 Xiaolin Jieda
Xiaolin Jieda Chaoge Yang
Chaoge Yang Yue Wu1,2
Yue Wu1,2 Wanting Xu
Wanting Xu Wenbin Dong
Wenbin Dong- 1Department of Neonatology, Children’s Medical Center, The Affiliated Hospital of Southwest Medical University, Luzhou, Sichuan, China
- 2Department of Pediatrics, Southwest Medical University, Luzhou, China
- 3Department of Neurosurgery, The Affiliated Hospital of Southwest Medical University, Luzhou, China
Spinal muscular atrophy (SMA) is an autosomal recessive genetic disorder marked by progressive, symmetrical muscle weakness and atrophy. While only a limited number of studies on human SMA have demonstrated brain involvement, there are also few reports detailing early brain MRI changes in SMA patients. In this paper, we present the case of a child whose initial symptom was limb hypotonia. The child's brain MRI revealed abnormal signal changes and genetic testing ultimately confirmed the diagnosis of SMA. By reviewing relevant literature, we aim to summarize the brain MRI signal changes observed in SMA patients and explore their possible mechanisms, with the goal of enhancing clinicians' ability to identify and treat neonatal SMA at an early stage.
Introduction
SMA is a degenerative neuromuscular disease affecting lower motor neurons in the anterior horn of the spinal cord, primarily caused by a homozygous deletion of SMN1 on chromosome 5q13 (1, 2). Its incidence ranges from 1/6,000 to 1/10,000, with a carrier frequency of 1/40 to 1/60 (1, 2). This case initially presented with decreased muscle tone in the extremities. Although some structures of the brain MRI showed abnormal signal changes similar to bilirubin encephalopathy, clinical manifestations and additional examinations did not support this diagnosis, leading to a final diagnosis of SMA through genetic testing. Characteristic brain imaging findings in neonatal SMA are rarely reported and are often confused with conditions like bilirubin encephalopathy, hypoxic-ischemic encephalopathy, and hypoglycemic encephalopathy, making diagnosis challenging. Further research is needed to clarify the diagnostic value of brain imaging changes in SMA.
Case report
We report a neonate presenting with hypotonia one day after birth. Born at 40+5 weeks with Apgar scores of 1–5–10 min are all rated at 10 and a weight of 2,700 g, the child showed no abnormalities in the amniotic fluid, umbilical cord, or placenta, nor signs of intrauterine distress or premature rupture of membranes. The child did not show any decrease in fetal movement or fetal heart rate during pregnancy. The mother's gestational age was 32 years. His parents had no history of smoking, alcohol, drug use, or inherited metabolic diseases. The child exhibited good development, alertness, and mild jaundice. Physical examination showed no abnormalities in the heart, lungs, or abdomen and no barrel chest or paradoxical breathing exercises. The child's upper limbs could move horizontally but could not be lifted off the bed, both lower limbs could be lifted slightly off the bed but could not resist resistance, the limbs were floppy and could not be flexed naturally in the supine position, and they were in a “frog position” in the prone position. None of the tendon reflexes were elicited, the feeding and sucking reflex could be elicited, whereas the grip and hug reflexes were markedly diminished. He had difficulty in erecting his head, and he had no myoclonus, tongue spasms, or finger contractures. The “Medical Research Council Scale for Muscle Strength” suggests grade 2 muscle strength in the upper limbs and grade 3 in the lower limbs. Upon admission, blood glucose was 3.4 mmol/L, serum total bilirubin was 9.5 mg/dl, and blood gas analysis was normal. During hospitalization, results for NSE (35 ng/ml), blood cell analysis + CRP, urine analysis, stool routine, liver and kidney function tests, electrolytes, blood culture, TORCH tests, procalcitonin, pre-blood transfusion, and Epstein–Barr virus tests were all normal. A spine MRI was unremarkable, while a brain MRI on postnatal day 3 showed symmetrical patchy T1W1 hyperintensity in the bilateral basal ganglia, thalamus, periventricular area, and brainstem (Figure 1). We did genetic testing, which showed a homozygous deletion of exon 7 of the SMN1 gene, with a heterozygous deletion in both parents, confirming a diagnosis of spinal muscular atrophy. Sadly, the child's parents withdrew treatment, and he died of respiratory failure a week later.
