 Jiaojiao Yin
Jiaojiao Yin Dan Huang2
Dan Huang2- 1Department of Clinical Laboratory, Gansu Provincial Maternity and Child-care Hospital, Lanzhou, Gansu, China
- 2School of Public Health, Gansu University of Chinese Medicine, Lanzhou, Gansu, China
- 3Department of Clinical Laboratory, 940th Hospital of Chinese People’s Liberation Army Joint Support Force, Lanzhou, Gansu, China
Pelger-Huët anomaly (PHA), an autosomal dominant disorder characterized by abnormal granulocyte morphology, was first described in 1928. Mutations in the lamin B receptor (LBR) gene cause a phenotypic spectrum ranging from isolated PHA, PHA with mild skeletal abnormalities, to the embryonic-lethal Greenberg skeletal dysplasia. We report a Chinese boy presenting peripheral blood granulocyte abnormalities associated with a novel LBR gene mutation. Whole-exome sequencing uncovered the LBR gene heterozygous mutation, NM_194442.2: c.561C > G (p.Tyr187*). Notably, the patient exhibited scoliosis secondary to hemivertebrae, potentially representing a previously unreported skeletal manifestation of mutations in the LBR gene. Analyzing the differential diagnosis between PHA, immature granulocytes, and pseudo-PHA, along with elucidating genotype-phenotype correlations for LBR mutations, is crucial for advancing our understanding of PHA and related disorders.
1 Introduction
Pelger-Huët anomaly (PHA) is an inherited morphologic disorder characterized by granulocyte nuclear hyposegmentation, typically manifesting as dumbbell-shaped or spectacle-like bilobed nuclei with coarse chromatin clumping (1). This autosomal dominant condition has been molecularly linked to chromosome 1q41-43 (2), specifically attributed to pathogenic variants in the lamin B receptor (LBR, MIM 600024) gene at 1q42.1 (3). While PHA is classically considered a benign hematologic finding, emerging evidence suggests potential associations with mild skeletal dysplasias (4). Herein, we report a pediatric case of genetically confirmed PHA exhibiting severe congenital scoliosis secondary to hemivertebrae formation, coupled with a novel heterozygous missense mutation in the LBR gene (c.561C > G).
2 Case report
A 2-year-old boy was admitted to our hospital for physical examination. Routine peripheral blood examination showed the white blood cell count of 6.13 × 109/L with normal differential counts (neutrophils 29.2%, eosinophils 8.4%, basophils 0.8%, monocytes 8.4%, and lymphocytes 53.2%). However, the percentage of immature granulocytes (IG%) is 2.4% that violated the routine blood retest rules and the Sysmex DI-60 analyzer was rechecked with an automatic pusher. A total of 110 cells were randomly analyzed including 17 neutrophils (5 mono-lobed and 12 bi-lobed nuclei) and 11 eosinophils (6 mono-lobed and 5 bi-lobed nuclei) (Figure 1A). To rule out hereditary PHA, we contacted the boy's biological parents for a peripheral blood smear and confirmed the existence of similar neutrophil-related morphological abnormalities in his biological mother while the boy's biological father had normal granulocytes (Figure 1A). Based on the physical examination and medical history, the boy was found to have scoliosis. Spinal radiographs showed that he had lumbar scoliosis and lumbar 2-vertebral hemivertebral deformity (Figure 1B). His mother was found fetal scoliosis at 23 weeks of pregnancy by systematic color Doppler ultrasound screening (Figure 1C).
