 Christy A. Barlow
Christy A. Barlow Brooke T. Mossman
Brooke T. Mossman- 1Aetia Scientific Collaborative, Boulder, CO, United States
- 2Department of Pathology and Laboratory Medicine, Larner College of Medicine, University of Vermont, Burlington, VT, United States
Although inhalation of sufficient doses and dimensions of airborne asbestos dusts in an occupational setting can produce cancer in the lungs, pleura and peritoneum, tumors occur in <5–10% of exposed individuals, even among persons with considerable historical exposures. In this perspective, we review cell defense mechanisms that are involved in protective and adaptive responses to asbestos exposure. These adaptive responses are orchestrated through a multifaceted cellular program involving the concerted action of diverse stress response pathways, including antioxidant responses, DNA repair mechanisms, molecular mechanisms for intracellular signaling leading to proliferation, apoptosis, and inflammation, and cell cycle regulation. These cell defenses suggest that humans can adjust to moderate levels of stress or change without experiencing negative effects, implying the existence of a threshold dose. Likewise, reported no-observed adverse-effect levels (NOAELs) for various asbestos fiber types and asbestos-related cancers in experimental and epidemiological data further support the existence of a threshold dose and are discussed here.
1 Introduction
Asbestos is a generic term for a family of naturally occurring hydrated silicate minerals (1, 2). It encompasses a group of six chemically and physically diverse types of asbestiform minerals that are characterized according to morphology as serpentine (chrysotile) or amphibole (crocidolite, amosite, tremolite, anthophyllite, and actinolite). Chrysotile, the sole member of the serpentine group, is a hydrated magnesium silicate that is represented by the chemical formula Mg3[Si2O5](OH)4, whereas the two most commercially important amphiboles, crocidolite and amosite, have a high iron content and are represented as and , respectively (3). It should be acknowledged that increased risks of both lung cancers and malignant mesotheliomas (MMs) have been observed in workers exposed to asbestos fibers occupationally, but the carcinogenic potency of commercially used fibers (crocidolite > amosite > chrysotile) is vastly different, especially in the development of MMs (4–10).
Asbestos-related disease conforms to standard toxicological principles for human disease and exhibits a non-linear dose-response relationship (8). Scientific data increasingly suggest that promoters and other non-genotoxic agents will have a non-linear dose-response relationship at low doses (11, 12). Non-linear relationships and the assumption that no increase in risk occurs at low doses of agents below a certain level have been endorsed for ionizing radiation and chemicals in general, providing evidence for thresholds for both DNA interacting and epigenetic chemical carcinogens (13–19).
Humans have evolved multiple defense mechanisms against the external environment to maintain homeostasis within their internal environment. In response to asbestos and other carcinogens, defensive and adaptive cellular responses exist to prevent cellular injury and permit normal cell survival. Adaptive responses include metabolism and elimination of foreign agents, as well as damage repair that can occur at both the cellular and molecular level. Formation of cancers is thought to involve a loss in multiple adaptive cellular responses (20). In this perspective, we review cell defense mechanisms that are involved in protective and adaptive responses to asbestos fiber exposures in lung epithelial and mesothelial cells, target cells of asbestos-induced lung carcinomas and MMs. Then, we review experimental and epidemiological evidence suggesting thresholds for exposures to asbestos fibers. Knowledge of these defense responses are important in the regulation of asbestos exposures and the design of preventive and therapeutic approaches to both lung cancers and MMs.
2 Cell defense/adaptive responses
Researchers have used in vitro studies on isolated cells and organ cultures to address key events in carcinogenesis by asbestos (21). These in vitro models allow for the measurement of acute responses, including those important in defense and adaptation, after exposure to known concentrations of fibers over a range of doses. In this perspective, in vitro data, have been correlated, whenever possible, to events occurring after inhalation, the natural route of access to the body, of various asbestos fiber types in rodents and human lungs (22). There are several limitations in many of the studies described below including a general lack of dose-response experiments and the lack of inclusion of non-disease causing particles or fibers (negative controls) to determine the specificity of asbestos-induced responses. Additionally, most animal studies evaluated effects attributed to asbestos on an equal mass or weight basis even though the number, surface area and reactivity of fibers at equal weight concentrations may be vastly different, thereby rendering comparisons between different experiments and human exposure difficult.
Adaptive cellular responses are of specific interest in this perspective and include multiple DNA repair mechanisms; a battery of antioxidant responses; proteins that monitor cellular proliferation, growth, and adhesion; and cell cycle check point mechanisms that pause cell proliferation to allow for DNA repair.
2.1 DNA repair mechanisms
Since a consequence of asbestos-induced oxidative damage may include DNA single strand breaks (SSB) and oxidant-induced 8-hydroxydeoxyguanosine (8-OHdG) DNA adducts (23, 24), it is important to discuss the human physiological response to such insult. In mammalian cells, four major DNA damage repair mechanisms are known to be responsible for repairing different DNA lesions: base excision repair (BER), nucleotide excision repair, mismatch repair, and recombinational system repair (25). Polymorphisms in genes encoding DNA repair proteins, such as XRCC1 and BRCA1, have also been suggested to be associated with the risk of MM (26, 27). Moreover, germline mutations in DNA repair genes may induce MMs (28) or predispose asbestos-exposed individuals to MMs (29, 30).
Different types of asbestos have been shown to produce 8-OHdG DNA adducts, DNA damage, and/or chromosomal aberrations in mesothelial cells in vitro and in vivo (23, 24). Adduct formation, oxidative damage, alkylation, and deamination can all create DNA base damage that is repaired by the same base excision repair mechanism. This pathway is also responsible for repairing DNA SSBs generated by reactive oxygen species (ROS) (31). DNA glycosylases recognize and remove the damaged base leaving a site that that is absent of a purine and pyrimidine (AP site) (32). The AP site is repaired by an AP-endonuclease and ligase which initially creates a gap in the DNA strand that is eventually filled in by DNA polymerase and DNA ligase (33). AP-endonuclease co-localizing with mitochondria is increased in mesothelial cells after exposure to crocidolite asbestos, suggesting that repair mechanisms exist (34). Moreover, mitochondrial 8-oxoguanine DNA glycosylase, a mtDNA BER enzyme, reduces mtDNA damage and apoptosis by amosite in alveolar epithelial cells and mitigates lung fibrosis (35).
Arguably, DSB are the most significant form of DNA damage and could lead to cell lethality and transformation if left unrepaired (36). As few as one DSB is sufficient to kill a cell if it inactivates an essential gene or induces apoptosis. Mammalian cells have two somewhat redundant mechanisms for repairing DSBs: homology-directed repair and non-homologous end-joining repair (33). The pathway that the cell uses to repair the DSB depends on the phase of cell cycle when the error is detected and the type of DNA lesion. The non-homologous end-joining mechanism is used if the cell is in the G1 phase of cell cycle. If the cells are in S or G2 phase of the cell cycle, then the cell repairs the DSB by the homology-directed repair mechanism (37). Marczynski et al. reported that there are higher incidences of DSBs in white blood cells of occupationally exposed asbestos workers as compared with the non-exposed control population (38).
The upregulation of DNA repair genes after exposure to asbestos is a strong indication of an adaptive response, meaning that a cell is actively increasing its ability to repair DNA damage in response to a stressful environment, allowing it to better tolerate and survive the damaging conditions. This upregulation is often part of a broader cellular response to stress, where the cell activates various mechanisms to mitigate damage and maintain homeostasis.
2.2 Antioxidant responses
Despite decades of research on the health effects of asbestos, the underlying mechanisms leading to asbestos-induced pulmonary toxicity and cancers are not completely understood; however, much research has focused on the importance of generation of ROS and reactive nitrogen species (RNS). Multiple mechanisms of generation of oxidant species occur in lung epithelial cells, mesothelial cells, and phagocytes, such as alveolar and pleural macrophages, after exposure to amphibole asbestos and other fibers (39, 40). For example, the high iron content and iron availability of amosite and crocidolite asbestos generates a Haber-Weiss reaction producing superoxide and hydrogen peroxide that may function extracellularly or within cells after fiber uptake. Whereas, cells of a variety of types can internalize fibers less than their cell diameters, longer fibers cause frustrated phagocytosis which stimulates NADPH (nicotinamide adenine dinucleotide phosphate) oxidase in cell membranes, a potent enzyme in production of ROS. Once internalized into an acidic phagolysosome, shorter fibers undergo changes in composition and structure (41) and exist in perinuclear vesicles or free in the cytoplasm (42).
Mitochondria are both a source and target of ROS generated by crocidolite asbestos in mesothelial and pulmonary epithelial cells. Oxidant elaboration results in damage to mitochondrial DNA (mtDNA) and dose-related alterations in mitochondrial gene expression (43–45). At high toxic fiber concentrations, apoptosis occurs. The importance of oxidative stress in asbestos-induced toxicity and cell death has been discussed previously (39, 40).
In addition to ROS- and RNS-induced protein modifications in vitro and in vivo, DNA and lipids undergo structural changes that are linked to damage of macromolecules and cell death at high concentrations of asbestos and initiation of multiple signaling pathways linked to carcinogenic events at low concentrations. Signatures of oxidative RNA and DNA damage have been observed for as long as 72 h in human mesothelial cells at growth inhibitory concentrations of crocidolite (34, 46), in isolated rat pleural mesothelial cells at both toxic and non-toxic concentrations of crocidolite (34), and in rats after intraperitoneal injection of crocidolite (47).
Since amosite and crocidolite asbestos are high iron-containing and the most potent in the induction of human mesotheliomas, most studies have focused on their mechanisms of oxidant stress and defense (40, 48, 49). Surface active sites reducing O2 and catalyzing the decomposition of H2O2 to reactive radicals, such as •OH, or lipid radicals are largely related to the presence and ionic state of iron. Moreover, pre-addition or co-administration of iron chelators or catalase inhibits cell injury, inflammation, and asbestosis in vivo (50, 51), supporting a direct role of iron in lung disease.
One well-documented protective response to longer (generally > 10 or 20 microns) amphibole fibers is the formation of asbestos (ferruginous) bodies in the lung that are comprised of predominately ferric iron with minor amounts of protein and mucopolysaccharides. The sequestration of fibers in a non-reactive core is an important defense mechanism as the valence sites of ferric moieties are not available for electron exchange and generation of ROS. In addition, asbestos bodies, in comparison to native fibers, show both diminished formation of ROS and reduced toxicity (49).
Upregulation and increased expression of a cadre of antioxidant enzymes have also been demonstrated in vitro and in vivo after exposures to asbestos fibers and inflammatory particles (52). Conventional antioxidant enzymes include copper–zinc containing superoxide dismutase (CuZnSOD or SOD1), manganese-containing superoxide dismutase (MnSOD or SOD2), catalase, and glutathione peroxidase (GPX) and may act alone or in combination to prevent asbestos-associated injury (51, 53). Other proteins, including heme oxygenase-1 (HO-1), GRP78, and heat shock proteins (HSP70), are linked to prevention of oxidant stress and increase after exposures of lung epithelial and mesothelial cells to crocidolite fibers in vitro (54, 55).
