 Michał Krawiec1
Michał Krawiec1 Joanna Śliwka2Szymon Pawlak2
Joanna Śliwka2Szymon Pawlak2 Michał Kapałka1*
Michał Kapałka1* Paulina Hamerling1Weronika Grzywacz1Wiktoria Hawel1Gabriela Skórska3Dorota Piekutowska-Abramczuk4Tomasz Hrapkowicz2
Paulina Hamerling1Weronika Grzywacz1Wiktoria Hawel1Gabriela Skórska3Dorota Piekutowska-Abramczuk4Tomasz Hrapkowicz2
- 1Faculty of Medical Sciences in Zabrze, Medical University of Silesia in Katowice, Zabrze, Poland
- 2Department of Cardiac, Vascular and Endovascular Surgery and Transplantology, Faculty of Medical Sciences in Zabrze, Medical University of Silesia in Katowice, Silesian Center for Heart Diseases in Zabrze, Zabrze, Poland
- 3Faculty of Medical Sciences in Katowice, Medical University of Silesia in Katowice, Katowice, Poland
- 4Department of Medical Genetics, The Children’s Memorial Health Institute, Warsaw, Poland
Introduction: Barth syndrome (BTHS) is an ultra-rare genetic disease caused by a mutation in the TAFAZZIN gene, located on the X chromosome. This gene codes for the protein tafazzin, which is involved in the metabolism of the mitochondrial phospholipid - cardiolipin. Symptoms of this genetic defect include dilated cardiomyopathy (DCM), skeletal myopathy, neutropenia, growth retardation, reduced cholesterol levels, increased serum lactic acid levels, and hypoglycemia in the neonatal period.
Case description: A Caucasian boy with DCM and left ventricular non-compaction associated with BTHS, caused by a previously unreported variant in the TAFAZZIN gene: NM_000116.4:c.525_533del; NP_000107.1:p.(His176_Phe178del) at NC_000023.11:g.154419607_154419615del, in the exon 6. Due to the patient's heart failure, a mechanical circulatory support (MCS) system was required, followed by orthotopic heart transplantation (OHT). Because of the presence of neutropenia, standard immunosuppressive therapy had to be modified in the postoperative period.
Conclusions: A previously unreported mutation is presented, leading to BTHS. This disease can have severe cardiovascular manifestations, requiring MCS and OHT.
Introduction
Barth syndrome (BTHS; MIM 302060) is an ultra-rare genetic disorder with an incidence of 1 in 400,000 to 1 in 1,000,000 births (1). Approximately 250 cases of the disease have been described worldwide since 1983 (1). The cause of BTHS is a mutation in the TAFAZZIN gene, located on the X chromosome (1, 2). This gene encodes the protein tafazzin, and its mutation results in impaired mitochondrial metabolism of the phospholipid cardiolipin. Tafazzin is a non-specific phospholipid-lysophospholipid transacylase responsible for modifying the structure of cardiolipin, a key component of the mitochondrial membrane. This leads to severe abnormalities, like a disruption of the electron transport chain, increased mitochondrial reactive oxygen species (both in the mechanism of enhanced production and accumulation), dysregulation of CoA-dependent metabolism, and dysfunction of the citric acid cycle (1–4).
Symptoms of this genetic disease include dilated cardiomyopathy (DCM), skeletal myopathy, neutropenia, growth retardation, reduced cholesterol level, increased serum lactic acid levels, and hypoglycemia in the neonatal period (1–5). This case study presents a male patient with DCM and left ventricular non-compaction (LVNC) attributed to BTHS, caused by a previously unreported variant of the c.525_533del p.(His176_Phe178del) in the TAFAZZIN gene.
Case description
A Caucasian boy was delivered by cesarean section at 36 weeks' gestation due to fetal growth restriction and placental insufficiency. Patient's birth weight was 2,450 g and he was 50 cm in length. The patient was from his mother's third pregnancy and second delivery. The mother had previously one miscarriage, and the boy's older brother had died at 5 months of age due to DCM with myocardial non-compaction.