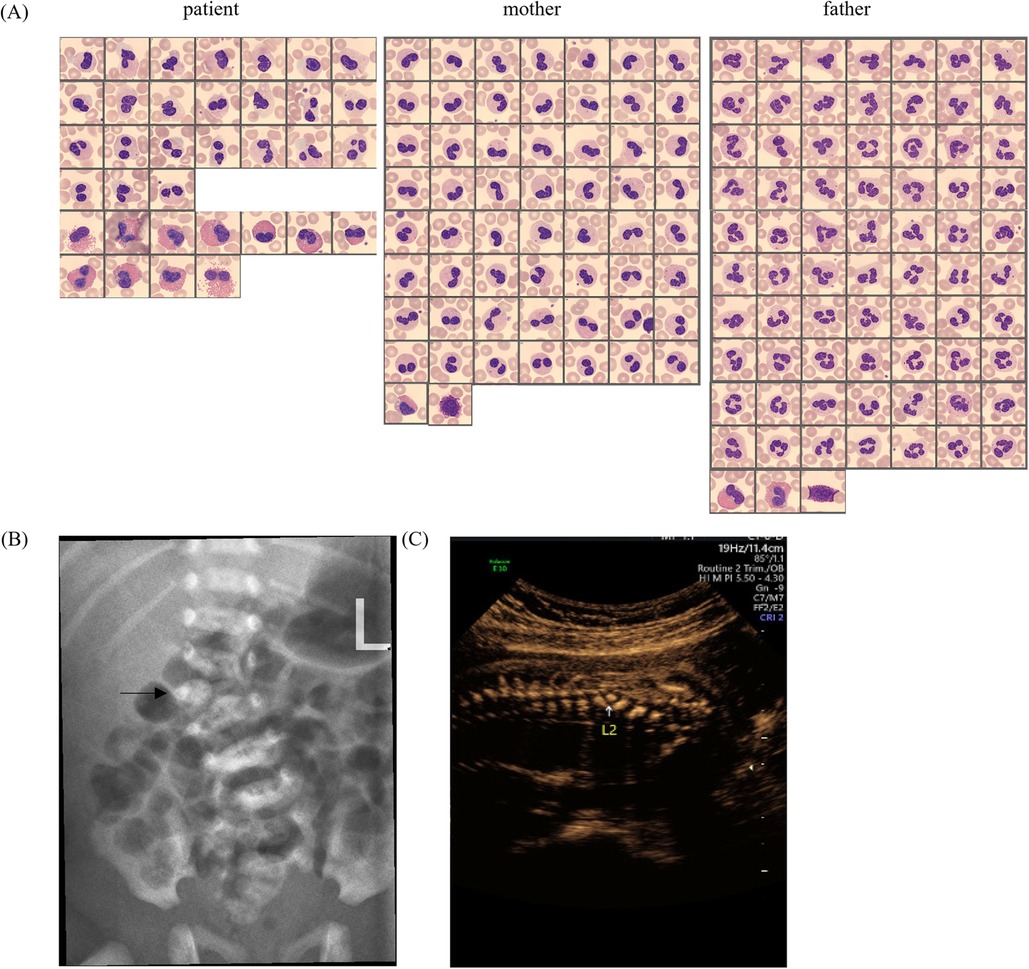
Figure 1. (A) Peripheral blood smear micrographs from the patient, mother, and father, showing Giemsa-stained granulocytes. (B) Lateral spinal radiograph (X-ray) with an arrow indicating a specific vertebral anomaly. (C) Prenatal ultrasound image focusing on the L2 vertebra, demonstrating abnormal ossification.
From birth to present, the child had a regular health check-up almost every three months. Clinical features assessed showed slightly lower head circumference, length, and weight when plotted on a standard WHO chart (Supplementary Figures S1A–C). The serum thyroid-stimulating hormone and genetic metabolites levels are within the normal ranges (Supplementary Table S1). The levels of 25-hydroxyvitamin D, serum ferritin and trace elements (including calcium, iron, zinc, lead, copper, cadmium, potassium, sodium, magnesium) were almost within the normal ranges (Supplementary Table S2). At postnatal 9 and 18 months, the Gesell Developmental Schedules (GDS) score (5) were under 86 and more than 85, respectively (Supplementary Figure S1D). Overall, the boy had slightly stunted growth but normal mental development.
To identify the potential gene mutation, the Whole-exome high-throughput sequencing is used, including exons of approximately 20,000 genes in the human genome and the mitochondrial genome. A new mutation site in the LBR gene was discovered: c.561C > G (p. Tyr187*). Sanger sequencing of the boy and his parents validated the candidate LBR gene variant that both the child and his mother were found to have heterozygous mutations at the locus (Figure 2). According to the ACMG (American College of Medical Genetics and Genomics) guidelines, this variant has been preliminarily classified as pathogenic: PVS1 + PP4 + PM2.
