 Jacek Kobak1*
Jacek Kobak1* Mateusz Szczupak
Mateusz Szczupak Karolina Czerkiewicz
Karolina Czerkiewicz Sabina Krupa-Nurcek
Sabina Krupa-Nurcek- 1Department of Otolaryngology, Faculty of Medicine, Medical University of Gdańsk, Gdańsk, Poland
- 2Department of Anesthesiology and Intensive Care, Copernicus Hospital, Gdańsk, Poland
- 3Student of Department of Surgery, Institute of Medical Sciences, Medical College of Rzeszów University, Gdańsk, Poland
- 4Department of Orthopedic and Spinal Surgery, Medical University of Gdańsk, Rzeszów, Poland
- 5Department of Surgery, Institute of Medical Sciences, Medical College of Rzeszów University, Rzeszów, Poland
Introduction: PURA syndrome is a rare genetic disorder first described in the medical literature in 2014. It is caused by pathogenic variants in the PURA gene, which is located on chromosome 5. The PURA gene is crucial for the production of the pur-α protein, which is expressed in all tissues, including the nervous system, muscles, and blood. The pur-α protein plays a vital role in normal brain development. The estimated incidence of PURA syndrome is 1 in 1,000,000, and as of 2024, approximately 706 cases of the syndrome have been identified worldwide.
Aim of study: The aim of the study was to present a case description of PURA syndrome and the genetic basis of the neurodevelopmental disorder in a 15-year-old girl.
Case report: This manuscript presents the case of a 15-year-old girl of Polish descent diagnosed with PURA syndrome through genetic testing. She was admitted to the Department of Orthopedics and Spine Surgery at the Medical University of Gdansk for surgical treatment of advanced idiopathic scoliosis caused by a postural defect.
Conclusion: PURA syndrome is a rare genetic condition that requires further research and observation. Although it shares many clinical features with other neurological disorders, certain symptoms—such as speech disorders, the ability to follow and execute simple commands, and an excessive acoustic reaction to surprises—should raise suspicion of this condition. These indicators should prompt genetic testing for confirmation and the implementation of appropriate multidisciplinary care for the patient.
1 Introduction
PURA syndrome is a neurodevelopmental disorder inherited in an autosomal dominant manner, classified as a rare disease (1). It is listed in the OMIM (Online Mendelian Inheritance in Man) database with the identifier #616158 (2). The PURA gene (Purine-rich element-binding protein A) encodes the Pur-α protein, which has regulatory functions in processes such as DNA repair and replication, as well as mRNA transport and translation (3, 4). This protein plays a crucial role in proper brain development after birth, including the formation of new synaptic connections, maturation of dendrites, and the proliferation of neural cells (5–8). PURA syndrome is caused by a heterozygous pathogenic sequence variant in the PURA gene, which is located on chromosome 5q31 (9). As of October 2024, approximately 706 cases of PURA syndrome have been confirmed worldwide across 60 countries (10). A total of 317 pathogenic variants of the PURA gene have been identified, with the most common variant being p.Phe233del (10). de novo mutations are most common (9, 11). PURA syndrome is characterized by moderate to severe developmental delays, including challenges in motor skills and speech. Individuals with this syndrome may experience hypothermia, hypotonia (reduced muscle tone), apnea (breathing interruptions), feeding difficulties, excessive hiccups, uncoordinated eye movements, visual disturbances, dystonia (involuntary muscle contractions), dyskinesia (difficulty with movement) and scoliosis, which occurs in 48% of cases (1, 9, 12–14). The individual displays facial features that may be considered dysmorphic (15). Less commonly, PURA syndrome may be associated with congenital heart defects, endocrine disorders, genitourinary malformations, and skeletal abnormalities (1, 9, 12, 13). A mutation in the PURA gene is linked to an autosomal dominant form of intellectual disability type 31 (OMIM:616158).
The majority of individuals affected by PURA syndrome are nonverbal, and many are unable to move independently (9). Approximately half of these patients experience epilepsy, with seizures typically beginning in infancy or early childhood; however, the age of onset can vary significantly (5). The syndrome is associated with a high incidence of epilepsy. There is an unclear relationship between genotype and phenotype in PURA syndrome, as patients with identical genetic variants can exhibit a wide range of symptoms and severity. Despite significant advances in molecular genetics, the complete clinical characteristics and epidemiological profile of PURA syndrome are still not fully understood (14, 16, 17). It is important to note that no cases from several continents have been documented in the existing scientific literature (14, 16, 17). The aim of the study was to present a case description of PURA syndrome and the genetic basis of the neurodevelopmental disorder in a 15-year-old girl.
Taniguchi et al. conducted a systematic review to explore the genotype-phenotype correlations in neurodevelopmental disorders associated with PURA syndrome. The authors found that patients with the 5q31.3 deletion syndrome experienced a higher incidence of congenital malformations, respiratory difficulties, and gait issues. In the case of PURA syndrome, variants that cause protein shortening, such as nonsense or frameshift mutations, were linked to increased speech deficits. Interestingly, the location of the PURA variant did not influence the occurrence of congenital defects or neurodevelopmental outcomes (18). Symptoms in PURA are presented on Figure 1.