Prenatal testing had shown thickened right ventricular myocardium. On the second day of life, the patient was admitted to the cardiology unit due to bradycardia (heart rate dropping to about 76 beats per minute). At that time, decreased muscle tone and a heart rate of 90–100 bpm with drops to 80 bpm were observed. Blood tests revealed leukopenia (7.59 × 103/µl; normal range: 9.40–34.00 × 103/µl) with a low number of eosinophils (0.04 × 103/µl; normal range: 0.2–0.4 × 103/µl) and an elevated number of monocytes (1.55 × 103/µl; normal range: 0.10–1.10 × 103/µl). Echocardiography revealed right ventricular myocardial thickening and decreased contractility of both ventricles, with a reduced left ventricular ejection fraction (LVEF) of approximately 55%. The left ventricular diameter measured 1.79 cm in diastole (LVDd) and 1.34 cm in systole (LVDs). A patent foramen ovale (PFO) and increased LV trabeculation was also identified. Based on these findings, DCM was diagnosed, and LVNC was suspected.
After treatment with captopril, spironolactone, and carvedilol, normalization of heart rate and an increase in LVEF to about 80% were achieved.
Based on the clinical symptoms, BTHS was suspected, and a urine test for organic acids using gas chromatography coupled to mass spectrometry (GC-MS) was ordered. The test showed elevated concentrations of lactic acid, 2-ketoglutaric acid, p-hydroxyphenyllactic acid, and p-hydroxyphenylpyruvic acid; no succinylacetone was found.
In the first months of the patient's life, delayed motor and speech development were observed. At the age of 14 months, he was hospitalized for sepsis of staphylococcal etiology and agranulocytosis.
During the diagnostic process, genomic DNA was isolated from blood leukocytes using standard procedures, including phenol/chloroform extraction and automated DNA extraction (MagNA Pure LC 2.0, Roche). Mutation analysis was performed using Sanger sequencing of PCR-amplified exons 2–11 of the TAFAZZIN gene, including exon/intron boundaries, on a 3130 Genetic Analyzer (Applied Biosystems/Life Technologies, Foster City, CA).
During the diagnostic process, DNA sequencing was performed using the Sanger method with analysis of exons 2–11 of the TAFAZZIN gene. Numbering of revealed nucleotide changes was based on the reference sequence for the TAFAZZIN gene (hg38; NM_000116.5; NP_000107.1); position +1 corresponded to the A of the ATG translation initiation codon. The study revealed a deletion of (NM_000116.4:c.525_533del; NP_000107.1:p.(His176_Phe178del); NC_000023.11:g.154419607_154419615del) in exon 6 of the TAFAZZIN gene. This molecular variant c.525_533del.p(His176_Phe178del) had not been previously reported in The Human Gene Mutation Database (HGMD). The same mutation was found in the patient's mother, who was heterozygous (a carrier), and was subsequently inherited by the child.
At the age of 7, the boy was admitted to a cardiac center due to a sudden deterioration in his condition. On admission, the patient's weight was 17.4 kg and he was 115 cm tall (both values were <3 percentile). There was tachycardia (approximately 160 bpm), profuse vomiting, and a quiet systolic murmur on the left side of the sternum. Blood tests showed elevated NT-proBNP (10,064 pg/ml; normal <83 pg/ml) and troponin T (86 pg/ml; normal <14 pg/ml). Chest X-ray revealed an increased cardiac silhouette (Figure 1). An echocardiogram revealed a dilated left ventricle (LVDd: 4.73 cm – Z-score: 2.7; LVDs: 4.33 cm – Z-score: 5.22) with features of non-compaction, PFO, and small regurgitations on the mitral and tricuspid valves (Figure 2). The LVEF was about 18%. The electrocardiograms (ECG) showed non-specific intraventricular conduction abnormalities, ST-T segment inversion in the V5, V6 leads and a borderline corrected QT interval (QTc) of up to 453 ms (normal: <440 ms). The Holter ECG showed ten ventricular extrasystoles. Due to the patient's deteriorating condition, a left ventricular assist device (LVAD) was implanted as a bridge to orthotopic heart transplant (OHT).
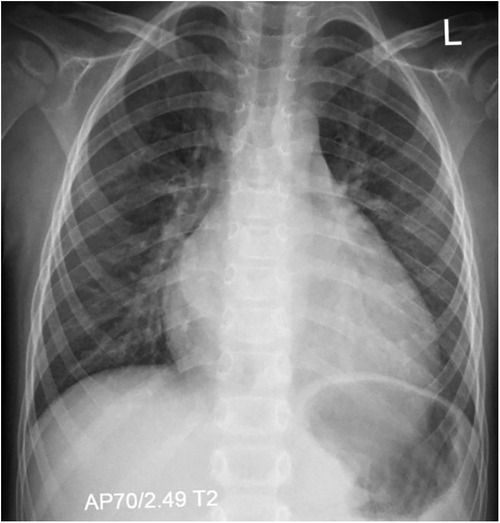
Figure 1. Chest x-ray (AP view) showing marked cardiomegaly consistent with dilated cardiomyopathy, without signs of pulmonary congestion.