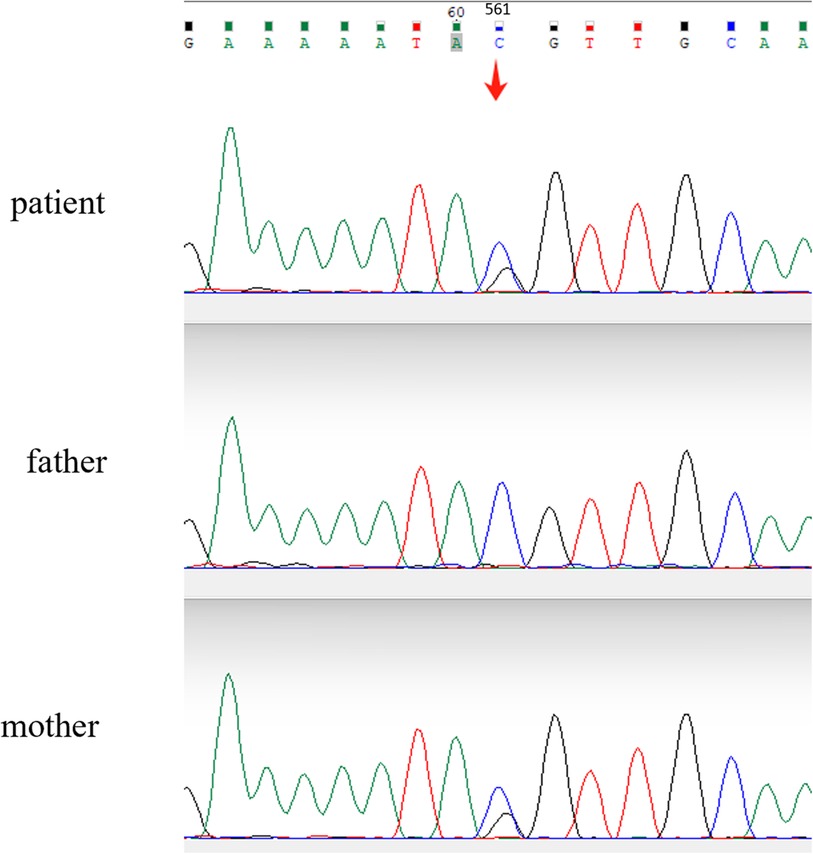
Figure 2. Sanger sequencing chromatograms comparing the LBR gene sequences of the patient and parents. A heterozygous mutation is evident in the patient's sequence (arrow), where a nucleotide substitution from cytosine (C) to guanine (G) occurs at position c.561, resulting in a missense variant.
3 Discussion
PHA was first discovered by Pelger in the peripheral blood of two tuberculosis patients, mistakenly believing that the nuclear was shifted to the left due to infection (6). The IG fraction includes promyelocytes, myelocytes and metamyelocytes (blasts and band cells are not included). Elevated IG% is common in the peripheral blood of patients with bacterial infections, neonates, pregnant women, and patients taking recombinant human granulocyte colony-stimulating factor(rhG-CSF) (7–9). When IG% is elevated, mild- to late myelocytes are seen on peripheral blood smear but not in PHA. Secondly, the large amount of chromatin condensation observed in the nucleus of PHA granulocytes can help distinguish PHA from the “left shift” of neutrophils. The Figure 1A shows the neutrophils of the child and his mother have a large amount of chromatin condensation compared to the father.
PHA needs to be differentiated from pseudo-PHA which is an acquired neutrophil dysplasia similar to PHA. This abnormality is frequently associated with myelodysplastic syndromes and also described in some clinical situations, especially under the effect of certain drugs (10). Congenital PHA is associated with 55% to 95% of circulating neutrophils exhibiting classic dysmorphology (11) and the frequency of pseudo-PHA neutrophils has ranged between 77% and 99% (12). We cannot distinguish PHA from pseudo-PHA by abnormal granulocyte ratios, however, pseudo-PHA has morphologically normal eosinophils and basophils (12), which are abnormal in PHA (Figure 1A). Additionally, differentiation can be achieved by taking a detailed patient medical history and performing morphological analysis of neutrophils in first-degree relatives.