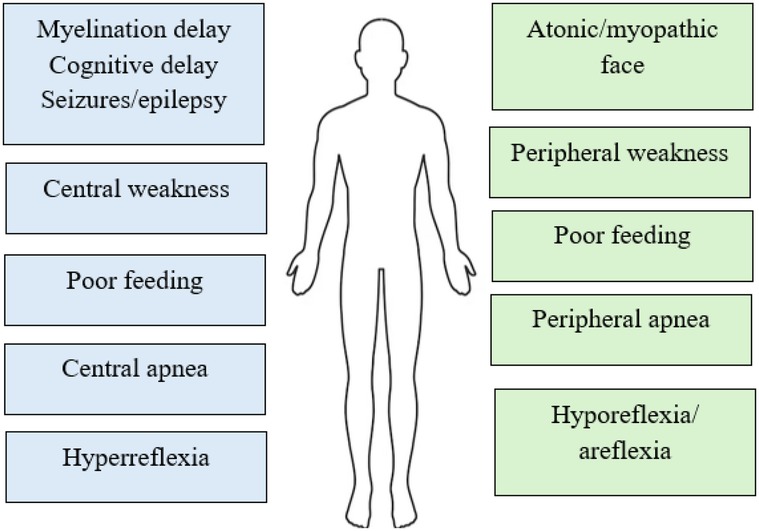
Figure 1. Central vs. peripheral symptoms in PURA (30).
2 PURA protein structure
PURA protein (purine-rich element binding protein A) is a protein consisting of 322 amino acids with repeated nucleic acid-binding domains (15). It is encoded by the PURA gene located on chromosome 5 (5q31) (4, 19). The PURA protein is primarily located in the cell nucleus and cytoplasm of neurons (4, 6, 20–22). The molecular weight of the PURA protein is 35–37 kDa. There are three domains of this protein, the so-called PUR repeats, namely PUR I, PUR II and PUR III (4). In humans, the PUR domain typically consists of 55–70 amino acids. Each of these domains can bind both DNA and RNA (23). Additionally, PUR they domains participate in dimer formation and interact with other proteins (24) Notably, the structure of the PURA protein lacks classical domains such as the helix-turn-helix (HTH) motif or a zinc finger (Figure 2). One of its key structural features is a high content of α-helical motifs, along with a tertiary structure that enables flexible nucleotide binding and involvement in various cellular processes (25). Pur-alpha has been demonstrated to bind to both single- and double-stranded nucleic acids that contain GGN motifs (4). Neuronal DNA/RNA binding protein Pur-alpha is a regulator of transcription and a major factor in mRNA localization (4). In addition to its DNA and RNA binding abilities, Pur-alpha also exhibits dsDNA destabilizing activity in an ATP-independent manner (26). Pur-alpha-mediated binding of nucleic acids involves three central PUR repeats, which are flanked on the N-terminal side by unstructured glycine-rich sequences and on the C-terminal side by regions rich in glutamine and glutamate (27). PUR-alpha interacts with the expanded CGG repeats of FMR1 RNA (28). It is assumed that FMR1 mRNA expression with abnormal trinucleotide repeat expansions is a major cause of neurodegenerative tremor/ataxia syndrome associated with a fragile X chromosome (11).PUR-alpha also interacts with the expanded GGGGCC repeat RNAs arising from hexanucleotide repeat expansions in the first intron of the c9orf72 transcript (29). These interactions of PUR-alpha protein with repeat RNAs lead to sequesteration and loss of PUR-alpha protein function. This loss of function contributes to the pathogenesis of Fragile X-associated tremor/ataxia syndrome (FXTAS) and amyotrophic lateral sclerosis/frontotemporal dementia (ALS/FTD), respectively (4, 22).
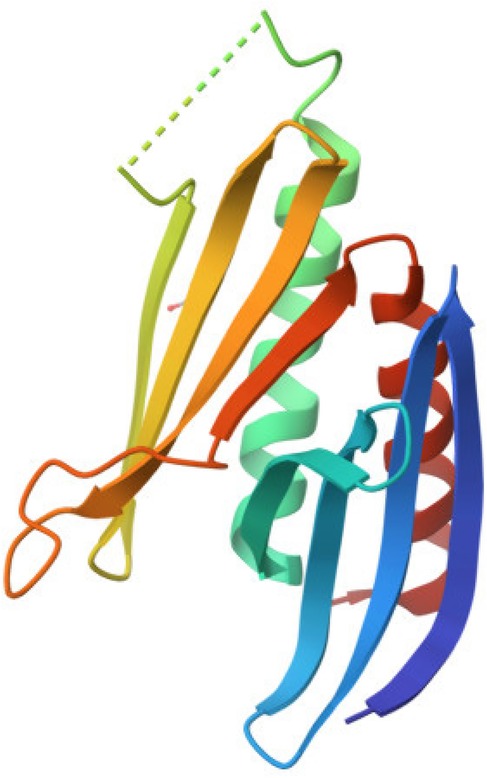
Figure 2. Graphic diagram of the PURA protein structure (44).