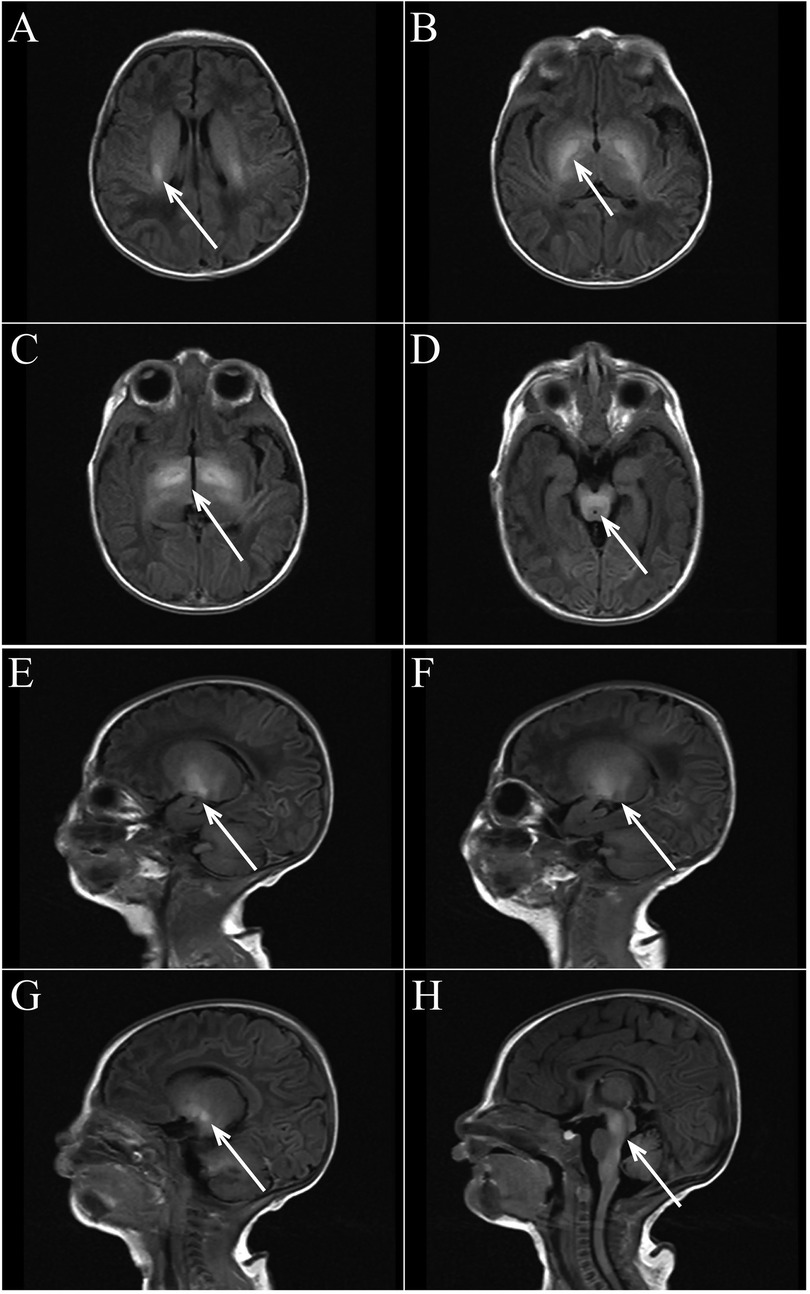
Figure 1. (A–D axial view) bilateral basal ganglia, thalamus, lateral ventricle, brain stem symmetrical patchy T1WI high signal shadow. (E–H sagittal view) Bilateral basal ganglia, thalamus, lateral ventricle, brain stem symmetrical patchy T1WI high signal shadow.
Discussion
In this case, the child was admitted to the hospital with reduced muscle tone as the primary symptom. His brain MRI showed high signal changes in multiple regions on T1WI. Neonatal brain MRI signal changes can have various causes, we excluded the diagnosis of bilirubin encephalopathy and the fact that the child had no postnatal hypoxic-ischemic events, such as asphyxia, intrauterine distress, severe hypoglycemia, or significant infections, making the origin of the MRI changes unclear. Ultimately, a diagnosis of SMA was confirmed through genetic testing.