The LBR gene contains 35 kb bases, encodes 615 amino acids, and has 13 coding regions, of which 1–4 are N-terminus, nucleoplasmic domain, and 5–13 are C-terminus, hydrophobic domain. Two seemingly unrelated functions have been attributed to LBR. The nucleoplasmic domain is associated with lamin B, chromatin, and other proteins causing crucial changes in nuclear architecture (3), while its transmembrane domains exhibit sterol reductase activity (13). Deletion of LBR N-terminal domains using CRISPR/Cas-9 gene editing technology in the mouse affected the morphology and chromatin organization of white blood cells but not skin or skeletal defects (14). However, in human there is no evidence that different phenotypes are associated with different LBR domains (Table 1). Genetic variation in LBR affects the expression of LBR protein, which in turn correlates with a continuum of clinical manifestations ranging from no phenotype to isolated PHA through PHA with mild skeletal dysplasia to Greenberg skeletal dysplasia (Table 1). Mutations in the LBR gene is a form of nuclear envelopathies and exhibit the characteristic phenotype of causing PHA (15). So far, we have found point mutation, splice site, frameshift and nonsense mutations in LBR (16). Heterozygous LBR mutations lead to nuclear hyposegmentation of neutrophils without causing disease, except for missense mutations such as p. Arg583Gln which did not alter nuclear shape in neutrophils (17). However, patients with homozygous/compound heterozygous mutations in LBR can lead to severe perinatal fatal autosomal recessive skeletal dysplasia, Greenberg skeletal dysplasia, and even those who survive are accompanied by severe skeletal dysplasia (Table 1). In this respect, heterozygote for mutation p. Tyr187* was associated with PHA with scoliosis. Notably, we also found that nonsense mutations, base pair insertions or deletions causing frame shifts that create premature stop codons can all lead to PHA (Table 1).
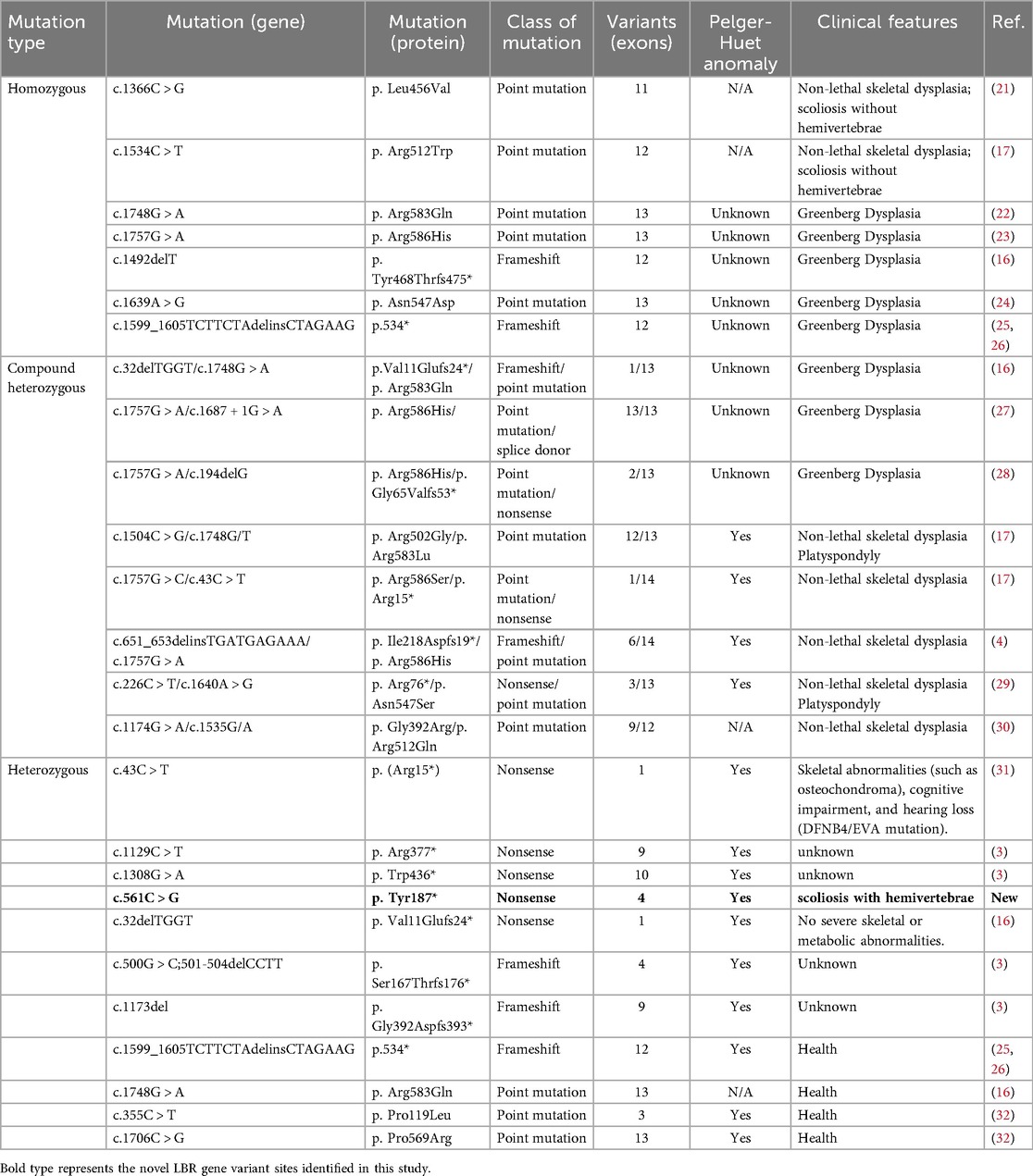
Table 1. Congenital disorders associated with mutations in the lamin B receptor gene.
In previous reports, skeletal abnormalities associated with LBR gene mutations included short stature, short upper extremities, short metacarpal bones, postaxial polydactyly, and kyphosis, with some cases showing reduced severity and spontaneous resolution of skeletal manifestations and imaging features with age (18). Although scoliosis has been reported in previous cases, scoliosis due to the hemivertebrae has been reported for the first time. In this case, the skeletal abnormality manifests as congenital scoliosis, a form of vertebral malformation with a genetic susceptibility (19). The genetic basis of congenital scoliosis is complex, involving mutations in multiple genes, particularly those related to the Notch signaling pathway, such as TBX6 and LFNG (20). This report provides additional evidence of variability for LBR-related disorders associated with Pelger-Huët anomaly, i.e., congenital scoliosis caused by hemivertebrae, which is not spontaneous regression.