The table below provides information on which selected RNA the Pur-alpha protein binds to and its function (Table 1).
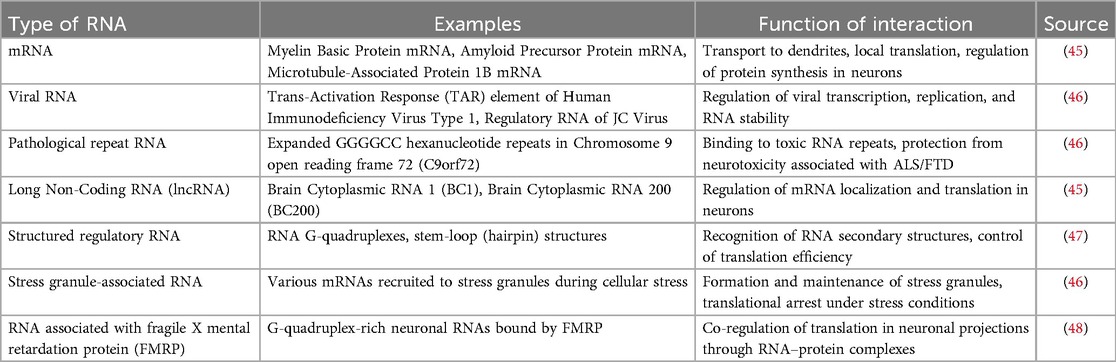
Table 1. The selected RNA binds to the Pur-alpha protein, and it is important to understand what function this interaction serves.
3 Aim of the study
The aim of the study was to describe the physical and neurodevelopmental presentation of a 15 year old with PURA syndrome.
4 Material and methods
To write this manuscript, a review of articles was conducted using the PubMed, Google Scholar, and Mendeley search engines. The keywords used were “Pura syndrome,” “intellectual disability,” and “scoliosis.” From the articles found and analyzed, those deemed relevant to the topic of this manuscript and valuable as sources of information were selected. The manuscript cites 54 publications and scientific reports. Additionally, it presents the case of a 15-year-old female patient who required surgical treatment in the Department of Orthopedics and Spine Surgery at the Medical University of Gdańsk due to an advanced postural defect, specifically scoliosis.
5 Case presentation
A 15-year-old girl was admitted to the Department of Orthopedics and Spine Surgery at the Medical University of Gdansk, where she was diagnosed with PURA syndrome, confirmed by genetic testing. She was admitted due to an advanced postural defect in the form of idiopathic scoliosis (Figures 3–5). This condition significantly impacted her quality of life and caused considerable difficulties in maintaining proper posture and movement.
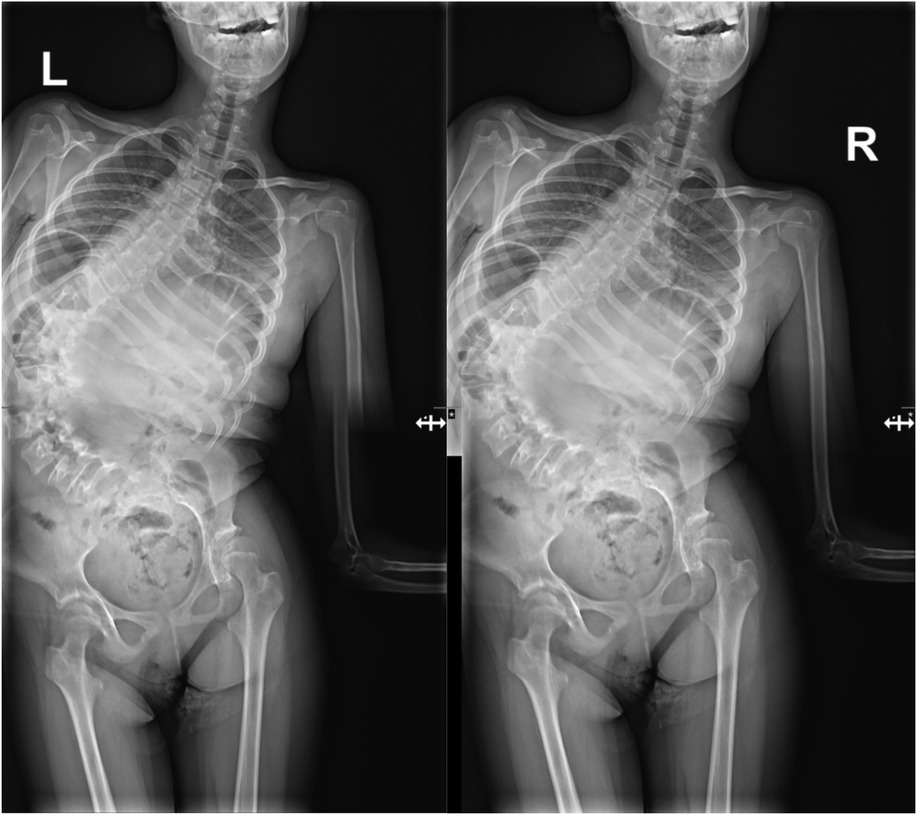
Figures 3 and 4. Radiological imaging study showing spinal deformity before surgical treatment.