SMA is the second most common fatal autosomal recessive disease in infancy and is classified into five types based on symptom onset and highest motor milestones achieved (2, 3). Patients with type 0, I, and II are more severe, and they mostly die from respiratory failure, while type III and IV life expectancy are generally not affected (1). Two SMA-related genes, SMN1 and SMN2, are located on chromosome 5, differing by a single nucleotide (C-to-T transition in exon 7); SMN1 produces SMN protein normally, whereas SMN2 produces only 10% of the total SMN protein (1). When a homozygous deletion of SMN1 exon 7 occurs on 5q13, SMN2 alone produces insufficient SMN protein, leading to SMA. Therefore, its severity and prognosis are closely linked to the number of SMN2 copies, with fewer copies indicating a more severe disease.
SMN protein levels in the brain have been found to decrease during development, especially in the fetal and postnatal stages (4), suggesting a critical role for SMN protein in the early stages of brain development. Neuropathological data show that severe forms of SMA affect the brain (5), and reduced brain structural size is observed in mouse models of severe SMA, especially in regions associated with high SMN protein levels, suggesting that high SMN protein levels are required for brain development (6). We speculate that the brain MRI changes may be related to SMA, although few studies have explored this. To further explore the characteristics of MRI changes in the brain of SMA, we reviewed the previously published English literature. Three case-control studies showed abnormal gray matter changes in patients with SMA, and seven individual studies reported a total of 16 patients with different types of SMA. They mention the presence of symmetrical high signal in the white matter, putamen and thalamus in patients with SMA type 0 (7); periventricular posterior horn of the lateral ventricle and bilateral anterolateral thalamus high-signal-intensity lesions in children with SMA type I (8); periventricular high intensity around the posterior horns of lateral ventricles and delayed myelin formation in patients with SMA type II (9) (Table 1).
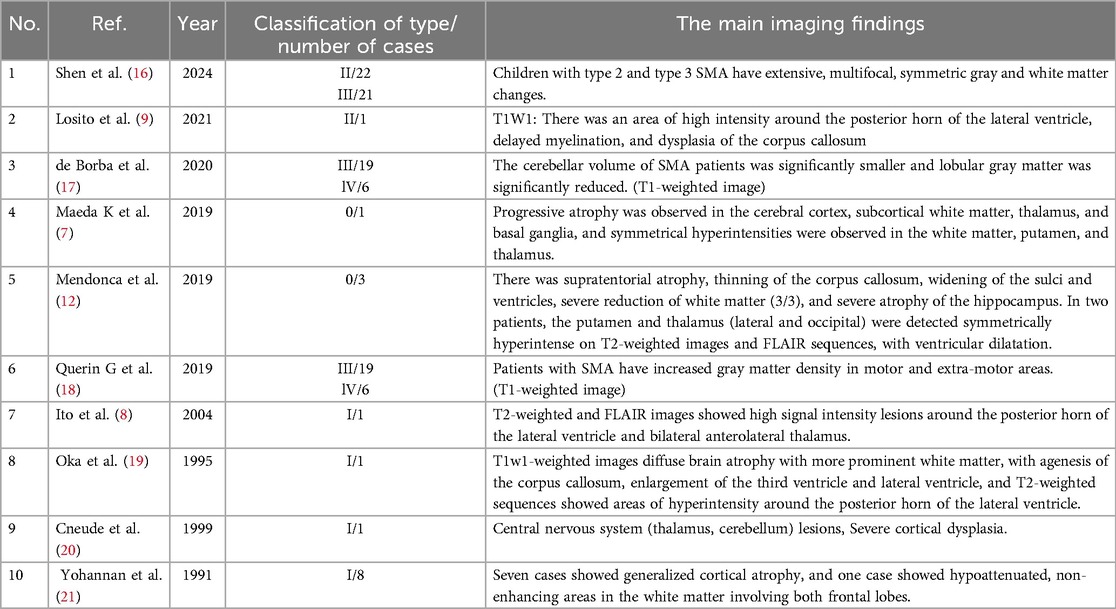
Table 1. Review the literature on brain MRI signal changes in SMA patients.