4 Conclusion
This case report describes a novel LBR mutation identified in a child presenting with PHA and hemivertebrae, further expanding the known phenotypic spectrum associated with LBR mutations. Our report details a heterozygous mutation. Analysis of current literature on congenital disorders linked to LBR gene mutations reveals that individuals with heterozygous LBR mutations are generally healthy apart from the characteristic features of PHA. This finding highlights the necessity of early PHA diagnosis, which facilitates preimplantation genetic testing (PGT) implementation to block intergenerational transmission of the pathogenic variant. Whether the occurrence of congenital scoliosis in this child is associated with the LBR gene mutation requires additional cases and functional studies to elucidate the underlying mechanisms and refine therapeutic strategies.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Institutional Review Boards of Gansu Provincial Maternity and Child-care Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
JY: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. DH: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review & editing. ZL: Conceptualization, Data curation, Formal analysis, Resources, Software, Supervision, Validation, Writing – review & editing. EZ: Formal analysis, Methodology, Software, Validation, Visualization, Writing – review & editing. CZ: Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing. LW: Funding acquisition, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing. BL: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Natural Science Foundation of Gansu Province, China (Grant 25JRRA424).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issue please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2025.1587175/full#supplementary-material
References
1. Shah SS, Parikh RS, Vaswani LP, Divkar R. Familial pelger–huet anomaly. Indian J Hematol Blood Transfus. (2015) 32(S1):347–50. doi: 10.1007/s12288-015-0508-3
2. Kalfa TA, Zimmerman SA, Goodman BK, McDonald MT, Ware RE. Pelger–Huët anomaly in a child with 1q42.3-44 deletion. Pediatr Blood Cancer. (2005) 46(5):645–8. doi: 10.1002/pbc.20504
3. Hoffmann K, Dreger CK, Olins AL, Olins DE, Shultz LD, Lucke B, et al. Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger–Huët anomaly). Nat Genet. (2002) 31(4):410–4. doi: 10.1038/ng925
4. Borovik L, Modaff P, Waterham HR, Krentz AD, Pauli RM. Pelger–Huet anomaly and a mild skeletal phenotype secondary to mutations in LBR. Am J Med Genet A. (2013) 161(8):2066–73. doi: 10.1002/ajmg.a.36019
5. Li Z, Pan L, Chen Y, Meng D, Liu Y, Li L, et al. The value of prenatal magnetic resonance imaging and postnatal follow-up using gesell developmental schedules score for mild-to-moderate simple bilateral fetal ventriculomegaly. J Matern Fetal Neonatal Med. (2021) 35(25):6229–35. doi: 10.1080/14767058.2021.1910657
6. Haverkamp Begemann N, van Lookeren Campagne A. Homozygous form of Pelger-Huët’s nuclear anomaly in man. Acta Haematol. (1952) 7(5):295–303. doi: 10.1159/000204063