Supplementation of antioxidant enzymes also inhibits asbestos-induced toxicity and signatures of carcinogenesis. For example, in tracheobronchial epithelial cells, minimally toxic concentrations of crocidolite or chrysotile asbestos caused protracted increases in total SOD levels, and toxicity was ameliorated by addition of SOD (56). In rat lungs and human bronchi, CuZnSOD was prominent in macrophages and bronchiolar epithelial cells, suggesting protective mechanisms in both target cells of lung cancers and effector cells of the immune system. Administration of antioxidant enzymes inhibited crocidolite-induced cell injury, inflammation, and fibrotic changes (51). In comparison to other antioxidant enzymes, mitochondrial MnSOD was most strikingly elevated, and linked to injury and inflammation of other fibrogenic minerals such as silica (57–59). Transfection of MnSOD into tracheal epithelial cells exposed to asbestos inhibited asbestos-induced toxicity (60). Individuals with a GSTM1 null allele and Ala/Ala genotypes of codon 16 within the MNSOD gene exhibit increased risk of MM (61).
Cell glutathione and thiol levels are critical defense mechanisms after oxidant stress by asbestos (62, 63). Depletion of total cell glutathione pools occurs after addition of crocidolite to mesothelial cells and is accompanied by increased levels of c-fos and c-jun early response prot(o)oncogenes (64). Addition of N-acetyl-cysteine to boost thiol levels decreased asbestos-mediated gene expression in a dose-dependent fashion (64).
The thioredoxin system is composed of NADPH, cytosolic and mitochondrial thioredoxins, and thioredoxin reductases. Crocidolite asbestos oxidized the pool of the antioxidant, Thioredoxin-1 (TRX1), in human mesothelial cells via production of ROS (65). Modulation of thioredoxins and thioredoxin interacting protein (TXNIP) showed that TRX1 overexpression or knockdown of TXNIP attenuated NLRP3 inflammasome activation, reinforcing the role of inflammasome activation by oxidants and subsequent generation of proteins, IL-1B and HMGB1, linked to mesothelioma development (66, 67).
Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) is a transcription factor that plays a key role in controlling the inducible expression of enzymes linked to the synthesis of glutathione and other antioxidants. As such, it is important in the control of inflammation (68). When exposed to ROS, Nrf2 migrates from the cytoplasm to the nucleus where it leads to the upregulation of expression of several antioxidant and detoxification genes (e.g., GSTs, SODs, and HO-1), the downregulation of NF-κB, and reduction in proinflammatory cytokines (e.g., IL-6 and IL-1β). This is an adaptive mechanism that enhances resiliency in response to subthreshold doses of toxins.
In summary, cells of the immune system and target cells of asbestos-associated diseases exhibit oxidative stress that can be counteracted by antioxidant responses at low concentrations of asbestos fibers that are non-toxic but are overwhelmed at high fiber concentrations that may be carcinogenic or cause cell death.
2.3 Molecular mechanisms of proliferation and inflammation
Both Activator Protein-1 (AP-1) and NF-κB are redox-acivated transcription factors that play crucial roles in regulating cellular processes such as proliferation and inflammation by controlling gene expression. These events are hallmarks of the cancer process (69). Generally, frustrated phagocytosis of asbestos fibers triggers the production of ROS within cells, which in turn activates cellular signaling pathways leading to the phosphorylation and nuclear translocation of AP-1 and NF-κB subunits (Figure 1). These events can lead to altered gene expression affecting cellular processes linked to proliferation, altered cell function (metaplasia), cell death, and inflammation. In general, a relatively low concentration of chrysotile or amphibole asbestos exposure (<1 μm/cm2) results in proliferative signaling in mesothelial cells, while higher asbestos concentrations result in apoptosis and block proliferation (70–74). Proliferation of mesothelial cells by crocidolite asbestos, the phorbol ester tumor promoter, TPA (12-O-tetradecanoylphorbol-13-acetate), and TNFα (Tumor Necrosis Factor alpha) suggest multiple cell pathways leading to tumor promotion in MMs (75). In contrast, apoptosis is an important protective mechanism by which DNA damaged cells are eliminated without initiating an inflammatory response (76). Likewise, acute inflammation is considered a vital defense mechanism as it is the immune system's response to harmful stimuli, like infections or injury, by ultimately restoring tissue homeostasis.
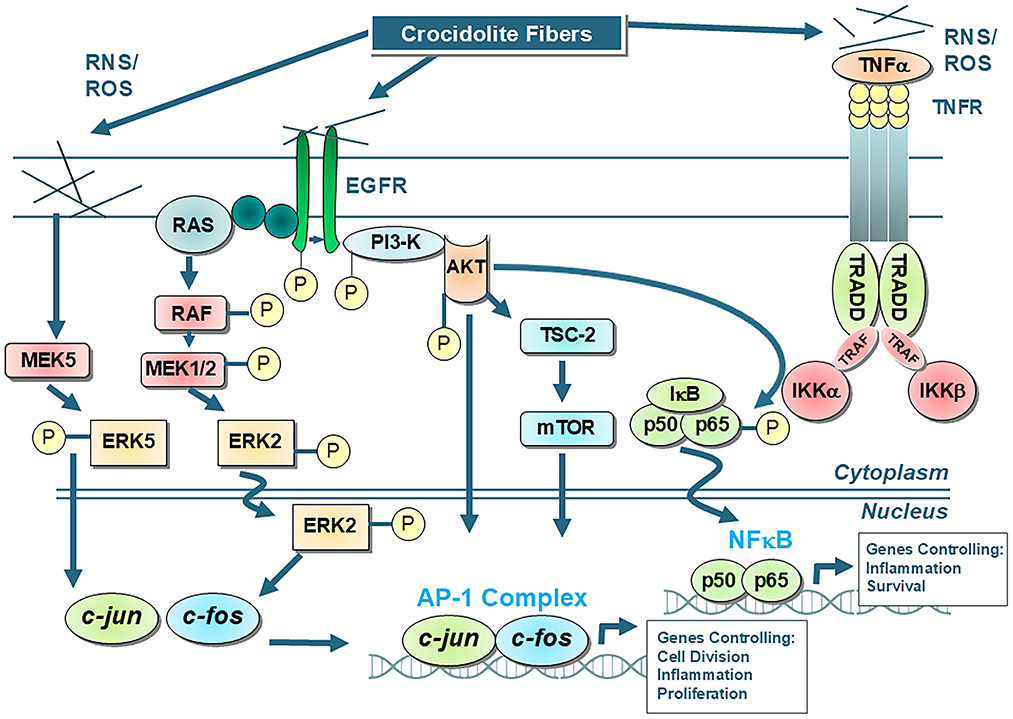
Figure 1. Shows the main AP-1 and NF-kB cell signaling pathways that occur in tracheobronchial/lung epithelial and mesothelial cells after exposures to asbestos fibers, such as amosite and crocidolite asbestos, that initially interact with the external cell membrane. Interactions of fibers or elaboration of oxidants then result in activation of protein cascades that cause alterations in gene expression. It is unlikely that asbestos and other elongated mineral fibers at low concentrations induce cancerous changes by interacting directly with the DNA of cells. For example, the mutagenic changes reported using chrysotile asbestos in a human/hybrid cell model are observed at high concentrations causing large scale deletions incompatible with cell survival (40, 140). Moreover, crocidolite and erionite induce polyploidy and clastogenic effects in cells, but not mutations (183). In contrast to chrysotile asbestos which dissolves over time in rodent and human lungs (139, 184), durable amosite and crocidolite fibers produce many of the hallmarks of cancers, most notably genomic instability, sustained cell proliferation and chronic inflammation (13, 69, 185). These and other characteristics of cancer observed in asbestos-associated cell outcomes are attributed to long, iron-rich amphibole fibers, altered cell signaling events, and production of reactive oxygen and nitrogen species (ROS, RNS) (69).
2.3.1 TP53
TP53 (p53) acts as a tumor suppressor protein that generally functions to suppress inflammatory responses by negatively regulating NF-κB signaling (77). NF-κB can suppress p53 levels by upregulating the expression of MDM2, a target gene of NF-kB.
Studies have shown increased p53 protein expression occurs in lung epithelial and mesothelial cells and in rodents exposed to asbestos (78–80). In chrysotile-exposed mice and rats, p53 increases are observed prior to reversible increases in lung epithelial cell proliferation (81, 82). p53 primarily functions as a transcription factor that regulates a wide spectrum of genes including apoptosis, cell cycle arrest, senescence, autophagy, DNA repair, and angiogenesis (83). Broadly, p53 is a tumor suppressor that limits cellular proliferation by inducing cell cycle arrest and apoptosis in response to cellular stresses, such as DNA damage.
Beyond its traditional role as a transcription factor, recent research has shown that p53 can also directly translocate to the mitochondria under stress conditions, where it can interact with mitochondrial proteins to disrupt their function and initiate apoptosis (84). Amphibole asbestos exposure causes mitochondrial dysfunction, which leads to cell oxidative stress and apoptosis through the p53-regulated mitochondrial pathway (85). Moreover, spontaneous p53 mutations in murine mesothelial cells increase their sensitivity to crocidolite asbestos and ionizing radiation (86).
Loss of function mutations in p53 have been associated with the development of MMs in humans and rodents (87, 88). A select group of MM patients demonstrate mutations in the p53 gene (89).
2.3.2 Activator protein-1 (AP-1)
Members of the Mitogen-Activated Protein Kinase (MAPK) family are critical to most aspects of AP-1 regulation. In vitro experiments have shown that the MAPK cascade is involved in both apoptotic and proliferative responses to abestos. Although the mechanism of action remains elusive, it has been shown that after interaction with cells, asbestos fibers trigger a series of multiple protein phosphorylation events occurring after asbestos-associated oxidant generation. After dimerization and rapid dissociation or phosphorylation of the the epidermal growth factor receptor (EGFR) (90), crocidolite asbestos fibers trigger many signaling cascades, including the AKT pathway and MAPK (73, 91–96). MAPKs consist of three major families: extracellular signal-regulated kinases (ERKs), c-Jun-NH2-terminal kinases (JNKs), and p38 kinases. ERKs (ERK1, 2, and 5) are effectors of the Ras proto-oncoprotein and are activated in response to mitogenic stimuli whereas JNKs and p38s are stress activated protein kinases that are activated preferentially by environmental stresses and inflammatory cytokines of the TNF family (97, 98). Asbestos has been shown to stimulate predominately ERKs, but also p38, and JNK pathways in alveolar type II epithelial cells and mesothelial cells (74, 99, 100). These oxidant regulated signaling pathways regulate gene expression of early response proto-oncogenes (fos/jun family) in mesothelial and lung epithelial cells (101, 102). Their proteins can dimerize, forming the transcription factor, AP-1 that interacts with DNA (103). These events may be linked to increases in early-response genes which govern cellular responses, such as proliferation and apoptosis (71). AP-1 activation also leads to increased production of pro-inflammatory cytokines (e.g., TNFα and IL-6) in certain cell types (104).
Since MAPKs are regulated by reversible phosphorylation on threonine and tyrosine residues, deactivation of MAPKs can also occur via dephosphorylation at these residues. This is the role and function of the MAPK phosphatases (MKPs) (105). MKP-1 specifically targets the phosphorylated forms of p38 and JNK, removing the phosphate group and rendering them inactive. By controlling p38 and JNK signaling, MKP-1 plays a crucial role in cell survival by regulating p38 and JNK in response to cellular stress. MKP-1 is upregulated in response to exposure to asbestos in human mesothelial cells (106).
2.3.3 Nuclear factor-kappa B (NF-κB)
NF-κB is a redox-sensitive transcription factor and can be a downstream target of MAPK signaling pathways. Studies have shown that asbestos fibers activate NF-κB in tracheal epithelial and mesothelial cells in vitro and in rat lungs in vivo after asbestos inhalation (107–110). NF-κB regulates multiple aspects of innate and adaptive immune functions and serves as a pivotal mediator of inflammatory responses (111–113). NF-κB is comprised of protein dimers, including the transcription-activating heterodimer consisting of p50 and p65 (RelA) subunits. It is a ubiquitous redox-regulated transcription factor that is retained in the cytoplasm by forming an inactive complex with its cytosolic repressor, IkB. Oxidative and pro-inflammatory stimuli activate NF-κB through phosphorylation-dependent proteasomal degradation of IkBα, ultimately allowing NF-κB to move into the nucleus where it can influence gene expression. NF-κB induces the expression of various pro-inflammatory genes, including those encoding cytokines and chemokines (e.g., TNFα, IL-6, COX-2). It also participates in inflammasome regulation (114). In addition, NF-κB plays a critical role in regulating the survival, activation and differentiation of innate immune cells and inflammatory T cells.
2.3.4 NLRP3 inflammasome
The NLRP3 inflammasome is a multiprotein complex that plays a pivotal role in regulating the innate immune system and inflammatory responses by interacting with various cell death pathways like apoptosis, pyroptosis, and necroptosis (115). By modulating these pathways, inflammation can be mitigated, and tissue repair and regeneration can be promoted.
The activation of the NLRP3 inflammasome is a two-step process, requiring a priming signal that upregulates NLRP3 expression and a subsequent activation signal that triggers the assembly of the inflammasome complex, leading to the maturation and release of pro-inflammatory cytokines like IL-1β and IL-18 (111, 115). Multiple studies have shown that crocidolite asbestos fibers prime and activate the NLRP3 inflammasome in human mesothelial and macrophage cells in vitro and in mice using inhalation models (66, 116, 117). Both AP-1 and NF-κB have been shown to play a role in regulating the NLRP3 inflammasome through transcription of its components, particularly by upregulating the expression of NLRP3 and pro-IL-1β, essentially acting as “priming” signals for NLRP3 activation in response to inflammatory stimuli.
Some studies suggest that prolonged NLRP3 inflammasome activation in response to crocidolite asbestos exposure can contribute to early and chronic inflammation (114). Others postulate that HMGB1, that both stimulates and is a consequence of inflammasome activation, contributes to MM development in rodents (67). However, NLRP3 deficient mice show a similar incidence of MMs when compared to wild-type mice, suggesting that NLRP3 activation may not be critical to the development of MMs (118).
2.3.5 Activator transcription factor 3 (ATF3)
Activator Transcription Factor 3 (ATF3) is a stress-induced transcription factor that acts as a hub of adaptive responses in cells (119). It has been shown using human mesothelial cells that crocidolite asbestos induced increased expression of ATF3, indicating its participation in cell defense from fibers and particles (120). Moreover, in cells exposed to asbestos, silencing of ATF3 increased production of inflammatory cytokines and growth factors such as IL-1B, PDGFBB, VEGF and IL-13.
2.4 Epigenetic control mechanisms
Asbestos fibers have been historically regarded as epigenetic as they do not act directly with DNA to form adducts or metabolites (121, 122). The most frequently studied epigenetic marker in cells is DNA methylation, a process catalyzed by DNA methyl transferases that results in covalent attachment of a methyl group to cytosine. Methylation also occurs at sites of CpG dinucleotides within the promoter regions of genes. Whereas, methylation causes condensation of chromatin, making it inaccessible for transcription, histone acetylation (addition of –COCH3) causes increased accessibility of DNA for transcription. The dynamic reversible processes of acetylation, deacetylation and methylation/demethylation control gene expression in normal and tumor cells (123).
DNA methylation profiling of MET5A mesothelial cells exposed to crocidolite or chrysotile revealed methylation at CpG sites located in genes related to migration and cell adhesion (124). Global and gene-specific DNA methylation effects of crocidolite, amosite and chrysotile fibers have been studied in immortalized human bronchial epithelial cells over a range of asbestos concentrations (125). Global DNA methylation was observed after exposures to crocidolite or amosite, but not chrysotile asbestos. Moreover, no significant changes were observed at the lowest concentrations of amphibole fibers, illustrating a threshold effect. Hierarchical clustering of gene-specific DNA methylation patterns also showed different patterns in chrysotile-exposed cells as compared to amphiboles. Examination of genome-wide methylation changes in lung cancers from smokers and individuals exposed to asbestos revealed unique changes, suggesting that methylation changes may be predictive of these risk factors (126).
Loss of function mutations of tumor suppressor genes that have been associated with cell cycle control have been reported in human and rodent MMs as discussed earlier in this perspective. For example, methylation of the CDKN2A/p16INK4A gene promoter region occurs in human MMs (127). The CDKN2A locus encodes the tumor suppressor proteins, p16INK4 and p14ARF, which regulate the Rb and p53 cell cycle pathways. Loss of CDK2NKB function has also been noted in lung cancers, MMs, and experimental models of mesothelioma where loss of function reflected increased numbers of tumors with decreased latency periods (128).
The methylation status and silencing of the CDKN2A gene has been studied in precancerous bronchial lesions from a series of 37 patients at high risk for lung cancer (129). Increases in methylation occurred with the severity of lesions, suggesting a relationship to the development of lung cancers.
The studies above illustrate the complexity of methylation changes that could be linked to gene expression governing cell defense or initiation of carcinogenic changes by asbestos fibers. The interplay between these epigenetic events and non-coding RNAs may exert protective effects or participate in oxidant-dependent signaling cascades (130).
3 Discussion
The likelihood and magnitude of a biologically relevant response is related to the dose of the substance to which one is exposed. In addition, other factors such as immunologic status, age of exposure, and the microenvironment, etc., influence the vulnerability and severity of exposure. A unifying concept in the biological sciences, and a fundamental tenet in toxicology, is the dose-response relationship. This principle, which has been recognized since at least the sixteenth century, holds that the likelihood and degree of a biologic response is related to the amount of the toxicant administered, and is often described by the oldest and most venerated axiom in toxicology: the dose makes the poison (131, 132). As described in Casarett and Doull's Toxicology: The Basic Science of Poisons, “[i]t is generally recognized that, for most types of toxic responses, a threshold exists such that at doses below the threshold, no toxicity is evident” [(133), p. 22].
Although in vitro or in vivo experimental studies do not provide precise estimates of the biologically effective dose (the actual dose that contributes to the risk of disease) that occurs under realistic exposure scenarios in humans, they do provide evidence for the existence of a threshold dose (a minimum dose that triggers minimal detectable biological effect). More specifically, in vitro studies using asbestos fibers over a range of concentrations demonstrated levels below which no increases in c-jun/c-fos gene expression and/or cell division occurred (70, 101, 134, 135).
3.1 Thresholds in genotoxicity
There is an obvious disconnect between toxicity, genotoxicity, and carcinogenic effects of asbestos fibers. In the majority of toxicity and genotoxicity studies, chrysotile asbestos at equal weight concentrations is more active than amphibole asbestos (23, 136). This is attributed to the positive surface charge of chrysotile rendered by Mg2+ interacting with negative sialic acid residues on the cell surface whereas amphiboles had a neutral or slightly negative charge (137, 138). In contrast, chrysotile asbestos is much less pathogenic in the causation of MMs than amphibole asbestos, in part because Mg2+ is leached from chrysotile over time resulting in its conversion to a non-reactive amorphous particle (139). In fact, its ability to cause genotoxicity, aneuploidy, and cytotoxicity due to large scale deletions in DNA (140) might explain why chrysotile asbestos is not a potent carcinogen in the development of MMs, as a dead cell cannot give rise to tumors.
Throughout the last two decades, numerous independent studies demonstrate the potential for asbestos fibers to act as genotoxic agents by inducing DNA and chromosomal damage in lung and pleural cells [reviewed in Barlow et al. (23)]. DNA damage induced by asbestos is an early event in vitro that may result in genetic instability, necrosis, or apoptosis at high concentrations of fibers and cell transformation at low doses (23, 141). All types of asbestos fibers are capable of mediating chromosomal and DNA damage, such as DNA breaks, cross-linking, and base lesions at high, toxic concentrations (142). Despite the overall lack of dose-response in genotoxic studies in the literature, dose-response relationships are observed in some genotoxicity studies, suggesting cell defense mechanisms such as DNA repair mechanisms and antioxidant responses. For example, non-toxic concentrations of crocidolite asbestos (1.25 and 2.5 μg/cm2) causes increased expression of AP-endonuclease in a dose-dependent manner in isolates of rat pleural mesothelial cells with persistent increases over a 72-h time frame (143). AP-endonuclease was induced at both non-toxic and toxic concentrations of crocidolite. Both transformed and normal cells exhibit dose-related responses to asbestos fibers as indicated by markers of DNA and chromosomal damage, including a lack of effects at lowest concentrations (21). These studies suggest NOAELs.
More recent studies of mechanisms of asbestos-induced injury and disease have focused on indirect effects that lead to DNA damage, and in some cases, the development of lung cancer or MM. These are different from the direct assault on DNA described above in that the focus is on the importance of generation of ROS. As outlined earlier, asbestos fibers were found to stimulate production of ROS though Fe-mediated and cell-mediated mechanisms in vitro (39, 49). Therefore, when discussing the genotoxic potential of asbestos, it is important to distinguish between primary and secondary genotoxicity. The surface properties associated with the different forms of asbestos are believed to play a major role in the primary genotoxicity of asbestos, while the excessive and persistent formation of ROS from inflammatory cells are postulated to play a role in secondary genotoxicity (23, 144). Inflammation is known to persist only at a sufficient dose, and therefore secondary genotoxicity is believed to occur at a threshold dose.
3.2 Thresholds in inflammation
Research has shown that exposure to long fibers of amphibole asbestos and carbon nanotubes significantly causes inflammatory responses in laboratory studies (145–148). Moreover, chronic inflammation is a critical process in the development of human MMs (149–151). Asbestos dose is a crucial determinant for triggering inflammation as high doses over short periods promote an acute neutrophil predominant inflammation whereas low doses over prolonged periods promote an alveolar macrophage predominant chronic inflammation. Of note, several studies using chrysotile and crocidolite have demonstrated levels below which no increases in gene and protein markers of inflammation and disease occur (152). The data inherently negate the legitimacy of a no threshold model as the induction of the inflammatory response contains a natural threshold for inflammatory response activation (153–155).
It is widely accepted that inflammation is a significant driver of carcinogenesis, particularly during the tumor promotion phase (156). Some of the proposed pathobiological processes for inflammation-induced carcinogenesis are through indirect DNA damage due to generation of ROS/RNS within target cells or from macrophages and other immune cells, changes in metabolism, and disruption of immune system homeostasis and function.
Inflammation is the main rate-limiting mode of action for increasing risk in inflammatory-mediated diseases, including MMs (156–160). As described above, the NLRP3 inflammasome plays a key role in initiating inflammation. The two-step process of priming and activation of the NLRP3 inflammasome are governed by several threshold mechanisms (153). Activation of the NLRP3 inflammasome requires a certain intensity and duration of stimulus to trigger its assembly and subsequent inflammatory cytokine release (161). Moreover, these triggering events, which may include sustained generation of intracellular and mitochondrial ROS, depletion of antioxidant pools, and lysosomal destabilization and rupture, also require a certain intensity and duration of stimulus. Both the priming (NFκB signaling and MAPK activation) and triggering event (the activating stimulus) must reach these threshold levels. As such, the NLRP3 inflammasome is activated by sufficiently high and prolonged exposures. This threshold system acts as a protective mechanism, preventing the body from overreacting to minor irritants encountered daily. These thresholds prevent small and brief exposures from triggering an inflammatory response while allowing sufficiently high and prolonged exposures to do so.
3.3 Thresholds in carcinogenicity
The existence of a threshold dose of asbestos at which no increased risk of asbestos-related disease can be observed is supported by occupational and environmental studies. Specifically, a number of published epidemiology studies have suggested that exposures to ambient asbestos concentrations of any fiber type are not associated with a significantly increased incidence of asbestos-related disease (162–170). For example, Price and Ware stated that although women's environmental exposures would likely have increased since the 1930s, with the increasing use of asbestos in the U.S., “the mesothelioma risk for women has not increased” [162, p. 111]. They noted that “[e]nvironmental exposure levels, although increasing, have not triggered a risk response in women. Therefore, those exposure levels must have been below a threshold for mesothelioma” [162, p. 111]. Similarly, Glynn et al. found that there was no increase in incidence rates of pleural mesothelioma among females in urban vs. rural areas in the U.S. between 1973 and 2012, despite measured differences of up to 10-fold or more in ambient airborne asbestos concentrations between these different geographical areas (171). According to the authors, these results suggested that ambient exposures to asbestos over a wide range of background concentrations have not significantly affected the incidence of pleural mesothelioma in the U.S. over the past 40 years.
Epidemiology studies of predominately chrysotile-exposed cohorts suggest that there is a cumulative chrysotile exposure below which there is negligible risk of asbestos-related diseases. Pierce et al. summarized NOAELs reported in the literature for predominantly chrysotile-exposed cohorts and found that the preponderance of studies showed cumulative chrysotile NOAELs for both lung cancers and MMs (172, 173). In an updated analysis incorporating epidemiologic studies published through 2022, Beckett et al. reported the lower- and upper-bound for the chrysotile NOAELs of 97–175 f/cc–yr for lung cancer and 250–379 f/cc-yr for mesothelioma (174). Conversely, epidemiological data have demonstrated a substantially elevated risk of all asbestos-related diseases, including mesothelioma, for occupations involving high cumulative exposures to amphibole asbestos or a combination of amphibole and chrysotile asbestos (175–182). Beckett et al. applied published relative potency factors for mesothelioma to the chrysotile NOAEL for mesothelioma reported to derive the best estimate NOAELs for predominately amosite- and crocidolite-exposed populations of 2–5 f/cc-yr and 0.6–1 f/cc-yr, respectively (174).
4 Summary and conclusions
We review here the mechanisms of asbestos-induced carcinogenicity with a focus on molecular pathways that are inhibited or modulated in cell defense from asbestos fiber exposures. These protective mechanisms are summarized within and are consistent with observations reported by others after exposures to chemical carcinogens and radiation (14–16). Simply put, humans have a cadre of defense mechanisms at the cellular and host level that maintain homeostasis and combat deleterious exposures to asbestos and other carcinogens. However, under certain conditions, such as increased vulnerability or exceeding critical response thresholds, these homeostatic mechanisms can be overwhelmed. This information and a review of the animal and human literature strongly suggest the existence of thresholds for MMs and lung cancers promoted by asbestos. Studies also illustrate the importance of asbestos type in calculation of NOAELs based upon different cellular responses to the commercial types of asbestos (chrysotile, amosite, and crocidolite) as well as their individual biodurability and size characteristics.
Author contributions
CB: Writing – original draft, Writing – review & editing. BM: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
This article was prepared and written exclusively by the authors. No outside entities provided input to the study or reviewed it prior to publication. CB and BM have served as experts in talc, silica, and/or asbestos-related litigation.
Correction note
A correction has been made to this article. Details can be found at: 10.3389/fpubh.2025.1713436.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mossman BT, Bignon J, Corn M, Seaton A, Gee JB. Asbestos: scientific developments and implications for public policy. Science. (1990) 247:294–301. doi: 10.1126/science.2153315
2. Guthrie Jr. GD, Mossman BT. Health Effects of Mineral Dusts, Volume 28: Proceedings. Washington, D.C.: Mineralogical Society of America (1993).
3. Virta RL. Mineral Commodity Profiles—Asbestos. Washington, D.C.: Department of the Interior, U.S. Geological Society (2005). Contract No.: USGS Circular 1255-KK.
4. Berman DW, Crump KS. Update of potency factors for asbestos-related lung cancer and mesothelioma. Crit Rev Toxicol. (2008) 38:1–47. doi: 10.1080/10408440802276167
5. Hodgson JT, Darnton A. The quantitative risks of mesothelioma and lung cancer in relation to asbestos exposure. Ann Occup Hyg. (2000) 44:565–601. doi: 10.1016/S0003-4878(00)00045-4
6. Hodgson JT, Darnton A. Mesothelioma risk from chrysotile. Occup Environ Med. (2010) 67:432. doi: 10.1136/oem.2009.052860
7. Berman DW, Crump KS, Chatfield EJ, Davis JM, Jones AD. The sizes, shapes, and mineralogy of asbestos structures that induce lung tumors or mesothelioma in AF/HAN rats following inhalation. Risk Anal. (1995) 15:181–95. doi: 10.1111/j.1539-6924.1995.tb00312.x
8. Darnton L. Quantitative assessment of mesothelioma and lung cancer risk based on Phase Contrast Microscopy (PCM) estimates of fibre exposure: an update of 2000 asbestos cohort data. Environ Res. (2023) 230:114753. doi: 10.1016/j.envres.2022.114753
9. Garabrant DH, Pastula ST, A. comparison of asbestos fiber potency and elongate mineral particle (EMP) potency for mesothelioma in humans. Toxicol Appl Pharmacol. (2018) 361:127–36. doi: 10.1016/j.taap.2018.07.003
10. Korchevskiy A, Rasmuson JO, Rasmuson EJ. Empirical model of mesothelioma potency factors for different mineral fibers based on their chemical composition and dimensionality. Inhal Toxicol. (2019) 31:180–91. doi: 10.1080/08958378.2019.1640320
11. Butterworth BE. Consideration of both genotoxic and nongenotoxic mechanisms in predicting carcinogenic potential. Mutat Res. (1990) 239:117–32. doi: 10.1016/0165-1110(90)90033-8
12. Klaunig JE, Kamendulis LM. Chemical carcinogenesis. In:Casarett LJ, Doull J, Klaassen CD, , editors. Casarett and Doull's Toxicology: The Basic Science of Poisons, (Seventh Edn). New York: McGraw-Hill (2008). p. 329–79.
13. Bogen KT, Sheehan PJ, Valdez-Flores C, Li AA. Reevaluation of Historical Exposures to Ethylene Oxide Among U.S. Sterilization Workers in the National Institute of Occupational Safety and Health (NIOSH) Study Cohort. Int J Environ Res Public Health. (2019) 16:1738. doi: 10.3390/ijerph16101738
14. Gueguen Y, Bontemps A, Ebrahimian TG. Adaptive responses to low doses of radiation or chemicals: their cellular and molecular mechanisms. Cell Mol Life Sci. (2019) 76:1255–73. doi: 10.1007/s00018-018-2987-5
15. Kobets T, Williams GM. Review of the evidence for thresholds for DNA-Reactive and epigenetic experimental chemical carcinogens. Chem Biol Interact. (2019) 301:88–111. doi: 10.1016/j.cbi.2018.11.011
16. Calabrese EJ, Priest ND, Kozumbo WJ. Thresholds for carcinogens. Chem Biol Interact. (2021) 341:109464. doi: 10.1016/j.cbi.2021.109464
17. Albert RE. Carcinogen risk assessment in the U.S Environmental Protection Agency. Crit Rev Toxicol. (1994) 24:75–85. doi: 10.3109/10408449409017920
18. USFDA. M7(R2) Assessment and Control of DNA Reactive (mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk Guidance for Industry. Washington, D.C.: U.S. Department of Health and Human Services, Food and Drug Administration (2023).
19. USFDA. M7(R1) Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk Guidance for Industry. Washington, D.C.: U.S. Department of Health and Human Services, Food and Drug Administration (2018).
20. Clapp RW, Jacobs MM, Loechler EL. Environmental and occupational causes of cancer: new evidence 2005-2007. Rev Environ Health. (2008) 23:1–37. doi: 10.1515/REVEH.2008.23.1.1
21. Mossman BT. Mechanistic in vitro studies: What they have told us about carcinogenic properties of elongated mineral particles (EMPs). Toxicol Appl Pharmacol. (2018) 361:62–7. doi: 10.1016/j.taap.2018.07.018
22. Mossman BT, Lippmann M, Hesterberg TW, Kelsey KT, Barchowsky A, Bonner JC. Pulmonary endpoints (lung carcinomas and asbestosis) following inhalation exposure to asbestos. J Toxicol Environ Health B Crit Rev. (2011) 14:76–121. doi: 10.1080/10937404.2011.556047
23. Barlow CA, Lievense L, Gross S, Ronk CJ, Paustenbach DJ. The role of genotoxicity in asbestos-induced mesothelioma: an explanation for the differences in carcinogenic potential among fiber types. Inhal Toxicol. (2013) 25:553–67. doi: 10.3109/08958378.2013.807321
24. Huang SX, Jaurand MC, Kamp DW, Whysner J, Hei TK. Role of mutagenicity in asbestos fiber-induced carcinogenicity and other diseases. J Toxicol Environ Health B Crit Rev. (2011) 14:179–245. doi: 10.1080/10937404.2011.556051
25. Toumpanakis D, Theocharis SE, DNA. repair systems in malignant mesothelioma. Cancer Lett. (2011) 312:143–9. doi: 10.1016/j.canlet.2011.08.021
26. Jiang J, Liang X, Zhou X, Huang R, Chu Z, Zhan Q, et al. DNA repair gene X-ray repair cross complementing group 1 Arg194Trp polymorphism on the risk of lung cancer: a meta-analysis on 22 studies. J Thorac Oncol. (2010) 5:1741–7. doi: 10.1097/JTO.0b013e3181f0c409
27. Betti M, Ferrante D, Padoan M, Guarrera S, Giordano M, Aspesi A, et al. XRCC1 and ERCC1 variants modify malignant mesothelioma risk: a case-control study. Mutat Res. (2011) 708:11–20. doi: 10.1016/j.mrfmmm.2011.01.001
28. Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. (2011) 43:1022–5. doi: 10.1038/ng.912
29. Betti M, Aspesi A, Ferrante D, Sculco M, Righi L, Mirabelli D, et al. Sensitivity to asbestos is increased in patients with mesothelioma and pathogenic germline variants in BAP1 or other DNA repair genes. Genes Chromosomes Cancer. (2018) 57:573–83. doi: 10.1002/gcc.22670
30. Betti M, Casalone E, Ferrante D, Aspesi A, Morleo G, Biasi A, et al. Germline mutations in DNA repair genes predispose asbestos-exposed patients to malignant pleural mesothelioma. Cancer Lett. (2017) 405:38–45. doi: 10.1016/j.canlet.2017.06.028
31. Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. (2008) 18:27–47. doi: 10.1038/cr.2008.8
32. Robertson AB, Klungland A, Rognes T, Leiros I, DNA. repair in mammalian cells: base excision repair: the long and short of it. Cell Mol Life Sci. (2009) 66:981–93. doi: 10.1007/s00018-009-8736-z
33. Jensen NM, Dalsgaard T, Jakobsen M, Nielsen RR, Sorensen CB, Bolund L, et al. An update on targeted gene repair in mammalian cells: methods and mechanisms. J Biomed Sci. (2011) 18:10. doi: 10.1186/1423-0127-18-10
34. Fung H, Kow YW, Van Houten B, Mossman BT. Patterns of 8-hydroxydeoxyguanosine formation in DNA and indications of oxidative stress in rat and human pleural mesothelial cells after exposure to crocidolite asbestos. Carcinogenesis. (1997) 18:825–32. doi: 10.1093/carcin/18.4.825
35. Kim SJ, Cheresh P, Jablonski RP, Williams DB, Kamp DW. The role of mitochondrial DNA in mediating alveolar epithelial cell apoptosis and pulmonary fibrosis. Int J Mol Sci. (2015) 16:21486–519. doi: 10.3390/ijms160921486
36. Chu G. Double strand break repair. J Biol Chem. (1997) 272:24097–100. doi: 10.1074/jbc.272.39.24097
37. Bernstein KA, Rothstein R. At loose ends: resecting a double-strand break. Cell. (2009) 137:807–10. doi: 10.1016/j.cell.2009.05.007
38. Marczynski B, Czuppon AB, Marek W, Reichel G, Baur X. Increased incidence of DNA double-strand breaks and anti-ds DNA antibodies in blood of workers occupationally exposed to asbestos. Hum Exp Toxicol. (1994) 13:3–9. doi: 10.1177/096032719401300102
39. Benedetti S, Nuvoli B, Catalani S, Galati R. Reactive oxygen species a double-edged sword for mesothelioma. Oncotarget. (2015) 6:16848–65. doi: 10.18632/oncotarget.4253
40. Shukla A, Gulumian M, Hei TK, Kamp D, Rahman Q, Mossman BT. Multiple roles of oxidants in the pathogenesis of asbestos-induced diseases. Free Radic Biol Med. (2003) 34:1117–29. doi: 10.1016/S0891-5849(03)00060-1
41. Vigliaturo R, Jamnik M, Drazic G, Podobnik M, Znidaric MT, Ventura GD, et al. Nanoscale transformations of amphiboles within human alveolar epithelial cells. Sci Rep. (2022) 12:1782. doi: 10.1038/s41598-022-05802-x
42. Mossman BT, Kessler JB, Ley BW, Craighead JE. Interaction of crocidolite asbestos with hamster respiratory mucosa in organ culture. Lab Invest. (1977) 36:131–9.
43. Janssen YM, Driscoll KE, Timblin CR, Hassenbein D, Mossman BT. Modulation of mitochondrial gene expression in pulmonary epithelial cells exposed to oxidants. Environ Health Perspect. (1998) 106:1191–5. doi: 10.1289/ehp.98106s51191
44. Kamp DW, Panduri V, Weitzman SA, Chandel N. Asbestos-induced alveolar epithelial cell apoptosis: role of mitochondrial dysfunction caused by iron-derived free radicals. Mol Cell Biochem. (2002) 234–235:153–60. doi: 10.1023/A:1015949118495
45. Shukla A, Jung M, Stern M, Fukagawa NK, Taatjes DJ, Sawyer D, et al. Asbestos induces mitochondrial DNA damage and dysfunction linked to the development of apoptosis. Am J Physiol. (2003) 285:L1018–25. doi: 10.1152/ajplung.00038.2003
46. Chen Q, Marsh J, Ames B, Mossman B. Detection of 8-oxo-2′-deoxyguanosine, a marker of oxidative DNA damage, in culture medium from human mesothelial cells exposed to crocidolite asbestos. Carcinogenesis. (1996) 17:2525–7. doi: 10.1093/carcin/17.11.2525
47. Schurkes C, Brock W, Abel J, Unfried K. Induction of 8-hydroxydeoxyguanosine by man made vitreous fibres and crocidolite asbestos administered intraperitoneally in rats. Mutat Res. (2004) 553:59–65. doi: 10.1016/j.mrfmmm.2004.06.021
48. Upadhyay D, Kamp DW. Asbestos-induced pulmonary toxicity: role of DNA damage and apoptosis. Exp Biol Med. (2003) 228:650–9. doi: 10.1177/153537020322800602
49. Ghio AJ, Stewart M, Sangani RG, Pavlisko EN, Roggli VL. Asbestos and iron. Int J Mol Sci. (2023) 24:12390. doi: 10.3390/ijms241512390
50. Kamp DW, Israbian VA, Yeldandi AV, Panos RJ, Graceffa P, Weitzman SA. Phytic acid, an iron chelator, attenuates pulmonary inflammation and fibrosis in rats after intratracheal instillation of asbestos. Toxicol Pathol. (1995) 23:689–95. doi: 10.1177/019262339502300606
51. Mossman BT, Marsh JP, Sesko A, Hill S, Shatos MA, Doherty J, et al. Inhibition of lung injury, inflammation, and interstitial pulmonary fibrosis by polyethylene glycol-conjugated catalase in a rapid inhalation model of asbestosis. Am Rev Respir Dis. (1990) 141:1266–71. doi: 10.1164/ajrccm/141.5_Pt_1.1266
52. Quinlan T, Spivack S, Mossman BT. Regulation of antioxidant enzymes in lung after oxidant injury. Environ Health Perspect. (1994) 102:79–87. doi: 10.1289/ehp.9410279
53. Shatos MA, Doherty JM, Marsh JP, Mossman BT. Prevention of asbestos-induced cell death in rat lung fibroblasts and alveolar macrophages by scavengers of active oxygen species. Environ Res. (1987) 44:103–16. doi: 10.1016/S0013-9351(87)80090-7
54. Janssen YM, Marsh JP, Absher MP, Gabrielson E, Borm PJ, Driscoll K, et al. Oxidant stress responses in human pleural mesothelial cells exposed to asbestos. Am J Respir Crit Care Med. (1994) 149:795–802. doi: 10.1164/ajrccm.149.3.8118652
55. Timblin CR, Janssen YM, Goldberg JL, Mossman BT. GRP78, HSP72/73, and cJun stress protein levels in lung epithelial cells exposed to asbestos, cadmium, or H2O2. Free Radic Biol Med. (1998) 24:632–42. doi: 10.1016/S0891-5849(97)00325-0
56. Mossman BT, Marsh JP, Shatos MA. Alteration of superoxide dismutase activity in tracheal epithelial cells by asbestos and inhibition of cytotoxicity by antioxidants. Lab Invest. (1986) 54:204–12.
57. Janssen YM, Marsh JP, Absher M, Borm PJ, Mossman BT. Increases in endogenous antioxidant enzymes during asbestos inhalation in rats. Free Radic Res Commun. (1990) 11:53–8. doi: 10.3109/10715769009109667
58. Janssen YM, Marsh JP, Absher MP, Hemenway D, Vacek PM, Leslie KO, et al. Expression of antioxidant enzymes in rat lungs after inhalation of asbestos or silica. J Biol Chem. (1992) 267:10625–30. doi: 10.1016/S0021-9258(19)50063-1
59. Janssen YM, Marsh JP, Driscoll KE, Borm PJ, Oberdorster G, Mossman BT. Increased expression of manganese-containing superoxide dismutase in rat lungs after inhalation of inflammatory and fibrogenic minerals. Free Radic Biol Med. (1994) 16:315–22. doi: 10.1016/0891-5849(94)90032-9
60. Mossman BT, Surinrut P, Brinton BT, Marsh JP, Heintz NH, Lindau-Shepard B, et al. Transfection of a manganese-containing superoxide dismutase gene into hamster tracheal epithelial cells ameliorates asbestos-mediated cytotoxicity. Free Radic Biol Med. (1996) 21:125–31. doi: 10.1016/0891-5849(96)00014-7
61. Landi S, Gemignani F, Neri M, Barale R, Bonassi S, Bottari F, et al. Polymorphisms of glutathione-S-transferase M1 and manganese superoxide dismutase are associated with the risk of malignant pleural mesothelioma. Int J Cancer. (2007) 120:2739–43. doi: 10.1002/ijc.22590
62. Rahman Q, Abidi P, Afaq F, Schiffmann D, Mossman BT, Kamp DW, et al. Glutathione redox system in oxidative lung injury. Crit Rev Toxicol. (1999) 29:543–68. doi: 10.1080/10408449991349276
63. Shukla A, Flanders T, Lounsbury KM, Mossman BT. The gamma-glutamylcysteine synthetase and glutathione regulate asbestos-induced expression of activator protein-1 family members and activity. Cancer Res. (2004) 64:7780–6. doi: 10.1158/0008-5472.CAN-04-1365
64. Janssen YM, Heintz NH, Mossman BT. Induction of c-fos and c-jun proto-oncogene expression by asbestos is ameliorated by N-acetyl-L-cysteine in mesothelial cells. Cancer Res. (1995) 55:2085–9.
65. Thompson JK, Westbom CM, MacPherson MB, Mossman BT, Heintz NH, Spiess P, et al. Asbestos modulates thioredoxin-thioredoxin interacting protein interaction to regulate inflammasome activation. Part Fibre Toxicol. (2014) 11:24. doi: 10.1186/1743-8977-11-24
66. Hillegass JM, Miller JM, MacPherson MB, Westbom CM, Sayan M, Thompson JK, et al. Asbestos and erionite prime and activate the NLRP3 inflammasome that stimulates autocrine cytokine release in human mesothelial cells. Part Fibre Toxicol. (2013) 10:39. doi: 10.1186/1743-8977-10-39
67. Suarez JS, Novelli F, Goto K, Ehara M, Steele M, Kim JH, et al. HMGB1 released by mesothelial cells drives the development of asbestos-induced mesothelioma. Proc Natl Acad Sci U S A. (2023) 120:e2307999120. doi: 10.1073/pnas.2307999120
68. Ahmed SM, Luo L, Namani A, Wang XJ, Tang X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:585–97. doi: 10.1016/j.bbadis.2016.11.005
69. Smith MT, Guyton KZ, Kleinstreuer N, Borrel A, Cardenas A, Chiu WA, et al. The key characteristics of carcinogens: relationship to the hallmarks of cancer, relevant biomarkers, and assays to measure them. Cancer Epidemiol Biomarkers Prev. (2020) 29:1887–903. doi: 10.1158/1055-9965.EPI-19-1346
70. Shukla A, Vacek P, Mossman BT. Dose-response relationships in expression of biomarkers of cell proliferation in in vitro assays and inhalation experiments. Nonlinear Biol Toxicol Med. (2004) 2:117–28. doi: 10.1080/15401420490464420
71. Shukla A, Ramos-Nino M, Mossman B. Cell signaling and transcription factor activation by asbestos in lung injury and disease. Int J Biochem Cell Biol. (2003) 35:1198–209. doi: 10.1016/S1357-2725(02)00315-1
72. Yuan Z, Taatjes DJ, Mossman BT, Heintz NH. The duration of nuclear extracellular signal-regulated kinase 1 and 2 signaling during cell cycle reentry distinguishes proliferation from apoptosis in response to asbestos. Cancer Res. (2004) 64:6530–6. doi: 10.1158/0008-5472.CAN-04-0946
73. Buder-Hoffmann S, Palmer C, Vacek P, Taatjes D, Mossman B. Different accumulation of activated extracellular signal-regulated kinases (ERK 1/2) and role in cell-cycle alterations by epidermal growth factor, hydrogen peroxide, or asbestos in pulmonary epithelial cells. Am J Respir Cell Mol Biol. (2001) 24:405–13. doi: 10.1165/ajrcmb.24.4.4290
74. Jimenez LA, Zanella C, Fung H, Janssen YM, Vacek P, Charland C, et al. Role of extracellular signal-regulated protein kinases in apoptosis by asbestos and H2O2. Am J Physiol. (1997) 273:L1029–35. doi: 10.1152/ajplung.1997.273.5.L1029
75. Goldberg JL, Zanella CL, Janssen YM, Timblin CR, Jimenez LA, Vacek P, et al. Novel cell imaging techniques show induction of apoptosis and proliferation in mesothelial cells by asbestos. Am J Respir Cell Mol Biol. (1997) 17:265–71. doi: 10.1165/ajrcmb.17.3.2991
76. Coultas L, Strasser A. The molecular control of DNA damage-induced cell death. Apoptosis. (2000) 5:491–507. doi: 10.1023/A:1009617727938
77. Shi D, Jiang P, A. Different Facet of p53 Function: regulation of Immunity and Inflammation During Tumor Development. Front Cell Dev Biol. (2021) 9:762651. doi: 10.3389/fcell.2021.762651
78. Levresse V, Renier A, Fleury-Feith J, Levy F, Moritz S, Vivo C, et al. Analysis of cell cycle disruptions in cultures of rat pleural mesothelial cells exposed to asbestos fibers. Am J Respir Cell Mol Biol. (1997) 17:660–71. doi: 10.1165/ajrcmb.17.6.2854
79. Mishra A, Liu JY, Brody AR, Morris GF. Inhaled asbestos fibers induce p53 expression in the rat lung. Am J Respir Cell Mol Biol. (1997) 16:479–85. doi: 10.1165/ajrcmb.16.4.9115760
80. Paakko P, Ramet M, Vahakangas K, Korpela N, Soini Y, Turunen S, et al. Crocidolite asbestos causes an induction of p53 and apoptosis in cultured A-549 lung carcinoma cells. Apoptosis. (1998) 3:203–12. doi: 10.1023/A:1009655007284
81. Brody AR, Overby LH. Incorporation of tritiated thymidine by epithelial and interstitial cells in bronchiolar-alveolar regions of asbestos-exposed rats. Am J Pathol. (1989) 134:133–40.
82. McGavran PD, Butterick CJ, Brody AR. Tritiated thymidine incorporation and the development of an interstitial lesion in the bronchiolar-alveolar regions of the lungs of normal and complement deficient mice after inhalation of chrysotile asbestos. J Environ Pathol Toxicol Oncol. (1989) 9:377–91.
83. Carra G, Lingua MF, Maffeo B, Taulli R, Morotti A. p53 vs NF-kappaB: the role of nuclear factor-kappa B in the regulation of p53 activity and vice versa. Cell Mol Life Sci. (2020) 77:4449–58. doi: 10.1007/s00018-020-03524-9
84. Zhang X, Li CF, Zhang L, Wu CY, Han L, Jin G, et al. TRAF6 restricts p53 mitochondrial translocation, apoptosis, and tumor suppression. Mol Cell. (2016) 64:803–14. doi: 10.1016/j.molcel.2016.10.002
85. Panduri V, Surapureddi S, Soberanes S, Weitzman SA, Chandel N, Kamp DW. p53 mediates amosite asbestos-induced alveolar epithelial cell mitochondria-regulated apoptosis. Am J Respir Cell Mol Biol. (2006) 34:443–52. doi: 10.1165/rcmb.2005-0352OC
86. Cistulli CA, Sorger T, Marsella JM, Vaslet CA, Kane AB. Spontaneous p53 mutation in murine mesothelial cells: increased sensitivity to DNA damage induced by asbestos and ionizing radiation. Toxicol Appl Pharmacol. (1996) 141:264–71. doi: 10.1016/S0041-008X(96)80032-9
87. Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet. (2016) 48:407–16. doi: 10.1038/ng.3520
88. Jean D, Jaurand MC. Mesotheliomas in genetically engineered mice unravel mechanism of mesothelial carcinogenesis. Int J Mol Sci. (2018) 19:2191. doi: 10.3390/ijms19082191
89. Andujar P, Pairon JC, Renier A, Descatha A, Hysi I, Abd-Alsamad I, et al. Differential mutation profiles and similar intronic TP53 polymorphisms in asbestos-related lung cancer and pleural mesothelioma. Mutagenesis. (2013) 28:323–31. doi: 10.1093/mutage/get008
90. Taylor ES, Wylie AG, Mossman BT, Lower SK. Repetitive dissociation from crocidolite asbestos acts as persistent signal for epidermal growth factor receptor. Langmuir. (2013) 29:6323–30. doi: 10.1021/la400561t
91. Carter AB, Tephly LA, Venkataraman S, Oberley LW, Zhang Y, Buettner GR, et al. High levels of catalase and glutathione peroxidase activity dampen H2O2 signaling in human alveolar macrophages. Am J Respir Cell Mol Biol. (2004) 31:43–53. doi: 10.1165/rcmb.2003-0377OC
92. de Melo M, Gerbase MW, Curran J, Pache JC. Phosphorylated extracellular signal-regulated kinases are significantly increased in malignant mesothelioma. J Histochem Cytochem. (2006) 54:855–61. doi: 10.1369/jhc.5A6807.2006
93. Manning CB, Sabo-Attwood T, Robledo RF, Macpherson MB, Rincon M, Vacek P, et al. Targeting the MEK1 cascade in lung epithelium inhibits proliferation and fibrogenesis by asbestos. Am J Respir Cell Mol Biol. (2008) 38:618–26. doi: 10.1165/rcmb.2007-0382OC
94. Shukla A, Barrett TF, MacPherson MB, Hillegass JM, Fukagawa NK, Swain WA, et al. An extracellular signal-regulated kinase 2 survival pathway mediates resistance of human mesothelioma cells to asbestos-induced injury. Am J Respir Cell Mol Biol. (2011) 45:906–14. doi: 10.1165/rcmb.2010-0282OC
95. Carbone M, Yang H. Molecular pathways: targeting mechanisms of asbestos and erionite carcinogenesis in mesothelioma. Clin Cancer Res. (2012) 18:598–604. doi: 10.1158/1078-0432.CCR-11-2259
96. Ramos-Nino ME, Haegens A, Shukla A, Mossman BT. Role of mitogen-activated protein kinases (MAPK) in cell injury and proliferation by environmental particulates. Mol Cell Biochem. (2002) 234–235:111–8. doi: 10.1023/A:1015924413043
97. Kyriakis JM. Activation of the AP-1 transcription factor by inflammatory cytokines of the TNF family. Gene Expr. (1999) 7:217–31.
98. Guo R, DuBoff M, Jayakumaran G, Kris MG, Ladanyi M, Robson ME, et al. Novel germline mutations in DNA damage repair in patients with malignant pleural mesotheliomas. J Thorac Oncol. (2020) 15:655–60. doi: 10.1016/j.jtho.2019.12.111
99. Zanella CL, Posada J, Tritton TR, Mossman BT. Asbestos causes stimulation of the extracellular signal-regulated kinase 1mitogen-activated protein kinase cascade after phosphorylation of theepidermal growth factor receptor. Cancer Res. (1996) 56:5334–8.
100. Robledo RF, Buder-Hoffmann SA, Cummins AB, Walsh ES, Taatjes DJ, Mossman BT. Increased phosphorylated extracellular signal-regulated kinase immunoreactivity associated with proliferative and morphologic lung alterations after chrysotile asbestos inhalation in mice. Am J Pathol. (2000) 156:1307–16. doi: 10.1016/S0002-9440(10)65001-8
101. Heintz NH, Janssen YM, Mossman BT. Persistent induction of c-fos and c-jun expression by asbestos. Proc Natl Acad Sci U S A. (1993) 90:3299–303. doi: 10.1073/pnas.90.8.3299
102. Heintz NH, Janssen-Heininger YM, Mossman BT. Asbestos, lung cancers, and mesotheliomas: from molecular approaches to targeting tumor survival pathways. Am J Respir Cell Mol Biol. (2010) 42:133–9. doi: 10.1165/rcmb.2009-0206TR
103. Mossman BT, Shukla A, Heintz NH, Verschraegen CF, Thomas A, Hassan R. New insights into understanding the mechanisms, pathogenesis, and management of malignant mesotheliomas. Am J Pathol. (2013) 182:1065–77. doi: 10.1016/j.ajpath.2012.12.028
104. Liu X, Yin S, Chen Y, Wu Y, Zheng W, Dong H, et al. LPS-induced proinflammatory cytokine expression in human airway epithelial cells and macrophages via NF-kappaB, STAT3 or AP-1 activation. Mol Med Rep. (2018) 17:5484–91. doi: 10.3892/mmr.2018.8542
105. Moosavi SM, Prabhala P, Ammit AJ. Role and regulation of MKP-1 in airway inflammation. Respir Res. (2017) 18:154. doi: 10.1186/s12931-017-0637-3
106. Shukla A, Bosenberg MW, MacPherson MB, Butnor KJ, Heintz NH, Pass HI, et al. Activated cAMP response element binding protein is overexpressed in human mesotheliomas and inhibits apoptosis. Am J Pathol. (2009) 175:2197–206. doi: 10.2353/ajpath.2009.090400
107. Janssen YM, Barchowsky A, Treadwell M, Driscoll KE, Mossman BT. Asbestos induces nuclear factor kappa B (NF-kappa B) DNA-binding activity and NF-kappa B-dependent gene expression in tracheal epithelial cells. Proc Natl Acad Sci U S A. (1995) 92:8458–62. doi: 10.1073/pnas.92.18.8458
108. Janssen YM, Driscoll KE, Howard B, Quinlan TR, Treadwell M, Barchowsky A, et al. Asbestos causes translocation of p65 protein and increases NF-kappa B DNA binding activity in rat lung epithelial and pleural mesothelial cells. Am J Pathol. (1997) 151:389–401.
109. Simeonova PP, Toriumi W, Kommineni C, Erkan M, Munson AE, Rom WN, et al. Molecular regulation of IL-6 activation by asbestos in lung epithelial cells: role of reactive oxygen species. J Immunol. (1997) 159:3921–8. doi: 10.4049/jimmunol.159.8.3921
110. Sartore-Bianchi A, Gasparri F, Galvani A, Nici L, Darnowski JW, Barbone D, et al. Bortezomib inhibits nuclear factor-kappaB dependent survival and has potent in vivo activity in mesothelioma. Clin Cancer Res. (2007) 13:5942–51. doi: 10.1158/1078-0432.CCR-07-0536
111. Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
112. Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. (2006) 25:6680–4. doi: 10.1038/sj.onc.1209954
113. Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene. (1999) 18:6867–74. doi: 10.1038/sj.onc.1203219
114. Sayan M, Mossman BT. The NLRP3 inflammasome in pathogenic particle and fibre-associated lung inflammation and diseases. Part Fibre Toxicol. (2016) 13:51. doi: 10.1186/s12989-016-0162-4
115. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. (2019) 20:3328. doi: 10.3390/ijms20133328
116. Li M, Gunter ME, Fukagawa NK. Differential activation of the inflammasome in THP-1 cells exposed to chrysotile asbestos and Libby “six-mix” amphiboles and subsequent activation of BEAS-2B cells. Cytokine. (2012) 60:718–30. doi: 10.1016/j.cyto.2012.08.025
117. Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. (2008) 320:674–7. doi: 10.1126/science.1156995
118. Chow MT, Sceneay J, Paget C, Wong CS, Duret H, Tschopp J, et al. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res. (2012) 72:5721–32. doi: 10.1158/0008-5472.CAN-12-0509
119. Ku HC, Cheng CF. Master regulator activating transcription factor 3 (ATF3) in metabolic homeostasis and cancer. Front Endocrinol. (2020) 11:556. doi: 10.3389/fendo.2020.00556
120. Shukla A, MacPherson MB, Hillegass J, Ramos-Nino ME, Alexeeva V, Vacek PM, et al. Alterations in gene expression in human mesothelial cells correlate with mineral pathogenicity. Am J Respir Cell Mol Biol. (2009) 41:114–23. doi: 10.1165/rcmb.2008-0146OC
121. Williams GM. Review of in vitro test systems using DNA damage and repair for screening of chemical carcinogens. J Assoc Off Anal Chem. (1979) 62:857–63. doi: 10.1093/jaoac/62.4.857
122. Shelby MD. The genetic toxicity of human carcinogens and its implications. Mutat Res. (1988) 204:3–15. doi: 10.1016/0165-1218(88)90113-9
123. Mossman BT. Cell Signaling and Epigenetic Mechanisms in Mesothelioma. Asbestos Mesothelioma. (2017) 2017:211–35. doi: 10.1007/978-3-319-53560-9_10
124. Casalone E, Allione A, Viberti C, Pardini B, Guarrera S, Betti M, et al. DNA methylation profiling of asbestos-treated MeT5A cell line reveals novel pathways implicated in asbestos response. Arch Toxicol. (2018) 92:1785–95. doi: 10.1007/s00204-018-2179-y
125. Oner D, Ghosh M, Moisse M, Duca RC, Coorens R, Vanoirbeek JAJ, et al. Global and gene-specific DNA methylation effects of different asbestos fibres on human bronchial epithelial cells. Environ Int. (2018) 115:301–11. doi: 10.1016/j.envint.2018.03.031
126. Kettunen E, Hernandez-Vargas H, Cros MP, Durand G, Le Calvez-Kelm F, Stuopelyte K, et al. Asbestos-associated genome-wide DNA methylation changes in lung cancer. Int J Cancer. (2017) 141:2014–29. doi: 10.1002/ijc.30897
127. Christensen BC, Marsit CJ, Houseman EA, Godleski JJ, Longacker JL, Zheng S, et al. Differentiation of lung adenocarcinoma, pleural mesothelioma, and nonmalignant pulmonary tissues using DNA methylation profiles. Cancer Res. (2009) 69:6315–21. doi: 10.1158/0008-5472.CAN-09-1073
128. Altomare DA, Menges CW, Xu J, Pei J, Zhang L, Tadevosyan A, et al. Losses of both products of the Cdkn2a/Arf locus contribute to asbestos-induced mesothelioma development and cooperate to accelerate tumorigenesis. PLoS ONE. (2011) 6:e18828. doi: 10.1371/journal.pone.0018828
129. Lamy A, Sesboue R, Bourguignon J, Dautreaux B, Metayer J, Frebourg T, et al. Aberrant methylation of the CDKN2a/p16INK4a gene promoter region in preinvasive bronchial lesions: a prospective study in high-risk patients without invasive cancer. Int J Cancer. (2002) 100:189–93. doi: 10.1002/ijc.10474
130. Gandhi M, Nair S. New vistas in malignant mesothelioma: MicroRNA architecture and NRF2/MAPK signal transduction. Life Sci. (2020) 257:118123. doi: 10.1016/j.lfs.2020.118123
131. Gallo MA. History and scope of toxicology. In:Klaassen CD, , editor. Casarett and Doull's Toxicology: The Basic Science of Poisons (Seventh Edn). New York: McGraw-Hill (2008). p. 3–10.
132. Rozman K, Klaassen CD. Absorption, distribution, and excretion of toxicants. In:Klaassen C, , editor. Casarett and Doull's Toxicology The Basic Science of Poisons (Sixth Edn). New York: McGraw-Hill Medical Publishing Division (2001). p. 107–32.
133. Eaton DL, Gilbert SG. Principles of toxicology. In:Klaassen CD, , editor. Casarett and Doull's Toxicology: The Basic Science of Poisons (Seventh Edn). New York: McGraw-Hill (2008). p. 11–43.
134. Sesko AM, Mossman BT. Sensitivity of hamster tracheal epithelial cells to asbestiform minerals modulated by serum and by transforming growth factor beta 1. Cancer Res. (1989) 49:2743–9.
135. Lemaire I, Beaudoin H, Dubois C. Cytokine regulation of lung fibroblast proliferation. Pulmonary and systemic changes in asbestos-induced pulmonary fibrosis. Am Rev Respir Dis. (1986) 134:653–8.
136. Woodworth CD, Mossman BT, Craighead JE. Comparative effects of fibrous and nonfibrous minerals on cells and liposomes. Environ Res. (1982) 27:190–205. doi: 10.1016/0013-9351(82)90070-6
137. Mossman B, Light W, Wei E. Asbestos: mechanisms of toxicity and carcinogenicity in the respiratory tract. Annu Rev Pharmacol Toxicol. (1983) 23:595–615. doi: 10.1146/annurev.pa.23.040183.003115
138. Mossman BT, Churg A. Mechanisms in the pathogenesis of asbestosis and silicosis. Am J Respir Crit Care Med. (1998) 157:1666–80. doi: 10.1164/ajrccm.157.5.9707141
139. Giacobbe C, Di Giuseppe D, Zoboli A, Lassinantti Gualtieri M, Bonasoni P, Moliterni A, et al. Crystal structure determination of a lifelong biopersistent asbestos fibre using single-crystal synchrotron X-ray micro-diffraction. IUCrJ. (2021) 8:76–86. doi: 10.1107/S2052252520015079
140. Hei TK, Piao CQ, He ZY, Vannais D, Waldren CA. Chrysotile fiber is a strong mutagen in mammalian cells. Cancer Res. (1992) 52:6305–9.
141. Nymark P, Wikman H, Hienonen-Kempas T, Anttila S. Molecular and genetic changes in asbestos-related lung cancer. Cancer Lett. (2008) 265:1–15. doi: 10.1016/j.canlet.2008.02.043
142. Liu G, Beri R, Mueller A, Kamp DW. Molecular mechanisms of asbestos-induced lung epithelial cell apoptosis. Chem Biol Interact. (2010) 188:309–18. doi: 10.1016/j.cbi.2010.03.047
143. Fung H, Kow YW, Van Houten B, Taatjes DJ, Hatahet Z, Janssen YM, et al. Asbestos increases mammalian AP-endonuclease gene expression, protein levels, and enzyme activity in mesothelial cells. Cancer Res. (1998) 58:189–94.
144. Schins RP, Knaapen AM. Genotoxicity of poorly soluble particles. Inhal Toxicol. (2007) 19:189–98. doi: 10.1080/08958370701496202
145. Hillegass JM, Shukla A, Lathrop SA, MacPherson MB, Beuschel SL, Butnor KJ, et al. Inflammation precedes the development of human malignant mesotheliomas in a SCID mouse xenograft model. Ann N Y Acad Sci. (2010) 1203:7–14. doi: 10.1111/j.1749-6632.2010.05554.x
146. Mossman BT, Craighead JE, MacPherson BV. Asbestos-induced epithelial changes in organ cultures of hamster trachea: inhibition by retinyl methyl ether. Science. (1980) 207:311–3. doi: 10.1126/science.7350661
147. Schinwald A, Murphy FA, Prina-Mello A, Poland CA, Byrne F, Movia D, et al. The threshold length for fiber-induced acute pleural inflammation: shedding light on the early events in asbestos-induced mesothelioma. Toxicol Sci. (2012) 128:461–70. doi: 10.1093/toxsci/kfs171
148. Murphy FA, Poland CA, Duffin R, Al-Jamal KT, Ali-Boucetta H, Nunes A, et al. Length-dependent retention of carbon nanotubes in the pleural space of mice initiates sustained inflammation and progressive fibrosis on the parietal pleura. Am J Pathol. (2011) 178:2587–600. doi: 10.1016/j.ajpath.2011.02.040
149. Kraynie A, de Ridder GG, Sporn TA, Pavlisko EN, Roggli VL. Malignant mesothelioma not related to asbestos exposure: analytical scanning electron microscopic analysis of 83 cases and comparison with 442 asbestos-related cases. Ultrastruct Pathol. (2016) 40:142–6. doi: 10.3109/01913123.2016.1154633
150. Mujahed T, Tazelaar HD, Sukov WR, Halling KC, Davila JI, Glass C, et al. Malignant peritoneal mesothelioma arising in young adults with long-standing indwelling intra-abdominal shunt catheters. Am J Surg Pathol. (2021) 45:255–62. doi: 10.1097/PAS.0000000000001574
151. Vogl M, Rosenmayr A, Bohanes T, Scheed A, Brndiar M, Stubenberger E, et al. Biomarkers for malignant pleural mesothelioma—a novel view on inflammation. Cancers. (2021) 13:658. doi: 10.3390/cancers13040658
152. Mossman BT. Assessment of the pathogenic potential of asbestiform vs. nonasbestiform particulates (cleavage fragments) in in vitro (cell or organ culture) models and bioassays. Regul Toxicol Pharmacol. (2008) 52:S200–3. doi: 10.1016/j.yrtph.2007.10.004
153. Cox LA. Nonlinear dose-time-response functions and health-protective exposure limits for inflammation-mediated diseases. Environ Res. (2020) 182:109026. doi: 10.1016/j.envres.2019.109026
154. Cox LA, Bogen KT, Conolly R, Graham U, Moolgavkar S, Oberdorster G, et al. Mechanisms and shapes of causal exposure-response functions for asbestos in mesotheliomas and lung cancers. Environ Res. (2023) 230:115607. doi: 10.1016/j.envres.2023.115607
155. Cox LA. Dose-response modeling of NLRP3 inflammasome-mediated diseases: asbestos, lung cancer, and malignant mesothelioma as examples. Crit Rev Toxicol. (2019) 49:614–35. doi: 10.1080/10408444.2019.1692779
156. Liu W, Deng Y, Li Z, Chen Y, Zhu X, Tan X, et al. Cancer Evo-Dev: a theory of inflammation-induced oncogenesis. Front Immunol. (2021) 12:768098. doi: 10.3389/fimmu.2021.768098
157. Bogen KT. Ultrasensitive dose-response for asbestos cancer risk implied by new inflammation-mutation model. Environ Res. (2023) 230:115047. doi: 10.1016/j.envres.2022.115047
158. Comar M, Zanotta N, Zanconati F, Cortale M, Bonotti A, Cristaudo A, et al. Chemokines involved in the early inflammatory response and in pro-tumoral activity in asbestos-exposed workers from an Italian coastal area with territorial clusters of pleural malignant mesothelioma. Lung Cancer. (2016) 94:61–7. doi: 10.1016/j.lungcan.2016.01.020
159. Kadariya Y, Menges CW, Talarchek J, Cai KQ, Klein-Szanto AJ, Pietrofesa RA, et al. Inflammation-related IL1beta/IL1R signaling promotes the development of asbestos-induced malignant mesothelioma. Cancer Prev Res. (2016) 9:406–14. doi: 10.1158/1940-6207.CAPR-15-0347
160. Kumagai-Takei N, Yamamoto S, Lee S, Maeda M, Masuzzaki H, Sada N, et al. Inflammatory alteration of human T cells exposed continuously to asbestos. Int J Mol Sci. (2018) 19:504. doi: 10.3390/ijms19020504
161. Gottschalk RA, Martins AJ, Angermann BR, Dutta B, Ng CE, Uderhardt S, et al. Distinct NF-kappaB and MAPK Activation Thresholds Uncouple Steady-State Microbe Sensing from Anti-pathogen Inflammatory Responses. Cell Syst. (2016) 2:378–90. doi: 10.1016/j.cels.2016.04.016
162. Price B, Ware A. Mesothelioma trends in the United States: an update based on surveillance, epidemiology, and end results program data for 1973 through 2003. Am J Epidemiol. (2004) 159:107–12. doi: 10.1093/aje/kwh025
163. Price B, Ware A. Mesothelioma: risk apportionment among asbestos exposure sources. Risk Anal. (2005) 25:937–43. doi: 10.1111/j.1539-6924.2005.00643.x
164. Teta MJ, Mink PJ, Lau E, Sceurman BK, Foster ED, US. mesothelioma patterns 1973-2002: indicators of change and insights into background rates. Eur J Cancer Prev. (2008) 17:525–34. doi: 10.1097/CEJ.0b013e3282f0c0a2
165. Moolgavkar SH, Meza R, Turim J. Pleural and peritoneal mesotheliomas in SEER: age effects and temporal trends, 1973-2005. Cancer Causes Control. (2009) 20:935–44. doi: 10.1007/s10552-009-9328-9
166. Antman KH, Schiff PB, Pass HI. Benign and malignant mesothelioma. In:DeVita VT, S. Hellman S, Rosenberg SA, , editors. Cancer: Principles and Practice of Oncology. 2. 5th ed. Philadelphia: Lippincott-Raven Publishers (1997). p. 1853–78.
167. McDonald JC. Health implications of environmental exposure to asbestos. Environ Health Perspect. (1985) 62:319–28. doi: 10.1289/ehp.8562319
168. McDonald JC, McDonald A. Mesothelioma: Is there a background? In:Jaurand M, Bignon J, , editors. Mesothelial Cell Mesothelioma. New York: Marcel Decker (1994). p. 37–45.
169. Moore AJ, Parker RJ, Wiggins J. Malignant mesothelioma. Orphanet J Rare Dis. (2008) 3:34. doi: 10.1186/1750-1172-3-34
170. Price B. Projection of future numbers of mesothelioma cases in the US and the increasing prevalence of background cases: an update based on SEER data for 1975 through 2018. Crit Rev Toxicol. (2022) 52:317–24. doi: 10.1080/10408444.2022.2082919
171. Glynn ME, Keeton KA, Gaffney SH, Sahmel J. Ambient asbestos fiber concentrations and long-term trends in pleural mesothelioma incidence between urban and rural areas in the United States (1973–2012). Risk Anal. (2018) 38:454–71. doi: 10.1111/risa.12887
172. Pierce JS, McKinley MA, Paustenbach DJ, Finley BL. An evaluation of reported no-effect chrysotile asbestos exposures for lung cancer and mesothelioma. Crit Rev Toxicol. (2008) 38:191–214. doi: 10.1080/10408440701845609
173. Pierce JS, Ruestow PS, Finley BL. An updated evaluation of reported no-observed adverse effect levels for chrysotile asbestos for lung cancer and mesothelioma. Crit Rev Toxicol. (2016) 46:561–86. doi: 10.3109/10408444.2016.1150960
174. Beckett EM, Abelmann A, Roberts B, Lewis RC, Cheatham D, Miller EW, et al. An updated evaluation of reported no-observed adverse effect levels for chrysotile, amosite, and crocidolite asbestos for lung cancer and mesothelioma. Crit Rev Toxicol. (2023) 53:611–57. doi: 10.1080/10408444.2023.2283169
175. Cocco P, Dosemeci M. Peritoneal cancer and occupational exposure to asbestos: results from the application of a job-exposure matrix. Am J Ind Med. (1999) 35:9–14. doi: 10.1002/(sici)1097-0274(199901)35:1<9::aid-ajim2>3.0.co;2-v
176. Drever F. Occupational Health: Decennial Supplement: The Registrar General's Decennial Supplement For England and Wales. London: Office of Population Censuses and Surveys, Health and Safety Executive, Her Majesty's Stationery Office (1995). Contract No.: Series DS No. 10.
177. Engholm G, Englund A. Asbestos hazard in the Swedish construction industry—recent trends in mesothelioma incidence. Scand J Work Environ Health. (2005) 31:27–30.
178. Koskinen K, Pukkala E, Martikainen R, Reijula K, Karjalainen A. Different measures of asbestos exposure in estimating risk of lung cancer and mesothelioma among construction workers. J Occup Environ Med. (2002) 44:1190–6. doi: 10.1097/00043764-200212000-00015
179. Malker HS, McLaughlin JK, Malker BK, Stone BJ, Weiner JA, Erickson JL, et al. Occupational risks for pleural mesothelioma in Sweden, 1961-79. J Natl Cancer Inst. (1985) 74:61–6.
180. McDonald AD, McDonald JC. Malignant mesothelioma in North America. Cancer. (1980) 46:1650–6. doi: 10.1002/1097-0142(19801001)46:7<1650::aid-cncr2820460726>3.0.co
181. Menegozzo M, Belli S, Borriero S, Bruno C, Carboni M, Grignoli M, et al. [Mortality study of a cohort of insulation workers]. Epidemiol Prev. (2002) 26:71–5.
182. Ulvestad B, Kjaerheim K, Martinsen JI, Mowe G, Andersen A. Cancer incidence among members of the Norwegian trade union of insulation workers. J Occup Environ Med. (2004) 46:84–9. doi: 10.1097/01.jom.0000105981.46987.42
183. Kelsey KT, Yano E, Liber HL, Little JB. The in vitro genetic effects of fibrous erionite and crocidolite asbestos. Br J Cancer. (1986) 54:107–14. doi: 10.1038/bjc.1986.158
184. Jaurand MC, Gaudichet A, Halpern S, Bignon J. In vitro biodegradation of chrysotile fibres by alveolar macrophages and mesothelial cells in culture: comparison with a pH effect. Br J Ind Med. (1984) 41:389–95. doi: 10.1136/oem.41.3.389
Keywords: asbestos, elongated mineral fibers (EMPs), mesothelioma, lung cancer, threshold
Citation: Barlow CA and Mossman BT (2025) Cellular defense mechanisms against asbestos fibers. Front. Public Health 13:1566473. doi: 10.3389/fpubh.2025.1566473
Received: 24 January 2025; Accepted: 17 April 2025;
Published: 14 May 2025; Corrected: 29 October 2025.
Edited by:
Richard Attanoos, Cardiff University, United KingdomReviewed by:
Damaris Albores-Garcia, Department of Pharmacobiology—CINVESTAV, MexicoEmanuela Felley-Bosco, Université de Lausanne, Switzerland
Copyright © 2025 Barlow and Mossman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brooke T. Mossman, QnJvb2tlLk1vc3NtYW5AbWVkLnV2bS5lZHU=