Further exploration of the pathogenesis suggests that there is a modifier gene for SMA, the zinc finger protein (ZPR1) gene (10). It can interact with SMN sites to induce neuronal differentiation, stimulate axon growth in motor neuron-like cells, and increase SMN levels (10, 11). We hypothesized that previously reported cerebral atrophy may be related to chronic hypoxia in the brain (12), because defects in ZPR1 in critically ill SMA patients contribute to phrenic nerve axonal loss of function and myelin proliferation, leading to defects in diaphragmatic respiratory function causing respiratory muscle weakness, which ultimately can lead to chronic hypoxia in the brain (10), Hypoxia can cause brain MRI signal changes, and abnormal signals may appear in the basal ganglia, thalamus and surrounding cortex (13). Moreover, the low SMN levels in SMA can lead to motor neuron degeneration, potentially causing insufficient myelin maturation. This could explain white matter atrophy and high-signal intensity in the lateral ventricle and thalamus, which may reflect unmyelinated regions or abnormal myelin development (14). Obviously, these brain MRI changes are unusual, and the brain MRI changes in SMA patients are diverse, and we cannot conclude that the brain MRI changes in SMA patients are specific. The mechanism of brain MRI changes in patients with SMA is not fully understood, and the evidence that ZPR1 deficiency allows hypoxic episodes and reduced SMN protein levels to cause brain MRI signal alterations remains insufficient. However, abnormal MRI signal may indicate the presence of SMA, and clinicians should be alert to the occurrence of such diseases. This paper reports abnormal MRI signal changes in a patient with SMA, suggesting a possible connection to the disease. A limitation of this case is that the child did not undergo SMN2 gene copy number testing, but in combination with the time of onset of the disease and the characteristics of the child, we made a clinical diagnosis of SMN type 0.
SMN1 deletion and SMN2 copy number can be detected by a variety of techniques, with genetic testing serving as the gold standard for diagnosing SMA. Although brain MRI is not required for diagnosis, it can reveal structural brain changes and is a widely available, non-invasive tool that may assist in the diagnostic process. Early treatment has been shown to prevent the most severe forms of SMA, with its effectiveness highly dependent on early administration. Presymptomatic treatment can result in normal or mildly subnormal motor development that would otherwise progress to severe disease. Currently, several approaches are available for early treatment of SMA (15), which are based on the general principle of increasing SMN protein expression. Pharmacological or gene therapies that increase SMN2 expression, antisense oligonucleotide (ASO) -based therapies, and virus-mediated therapies are included (1). ZPR1 upregulates SMN2 protein transcription and promotes SMN protein activation for myelin regeneration, and upregulation of ZPR1 expression to increase SMN levels is a viable therapeutic target for the development of new approaches to SMA treatment (10, 11).
Conclusion
Brain MRI signal changes of SMA are rare, with high mortality and poor prognosis. So far, SMA can only delay the progression of the disease rather than completely cure it (1), which highlights the difficulty and importance of differential diagnosis of this disease. Genetic testing is the key to the diagnosis of SMA, and cranial MRI may be helpful for the diagnosis. Prenatal diagnosis and newborn screening are the most important prevention options, and early identification and treatment can help improve the prognosis of SMA patients.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
XJ: Writing – original draft. CY: Writing – original draft. YW: Writing – original draft. RZ: Writing – review & editing. WX: Writing – review & editing. WD: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kolb SJ, Kissel JT. Spinal muscular atrophy. Neurol Clin. (2015) 33:831–46. doi: 10.1016/j.ncl.2015.07.004
2. Burr P, Reddivari AKR. Spinal Muscle Atrophy, StatPearls, StatPearls Publishing Copyright © 2024. Treasure Island (FL): StatPearls Publishing LLC (2024). Ineligible companies. Disclosure: Anil Kumarch Reddy Reddivari declares no relevant financial relationships with ineligible companies.