7. Carulli G. Effects of recombinant human granulocyte colony-stimulating factor administration on neutrophil phenotype and functions. Haematologica. (1997) 82(5):606–16.9407734
8. Fernández-Suárez A, Pascual VT, Gimenez MTF, Hernández JFS. Immature granulocyte detection by the SE-9000 haematology analyser during pregnancy. Clin Lab Haematol. (2003) 25(6):347–51. doi: 10.1046/j.0141-9854.2003.00551.x
9. Shane AL, Sánchez PJ, Stoll BJ. Neonatal sepsis. Lancet. (2017) 390(10104):1770–80. doi: 10.1016/S0140-6736(17)31002-4
10. Dusse LMS, Moreira AMB, Vieira LM, Rios DRA, Morais e Silva RM, das Graças Carvalho M. Acquired Pelger–Huët: what does it really mean? Clin Chim Acta. (2010) 411(21–22):1587–90. doi: 10.1016/j.cca.2010.07.011
11. Roberts T, Linenberger M. A case of benign Pelger-Huët anomaly. Blood. (2015) 126(5):693. doi: 10.1182/blood-2015-04-640474
12. Savaşan S, Buck S, Gadgeel M, Poulik J. Persistent pseudo-Pelger-Huët anomaly. Ann Hematol. (2020) 100(10):2661–3. doi: 10.1007/s00277-020-04242-9
13. Nikolakaki E, Mylonis I, Giannakouros T. Lamin B receptor: interplay between structure, function and localization. Cells. (2017) 6(3):28. doi: 10.3390/cells6030028
14. Young AN, Perlas E, Ruiz-Blanes N, Hierholzer A, Pomella N, Martin-Martin B, et al. Deletion of LBR N-terminal domains recapitulates Pelger-Huet anomaly phenotypes in mouse without disrupting X chromosome inactivation. Commun Biol. (2021) 4(1):478. doi: 10.1038/s42003-021-01944-2
15. Worman HJ, Ostlund C, Wang Y. Diseases of the nuclear envelope. Cold Spring Harb Perspect Biol. (2010) 2(2):a000760. doi: 10.1101/cshperspect.a000760
16. Turner EM, Schlieker C. Pelger-Huët anomaly and greenberg skeletal dysplasia: lBR-associated diseases of cholesterol metabolism. Rare Diseases. (2016) 4(1):e1241363. doi: 10.1080/21675511.2016.1241363
17. Clayton P, Fisher B, Mann A, Mansour S, Rossier E, Veen M, et al. Mutations causing greenberg dysplasia but not pelger anomaly uncouple enzymatic from structural functions of a nuclear membrane protein. Nucleus. (2010) 1(4):354–66. doi: 10.4161/nucl.1.4.12435
18. Thompson E, Abdalla E, Superti-Furga A, McAlister W, Kratz L, Unger S, et al. Lamin B receptor-related disorder is associated with a spectrum of skeletal dysplasia phenotypes. Bone. (2019) 120:354–63. doi: 10.1016/j.bone.2018.11.006
19. Wu N, Ming X, Xiao J, Wu Z, Chen X, Shinawi M, et al. TBX6Null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med. (2015) 372(4):341–50. doi: 10.1056/NEJMoa1406829
20. Takeda K, Kou I, Mizumoto S, Yamada S, Kawakami N, Nakajima M, et al. Screening of known disease genes in congenital scoliosis. Mol Genet Genomic Med. (2018) 6(6):966–74. doi: 10.1002/mgg3.466
21. Collins M, Miranda V, Rousseau J, Kratz LE, Campeau PM. A homozygous variant in the lamin B receptor gene LBR results in a non-lethal skeletal dysplasia without Pelger-Huët anomaly. Bone. (2020) 141:115601. doi: 10.1016/j.bone.2020.115601