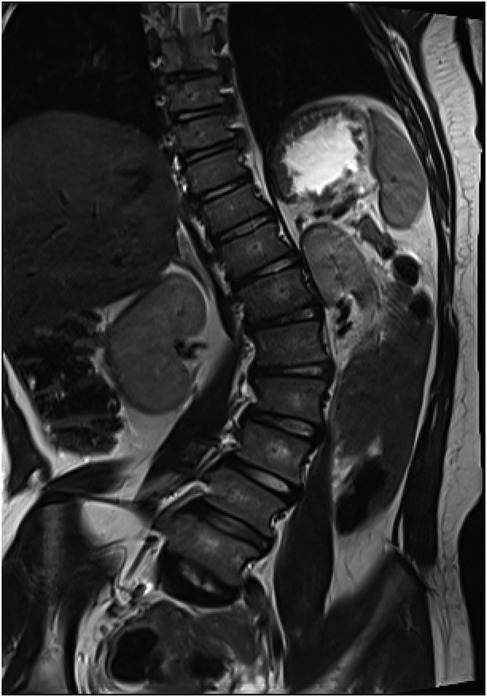
Figure 5. Computer tomography scan before surgical treatment.
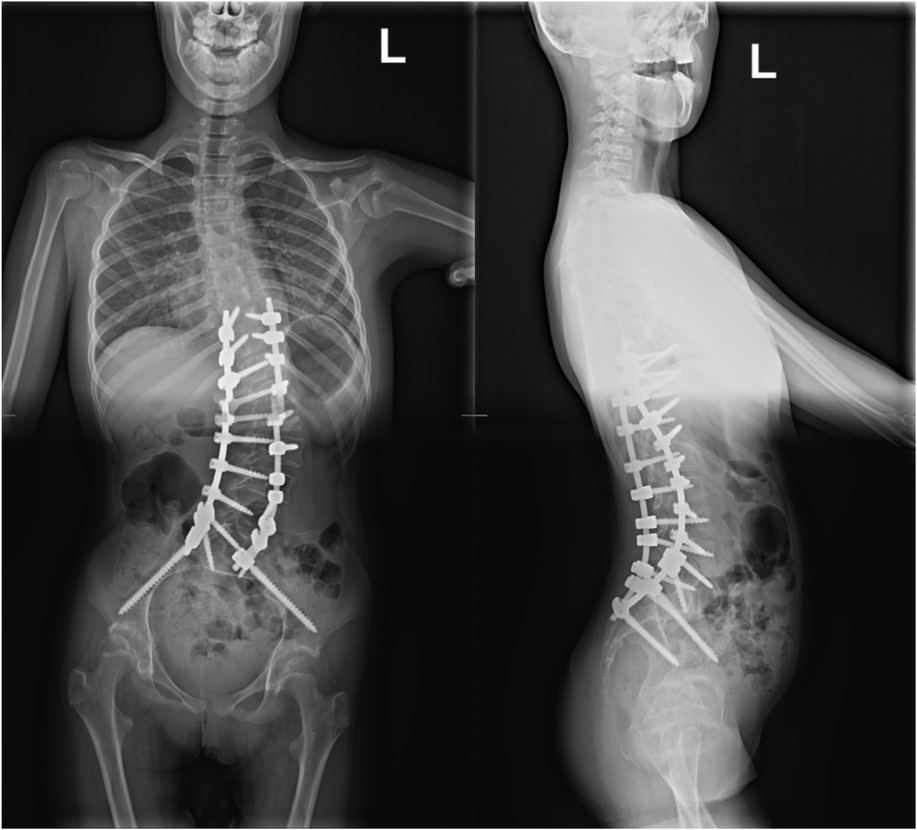
Figures 6 and 7. Toposcan after surgical treatment of idiopathic scoliosis.
A girl was born to young, healthy, and unrelated parents, with no history of genetic diseases in the family and no exposure to harmful environmental factors. She was a product of an uncomplicated first pregnancy, delivered naturally at 41 weeks, with a birth weight of 3,800 grams. She scored 8 points on the Apgar scale. In the early childhood period, the child showed symptoms in the form of motor development disorders (difficulty lifting the head in the neonatal period, upper and lower limbs), involuntary movements, unstable gait on a broad base, as well as verbal communication disorders (the child only utters sounds, does not speak single words). Facial dysmorphia, uncoordinated eye movements, and hypotonia of the trunk muscles were also observed. Additionally, feeding problems and symptoms of gastroesophageal reflux were present, including a persistent cough (without other features of an upper respiratory tract infection), regurgitation of gastric contents, and empty belching. Episodes of epileptic seizures were and still are present—for this reason, the girl is treated with valproic acid. Due to delays in psychomotor development and decreased muscle tone from infancy, she was referred to a genetic counseling center for further evaluation. During the diagnostic process, chromosomal abnormalities were examined, revealing a normal karyotype of 46, XX. Conditions such as Prader-Willi syndrome and spinal muscular atrophy were excluded. Molecular studies conducted using next-generation sequencing identified a mutation in one allele of the PURA gene when the girl was 8 years old. Specifically, there was a single nucleotide change (c.470T > G) in exon one, resulting in a missense mutation that alters methionine to arginine (p.Met157Arg). Bioinformatics analysis indicated that this mutation is pathogenic and of de novo origin.
In the Orthopedic and Spine Surgery clinic, after properly preparing the patient and obtaining the necessary consents from the girl's legal guardian for the proposed surgical treatment and anesthesia, the procedure of posterior correction and posterolateral stabilization was performed. The surgical procedure and anesthesia were completed without complications. During the recovery period, the child did not experience any neurological deficits and was rehabilitated successfully, resulting in a satisfactory postoperative outcome. The girl was discharged from the clinic on the fifth day after the operation for continued outpatient care. Nine weeks post-operation, the patient attended a follow-up visit at the trauma and orthopedic surgery clinic. An imaging study, a toposcan of the spine, was conducted (see Figures 6, 7). According to the consulting orthopedic surgeon, the results of the operation were satisfactory. The girl continued her rehabilitation program and gradually regained her mobility, which her parents noted significantly improved her quality of life.
6 Discussion
Our case illustrates the defining features of PURA syndrome, such as hypotonia, motor and speech delays, epilepsy, and scoliosis, as outlined in the literature (19, 30–34). However, it is important to note that symptoms can vary among individuals. For instance, Liu et al. (14) reported a newborn who experienced feeding difficulties, lethargy, and respiratory failure—symptoms that were not present in our patient. Additionally, while some patients may show signs of multi-organ involvement, this was not evident in our case. PURA syndrome is typically diagnosed in childhood, but the age at which individuals are diagnosed can vary from infancy to adulthood. One study indicated that the average age of diagnosis is 7.4 years, with the youngest patient being just 11 months old and the oldest being 27 years old (14). Conditions that should be considered in the differential diagnosis include Prader-Willi syndrome, myotonic dystrophy, spinal muscular atrophy, and congenital muscular dystrophy (35). Ongoing research suggests that there may be morphological abnormalities associated with the central nervous system (36) including inappropriate acoustic responses to unexpected sounds. For instance, Mroczek et al. (37) described variations in nerve fiber size and rapidly progressive muscular atrophy, which aligns with our patient's presentation of decreased muscle tone and muscular atrophy. Common characteristics of PURA syndrome include abnormal movements and varying severity of epilepsy over time (32). Epilepsy is a frequent occurrence in PURA syndrome, affecting around 50% of patients (33, 34). Our patient has been diagnosed with epilepsy and is currently being treated with valproic acid. The severity and types of seizure activity can differ significantly, highlighting the phenotypic diversity associated with PURA syndrome. Several studies have indicated that apnea is particularly prevalent among newborns with PURA syndrome (11–13) and may serve as a diagnostic indicator. However, apneas were not observed in our case. Skeletal abnormalities, such as scoliosis, are significant features of PURA syndrome (38). In our patient, the worsening scoliosis negatively impacted thoracic organ function, which was evident through shallow breathing and deterioration in gas exchange, as confirmed by acid-base balance analysis. Similar cases have reported gastrointestinal disorders, such as reflux and constipation, likely stemming from impaired motility (15, 39–41). Though diagnosing PURA syndrome can be challenging, the presence of characteristic symptoms should prompt appropriate testing to confirm the diagnosis.
7 Conclusion
PURA syndrome is a rare genetic disorder characterized by various systemic and organ-related symptoms. It includes specific features such as speech disorders, difficulty following simple commands, inadequate acoustic responses to unexpected sounds, and feeding difficulties starting from the neonatal period. These symptoms should prompt genetic testing to confirm the diagnosis and allow for appropriate multidisciplinary therapeutic management.
Early and rapid genetic testing in children with developmental disorders is important because it makes it possible to implement early psycho-motor rehabilitation and early detection of additional complications, which in some diseases coexist with a genetic disease. Thanks to genetic testing, we can predict whether the next child also has a risk of developing the disease. In addition, parents can prepare for the presence of additional diseases and faster intervention is possible (18, 38, 42, 43, 49–51).
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. Written informed consent from the patients/participants or patients/participants legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
JK: Writing – original draft, Methodology, Formal analysis. MS: Writing – original draft, Formal analysis, Resources, Methodology, Conceptualization. KC: Writing – original draft, Conceptualization. SB: Writing – original draft, Resources. SK-N: Methodology, Formal analysis, Conceptualization, Supervision, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rodríguez-García ME, Cotrina-Vinagre FJ, Arranz-Canales E, Aragón AM, Hernández-Sánchez L, Rodríguez-Fornés F, et al. A novel de novo mutation in the PURA gene associated with a new clinical finding: large brainstem. J Genet. (2020) 99:7. doi: 10.1007/s12041-019-1165-3
2. OMIM Entry - #616158—Neurodevelopmental Disorder with Neonatal Respiratory Insufficiency, Hypotonia, and Feeding Difficulties; NEDRIHF. (2021). Available online at: https://www.omim.org/entry/616158 (Accessed March 03, 2025).
3. Reijnders MRF, Janowski R, Alvi M, Self JE, van Essen TJ, Vreeburg M, et al. PURA syndrome: clinical delineation and genotype-phenotype study in 32 individuals with review of published literature. J Med Genet. (2018) 55(2):104–13. doi: 10.1136/jmedgenet-2017-104946
4. Weber J, Bao H, Hartlmüller C, Wang Z, Windhager A, Janowski R, et al. Structural basis of nucleic-acid recognition and double-strand unwinding by the essential neuronal protein pur-alpha. eLife. (2016) 5:e11297. doi: 10.7554/eLife.11297
5. Johannesen KM, Gardella E, Gjerulfsen CE, Bayat A, Rouhl RPW, Reijnders M, et al. PURA-Related Developmental and epileptic encephalopathy: phenotypic and genotypic Spectrum. Neurol Genet. (2021) 7(6):e613. doi: 10.1212/NXG.0000000000000613
6. Molitor L, Bacher S, Burczyk S, Niessing D. The molecular function of PURA and its implications in neurological diseases. Front Genet. (2021) 12:638217. doi: 10.3389/fgene.2021.638217
7. Khalili K, Del Valle L, Muralidharan V, Gault WJ, Darbinian N, Otte J, et al. Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol Cell Biol. (2003) 23(19):6857–75. doi: 10.1128/MCB.23.19.6857-6875.2003
8. Hokkanen S, Feldmann HM, Ding H, Jung CK, Bojarski L, Renner-Müller I, et al. Lack of pur-alpha alters postnatal brain development and causes megalencephaly. Hum Mol Genet. (2012) 21(3):473–84. doi: 10.1093/hmg/ddr476
9. Reijnders MRF, Leventer RJ, Lee BH, Baralle D, Selber P, Paciorkowski AR, et al. PURA-Related Neurodevelopmental disorders. In: Adam MP, editor. GeneReviews®. Seattle.: University of Washington (2017).
10. PURA Foundation Australia. (2025). PURA syndrome. Available online at: https://www.purafoundation.au/pura (Accessed April 03, 2025).
11. Tanaka AJ, Bai R, Cho MT, Anyane-Yeboa K, Ahimaz P, Wilson AL, et al. de novo mutations in PURA are associated with hypotonia and developmental delay. Cold Spring Harb Mol Case Stud. (2015) 1:a000356. doi: 10.1101/mcs.a000356
12. Lalani SR, Zhang J, Schaaf CP, Brown CW, Magoulas P, Tsai AC, et al. Mutations in PURA cause profound neonatal hypotonia, seizures, and encephalopathy in 5q31.3 microdeletion syndrome. Am J Hum Genet. (2014) 95(5):579–83. doi: 10.1016/j.ajhg.2014.09.014
13. Hunt D, Leventer RJ, Simons C, Taft R, Swoboda KJ, Gawne-Cain M, et al. Whole exome sequencing in family trios reveals de novo mutations in PURA as a cause of severe neurodevelopmental delay and learning disability. J Med Genet. (2014) 51(12):806–13. doi: 10.1136/jmedgenet-2014-102798
14. Liu Y, Liu R, Xu T, Zhou Y-X, Zhang S-C. Neonatal PURA syndrome: a case report and literature review. Transl Pediatr. (2021) 10:194–203. doi: 10.21037/tp-20-248
15. Crippa ACS, Bacheladenski EP, Rodrigues DCB, Ferreira LP, Meira AT, Franklin GL. Movement disorders in PURA syndrome: a video case series. Mov Disord Clin Pract. (2023) 10(10):1542–6. doi: 10.1002/mdc3.13804
16. Cinquina V, Ciaccio C, Venturini M, Masson R, Ritelli M, Colombi M. Expanding the PURA syndrome phenotype: a child with the recurrent PURA p.(Phe233del) pathogenic variant showing similarities with cutis laxa. Mol Genet Genomic Med. (2020) 9:e1562. doi: 10.1002/mgg3.1562
17. Fukuda Y, Kudo Y, Saito M, Kaname T, Oota T, Shoji R. Expanding the PURA syndrome phenotype with manifestations in a Japanese female patient. Hum Genome Var. (2022) 9:1–5. doi: 10.1038/s41439-022-00189-7
18. Taniguchi N, Watanuki K, Nakato D, Takenouchi T, Kosaki K, Koga H. PURA-related neurodevelopmental disorders: a systematic review on genotype-phenotype correlations. J Med Genet. (2025) 62(3):191–8. doi: 10.1136/jmg-2024-110379
19. Daniel DC, Johnson EM. PURA, the gene encoding pur-alpha, member of an ancient nucleic acid-binding protein family with mammalian neurological functions. Gene. (2018) 643:133–43. doi: 10.1016/j.gene.2017.12.004
20. Scott MP, Jung R, Muntz K, Nielsen NC. A protease responsible for post-translational cleavage of a conserved asn-gly linkage in glycinin, the major seed storage protein of soybean. N Proc Natl Acad Sci USA. (1992) 89(2):658–62. doi: 10.1073/pnas.89.2.658
21. Yu S, Jiang C, Yang Y, Cheng F, Liu F, Liu C, et al. Purine-rich element binding protein alpha: a DNA/RNA binding protein with multiple roles in cancers. Mol Med. (2025) 31:20. doi: 10.1186/s10020-025-01087-8
22. Graebsch A, Roche S, Niessing D. x-ray structure of pur-α reveals a whirly-like fold and an unusual nucleic-acid binding surface. Proc Natl Acad Sci USA. (2009) 106(44):18521–6. doi: 10.1073/pnas.0907990106
23. Proske M, Janowski R, Bacher S, Kang HS, Monecke T, Koehler T, et al. PURA syndrome-causing mutations impair PUR-domain integrity and affect P-body association. Elife. (2024) 13:RP93561. doi: 10.7554/eLife.93561
24. Janowski R, Niessing D. The large family of PC4-like domains - similar folds and functions throughout all kingdoms of life. RNA Biol. (2020) 17(9):1228–38. doi: 10.1080/15476286.2020.1761639
25. Miller JM, Enemark EJ. Fundamental characteristics of AAA+ protein family structure and function. Archaea. (2016) 2016:9294307. doi: 10.1155/2016/9294307
26. Darbinian N, Gallia GL, Khalili K. Helix-destabilizing properties of the human single-stranded DNA- and RNA-binding protein puralpha. J Cell Biochem. (2001) 80(4):589–95. doi: 10.1002/1097-4644(20010315)80:4%3C589::AID-JCB1013%3E3.0.CO;2-0
27. Johnson EM, Daniel DC, Gordon J. The pur protein family: genetic and structural features in development and disease. J Cell Physiol. (2013) 228(5):930–7. doi: 10.1002/jcp.24237
28. Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, et al. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron. (2007) 55(4):556–64. doi: 10.1016/j.neuron.2007.07.020
29. Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc Natl Acad Sci U S A. (2013) 110(19):7778–83. doi: 10.1073/pnas.1219643110
30. Mroczek M, Iyadurai S. Neuromuscular and neuromuscular junction manifestations of the PURA-NDD: a systematic review of the reported symptoms and potential treatment options. Int J Mol Sci. (2023) 24(3):2260. doi: 10.3390/ijms24032260
31. Kofoed AWS, Kristiansen SS, Miranda MJ, Rubboli G, Johannesen KM. Differences in manifestations of epilepsy and developmental delay in PURA syndrome and 5q31 microdeletions. Clin Genet. (2024) 106(4):386–93. doi: 10.1111/cge.14581
32. Le Meur N, Holder-Espinasse M, Jaillard S, Goldenberg A, Joriot S, Amati-Bonneau P, et al. MEF2C haploinsufficiency caused by either microdeletion of the 5q14.3 region or mutation is responsible for severe mental retardation with stereotypic movements, epilepsy and/or cerebral malformations. J Med Genet. (2010) 47:22–9. doi: 10.1136/jmg.2009.069732
33. McMahon KQ, Papandreou A, Ma M, Barry BJ, Mirzaa GM, Dobyns WB, et al. Familial recurrences of FOXG1-related disorder: evidence for mosaicism. Am J Med Genet A. (2015) 167:3096–102. doi: 10.1002/ajmg.a.37353
34. Lee BH, Reijnders MRF, Abubakare O, Tuttle E, Lape B, Minks KQ, et al. Expanding the neurodevelopmental phenotype of PURA syndrome. Am J Med Genet A. (2018) 176(1):56–67. doi: 10.1002/ajmg.a.38521
35. Zadeh N, Hudgins L. The genetic approach to hypotonia in the neonate. NeoReviews. (2009) 10:e600–7. doi: 10.1542/neo.10-12-e600
36. Trau SP, Pizoli CE. PURA syndrome and myotonia. Pediatr. Neurol. (2020) 104:62–3. doi: 10.1016/j.pediatrneurol.2019.09.008
37. Mroczek M, Zafeiriou D, Gurgel-Gianetti J, Vilela Morais De Azevedo B, Roos A, Bartels E, et al. Three individuals with PURA syndrome in a cohort of patients with neuromuscular disease. Neuropediatrics. (2020) 52:390–3. doi: 10.1055/s-0040-1715625
38. Beerhorst K, van der Kruijs SJM, Verschuure P, Tan IYF, Aldenkamp AP. Bone disease during chronic antiepileptic drug therapy: general versus specific risk factors. J Neurol Sci. (2013) 331:19–25. doi: 10.1016/j.jns.2013.05.005
39. Hildebrand MS, Braden RO, Lauretta ML, Kaspi A, Leventer RJ, Anderson M, et al. Inherited PURA pathogenic variant associated with a mild neurodevelopmental disorder. Neurol Genet. (2024) 10(5):e200181. doi: 10.1212/NXG.0000000000200181
40. Mora-Martinez S, Castaño-Giraldo N, Nati-Castillo HA, Barahona Machado L, Mora Arbeláez T, Gordillo-Gonzalez G, et al. Case report: expanding the phenotypic spectrum of PURA syndrome in South America with the first presentation of concurrent vitiligo. Front Pediatr. (2024) 12:1323014. doi: 10.3389/fped.2024.1323014
41. Reijnders MRF, Leventer RJ, Lee BH, Baralle D, Selber P, Paciorkowski AR, et al. PURA-Related Neurodevelopmental disorders. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews®. Seattle, WA: University of Washington, Seattle (2017). p. 1993–2025. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK426063/
42. Moeschler JB, Shevell M, American Academy of Pediatrics Committee on Genetics. Clinical genetic evaluation of the child with mental retardation or developmental delays. Pediatrics. (2006) 117(6):2304–16. doi: 10.1542/peds.2006-1006
43. Moeschler JB. Genetic evaluation of intellectual disabilities. Semin Pediatr Neurol. (2008) 15(1):2–9. doi: 10.1016/j.spen.2008.01.002
44. Available online at: https://www.rcsb.org/structure/8CHT (Accessed April 03, 2025).
45. Johnson EM, Kinoshita Y, Weinreb DB, Wortman MJ, Simon R, Khalili K, et al. Role of pur alpha in targeting mRNA to sites of translation in hippocampal neuronal dendrites. J Neurosci Res. (2006) 83(6):929–43. doi: 10.1002/jnr.20806
46. Daigle JG, Krishnamurthy K, Ramesh N, Casci I, Monaghan J, McAvoy K, et al. Pur-alpha regulates cytoplasmic stress granule dynamics and ameliorates FUS toxicity. Acta Neuropathol. (2016) 131(4):605–20. doi: 10.1007/s00401-015-1530-0
47. Asamitsu S, Yabuki Y, Matsuo K, Kawasaki M, Hirose Y, Kashiwazaki G, et al. RNA G-quadruplex organizes stress granule assembly through DNAPTP6 in neurons. Sci Adv. (2023) 9(8):eade2035. doi: 10.1126/sciadv.ade2035
48. Goering R, Hudish LI, Guzman BB, Raj N, Bassell GJ, Russ HA, et al. FMRP promotes RNA localization to neuronal projections through interactions between its RGG domain and G-quadruplex RNA sequences. Elife. (2020) 9:e52621. doi: 10.7554/eLife.52621
49. Geng Y, Cai Q. Role of C9orf72 hexanucleotide repeat expansions in ALS/FTD pathogenesis. Front Mol Neurosci. (2024) 17:1322720. doi: 10.3389/fnmol.2024.1322720
50. McCallister TX, Lim CKW, Terpstra WM, Alejandra Zeballos CM, Zhang S, Powell JE, et al. A high-fidelity CRISPR-Cas13 system improves abnormalities associated with C9ORF72-linked ALS/FTD. Nat Commun. (2025) 16:460. doi: 10.1038/s41467-024-55548-5
51. Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. (2013) 126(6):829–44. doi: 10.1007/s00401-013-1192-8
Keywords: PURA syndrome, PURA gene, neurodevelopment disorders, genetic disease, genetic defect
Citation: Kobak J, Szczupak M, Czerkiewicz K, Bielocerkowski S and Krupa-Nurcek S (2025) PURA syndrome—a genetic cause of a neurodevelopmental disorder—case report. Front. Pediatr. 13:1607213. doi: 10.3389/fped.2025.1607213
Received: 7 April 2025; Accepted: 19 June 2025;
Published: 10 July 2025.
Edited by:
Sandeep Kumar Singh, Banaras Hindu University, IndiaReviewed by:
Maria Gogou, Aristotle University of Thessaloniki, GreeceKelly King, University of Minnesota, United States
Sonali Vishal, Yale University, United States
Copyright: © 2025 Kobak, Szczupak, Czerkiewicz, Bielocerkowski and Krupa-Nurcek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jacek Kobak, amFjZWsua29iYWtAZ3VtZWQuZWR1LnBs