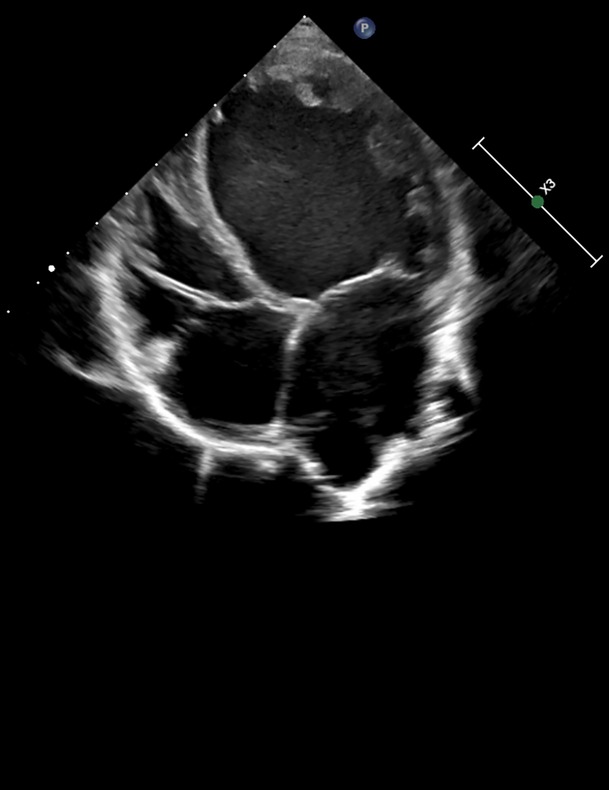
Figure 2. Echocardiographic image in a parasternal long-axis view showing the left ventricle, left atrium, and aortic root. Cardiac structures appear with clear wall delineation, suggesting preserved image quality for structural assessment.
During hospitalization, fluctuating levels of neutrophils (from 0.07 × 103/µl to 8.34 × 103/µl, often remaining below the lower limit of normal) and monocytes (from 0.60 × 103/µl to 2.74 × 103/µl, consistently above the upper limit of normal) were observed. Two times the LVAD chamber had to be exchanged due to fibrin deposits. Antibiotic therapy was required on several occasions: vancomycin due to Clostridioides difficile infection, ceftriaxone due to urinary tract infection and cloxacillin due to LVAD cannulation site infection. After 383 days of mechanical circulatory support, the patient underwent OHT.
After OHT, immunosuppression was implemented: methyloprednisone (in dose 50 mg twice daily), basiliximab (in dose 10 mg). On the day following the surgery, a neutropenia (2.19 × 103/µl) was observed, leading to a temporary decision to withhold mycophenolate mofetil. Two days later, normal neutrophil levels were achieved. The tacrolimus in dose 2 mg was ordered. The mycophenolate mofetil was introduced on the fifth post-operative day at a dose of 250 mg twice daily. Additionally, on the fifth day after surgery, methyloprednisolone was substituted with prednisone in dose 20 mg. The patient was discharged home in good general condition after three weeks. Twenty months after surgery, the patient is well and has not required further cardiac interventions. The function of the transplanted heart was normal with an LVEF of 55%. During follow-up, no significant haematological disorders or infections were observed.
Discussion
Cardiomyopathies are a heterogeneous group of myocardial disorders classified according to etiology, phenotypic features, and impact on cardiac function. In the category of primary genetic cardiomyopathies, hypertrophic cardiomyopathy (HCM), arrhythmogenic right ventricular cardiomyopathy (ARVC), and LVNC are distinguished. Cardiomyopathies of mixed etiology include DCM and restrictive cardiomyopathy (RCM), while peripartum and stress-induced cardiomyopathy (Tako-Tsubo syndrome) are classified as acquired cardiomyopathies. Secondary cardiomyopathies result from systemic diseases (6).
DCM, the most common cardiomyopathy in the general population (1:250–1:2500 people) with an incidence of 5–7 cases per 100,000 persons per year. DCM most often manifests itself between 20 and 60 years of age, but it is also the most common form of cardiomyopathy in children (more than 60% of childhood cardiomyopathies) (7). DCM is characterized by dilatation of the heart chambers with preserved wall thickness and systolic dysfunction, often leading to heart failure with reduced ejection fraction (8). The most common primary cardiomyopathy is HCM (9). Among primary cardiomyopathies, rare forms such as LVNC, which has an embryonic origin manifesting as significant trabeculation and development of intertrabecular spaces in the left ventricle, are also identified. The prevalence of LVNC in the general population is difficult to determine but is estimated to affect less than 1% of the population (6).