22. Gregersen PA, McKay V, Walsh M, Brown E, McGillivray G, Savarirayan R. A new case of greenberg dysplasia and literature review suggest that greenberg dysplasia, dappled diaphyseal dysplasia, and astley–kendall dysplasia are allelic disorders. Mol Genet Genomic Med. (2020) 8(6):e1173. doi: 10.1002/mgg3.1173
23. Shen X, Li Z, Pan X, Yao J, Shen G, Zhang S, et al. Prenatal diagnosis of recurrent moderate skeletal dysplasias in lamin B receptors. Front Genet. (2023) 13:1020475. doi: 10.3389/fgene.2022.1020475
24. Konstantinidou A, Karadimas C, Waterham HR, Superti-Furga A, Kaminopetros P, Grigoriadou M, et al. Pathologic, radiographic and molecular findings in three fetuses diagnosed with HEM/greenberg skeletal dysplasia. Prenat Diagn. (2008) 28(4):309–12. doi: 10.1002/pd.1976
25. Waterham HR, Koster J, Mooyer P, Noort G, Kelley RI, Wilcox WR, et al. Autosomal recessive HEM/greenberg skeletal dysplasia is caused by 3β-hydroxysterol Δ14-reductase deficiency due to mutations in the lamin B receptor gene. Am J Hum Genet. (2003) 72(4):1013–7. doi: 10.1086/373938
26. Tsai P-L, Zhao C, Turner E, Schlieker C. The lamin B receptor is essential for cholesterol synthesis and perturbed by disease-causing mutations. eLife. (2016) 5:e16011. doi: 10.7554/eLife.16011
27. Ding Y, Wang T, Xiang J. Genetic analysis of a fetus with rhizomelic skeletal dysplasia. Chin J Med Genet. (2024) 41(7):844–8. doi: 10.3760/cma.j.cn511374-20230523-00309
28. Peng Y, Yang S, Huang X, Pang J, Liu J, Hu J, et al. Whole exome sequencing analysis in fetal skeletal dysplasia detected by ultrasonography: an analysis of 38 cases. Front Genet. (2021) 12:728544. doi: 10.3389/fgene.2021.728544
29. Sobreira N, Modaff P, Steel G, You J, Nanda S, Hoover-Fong J, et al. An anadysplasia-like, spontaneously remitting spondylometaphyseal dysplasia secondary to lamin B receptor (LBR) gene mutations: further definition of the phenotypic heterogeneity of LBR-bone dysplasias. Am J Med Genet Part A. (2014) 167(1):159–63. doi: 10.1002/ajmg.a.36808
30. Zhang W, Taylor SP, Ennis HA, Forlenza KN, Duran I, Li B, et al. Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies. Hum Mutat. (2018) 39(1):152–66. doi: 10.1002/humu.23362
31. Cinleti T, Yılmaz Uzman C, Akyol Ş, Tüfekçi Ö, Erçal MD, Giray Bozkaya Ö. Blended phenotype of Pelger-Huet anomaly with osteochondroma and autosomal recessive deafness with enlarged vestibular aqueduct. Mol Syndromol. (2022) 13(3):200–5. doi: 10.1159/000519364
Keywords: Pelger-Huët anomaly, congenital scoliosis, hemivertebrae, lamin B receptor, nonsense mutation
Citation: Yin J, Huang D, Liu Z, Zhu E, Zhang C, Wang L and Li B (2025) Novel lamin B receptor mutation (c.561C > G) in a patient with Pelger-Huët anomaly: a case report. Front. Pediatr. 13:1587175. doi: 10.3389/fped.2025.1587175
Received: 4 March 2025; Accepted: 25 August 2025;
Published: 4 September 2025.
Edited by:
Hua Zhong, University of Hawaii at Manoa, United StatesCopyright: © 2025 Yin, Huang, Liu, Zhu, Zhang, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Linyan Wang, d2x5YW4xOTc0QDE2My5jb20=; Bing Li, bGliaW5nX2l3b3JrQDE2My5jb20=