Corina Zaharie, University of Medicine and Pharmacy Iuliu Hatieganu, RomaniaAdonis S. Ioannides, University of Nicosia, Cyprus
Copyright: © 2025 Wang, Peng, Lou, Ren and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shixiu Liao, eWNzMjQwM0AxMjYuY29t