DCM is the most common form of cardiomyopathy leading to orthotopic heart transplantation (OHT) and represents a major cardiac complication in BTHS (2, 3). In the present case, prenatal thickening of the right ventricular myocardium and postnatal deterioration of biventricular contractility, with a reduced ejection fraction, indicate early cardiac involvement in the disease process. These findings are consistent with previous reports describing BTHS manifestations as early as the fetal period (10). LVNC is also a common phenotype in patients with BTHS, affecting 20%–50% of these patients (1, 4). Unlike other etiologies of DCM, where ventricular dilatation is usually progressive and associated with continuous impairment of systolic function, in BTHS it can be fluctuating, which may be related to the underlying mitochondrial dysfunction characteristic of the syndrome. In BTHS, despite the presence of DCM, a preserved or slightly reduced ejection fraction is often observed, distinguishing it from other causes of DCM (2, 5). In BTHS, left ventricular dilatation is not directly related to left ventricular weakness but may result from impaired relaxation and filling of the ventricle. This condition may be partially reversible, as evidenced by cases of improvement after supportive treatment (1, 4).
In the presented case, initial management centered on the administration of captopril, spironolactone, and carvedilol. This standard pharmacotherapy, in accordance with current clinical guidelines, led to the normalization of heart rhythm and a significant improvement in LVEF, reaching approximately 80%. This outcome aligns with existing evidence suggesting that patients with BTHS may respond favorably to conventional heart failure therapies, despite the absence of specific clinical trials evaluating the efficacy of these treatments in this population (1, 11, 12).
However, due to an acute clinical deterioration characterized by left ventricular dilation and a marked reduction in LVEF to approximately 18%, more advanced therapeutic interventions became necessary. LVAD implantation is typically employed as a bridging therapy to OHT (13), which is considered the definitive treatment option when other therapeutic modalities have proven insufficient (1). Available data indicate that approximately 14% of patients with BTHS ultimately require OHT (1).
The molecular etiology of BTHS is attributed to mutations in the TAFAZZIN gene, which encodes the tafazzin protein. This mutation disrupts the regulation of cardiolipin biosynthesis and remodelling within the inner mitochondrial membrane (1–5). Given the high mitochondrial density in organs such as the heart and skeletal muscle, mitochondrial dysfunction typically manifests as cardiomyopathy or skeletal myopathy (1).
The described proband, along with his mother, was found to harbor a previously unidentified gene mutation, c.525_533del.p(His176_Phe178del), located in exon 6 of the TAFAZZIN gene (Figure 3). This variant has not been previously reported in the Human Gene Mutation Database (HGMD) (14), the ClinVar database maintained by the National Center for Biotechnology Information (15), the Human Tafazzin Gene Variants Database curated by the Barth Syndrome Foundation (16) (accessed on 14th April 2025) and also in GnomAD v.4.1.0 database (17).
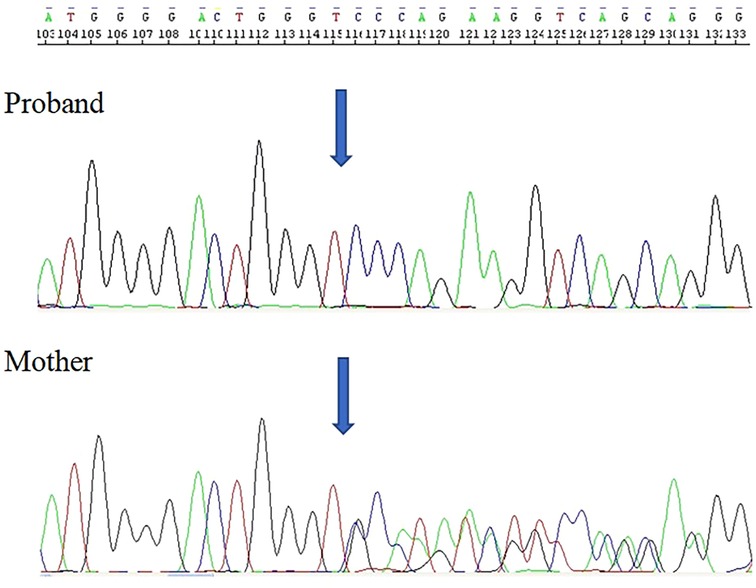
Figure 3. Electropherogram of the TAFAZZIN gene sequencing in the hemizygous patient revealed the in-frame deletion c.525_533del.p(His176_Phe178del).
Variant is located in a region where there are missense (likely) pathogenic changes reported (chrX: 154419614T>A and chrX: 154419614T>C), both affects protein function (acyltransferase domain). MutationTaster algorithm predicts its deleterious effect (18).
According to AlphaFold, the average missense pathogenicity scores were 0.995 for histidine, 0.876 for isoleucine, and 0.99 for phenylalanine, with a threshold of 0.564 above which pathogenicity is considered likely (19) (Figure 4).
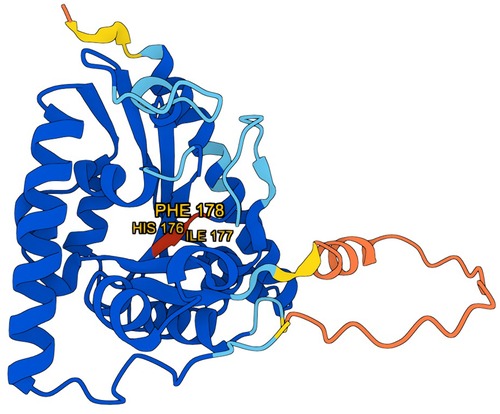
Figure 4. 3D model of the tafazzin protein generated using AlphaFold (DeepMind), with amino acids affected by mutation marked in red. Model visualization is based on data provided by AlphaFold, [used under the Creative Commons Attribution 4.0 (CC BY 4.0)].
The potential impact of the c.525_533del variant on splicing was assessed using SpliceAI and Pangolin. SpliceAI predicted a low-confidence donor site loss (score = 0.23 at position −2 bp relative to the variant), while Pangolin indicated minimal splice site loss (score = 0.05) and negligible splice site gain (score = 0.01). These findings suggest that the variant is unlikely to significantly disrupt canonical splicing or create cryptic splice sites (20).
According to Ensembl - all TAFAZZIN gene variants encoding the tafazzin protein contain the locus in which the mutation described in our study has occurred (21).
In silico conservation analysis using PhyloP100 revealed a score of 8.555 for the deleted region, indicating strong evolutionary conservation (22). This supports the pathogenic potential of the in-frame deletion.
According to American College of Medical Genetics and Genomics (ACMG) scoring this variant was classified as likely pathogenic: PM1 (located in a critical and well-established functional domain - active site of an enzyme); PM2 (absent from controls in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium); PM4 (protein length changes as a result of in-frame deletions in a nonrepeat region) and PP3 (can be used only once in any evaluation of a variant) (23).
Analysis of the c.525_533del.p(His176_Phe178del) mutation revealed its impact at the amino acid level, altering a conserved region within the tafazzin protein. This protein functions as a phospholipid–lysophospholipid transacylase, essential for cardiolipin remodeling. Disruption of tafazzin activity leads to an accumulation of monolysocardiolipin and a decrease in mature cardiolipin levels, resulting in increased mitochondrial reactive oxygen species (ROS) production. Elevated ROS levels activate Ca2+/calmodulin-dependent protein kinase II (CaMKII), which phosphorylates ryanodine receptor 2 (RyR2), enhancing Ca2+ leakage from the sarcoplasmic reticulum. This cascade elevates diastolic Ca2+ levels and depletes sarcoplasmic reticulum Ca2+ stores, impairing cardiomyocyte contractility, relaxation, and development, thereby contributing to the pathophysiology of BTHS and DCM (1, 24).
This finding is particularly significant given the previously reported novel mutation c.83T > A, p.Val28Glu, in a Polish family (25). The challenges encountered in diagnosing BTHS, as highlighted by the previously reported familial mosaicism necessitating multi-tissue genetic testing, further emphasize the critical need for increased genetic diagnostic efforts. The rarity of diagnosed cases, coupled with the recurrent discovery of novel pathogenic variants in the Polish population, strongly advocates for a proactive approach to genetic screening.
Additional mutations within this region have been documented, underscoring the significance of this genomic segment in the pathogenesis of BTHS. A case involving a c.526C>T mutation (p.His176Tyr), described by Hirono et al., concerned a neonate diagnosed on the second day of life. The patient presented with heart failure, marked by an LVEF of 17%, ventricular tachycardia, tachypnoea, neutropenia, motor retardation, and 3-methylglutaconic aciduria, with no relevant family history. The patient succumbed to the condition at 5 months of age (26). Similarly, Thompson et al. reported a 4-year-old patient with the same mutation, presenting with delayed growth (Z-scores for height and weight of −3.25 and −2.60, respectively), increased LVDd and LVSd (Z-scores of 1.54 and 2.60, respectively), reduced left ventricular fractional shortening (29.06%), and elevated cardiolipin ratio (17.3; normal <0.2) along with raised methylglutaconic acid levels (1,048.5 nmol/L; normal range 162 ± 68 nmol/L) (27). Wang C. et al. described a case of fetal death due to congestive heart failure, attributed to a double pathogenic mutation involving both p.His176Tyr in the TAFAZZIN gene and p.Arg99His in the KCNE3 gene, the latter being associated with Brugada syndrome (28).
Wang J. et al. reported the c.527A>G mutation (p.His176Arg) in twin brothers. These male infants were born with moderately low birth weights and presented with pneumonia and heart failure at 2.5 months of age, followed by a diagnosis of cardiomyopathy. Both exhibited hypotonia, motor delay, and growth retardation. Echocardiographic findings revealed reduced LVEF (45.6% and 36.2%), decreased left ventricular shortening fractions (22.1% and 16.7%), and LVDd Z-scores of 5.7 and 3.8, respectively. Additionally, a high, though sub-pathological, noncompaction/compaction (NC/C) ratio of 1.58 and 2.20 was observed. ECG revealed ST-T segment abnormalities and borderline QTc intervals (441 ms and 431 ms), alongside 3-methylglutaconic aciduria. Both patients died at 7 and 7.5 months of age (29).
Wang H. et al. reported a mutation at c.528A>C (p.His176Pro) (30).
The c.528_541 + 7del (r.spl) mutation is listed in the ClinVar database (31).
The c.532T>A mutation (p.Phe178Ile) was identified in a patient presenting with DCM, neutropenia, and growth retardation. The patient's brother was also affected, and the patient died at 9 months of age (32).
The c.532T>C mutation (p.Phe178Leu) is noted in the Human Tafazzin Gene Variants Database, based on an individual report (15).
The available data suggest that patients carrying mutations affecting at least one nucleotide within the region of the c.525_533del deletion, as observed in our patient, exhibit a similar constellation of symptoms: heart failure with reduced LVEF, DCM, tachypnea, prolonged QTc interval, ventricular extrasystoles, ST-T segment changes, growth retardation, motor delays, or neutropenia. Data regarding cardiolipin levels and 3-methylglutaconic aciduria in the described patient were not available. According to the literature, our patient with the c.525_533del deletion is the longest-lived patient (currently 10 years) and the only patient with a mutation in this region to have undergone OHT.
Neutropenia is a well-documented, dose-dependent adverse effect of mycophenolate mofetil (33). There are also reports describing abnormalities in both the number and morphology of neutrophils in heart transplant recipients treated with mycophenolate mofetil (34). It has been suggested that these morphological changes may result from inhibition of the enzyme inosine monophosphate dehydrogenase, leading to reduced de novo synthesis of guanosine nucleotides (35). Azathioprine is another immunosuppressive agent associated with a high risk of neutropenia (36); however, it was not administered in the case described. In patients with an increased risk of neutropenia, such as those with BTHS, immunosuppressive therapy should be managed with particular caution.
Conclusions
The deletion c.525_533del.p(His176_Phe178del) is strongly suspected to be responsible for the clinical manifestations of BTHS. OHT remains the gold standard of treatment for advanced cardiomyopathy in this syndrome. Therefore, our findings, coupled with the limited number of diagnosed cases and the prevalence of novel TAFAZZIN variants in the Polish/Caucasian population, strongly underscore the critical need for increased awareness and the routine implementation of genetic diagnostics in individuals presenting with pathognomonic symptoms or a family history of such cardiac manifestations.
Data availability statement
The original contributions presented in the study are included in the article and the Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the patient’s legal guardian for the publication of any potentially identifiable images or data included in this article.
Author contributions
MKr: Writing – original draft, Writing – review & editing. JŚ: Methodology, Writing – original draft. SP: Writing – original draft, Validation. MKa: Writing – review & editing, Formal analysis. PH: Writing – original draft. WG: Writing – original draft. WH: Writing – original draft. GS: Writing – original draft. DP-A: Data curation, Formal analysis, Investigation, Visualization, Writing – review & editing. TH: Funding acquisition, Writing – original draft, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Medical University of Silesia.
Acknowledgments
The authors would like to express their sincere gratitude to Professor Agnieszka Madej-Pilarczyk MD, PhD for her organizational support and Professor Tadeusz Osadnik, MD, PhD, for his valuable assistance in editing the manuscript and interpreting the results of the genetic analyses.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Taylor C, Rao ES, Pierre G. Clinical presentation and natural history of Barth syndrome: an overview. J Inherit Metab Dis. (2022) 45(1):7–16. doi: 10.1002/jimd.12422
2. Sabbah HN. Elamipretide for Barth syndrome cardiomyopathy: gradual rebuilding of a failed power grid. Heart Fail Rev. (2022) 27:1911–23. doi: 10.1007/s10741-021-10177-8
3. Dudek J, Maack C. Barth syndrome cardiomyopathy. Cardiovasc Res. (2017) 113:399–410. doi: 10.1093/cvr/cvx014
4. Reynolds S. Successful management of Barth syndrome: a systematic review highlighting the importance of a flexible and multidisciplinary approach. J Multidiscip Healthc. (2015) 8:345–58. doi: 10.2147/JMDH.S54802
5. Pang J, Bao Y, Mitchell-Silbaugh K, Veevers J, Fang X. Barth syndrome cardiomyopathy: an update. Genes (Basel). (2022) 13:656. doi: 10.3390/genes13040656
6. Brieler J, Breeden MA, Tucker J. Cardiomyopathy: an overview. Am Fam Physician. (2017) 96:640–6.29431384
7. Reichart D, Magnussen C, Zeller T, Blankenberg S. Dilated cardiomyopathy: from epidemiologic to genetic phenotypes: a translational review of current literature. J Intern Med. (2019) 286:362–72. doi: 10.1111/joim.12944
8. Pinto YM, Elliott PM, Arbustini E, Adler J, Anastasakis A, Böhm M, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. (2016) 37:1850–8. doi: 10.1093/eurheartj/ehv727
9. Maron BJ, Desai MY, Nishimura RA, Spirito P, Rakowski H, Towbin JA, et al. Diagnosis and evaluation of hypertrophic cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. (2022) 79:372–89. doi: 10.1016/j.jacc.2021.12.002
10. Zhao X, Li X, Sun W, Jia JA, Yu M, Tian R. Prenatal case report of Barth syndrome caused by novel TAFAZZIN mutation: clinical characteristics of fetal dilated cardiomyopathy with ascites. Front Pediatr. (2022) 10:1004485. doi: 10.3389/fped.2022.1004485
11. Jefferies JL. Barth syndrome. Am J Med Genet C Semin Med Genet. (2013) 163C:198–205. doi: 10.1002/ajmg.c.31372
12. Thompson R, Jefferies J, Wang S, Pu WT, Takemoto C, Hornby B, et al. Current and future treatment approaches for Barth syndrome. J Inherit Metab Dis. (2022) 45:17–28. doi: 10.1002/jimd.12453
13. Hanke SP, Gardner AB, Lombardi JP, Manning PB, Nelson DP, Towbin JA, et al. Left ventricular noncompaction cardiomyopathy in Barth syndrome: an example of an undulating cardiac phenotype necessitating mechanical circulatory support as a bridge to transplantation. Pediatr Cardiol. (2012) 33:1430–4. doi: 10.1007/s00246-012-0258-z
14. The Human Gene Mutation Database. (2025). Available online at: www.hgmd.cf.ac.uk/ (Accessed April 14, 2025)
15. National Library of Medicine. (2025). ClinVar. Available online at: https://www.ncbi.nlm.nih.gov/clinvar/ (Accessed April 14, 2025)
16. Barth Syndrome Foundation. (2025). Available online at: www.barthsyndrome.org/research/tafazzindatabase.html (Accessed April 14, 2025)
17. GnomAD v.4.1.0 database. (2025). Available online at: https://gnomad.broadinstitute.org/ (Accessed July 1, 2025)
18. MutationTaster2025. (2025). Available online at: https://www.mutationtaster.org/ (Accessed July 1, 2025)
19. AlphaFold Protein Structure Database. (2025). Available online at: https://alphafold.com/ (Accessed July 1, 2025)
20. SpliceAI. (2025). Available online at: https://spliceailookup.broadinstitute.org/ (Accessed July 1, 2025)
21. Ensembl. (2025). Available online at: https://www.ensembl.org/index.html (Accessed July 1, 2025)
22. VarSome The Human Genomics Community. (2025). Available online at: https://varsome.com (Accessed July 1, 2025)
23. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
24. Liu X, Wang S, Guo X, Li Y, Ogurlu R, Lu F, et al. Increased reactive oxygen species–mediated Ca2+/calmodulin-dependent protein kinase II activation contributes to calcium handling abnormalities and impaired contraction in Barth syndrome. Circulation. (2021) 143(19):1894–911. doi: 10.1161/CIRCULATIONAHA.120.048698
25. Zapała B, Płatek T, Wybrańska I. A novel TAZ gene mutation and mosaicism in a Polish family with Barth syndrome. Ann Hum Genet. (2015) 79(3):218–24. doi: 10.1111/ahg.12108
26. Hirono K, Hata Y, Nakazawa M, Momoi N, Tsuji T, Matsuoka T, et al. Clinical and echocardiographic impact of Tafazzin variants on dilated cardiomyopathy phenotype in left ventricular non-compaction patients in early infancy. Circulation J. (2018) 82:2609–18. doi: 10.1253/circj.CJ-18-0470
27. Thompson WR, DeCroes B, McClellan R, Rubens J, Vaz FM, Kristaponis K, et al. New targets for monitoring and therapy in barth syndrome. Genet Med. (2016) 18:1001–10. doi: 10.1038/gim.2015.204
28. Wang C, Hata Y, Hirono K, Takasaki A, Oława SW, Nakaoka H, et al. A wide and specific spectrum of genetic variants and genotype-phenotype correlations revealed by next-generation sequencing in patients with left ventricular noncompaction. J Am Heart Assoc. (2017) 6:006210. doi: 10.1161/JAHA.117.006210
29. Wang J, Guo Y, Huang M, Zhang Z, Zhu J, Liu T, et al. Identification of TAZ mutations in pediatric patients with cardiomyopathy by targeted next-generation sequencing in a Chinese cohort. Orphanet J Rare Dis. (2017) 12:26. doi: 10.1186/s13023-016-0562-4
30. Wang H, Westerfield BA, Pena LS, Do A, Rodriguez GA, Zou W, et al. Abstract 16329: ten novel mutations of the TAZ gene in patients with Barth syndrome. Circulation. (2011) 124(Number suppl_21):A16329. doi: 10.1161/circ.124.suppl_21.A16329
31. National Library of Medicine. (2024). ClinVar. Available online at: https://www.ncbi.nlm.nih.gov/clinvar/variation/855792/?oq=c.528_541+7del&m=NM_000116.5(TAFAZZIN):c.528_541%207del (Accessed September 14, 2024)
32. D'Adamo P, Fassone L, Gedeon A, Janssen EA, Bione S, Bolhuis PA, et al. The X-linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet. (1997) 61:862–7. doi: 10.1086/514886
33. Braghieri L, Jennings DL, Bohn B, Habal M, Pinsino A, Mondellini GM, et al. Temporal shifts in safety and efficacy profile of mycophenolate mofetil 2 g versus 3 g daily early after heart transplantation. Pharmacotherapy. (2022) 42:697–706. doi: 10.1002/phar.2724
34. Banerjee R, Halil O, Bain BJ, Cummins D, Banner NR. Neutrophil dysplasia caused by mycophenolate mofetil. Transplantation. (2000) 70:1608–10. doi: 10.1097/00007890-200012150-00012
35. Allison AC, Eugui EM. Purine metabolism and immunosuppressive effects of mycophenolate mofetil (MMF). Clin Transplant. (1996) 10:77–84. doi: 10.1111/j.1399-0012.1996.tb00651.x
Keywords: Barth syndrome, TAFAZZIN gene, tafazzin, cardiolipin, cardiomyopathy, heart transplant
Citation: Krawiec M, Śliwka J, Pawlak S, Kapałka M, Hamerling P, Grzywacz W, Hawel W, Skórska G, Piekutowska-Abramczuk D and Hrapkowicz T (2025) A novel TAFAZZIN gene variant c.525_533del causing Barth syndrome and leading to heart transplantation: a case report. Front. Pediatr. 13:1634258. doi: 10.3389/fped.2025.1634258
Received: 23 May 2025; Accepted: 14 July 2025;
Published: 18 August 2025.
Edited by:
Simona Lobasso, University of Bari Aldo Moro, ItalyReviewed by:
Mindong Ren, New York University, United StatesTeresa Platek, Jagiellonian University Medical College, Poland
Copyright: © 2025 Krawiec, Śliwka, Pawlak, Kapałka, Hamerling, Grzywacz, Hawel, Skórska, Piekutowska-Abramczuk and Hrapkowicz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michał Kapałka, bWljaGFsLmthcGFsa2EwMEBnbWFpbC5jb20=