3. Ojala KS, Reedich EJ, DiDonato CJ, Meriney SD. In search of a cure: the development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. (2021) 11(2):194. doi: 10.3390/brainsci11020194
4. Ramos DM, d'Ydewalle C, Gabbeta V, Dakka A, Klein SK, Norris DA, et al. Age-dependent SMN expression in disease-relevant tissue and implications for SMA treatment. J Clin Invest. (2019) 129:4817–31. doi: 10.1172/JCI124120
5. Masson R, Brusa C, Scoto M, Baranello G. Brain, cognition, and language development in spinal muscular atrophy type 1: a scoping review. Dev Med Child Neurol. (2021) 63:527–36. doi: 10.1111/dmcn.14798
6. Wishart TM, Huang JP, Murray LM, Lamont DJ, Mutsaers CA, Ross J, et al. SMN Deficiency disrupts brain development in a mouse model of severe spinal muscular atrophy. Hum Mol Genet. (2010) 19:4216–28. doi: 10.1093/hmg/ddq340
7. Maeda K, Chong PF, Yamashita F, Akamine S, Kawakami S, Saito K, et al. Global central nervous system atrophy in spinal muscular atrophy type 0. Ann Neurol. (2019) 86:801–2. doi: 10.1002/ana.25596
8. Ito Y, Kumada S, Uchiyama A, Saito K, Osawa M, Yagishita A, et al. Thalamic lesions in a long-surviving child with spinal muscular atrophy type I: MRI and EEG findings. Brain Dev. (2004) 26:53–6. doi: 10.1016/S0387-7604(03)00075-5
9. Losito L, Gennaro L, Lucarelli E, Trabacca A. Brain MRI abnormalities in a child with spinal muscular atrophy type II. Acta Neurol Belg. (2021) 121:1883–5. doi: 10.1007/s13760-020-01524-x
10. Ahmad S, Wang Y, Shaik GM, Burghes AH, Gangwani L. The zinc finger protein ZPR1 is a potential modifier of spinal muscular atrophy. Hum Mol Genet. (2012) 21:2745–58. doi: 10.1093/hmg/dds102
11. Kannan A, Jiang X, He L, Ahmad S, Gangwani L. ZPR1 Prevents R-loop accumulation, upregulates SMN2 expression and rescues spinal muscular atrophy. Brain. (2020) 143:69–93. doi: 10.1093/brain/awz373
12. Mendonça RH, Rocha AJ, Lozano-Arango A, Diaz AB, Castiglioni C, Silva AMS, et al. Severe brain involvement in 5q spinal muscular atrophy type 0. Ann Neurol. (2019) 86:458–62. doi: 10.1002/ana.25549
13. Parmentier CEJ, de Vries LS, Groenendaal F. Magnetic resonance imaging in (Near-)term infants with hypoxic-ischemic encephalopathy. Diagnostics. (2022) 12:645. doi: 10.3390/diagnostics12030645
14. Mugisha N, Oliveira-Carneiro A, Behlim T, Oskoui M. Brain magnetic resonance imaging (MRI) in spinal muscular atrophy: a scoping review. J Neuromuscul Dis. (2023) 10:493–503. doi: 10.3233/JND-221567
15. De Siqueira Carvalho AA, Tychon C, Servais L. Newborn screening for spinal muscular atrophy—what have we learned? Expert Rev Neurother. (2023) 23:1005–12. doi: 10.1080/14737175.2023.2252179
16. Shen W, Yan Z, Su S, Xiang P, Zhou Q, Zou M, et al. Gray and white matter abnormalities in children with type 2 and 3 SMA: a morphological assessment. Eur J Pediatr. (2024) 183:1381–8. doi: 10.1007/s00431-023-05397-z
17. de Borba FC, Querin G, França MC Jr., Pradat PF. Cerebellar degeneration in adult spinal muscular atrophy patients. J Neurol. (2020) 267:2625–31. doi: 10.1007/s00415-020-09875-4
18. Querin G, El Mendili MM, Lenglet T, Behin A, Stojkovic T, Salachas F, et al. The spinal and cerebral profile of adult spinal-muscular atrophy: a multimodal imaging study. Neuroimage Clin. (2019) 21:101618. doi: 10.1016/j.nicl.2018.101618
19. Oka A, Matsushita Y, Sakakihara Y, Momose T, Yanaginasawa M. Spinal muscular atrophy with oculomotor palsy, epilepsy, and cerebellar hypoperfusion. Pediatr Neurol. (1995) 12:365–9. doi: 10.1016/0887-8994(95)00058-N
20. Cneude F, Sukno S, Boidein F, Dehouck MB, Bourlet A, Vittu G. Cerebral agyria-pachygyria in a child with Werdnig-Hoffmann disease. Rev Neurol. (1999) 155:589–91.10486849
Keywords: neonatal, spinal muscular atrophy (SMA), SMN, brain MRI, case report
Citation: Jieda X, Yang C, Wu Y, Zhang R, Xu W and Dong W (2025) Neonatal spinal muscular atrophy with brain magnetic resonance imaging hypersignal: a case report. Front. Pediatr. 13:1508565. doi: 10.3389/fped.2025.1508565
Received: 9 October 2024; Accepted: 17 April 2025;
Published: 29 April 2025.
Edited by:
Shoulong Deng, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaReviewed by:
Lorenzo Pavone, University of Catania, ItalyElvio Della Giustina, University Hospital of Modena, Italy
Copyright: © 2025 Jieda, Yang, Wu, Zhang, Xu and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenbin Dong, ZG9uZ3dlbmJpbjIwMDBAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship