 Yi Ding
Yi Ding Qiliang Wan†
Qiliang Wan† Wenduo Liu
Wenduo Liu- Department of Sports Science, College of Natural Science, Jeonbuk National University, Jeonju, Republic of Korea
This study aims to explore the multifaceted impacts and mechanisms of fine particulate matter (PM2.5) exposure on skeletal muscle mitochondria. Evidence suggests that PM2.5 can penetrate the respiratory barrier and enter the circulatory system, spreading throughout the body and causing significant damage to the morphology, quantity, and function of skeletal muscle mitochondria. This is manifested by a decline in oxidative phosphorylation efficiency and mitochondrial dysfunction. Meanwhile, PM2.5 exposure induces excessive production of reactive oxygen species, triggering oxidative stress responses that impair mitochondrial dynamic regulation. This further disrupts the balance of glucose and lipid metabolism in skeletal muscle, exacerbating the development of metabolic diseases. The review underscores the systemic effects on skeletal muscle following mitochondrial dysfunction after PM2.5 exposure and the preventive and treatment potential of exercise.
1 Introduction
With the rapid economic development and continuous industrial expansion, the increase in particulate matter (PM) in the air has become increasingly severe (1), making particulate pollution a significant global issue (2). In the 2006 World Health Organization Air Quality Guidelines, PM was recommended as an indicator of particulate pollution in the air (3), representing a highly complex mixture of particles with varying sizes in the air (4), and typically categorized based on aerodynamic diameter into PM10 (2.5–10 μm) and PM2.5 (<2.5 μm) (5). PM2.5 penetrates deeply into the alveoli and can remain in the lungs (6). These particles may also cross the air–blood barrier, enter the bloodstream, and trigger systemic inflammation (7, 8). Furthermore, acute inhalation of high concentrations of PM2.5 can also impair athletic performance (9).
It has been reported that toxic substances and heavy metals in PM2.5 particles can induce excessive production of reactive oxygen species (ROS), thereby triggering oxidative stress responses in the body (10, 11). Under oxidative stress conditions, ROS disrupt normal cellular physiological pathways (11, 12). Furthermore, due to the microscopic characteristics of PM2.5 (13), it can penetrate cell membranes and enter mitochondria (14), leading to insufficient adenosine triphosphate (ATP) supply to meet cellular metabolic demands, thereby causing energy metabolism disorders (15). This disruption further contributes to chronic inflammation and insulin resistance (16, 17), ultimately increasing the incidence of diabetes and metabolic diseases (18, 19). In addition, PM2.5 exposure also causes severe damage to various tissues, including the lungs (20), liver (21), adipose tissue (22), heart (23), and skeletal muscle (24).
The aforementioned evidence indicates that PM2.5 exposure not only disrupts systemic metabolic homeostasis but may also exert profound effects on key metabolic organs. It is well known that skeletal muscle is the largest metabolic organ in the human body (25, 26), not only playing a crucial role in regulating overall metabolism (27), but also in maintaining glucose and lipid metabolic homeostasis (28), utilizing various metabolic pathways to ensure the stability and efficiency of energy supply within the body (29). Compared to other tissues, skeletal muscle has a high density of mitochondria and a dense network of capillaries, which makes it highly dependent on respiration and oxygen exchange (30). Previous studies have shown that PM2.5 inhaled through the lungs can increase oxidative damage levels in peripheral blood (31). Due to its high blood flow perfusion and active material exchange (32), skeletal muscle may become one of the primary target tissues for PM2.5 accumulation and damage. These physiological characteristics determine the vulnerability of skeletal muscle to PM2.5 exposure.
PM2.5 exposure leads to excessive ROS production (10), disrupting the redox balance in myocytes and resulting in insufficient energy supply (33). Since mitochondria are the “central regulators” of cellular energy metabolism (34, 35), the negative reactions induced by PM2.5 exposure may not only damage cells but also impair mitochondrial function (36). Under normal conditions, mitochondria provide substrates (NADH and FADH2) for ATP synthesis to oxidative phosphorylation (OXPHOS) through the tricarboxylic acid (TCA) cycle (37), followed by the transfer of electrons through the electron transport chain (ETC), creating a proton gradient (38). Ultimately, ATP is synthesized via ATP synthase (39). This entire process efficiently converts glucose and fatty acids into ATP (40). Moreover, it can be flexibly adjusted based on cellular environment and energy demands, providing a stable energy source for skeletal muscle (41). PM2.5 exposure inhibits the expression of citrate synthase activity in skeletal muscle mitochondria (42), thereby reducing the generation of TCA cycle substrates (NADH and FADH2) and ultimately leading to a decline in ATP production efficiency (43). Furthermore, an experimental study observed that short-term PM2.5 exposure causes damage to mitochondrial morphology and dynamics in skeletal muscle, leading to the disruption of mitochondrial homeostasis (24).
In summary, the impact of PM2.5 exposure on skeletal muscle mitochondria is profound, not only damaging mitochondrial morphology but also significantly affecting their functionality and metabolic capacity. The existing research on the effects of PM2.5 exposure on skeletal muscle mitochondria lacks a systematic perspective. Therefore, this review aims to explore the effects and mechanisms of PM2.5 exposure on mitochondrial morphology and function in skeletal muscle, as well as the potential threats these issues pose to the skeletal muscle system. For this purpose, this study included “original research conducted under PM2.5 particle exposure models,” excluding other pollutants or finer particles (such as PM0.1). The study subjects were limited to rodents or humans. The literature screening period was from 2000 to 2025. This work seeks to provide new insights and references for the prevention and treatment of related diseases in the context of contemporary environmental PM2.5 pollution.
2 The impact of PM2.5 exposure on skeletal muscle mitochondria
Studies have shown that PM2.5 exposure has long-term damaging effects on skeletal muscle mitochondria, including morphological damage, inhibition of mitochondrial biogenesis-related pathways, reduced mitochondrial enzyme expression levels and mitochondrial function. Table 1 summarizes studies on the effects of PM2.5 exposure on skeletal muscle mitochondria.

Table 1. Effects of PM2.5 exposure on skeletal muscle mitochondria.
2.1 Effects of PM2.5 exposure on mitochondrial morphology
Skeletal muscle mitochondria possess a unique double-membrane structure (44). The outer mitochondrial membrane (OMM) faces the cytoplasm, serving as a barrier and protective layer, while the inner mitochondrial membrane (IMM) is highly folded into cristae, forming a complex sac-like structure (45, 46). This unique architecture plays a pivotal role in energy production processes (47). The mitochondrial cristae are enriched with complexes required for the electron transport chain and oxidative phosphorylation (48). The extensive folding of the cristae significantly increases the surface area of the inner membrane, thereby enhancing the number of attachment sites for these complexes (49, 50). This structural adaptation facilitates the efficiency of OXPHOS, providing an optimal environment for ATP synthesis (51). The mitochondrial membranes ensure the functional integrity of mitochondria (52). Additionally, mitochondria exhibit dynamic properties by regulating their size and quantity through the processes of fission and fusion, allowing them to adapt to cellular energy demands (53, 54). This dynamic nature relies on the coordinated interplay of biogenesis, autophagy, and mitochondrial dynamics (55), forming a homeostatic network within the cell (56). The regulation of mitochondrial biogenesis primarily depends on a series of critical factors (57, 58). Among these, peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) serves as the primary driver of mitochondrial biogenesis (59–61). PGC-1α upregulates the expression of nuclear respiratory factor 1 (NRF-1), which subsequently activates mitochondrial transcription factor A (mTFA), promoting the replication and transcription of mitochondrial DNA. This process increases mitochondrial quantity and quality to meet cellular energy demands (62–64). Meanwhile, mitophagy ensures mitochondrial health and functional activity by marking and removing damaged mitochondria through the actions of PTEN-induced kinase 1 (PINK-1) and parkin genes (Parkin) (65, 66). Mitochondrial dynamics play a critical role in regulating morphology and energy homeostasis (67). Mitofusins (Mfn-1 and Mfn-2) and optic atrophy protein 1 (OPA-1) act as effector proteins for mitochondrial fusion (68), mediating the fusion of the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM), respectively, to form larger mitochondrial networks (69). In contrast, dynamin-related protein 1 (Drp-1) and mitochondrial fission factor 1 (Fis1) facilitate mitochondrial fission, aiding in the removal of damaged mitochondria (70). These processes ensure the uniform distribution of energy while maintaining intracellular quality control.
Existing studies have demonstrated that PM2.5 exposure can lead to alterations in skeletal muscle mitochondrial morphology, such as cristae loss and outer membrane damage (24, 71, 72). Further studies have revealed that the extent of damage to skeletal muscle mitochondrial morphology varies with different durations of PM2.5 exposure. A study on short-term PM2.5 exposure reported that male mice exposed to PM2.5 (50.1 ± 8.1 μg/m3) three times within 1 week exhibited mitochondrial abnormalities, such as cristae swelling or cristae loss, in skeletal muscle. Muscle samples were processed 48 h after the final exposure. Additionally, a decrease in the expression of the mitochondrial outer membrane fusion protein Mfn1 was observed, while no significant changes were detected in other proteins involved in mitochondrial fission and fusion (24).
Another short-term PM2.5 exposure study (50.9 ± 10.4 μg/m3), involving continuous exposure for 5 days with a follow-up period, revealed sex-dependent effects on mitochondrial morphology. In male mice, 1 month after PM2.5 exposure, mitochondrial outer membrane damage was observed, accompanied by increased expression of mitochondrial fusion proteins (Mfn-1 and Mfn-2). In contrast, female mice exhibited more severe outer membrane damage, yet no changes were detected in mitochondrial dynamics-related factors. By 3 months post-exposure, mitochondrial morphology in both male and female mice showed recovery, and mitochondrial dynamics stabilized. Interestingly, at 15 months post-exposure, male mice exhibited signs of ongoing recovery, with increased expression of the fission protein Fis-1, while other dynamics-related factors remained stable. Conversely, female mice displayed smaller mitochondria under transmission electron microscopy (TEM) observation, decreased expression of the fission protein Drp-1, and a significant increase in Fis-1 expression. These changes suggested excessive mitochondrial fission. Additionally, among fusion proteins, only OPA-1 expression increased. These findings indicate that female mice might remain in a state of sustained mitochondrial damage (71).
In an experimental study of long-term PM2.5 exposure (71.20 ± 45.01 μg/m3), a 4-month exposure led to compromised mitochondrial membrane integrity in skeletal muscle, mild cristae loss, small vacuoles, and slight expansion of the sarcoplasmic reticulum. Over time, after 6 months of exposure, mitochondrial damage worsened, characterized by the presence of numerous mitochondrial vacuoles. Interestingly, in terms of mitochondrial dynamics, both fusion and fission protein expression showed significant increases compared to the control group (72). Compared to short-term exposure studies, the increased expression of fusion proteins Mfn-1, Mfn-2, and OPA-1 may represent a compensatory mechanism in response to the elevated expression of fission proteins (72).
The above studies indicate that although most mitochondria survive after PM2.5 exposure, they fail to fully recover to their original state (Figure 1). The dose and duration of PM2.5 exposure may be positively correlated with the severity of mitochondrial damage (73). Higher concentrations or prolonged exposure can exacerbate the structural disruption of mitochondrial morphology (74). Notably, mitochondrial fusion and fission proteins may exhibit adaptive feedback mechanisms to maintain the balance of mitochondrial dynamics (71, 72, 75). Moreover, based on sex differences, the effects of PM2.5 exposure on mitochondrial morphology and dynamics exhibit significant variability (71).
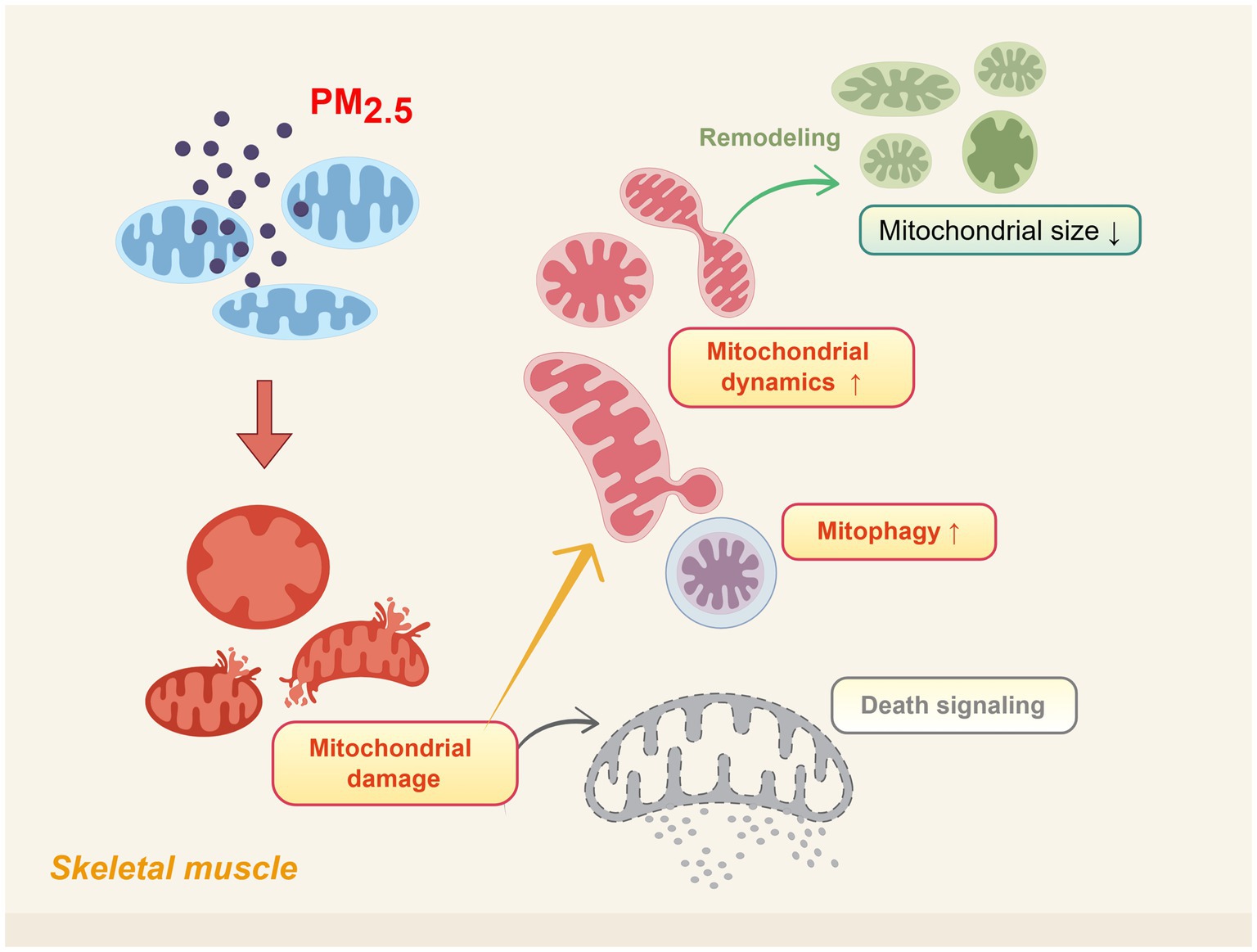
Figure 1. Schematic representation of skeletal muscle mitochondrial morphological damage induced by PM2.5 exposure. Including alterations in cristae structure, outer/inner membrane integrity, and increased mitochondrial fission activity. Created using Figdraw (ID: USTTAec5f8).
It is well established that mitochondrial dynamics—such as number, size, and volume—are regulated through the coordinated processes of mitochondrial biogenesis and mitophagy (76). However, exposure to PM2.5 may disrupt this dynamic balance. PM2.5 exposure inhibits the SIRT-1/AMPKα/PGC-1α/NRF-1 signaling pathway in skeletal muscle, leading to a reduction in mitochondrial DNA copy number (72), a key marker of mitochondrial biogenesis (77). This suppression weakens the capacity for mitochondrial growth and proliferation (78). PM2.5 exposure has been shown to activate autophagy pathways (24). Although the precise mechanisms remain unclear, PINK-1, a mitochondrial enzyme, plays a key role when mitochondrial membrane damage occurs. PINK-1 accumulates on the outer membrane of damaged mitochondria, recruiting Parkin to initiate the autophagic pathway, thereby promoting ubiquitination (79, 80). This process facilitates the clearance of damaged mitochondria, helping to maintain mitochondrial quality within the cell (81). When mitochondrial biogenesis is impaired, the loss of damaged mitochondria cannot be compensated by the generation of new ones. This disruption of dynamic balance may lead to a reduction in the size of the remaining mitochondria or even a decrease in their total number within the cell (82).
2.2 Effects of PM2.5 exposure on mitochondrial enzymes
Skeletal muscle mitochondria produce ATP through three enzymatic pathways, fatty acid beta oxidation, the TCA cycle, and the ETC, constituting a chain reaction of oxidative metabolism (83, 84). Fatty acid β-oxidation primarily occurs within mitochondria (85). Prior to this process, the expression of carnitine palmitoyltransferase 1 (CPT1) is regulated by peroxisome proliferator-activated receptor α (PPARα) (86, 87). CPT1 catalyzes the transfer of long-chain fatty acids from coenzyme A to carnitine, enabling fatty acids to be transported into the mitochondria in the form of carnitine esters for β-oxidation (88), thereby generating substrates (acetyl-CoA) required for the TCA cycle (89). Subsequently, acetyl-CoA enters the TCA cycle, where it combines with oxaloacetate to form citrate under the catalytic action of citrate synthase (90). This marks the formal entry of acetyl-CoA into the TCA cycle, during which a series of enzymatic reactions generate key electron donors (NADH and FADH2) (37). These electrons are transferred through complexes I to IV of the electron transport chain, where they combine with oxygen to form water (91), ultimately driving ATP synthesis via complex V (ATP synthase) (92). This chain reaction ensures a stable energy supply for skeletal muscle metabolism.
A substantial body of research has demonstrated that exposure to PM2.5 in tissues such as the heart and liver suppresses the expression of PPARα and CPT1, thereby hindering the transport of fatty acids into mitochondria for β-oxidation (93, 94). Similar to the heart and liver, skeletal muscle also exhibits suppressed PPARα expression following PM2.5 exposure (71). However, long-term observations after PM2.5 exposure reveal that the damage to mitochondrial fatty acid β-oxidation is persistent and varies slightly depending on gender differences (71). One month after PM2.5 exposure, a decrease in the expression of PPARα and long-chain acyl-CoA dehydrogenase (LCAD) was observed in male mice, whereas no similar changes were detected in female mice (71). The reduction in PPARα expression may impair the efficiency of fatty acid transport into mitochondria (95, 96). Meanwhile, LCAD, a key enzyme in mitochondrial fatty acid β-oxidation (97), plays a crucial role in the breakdown of fatty acids during this process. Its decreased expression hinders the metabolic breakdown of fatty acids, slows down the β-oxidation pathway, and suppresses this critical mitochondrial energy supply mechanism (98, 99). At 3 months, the expression levels of PPARα and LCAD in male mice tended to return to normal, whereas female mice exhibited a decrease in PPARα expression (71). This may suggest that the damage induced by PM2.5 exposure on mitochondria could persist even after exposure has ceased (100). After 15 months of PM2.5 exposure, when the mice entered the aging phase, the expression of mitochondrial fatty acid β-oxidation proteins in male mice returned to normal. In contrast, in female mice, the phosphorylation of acetyl-CoA carboxylase (ACC) was suppressed (71). When ACC phosphorylation is suppressed, ACC activity increases, leading to more acetyl-CoA being converted into malonyl-CoA for fatty acid synthesis. The reduction in acetyl-CoA availability may compromise the efficiency of the TCA cycle (101).
Although PM2.5 exposure disrupts fatty acid β-oxidation and, through a chain reaction, further impacts oxidative phosphorylation (102). Nevertheless, the impact of PM2.5 exposure on oxidative phosphorylation is not solely related to fatty acid metabolism but is also mediated by abnormalities in energy metabolism caused by disruptions in enzymes within the TCA cycle. This is primarily reflected in the decreased expression of succinate dehydrogenase subunit B (SDHB) (24), a key enzyme linking the TCA cycle and the ETC (103). When PM2.5 exposure leads to a reduction in the expression of enzymes in the TCA cycle, the central role of the TCA cycle means that any damage to it could significantly impair the efficiency of oxidative phosphorylation (104). However, long-term observations of skeletal muscle mitochondria following PM2.5 exposure reveal that the effects vary by gender. One month after exposure, differences were observed in the expression of electron transport chain complexes: male mice exhibited reduced expression of mitochondrial complexes I, II, and IV, whereas female mice showed suppressed expression of complexes I–IV (71). This may be related to the morphological damage caused during the early stages of PM2.5 exposure, where severe structural damage leads to a significant decline in the expression of complexes housed within the mitochondrial cristae (105). By 3 months post-exposure, the expression of succinate ubiquinone oxidoreductase (SUO) and cytochrome c oxidase subunit 4 (COX-4) increased in male mice, while female mice returned to baseline levels. At 15 months, as the mice entered the aging phase, the electron transport chain expression in male mice tended to normalize, whereas female mice exhibited an increase in complex V expression (71). These effects highlight the long-term impact of PM2.5 exposure and its gender-specific differences, suggesting that mitochondrial damage caused by PM2.5 exposure may be lifelong. Future research should focus on the influence of PM2.5 exposure on aging-related metabolic changes.
2.3 Effects of PM2.5 exposure on mitochondrial function
Current studies often define mitochondrial functional damage through the observation of mitochondrial morphological abnormalities using transmission electron microscopy (TEM) (106), as well as through the evaluation of mitochondrial protein expression levels (such as quantity and overall protein abundance) (107). In fact, these indicators can only indirectly reflect the integrity of mitochondrial function. The activity of mitochondrial proteins, which reflects the efficiency of their functional execution, provides a more precise measure of mitochondrial function compared to expression levels (108). Therefore, the evaluation of mitochondrial function should focus on functional assessments, such as complex activity (108), reactive oxygen species (ROS) levels (109), respiration rate (110), and membrane potential (ΔΨm) (111), which are critical indicators.
Mitochondria are the primary site of ROS production within cells (112, 113). During the operation of the electron transport chain, partial electron leakage may occur during electron transfer from Complexes I and III (114, 115), leading to the formation of superoxide (O₂−) through interaction with molecular oxygen. This superoxide is subsequently dismutated into hydrogen peroxide (H₂O₂) by mitochondrial superoxide dismutase (SOD-2) (116). Hydrogen peroxide can be decomposed into water and oxygen under the action of glutathione peroxidase (GPx) using reduced glutathione (GSH), thereby mitigating oxidative damage (117, 118). ROS exhibit a dual role, under physiological conditions, the levels of ROS generation and antioxidant capacity are maintained in a state of equilibrium (119). Conversely, excessive accumulation of ROS or insufficient clearance can result in oxidative stress (120), and oxidative stress in skeletal muscle may be one of the key determinants of mitochondrial dysfunction (121).
In general, directly measuring intracellular ROS levels presents certain challenges. Most studies instead utilize the detection of lipid peroxidation byproduct malondialdehyde (MDA) as a surrogate marker (122, 123), MDA is a marker of lipid peroxidation and has the potential to induce cytotoxicity and cellular stress (124). It indirectly reflects intracellular ROS levels and the oxidative stress state through the measurement of thiobarbituric acid reactive substances (TBARS) (125). Recent studies have demonstrated that exposure to PM2.5 increases levels of MDA, a byproduct of lipid peroxidation, and hydrogen peroxide (H₂O₂) in skeletal muscle, leading to oxidative stress responses (126). After 15 months of PM2.5 exposure, the expression of TBARS in skeletal muscle remains at a high level, exacerbating oxidative stress responses in skeletal muscle (71).
In fact, mitochondria possess an antioxidant enzyme system capable of scavenging reactive oxygen species and maintaining redox balance (127). PM2.5 exposure suppresses the expression of superoxide dismutase 2 (SOD-2) in skeletal muscle (126). The downregulation of antioxidant enzyme expression disrupts the redox balance in the body (128), leading to excessive accumulation of ROS within mitochondria. This persistent oxidative stress response reduces the expression of mitochondrial DNA (mtDNA) (42). Interestingly, it is an established fact that exposure to PM2.5 exacerbates oxidative stress responses in skeletal muscle mitochondria (71, 72). However, the expression of antioxidant enzymes shows variability. In another study on short-term PM2.5 exposure, an increase in SOD-2 levels was observed (24). Although the expression of SOD-2 in mitochondria varies, including compensatory mechanisms associated with SOD-2 upregulation, it is evident that PM2.5 exposure disrupts the homeostasis of antioxidant enzymes. This disruption leads to ROS accumulation, resulting in sustained oxidative damage and subsequently contributing to mitochondrial dysfunction (129).
Notably, PM2.5 exposure can induce mitochondrial dysfunction by inhibiting mitochondrial enzyme activity (130), including a reduction in citrate synthase activity and a decline in overall cytochrome c oxidase (COX) activity (42, 126). Citrate synthase activity is commonly used as a marker of skeletal muscle aerobic capacity and mitochondrial density (131). A study on pregnant mice exposed to PM2.5 demonstrated that the reduction in citrate synthase activity leads to a decline in skeletal muscle oxidative capacity and a decrease in mitochondrial DNA content (42). Critically, citrate synthase is a key enzyme in the TCA cycle. A decline in citrate synthase activity may reduce the efficiency of citrate production from acetyl-CoA and oxaloacetate, thereby impairing overall TCA cycle efficiency (37). This disruption leads to a decreased generation of reducing equivalents (NADH and FADH₂), limiting electron input into the ETC (132). The mitochondrial Complexes I–V play critical roles in ATP synthesis (133), and their activity is closely associated with the efficiency of mitochondrial ATP production (134). Notably, a decline in mitochondrial cytochrome c oxidase (mtCOX) activity can lead to reduced oxidative capacity in skeletal muscle mitochondria (135). PM2.5 exposure can continuously impair mitochondrial functional integrity through a cascade of effects, including abnormalities in the antioxidant system, ROS accumulation, and decreased mitochondrial enzyme activity (Figure 2).
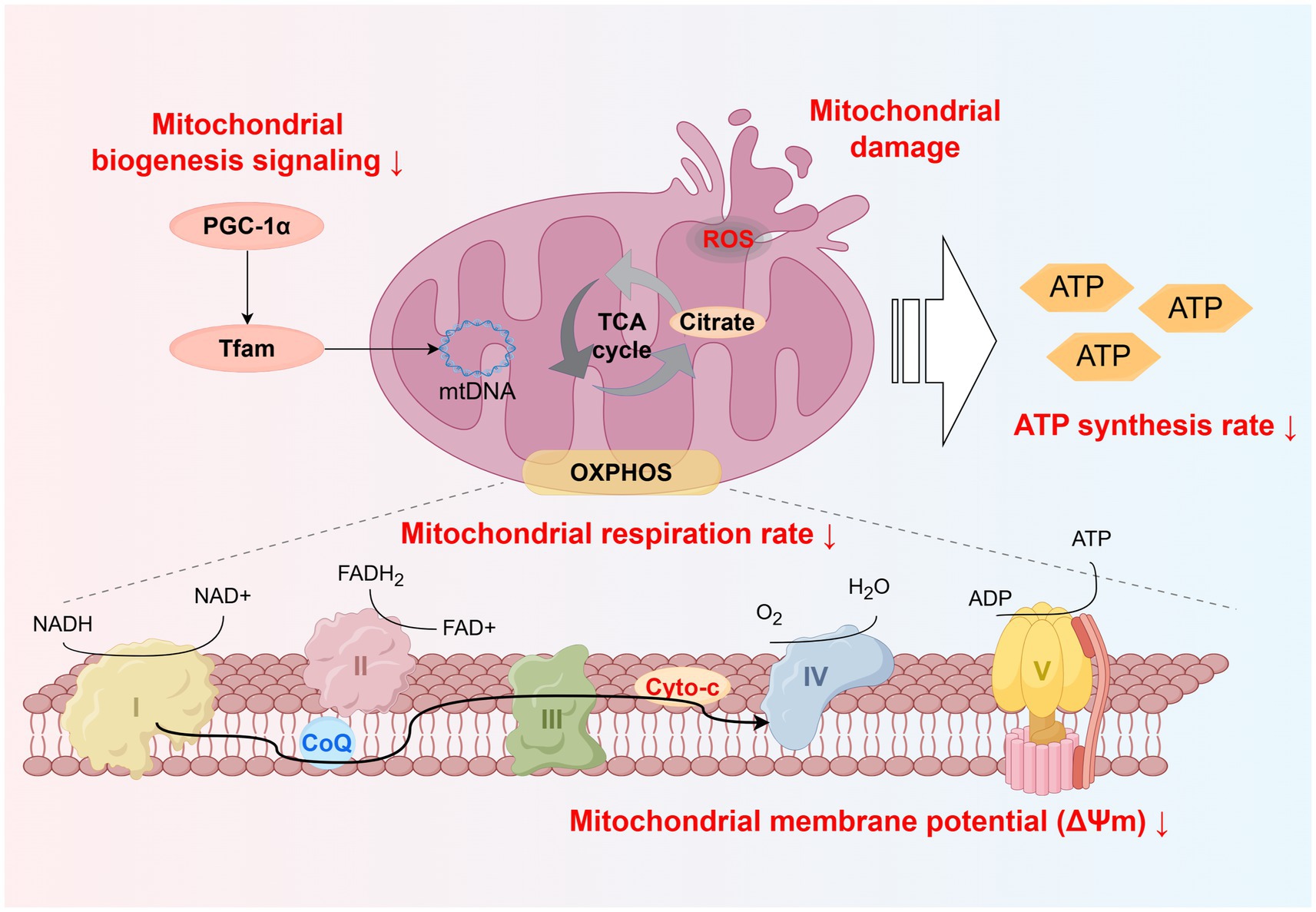
Figure 2. Schematic representation of skeletal muscle mitochondrial dysfunction induced by PM2.5 exposure. Including disrupted mitochondrial biogenesis signaling, increased ROS production, and reduced electron transport efficiency, leading to decreased mitochondrial respiration rate, membrane potential, and ATP synthesis. Created using Figdraw (ID: WSTWS9999d).
3 The impact of PM2.5-induced mitochondrial dysfunction on the skeletal muscle system
PM2.5-induced mitochondrial dysfunction has a variety of damaging effects on skeletal muscle, including oxidative stress, DNA damage, satellite stem cell senescence, muscle atrophy, and dysfunction of glycolipid metabolism. Table 2 summarizes studies on the effects of PM2.5 exposure on skeletal muscle system.

Table 2. Additional effects of PM2.5 exposure on skeletal muscle.
3.1 Skeletal muscle oxidative stress and DNA damage
Oxidative stress can be regarded as a critical factor in PM2.5 exposure-induced mitochondrial dysfunction (71, 72, 126). Under excessive oxidative stress, skeletal muscle proteins undergo degradation (136), and damage is inflicted on the genetic material (DNA) of cells (137). Notably, the significant elevation of 8-hydroxy-2′-deoxyguanosine (8-OHdG) levels is recognized as a critical marker of DNA oxidative damage (138). PM2.5 exposure activates the expression of 8-OHdG, leading to endogenous oxidative DNA damage in the body (139, 140). Mitochondrial DNA (mtDNA) is considered more susceptible to oxidative DNA damage (141), and the accumulation of damage to both mitochondrial and nuclear DNA is believed to ultimately impair cellular function, leading to the loss of muscle cells (142). An experimental study found that in animal models exposed to PM2.5, the apoptosis rate of skeletal muscle cells significantly increased, accompanied by a reduction in muscle fibers (71). The cumulative effects of DNA damage have a significant impact on the regenerative capacity of skeletal muscle (143). As the core of muscle repair, skeletal muscle satellite cells rely on DNA stability for their proliferation and differentiation capacity (144). DNA damage can inhibit the proliferation and differentiation functions of skeletal muscle stem cells, thereby slowing the repair of muscle fibers and exacerbating the process of muscle aging (145).
3.2 Skeletal muscle maintenance and regeneration
To date, cross-sectional studies have shown that for every 1.41 μg/m3 increase in PM2.5 concentration, skeletal muscle mass decreases by 0.4 kg (146). Additionally, for every 1 μg/m3 increase in PM2.5, the risk of sarcopenia increases by 11.1% (147), and every 10 μg/m3 increase in PM2.5 may result in a 0.7 kg reduction in grip strength (148). The above evidence indicates that PM2.5 exposure can lead to muscle atrophy and a decline in muscle strength.
An animal experimental study revealed that PM2.5 exposure can induce stem cell senescence and regenerative dysfunction, resulting in severe damage to muscle fibers. Moreover, the extent of the damage varies depending on age and gender (71). Specifically, PM2.5 exposure led to a significant reduction in the number of muscle fibers in juvenile male mice and decreased the expression of Myogenin (71). Myogenin, a critical transcription factor essential for muscle differentiation and regeneration (149), is suppressed, which weakens the regenerative capacity of muscle cells (150). In addition, PM2.5 exposure significantly upregulated the Bax/Bcl-2 ratio in juvenile male mice (71). The excessive expression of the Bax/Bcl-2 ratio is associated with increased muscle cell apoptosis (151). Exposure to PM2.5 during the juvenile period resulted in simultaneous muscle cell apoptosis and myostatin inhibition, further reducing the number of muscle fibers; although muscle fibers in adult male mice showed partial recovery, they failed to return to their original state, possibly due to the normalization of myostatin levels, but the recovery process remained slow with low regenerative efficiency owing to continued muscle cell apoptosis (71). In contrast, adult female mice exhibited significant muscle fiber damage at this stage, accompanied by simultaneous reductions in the expression of Myostatin and Myogenin (71). This disruption in the balance of skeletal muscle growth and differentiation likely further impairs muscle fiber regenerative capacity, increasing the risk of muscle atrophy (152, 153). Upon entering the aging stage, male mice exhibited persistent muscle fiber damage, which reflected a further decline in regenerative capacity, and although Myogenin expression was significantly elevated, indicating a compensatory attempt at regeneration, the repair outcomes remained limited (71). Interestingly, the number of muscle fibers in female mice returned to normal, which may be associated with the significant upregulation of Myogenin and Pax-7 expression (154). PM2.5 exposure also activated the expression of senescence markers β-galactosidase (β-Gal) and cyclin-dependent kinase inhibitor 2A (p16) in female mice, accelerating the aging of muscle stem cells (71).
In summary, the effects of PM2.5 exposure vary across different age groups, with juvenile male mice being more sensitive to PM2.5 exposure, while skeletal muscle fibers in the older adult stage experience the most severe damage. The muscle fiber damage caused by PM2.5 exposure is persistent, potentially linked to mitochondrial damage induced by PM2.5, which disrupts the energy supply required for skeletal muscle growth and regeneration. This process accelerates skeletal muscle atrophy, manifested as muscle fiber shrinkage and decreased muscle strength, thereby increasing the risk of conditions such as sarcopenia.
3.3 Skeletal muscle metabolic function
Skeletal muscle is a critical organ for glucose and fatty acid metabolism (155, 156). Mitochondrial dysfunction induced by PM2.5 exposure compromises ATP production, failing to meet the metabolic demands of cells (36). Consequently, cells may respond through adaptive changes in metabolic pathways, such as glucose and lipid metabolism (157, 158).
Skeletal muscle is the primary organ where insulin-mediated glucose uptake occurs through glucose transporter 4 (GLUT4) (159). A short-term PM2.5 exposure study demonstrated that although mitochondrial oxidative phosphorylation capacity in skeletal muscle declines, it compensates for the energy deficit by upregulating the expression of hexokinase 2 (HK2) (24). HK2, a key enzyme in the glycolytic pathway, enhances glucose uptake and utilization to sustain energy supply and adapt to metabolic stress (160). It has been reported that PM2.5 exposure reduces the expression of GLUT4 (71, 161), and this reduction impairs the ability of skeletal muscle to uptake glucose, leading to disruptions in glucose metabolism within skeletal muscle and the development of insulin resistance (162, 163). However, under the long-term effects of PM2.5 exposure, GLUT4 expression gradually increases over time, potentially linked to the decline in OXPHOS efficiency caused by mitochondrial dysfunction. This shift in cellular metabolism may progressively favor a glycolysis-dominant energy production mode, with upregulated GLUT4 expression enhancing glucose dependency (71).
Skeletal muscle is a crucial site for fatty acid metabolism (164). PM2.5 exposure affects the expression of lipid metabolic enzymes in skeletal muscle, with the downregulation of peroxisome proliferator-activated receptor alpha (PPARα) and long-chain acyl-CoA dehydrogenase (LCAD), leading to reduced fatty acid utilization and gradual lipid accumulation within the muscle (71). Abnormal lipid metabolism is more pronounced in females than in males after PM2.5 exposure, with females being more severely affected, which can lead to conditions such as obesity (71).
Changes in metabolic enzymes within skeletal muscle caused by mitochondrial dysfunction disrupt the homeostasis of glucose and lipid metabolism, impairing the metabolic function of skeletal muscle. Both glucose homeostasis imbalance and abnormal lipid metabolism are closely associated with insulin resistance (163, 165). This metabolic dysregulation not only exacerbates the energy metabolism burden on the body but also provides a critical pathological basis for the development of diabetes and related metabolic syndromes.
4 Regulatory effect of exercise on PM2.5-induced mitochondrial dysfunction
Exercise, as a non-pharmacological intervention, has been shown to mitigate the health risks associated with exposure to PM2.5 (126, 166). Studies have shown that long-term moderate exercise under conditions of low PM2.5 concentrations is beneficial to health (72). The Table 3 summarizes studies on the moderating effect of exercise on the damage caused by PM2.5 exposure.
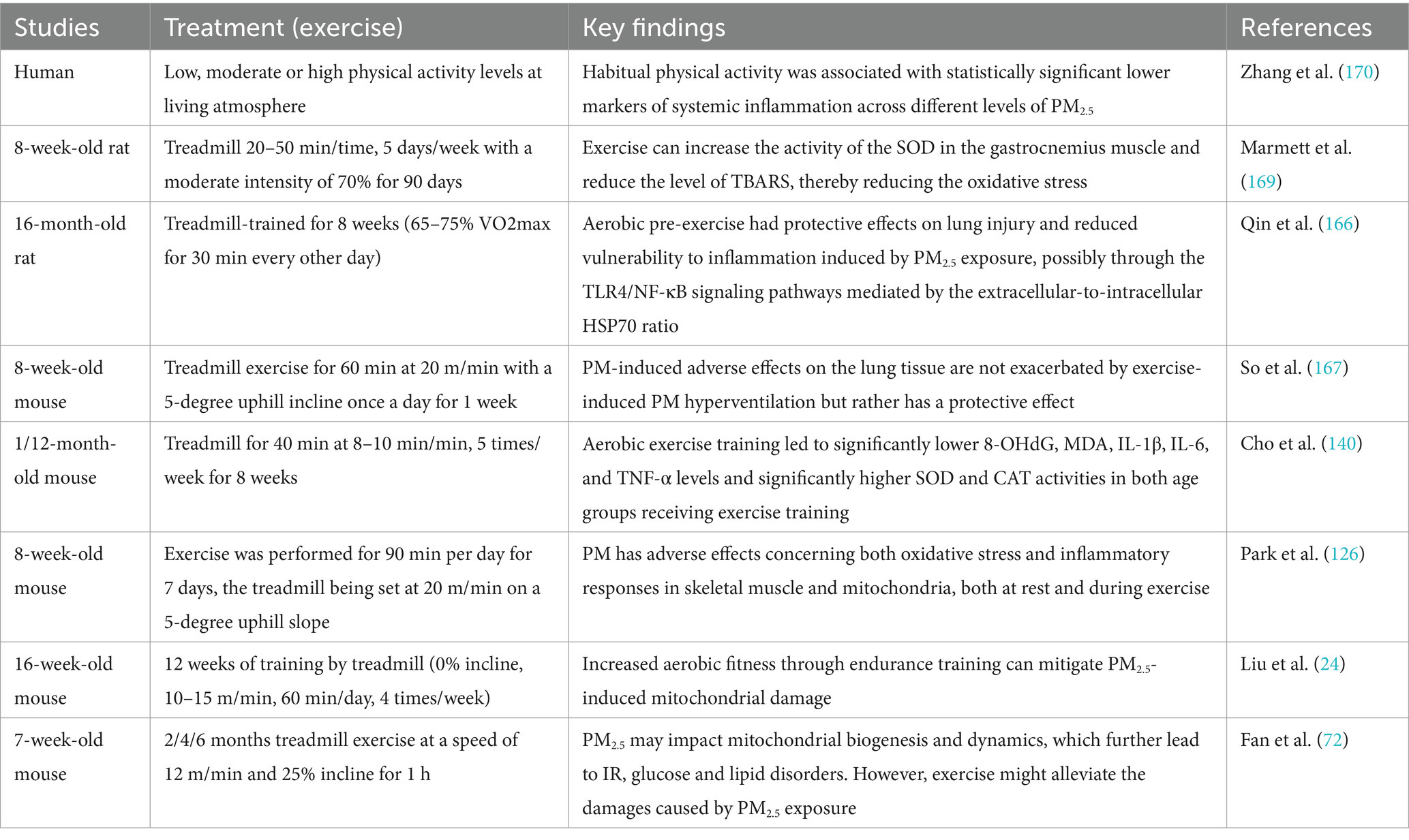
Table 3. The moderating effect of exercise on the damage caused by PM2.5 exposure.
4.1 Long-term endurance exercise can prevent PM2.5 exposure-induced mitochondrial dysfunction
Regular aerobic exercise can mitigate PM2.5-induced damage and oxidative stress by activating the SIRT-1/AMPKα/PGC1-α/NRF-1 signaling pathway (72). As a key regulator of mitochondrial biogenesis (59), PGC-1α enhances mtDNA replication and transcription by upregulating downstream factors such as mTFA, counteracting the suppression of mitochondrial biogenesis caused by PM2.5 exposure. This significantly improves the reduction in mitochondrial number and size induced by PM2.5 exposure, with the beneficial effects becoming more pronounced with prolonged exercise duration (72). In addition, exercise significantly reduced PM2.5-induced mitochondrial damage levels, representing another mechanism for preventing PM2.5-induced damage (167).
PM2.5 exposure leads to mitochondrial morphological damage and dynamics disruption. Although long-term endurance training cannot completely prevent acute skeletal muscle mitochondrial damage caused by acute exposure to PM2.5 (50.1 ± 8.1 μg/m3, 2 h/day, 5 days), a higher level of exercise adaptation can promote repair and regeneration following the damage (24). While endurance training can maintenance mitochondrial morphology, this adaptation does not prevent mitochondrial morphological damage under PM2.5 exposure or immediately following exercise after exposure (24). However, the activation of mitochondrial autophagy levels induced by long-term endurance exercise results in a rapid response to clear damaged mitochondria following exposure to PM2.5 (24). Endurance exercise enhances mitochondrial dynamics by activating mitophagy and biogenesis, thereby reinforcing the mitochondrial homeostasis (Figure 3).
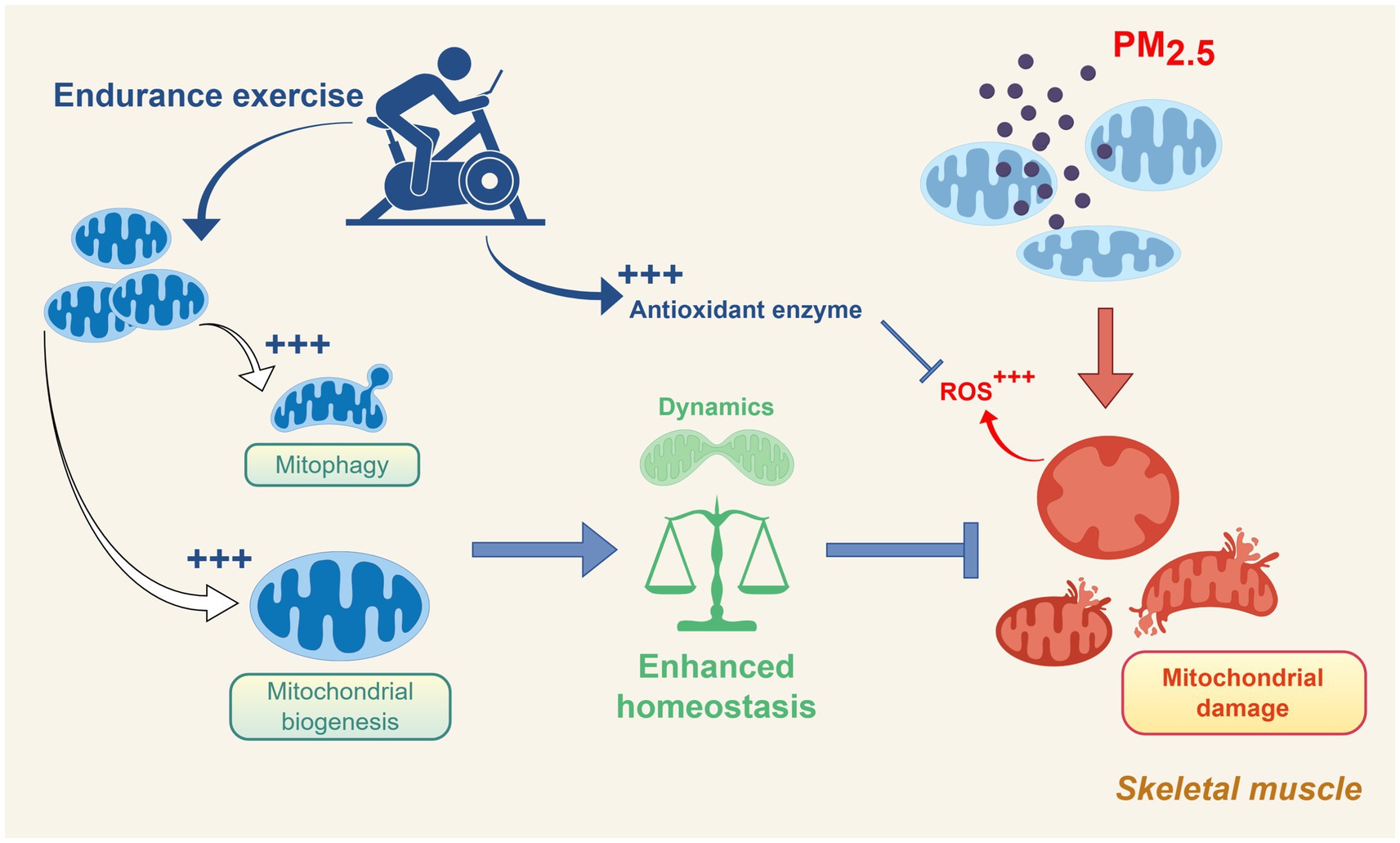
Figure 3. Schematic representation of the protective role of endurance exercise against skeletal muscle mitochondrial dysfunction induced by PM2.5 exposure. Exercise promotes mitochondrial biogenesis, mitophagy, and antioxidant defense, enhancing mitochondrial homeostasis and counteracting PM2.5-induced oxidative stress and damage. Created using Figdraw (ID: PWSAW4e4ee).
PM2.5 exposure induces mitochondrial dysfunction by inhibiting the expression and activity of mitochondrial respiratory chain complexes (71, 126). Long-term endurance exercise significantly enhances the expression of Complexes I, II, and IV, increasing their tolerance to oxidative damage caused by PM2.5 (24). Regular aerobic exercise further boosts the activity of Complex IV, thereby improving oxidative phosphorylation efficiency and reducing excessive ROS accumulation (126).
PM2.5-induced mitochondrial damage is often accompanied by a significant increase in ROS production, which subsequently triggers oxidative stress and inflammatory responses (167). Notably, endurance training enhances antioxidant capacity and alleviates inflammatory responses, which helps maintain health under conditions of PM2.5 exposure (168). Appropriate aerobic exercise can enhance the function of the body’s antioxidant system and increase the expression of superoxide dismutase (SOD) and catalase (140), with long-term regular aerobic exercise further improving SOD activity and reducing lipid peroxidation in skeletal muscle (169). In addition to the aforementioned effects, exercise can also mitigate systemic inflammatory responses by reducing inflammatory markers, such as leukocytes (170).
It is worth noting that PM2.5 (100 μg/m3, 1.5 h/day, 7 days) exposure during exercise can exacerbate oxidative stress responses in skeletal muscle, leading to more severe mitochondrial dysfunction (126). This is primarily because intense exercise in high-concentration environments increases respiratory volume, resulting in greater inhalation of PM2.5, which in turn triggers oxidative stress and inflammatory responses, adversely affecting skeletal muscle health (126).
The above results clarify the preventive and regulatory effects of long-term endurance exercise on PM2.5 exposure-induced skeletal muscle mitochondrial damage. The results consistently demonstrate the necessity of long-term endurance exercise, while also indicating that PM2.5 concentrations in the environment should not be too high during endurance exercise. When PM2.5 levels are higher, it is recommended to moderately reduce outdoor endurance exercise time to minimize PM2.5 intake. At the same time, awareness of using dust masks outdoors should be enhanced to avoid exposure to high concentrations. When PM2.5 concentrations persistently exceed the aforementioned minimum experimental concentration (50 μg/m3), it is recommended to relocate endurance exercises indoors and implement air purification measures (such as using air purifiers) to reduce indoor exposure levels. Therefore, appropriately arranging the intensity and location of endurance exercise may help reduce the potential damage to skeletal muscle mitochondria caused by PM2.5 exposure while enjoying the health benefits of exercise.
4.2 The preventive and treatment potential of low-intensity resistance exercise against PM2.5 exposure-induced skeletal muscle mitochondrial damage
The effects of exercise treatment in the current PM2.5 exposure models are all based on endurance exercise (treadmill) (Table 3). This may be due to the convenience of animal models for endurance exercise treatment and the significant specificity of endurance exercise in regulating mitochondrial function (171).
However, recent studies have shown that long-term low-intensity resistance exercise can also enhance mitochondrial biogenesis and regulate mitochondrial homeostasis (172). At the same time, low-intensity resistance training also has similar skeletal muscle protein synthesis gains to moderate- and high-intensity resistance training (173). Compared to endurance exercise, resistance training relies less on the aerobic oxidation function of mitochondria and mainly relies on the anaerobic glycolysis of muscle glycogen to rapidly provide ATP (29). Therefore, the level of ROS produced by mitochondria induced by resistance exercise stimulation is lower (174), while resistance exercise promotes the activation of lactate signaling, which also has multiple effects on the regulation of mitochondrial function (175). Thus, low-intensity resistance exercise may have a positive regulatory effect on skeletal muscle and mitochondria that differs from endurance exercise during periods of PM2.5 exposure or when PM2.5 exposure-induced mitochondrial damage occurs.
Moderate- to high-intensity resistance training causes extensive microdamage to skeletal muscle fibers and activates inflammation-related pathways (176). Moreover, the damage and remodeling process of the skeletal muscle system caused by moderate-to-high-intensity resistance exercise can induce sustained cellular stress and high ATP demand during the repair phase (176), which may result in more severe compound damage during periods of PM2.5 exposure.
In summary, low-intensity resistance exercise has the potential to prevent and regulate PM2.5 exposure-induced mitochondrial dysfunction and should be further investigated in future studies. On the other hand, high-intensity resistance training should be avoided when PM2.5 concentrations exceed the standard, even if training is conducted indoors.
4.3 Potential synergistic role of exercise combined with antioxidants
Antioxidants, as exogenous interventions, can effectively reduce the occurrence of oxidative stress (177). Although there are many types of antioxidants, and the mechanisms and targets of various antioxidants differ (177). However, studies have shown that multiple antioxidants have synergistic effects in alleviating oxidative stress while maintaining mitochondrial function (178, 179). Mitochondria-targeted antioxidant MitoQ can alleviate PM2.5-induced vascular fibrosis and related oxidative damage (178). Vitamin E and omega-3 fatty acids may reduce vascular endothelial cell inflammation and oxidative stress responses caused by exposure to PM2.5 (180). Although evidence linking skeletal muscle-targeted interventions to combined interventions remains limited, the effects of these independent interventions on other tissues provide important evidence for the development of synergistic strategies.
5 Limitations of the current research
Due to the current lack of human skeletal muscle targeting research in this field, the experimental studies included in this review are mainly based on rodent models. Although these models provide valuable insights into the mechanisms underlying PM2.5 exposure-induced skeletal muscle mitochondrial damage, their findings may have limitations in terms of applicability to humans. On the one hand, rodents and humans have certain differences in metabolic rate and movement adaptation patterns, which may lead to differences in their sensitivity and expression of stress responses and intervention effects caused by PM2.5. In addition, most animal studies use acute or high-dose exposure models, which differ from the long-term, low-dose exposure environment that humans encounter in real life.
In future studies, further exploration of population-based longitudinal observational and interventional studies should be conducted, combining real-life exposure scenarios to systematically assess the long-term effects of PM2.5 on human skeletal muscle mitochondrial function. And explore the dose–response relationship under PM2.5 exposure and the potential protective mechanisms under different types of exercise interventions. In addition, attention should be paid to the toxic effects of different components of PM2.5 and their specific impact on skeletal muscle damage.
6 Conclusion
Studies on the effects of PM2.5 exposure on skeletal muscle mitochondria have shown that exposure to PM2.5 induces significant alterations in mitochondrial morphology and disrupts the balance of mitochondrial dynamics, biogenesis, and autophagy processes. By inhibiting fatty acid β-oxidation and oxidative phosphorylation efficiency, PM2.5 exposure ultimately leads to a reduction in ATP production capacity and disruptions in energy metabolism. The oxidative stress response induced by PM2.5 exposure can reduce mitochondrial enzyme activity, exacerbating mitochondrial dysfunction. It may also cause DNA damage in skeletal muscle cells and disrupt the balance of skeletal muscle maintenance and regeneration. This accelerates muscle fiber atrophy and increases the risk of sarcopenia while profoundly affecting glucose and lipid metabolic homeostasis, further elevating the incidence of metabolic diseases. Although regular aerobic exercise has been shown to alleviate the negative effects of PM2.5 exposure by activating certain mitochondrial pathways, exercising in high-concentration PM2.5 environments may exacerbate oxidative damage, highlighting the double-edged nature of exercise interventions. When daily PM2.5 exceeds 50 μg/m3, endurance exercise should be moved indoors. In summary, future research should not only elucidate the molecular mechanisms underlying the detrimental effects of PM2.5 on mitochondrial function but also focus more on potential therapeutic intervention strategies, such as combined interventions involving exercise and antioxidants.
Author contributions
YD: Visualization, Writing – original draft, Writing – review & editing, Investigation. QW: Writing – original draft, Investigation, Visualization, Writing – review & editing. WL: Writing – original draft, Project administration, Writing – review & editing, Conceptualization, Methodology.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Landrigan, PJ. Air pollution and health. Lancet Public Health. (2017) 2:e4–5. doi: 10.1016/S2468-2667(16)30023-8
2. Sang, S, Chu, C, Zhang, T, Chen, H, and Yang, X. The global burden of disease attributable to ambient fine particulate matter in 204 countries and territories, 1990-2019: a systematic analysis of the global burden of disease study 2019. Ecotoxicol Environ Saf. (2022) 238:113588. doi: 10.1016/j.ecoenv.2022.113588
3. WHO. WHO’S global air-quality guidelines. Lancet. (2006) 368:1302. doi: 10.1016/S0140-6736(06)69530-5
4. Bu, X, Xie, Z, Liu, J, Wei, L, Wang, X, Chen, M, et al. Global PM2.5-attributable health burden from 1990 to 2017: estimates from the global burden of disease study 2017. Environ Res. (2021) 197:111123. doi: 10.1016/j.envres.2021.111123
5. Bernstein, JA, Alexis, N, Barnes, C, Bernstein, IL, Bernstein, JA, Nel, A, et al. Health effects of air pollution. J Allergy Clin Immunol. (2004) 114:1116–23. doi: 10.1016/j.jaci.2004.08.030
6. Pinkerton, KE, Green, FH, Saiki, C, Vallyathan, V, Plopper, CG, Gopal, V, et al. Distribution of particulate matter and tissue remodeling in the human lung. Environ Health Perspect. (2000) 108:1063–9. doi: 10.1289/ehp.001081063
7. Araujo, JA, and Nel, AE. Particulate matter and atherosclerosis: role of particle size, composition and oxidative stress. Part Fibre Toxicol. (2009) 6:24. doi: 10.1186/1743-8977-6-24
8. López-Villarrubia, E, Iñiguez, C, Peral, N, García, MD, and Ballester, F. Characterizing mortality effects of particulate matter size fractions in the two capital cities of the Canary Islands. Environ Res. (2012) 112:129–38. doi: 10.1016/j.envres.2011.10.005
9. Rundell, KW, and Caviston, R. Ultrafine and fine particulate matter inhalation decreases exercise performance in healthy subjects. J Strength Cond Res. (2008) 22:2–5. doi: 10.1519/JSC.0b013e31815ef98b
10. Liu, X, Zhao, X, Li, X, Lv, S, Ma, R, Qi, Y, et al. PM2.5 triggered apoptosis in lung epithelial cells through the mitochondrial apoptotic way mediated by a ROS-DRP1-mitochondrial fission axis. J Hazard Mater. (2020) 397:122608. doi: 10.1016/j.jhazmat.2020.122608
11. Shan, X, Liu, L, Li, G, Xu, K, Liu, B, and Jiang, W. PM2.5 and the typical components cause organelle damage, apoptosis and necrosis: role of reactive oxygen species. Sci Total Environ. (2021) 782:146785. doi: 10.1016/j.scitotenv.2021.146785
12. Ermak, G, and Davies, KJA. Calcium and oxidative stress: from cell signaling to cell death. Mol Immunol. (2002) 38:713–21. doi: 10.1016/s0161-5890(01)00108-0
13. Cheng, Z, Luo, L, Wang, S, Wang, Y, Sharma, S, Shimadera, H, et al. Status and characteristics of ambient PM2.5 pollution in global megacities. Environ Int. (2016) 89-90:212–21. doi: 10.1016/j.envint.2016.02.003
14. Gao, M, Liang, C, Hong, W, Yu, X, Zhou, Y, Sun, R, et al. Biomass-related PM2.5 induces mitochondrial fragmentation and dysfunction in human airway epithelial cells. Environ Pollut. (2022) 292:118464. doi: 10.1016/j.envpol.2021.118464
15. Song, Y, Li, R, Zhang, Y, Wei, J, Chen, W, Chung, CKA, et al. Mass spectrometry-based metabolomics reveals the mechanism of ambient fine particulate matter and its components on energy metabolic reprogramming in BEAS-2B cells. Sci Total Environ. (2019) 651:3139–50. doi: 10.1016/j.scitotenv.2018.10.171
16. Huttunen, K, Siponen, T, Salonen, I, Yli-Tuomi, T, Aurela, M, Dufva, H, et al. Low-level exposure to ambient particulate matter is associated with systemic inflammation in ischemic heart disease patients. Environ Res. (2012) 116:44–51. doi: 10.1016/j.envres.2012.04.004
17. Zhang, Z, Hu, S, Fan, P, Li, L, Feng, S, Xiao, H, et al. The roles of liver inflammation and the insulin signaling pathway in PM2.5 instillation-induced insulin resistance in Wistar rats. Dis Markers. (2021) 2021:2821673. doi: 10.1155/2021/2821673
18. Zanobetti, A, Dominici, F, Wang, Y, and Schwartz, JD. A national case-crossover analysis of the short-term effect of PM2.5 on hospitalizations and mortality in subjects with diabetes and neurological disorders. Environ Health. (2014) 13:38. doi: 10.1186/1476-069X-13-38
19. Pan, K, Jiang, S, Du, X, Zeng, X, Zhang, J, Song, L, et al. AMPK activation attenuates inflammatory response to reduce ambient PM2.5-induced metabolic disorders in healthy and diabetic mice. Ecotoxicol Environ Saf. (2019) 179:290–300. doi: 10.1016/j.ecoenv.2019.04.038
20. Ning, X, Ji, X, Li, G, and Sang, N. Ambient PM2.5 causes lung injuries and coupled energy metabolic disorder. Ecotoxicol Environ Saf. (2019) 170:620–6. doi: 10.1016/j.ecoenv.2018.12.028
21. Qiu, Y-N, Wang, G-H, Zhou, F, Hao, J-J, Tian, L, Guan, L-F, et al. PM2.5 induces liver fibrosis via triggering ROS-mediated mitophagy. Ecotoxicol Environ Saf. (2019) 167:178–87. doi: 10.1016/j.ecoenv.2018.08.050
22. Della Guardia, L, and Shin, AC. White and brown adipose tissue functionality is impaired by fine particulate matter (PM2.5) exposure. J Mol Med (Berl). (2022) 100:665–76. doi: 10.1007/s00109-022-02183-6
23. Li, R, Kou, X, Geng, H, Xie, J, Tian, J, Cai, Z, et al. Mitochondrial damage: an important mechanism of ambient PM2.5 exposure-induced acute heart injury in rats. J Hazard Mater. (2015) 287:392–401. doi: 10.1016/j.jhazmat.2015.02.006
24. Liu, W, Wang, Z, Gu, Y, So, H-S, Kook, S-H, Park, Y, et al. Effects of short-term exercise and endurance training on skeletal muscle mitochondria damage induced by particular matter, atmospherically relevant artificial PM2.5. Front Public Health. (2024) 12:1302175. doi: 10.3389/fpubh.2024.1302175
25. Meng, Z-X, Gong, J, Chen, Z, Sun, J, Xiao, Y, Wang, L, et al. Glucose sensing by skeletal myocytes couples nutrient signaling to systemic homeostasis. Mol Cell. (2017) 66:332–344.e4. doi: 10.1016/j.molcel.2017.04.007
26. Gheller, BJF, Riddle, ES, Lem, MR, and Thalacker-Mercer, AE. Understanding age-related changes in skeletal muscle Metabolism: differences between females and males. Annu Rev Nutr. (2016) 36:129–56. doi: 10.1146/annurev-nutr-071715-050901
27. Argilés, JM, Campos, N, Lopez-Pedrosa, JM, Rueda, R, and Rodriguez-Mañas, L. Skeletal muscle regulates Metabolism via Interorgan crosstalk: roles in health and disease. J Am Med Dir Assoc. (2016) 17:789–96. doi: 10.1016/j.jamda.2016.04.019
28. Merz, KE, and Thurmond, DC. Role of skeletal muscle in insulin resistance and glucose uptake. Compr Physiol. (2020) 10:785–809. doi: 10.1002/cphy.c190029
29. Hargreaves, M, and Spriet, LL. Skeletal muscle energy metabolism during exercise. Nat Metab. (2020) 2:817–28. doi: 10.1038/s42255-020-0251-4
30. Poole, DC, Copp, SW, Ferguson, SK, and Musch, TI. Skeletal muscle capillary function: contemporary observations and novel hypotheses. Exp Physiol. (2013) 98:1645–58. doi: 10.1113/expphysiol.2013.073874
31. Lin, Z-Q, Xi, Z-G, Yang, D-F, Chao, F-H, Zhang, H-S, Zhang, W, et al. Oxidative damage to lung tissue and peripheral blood in endotracheal PM2.5-treated rats. Biomed Environ Sci. (2009) 22:223–8. doi: 10.1016/S0895-3988(09)60049-0
32. Casey, DP, and Joyner, MJ. Local control of skeletal muscle blood flow during exercise: influence of available oxygen. J Appl Physiol. (2011) 111:1527–38. doi: 10.1152/japplphysiol.00895.2011
33. Powers, SK, Ji, LL, Kavazis, AN, and Jackson, MJ. Reactive oxygen species: impact on skeletal muscle. Compr Physiol. (2011) 1:941–69. doi: 10.1002/cphy.c100054
34. Mishra, P, and Chan, DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. (2016) 212:379–87. doi: 10.1083/jcb.201511036
35. Vakifahmetoglu-Norberg, H, Ouchida, AT, and Norberg, E. The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun. (2017) 482:426–31. doi: 10.1016/j.bbrc.2016.11.088
36. Wu, T, Liu, H, Xu, R, Li, Z, and Wei, Y. Differences in cellular and molecular processes in exposure to PM2.5 and O3. Environ Int. (2024) 192:109052. doi: 10.1016/j.envint.2024.109052
37. Martínez-Reyes, I, and Chandel, NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. (2020) 11:102. doi: 10.1038/s41467-019-13668-3
38. Vujic, A, Koo, ANM, Prag, HA, and Krieg, T. Mitochondrial redox and TCA cycle metabolite signaling in the heart. Free Radic Biol Med. (2021) 166:287–96. doi: 10.1016/j.freeradbiomed.2021.02.041
39. Burgoyne, JR, Mongue-Din, H, Eaton, P, and Shah, AM. Redox signaling in cardiac physiology and pathology. Circ Res. (2012) 111:1091–106. doi: 10.1161/CIRCRESAHA.111.255216
40. Johannsen, DL, and Ravussin, E. The role of mitochondria in health and disease. Curr Opin Pharmacol. (2009) 9:780–6. doi: 10.1016/j.coph.2009.09.002
41. Romanello, V, and Sandri, M. Mitochondrial quality control and muscle mass maintenance. Front Physiol. (2015) 6:422. doi: 10.3389/fphys.2015.00422
42. Stephenson, EJ, Ragauskas, A, Jaligama, S, Redd, JR, Parvathareddy, J, Peloquin, MJ, et al. Exposure to environmentally persistent free radicals during gestation lowers energy expenditure and impairs skeletal muscle mitochondrial function in adult mice. Am J Physiol Endocrinol Metab. (2016) 310:E1003–15. doi: 10.1152/ajpendo.00521.2015
43. Stremming, J, Chang, EI, Knaub, LA, Armstrong, ML, Baker, PR, Wesolowski, SR, et al. Lower citrate synthase activity, mitochondrial complex expression, and fewer oxidative myofibers characterize skeletal muscle from growth-restricted fetal sheep. Am J Physiol Regul Integr Comp Physiol. (2022) 322:R228–40. doi: 10.1152/ajpregu.00222.2021
44. Picard, M, White, K, and Turnbull, DM. Mitochondrial morphology, topology, and membrane interactions in skeletal muscle: a quantitative three-dimensional electron microscopy study. J Appl Physiol (1985). (2013) 114:161–71. doi: 10.1152/japplphysiol.01096.2012
45. Tilokani, L, Nagashima, S, Paupe, V, and Prudent, J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. (2018) 62:341–60. doi: 10.1042/EBC20170104
46. Giacomello, M, Pyakurel, A, Glytsou, C, and Scorrano, L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol. (2020) 21:204–24. doi: 10.1038/s41580-020-0210-7
47. Noone, J, O’Gorman, DJ, and Kenny, HC. OPA1 regulation of mitochondrial dynamics in skeletal and cardiac muscle. Trends Endocrinol Metab. (2022) 33:710–21. doi: 10.1016/j.tem.2022.07.003
48. Vogel, F, Bornhövd, C, Neupert, W, and Reichert, AS. Dynamic subcompartmentalization of the mitochondrial inner membrane. J Cell Biol. (2006) 175:237–47. doi: 10.1083/jcb.200605138
49. Frey, TG, and Mannella, CA. The internal structure of mitochondria. Trends Biochem Sci. (2000) 25:319–24. doi: 10.1016/s0968-0004(00)01609-1
50. Quintana-Cabrera, R, Mehrotra, A, Rigoni, G, and Soriano, ME. Who and how in the regulation of mitochondrial cristae shape and function. Biochem Biophys Res Commun. (2018) 500:94–101. doi: 10.1016/j.bbrc.2017.04.088
51. Ježek, P, Jabůrek, M, Holendová, B, Engstová, H, and Dlasková, A. Mitochondrial cristae morphology reflecting Metabolism, superoxide formation, redox homeostasis, and pathology. Antioxid Redox Signal. (2023) 39:635–83. doi: 10.1089/ars.2022.0173
52. Osellame, LD, Blacker, TS, and Duchen, MR. Cellular and molecular mechanisms of mitochondrial function. Best Pract Res Clin Endocrinol Metab. (2012) 26:711–23. doi: 10.1016/j.beem.2012.05.003
53. Taguchi, N, Ishihara, N, Jofuku, A, Oka, T, and Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. (2007) 282:11521–9. doi: 10.1074/jbc.M607279200
54. Hall, AR, Burke, N, Dongworth, RK, and Hausenloy, DJ. Mitochondrial fusion and fission proteins: novel therapeutic targets for combating cardiovascular disease. Br J Pharmacol. (2014) 171:1890–906. doi: 10.1111/bph.12516
55. Guo, Y, Zhang, H, Yan, C, Shen, B, Zhang, Y, Guo, X, et al. Small molecule agonist of mitochondrial fusion repairs mitochondrial dysfunction. Nat Chem Biol. (2023) 19:468–77. doi: 10.1038/s41589-022-01224-y
56. Ritenis, EJ, Padilha, CS, Cooke, MB, Stathis, CG, Philp, A, and Camera, DM. The acute and chronic influence of exercise on mitochondrial dynamics in skeletal muscle. Am J Physiol Endocrinol Metab. (2025) 328:E198–209. doi: 10.1152/ajpendo.00311.2024
57. Galluzzi, L, Baehrecke, EH, Ballabio, A, Boya, P, Bravo-San Pedro, JM, Cecconi, F, et al. Molecular definitions of autophagy and related processes. EMBO J. (2017) 36:1811–36. doi: 10.15252/embj.201796697
58. Liu, H, Zhu, S, Han, W, Cai, Y, and Liu, C. DMEP induces mitochondrial damage regulated by inhibiting Nrf2 and SIRT1/PGC-1α signaling pathways in HepG2 cells. Ecotoxicol Environ Saf. (2021) 221:112449. doi: 10.1016/j.ecoenv.2021.112449
59. Toyama, EQ, Herzig, S, Courchet, J, Lewis, TL, Losón, OC, Hellberg, K, et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. (2016) 351:275–81. doi: 10.1126/science.aab4138
60. Scarpulla, RC, Vega, RB, and Kelly, DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. (2012) 23:459–66. doi: 10.1016/j.tem.2012.06.006
61. Wenz, T. Regulation of mitochondrial biogenesis and PGC-1α under cellular stress. Mitochondrion. (2013) 13:134–42. doi: 10.1016/j.mito.2013.01.006
62. Lin, J, Handschin, C, and Spiegelman, BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. (2005) 1:361–70. doi: 10.1016/j.cmet.2005.05.004
63. Kang, C, and Li Ji, L. Role of PGC-1α signaling in skeletal muscle health and disease. Ann N Y Acad Sci. (2012) 1271:110–7. doi: 10.1111/j.1749-6632.2012.06738.x
64. Kang, C, Chung, E, Diffee, G, and Ji, LL. Exercise training attenuates aging-associated mitochondrial dysfunction in rat skeletal muscle: role of PGC-1α. Exp Gerontol. (2013) 48:1343–50. doi: 10.1016/j.exger.2013.08.004
65. Seabright, AP, Fine, NHF, Barlow, JP, Lord, SO, Musa, I, Gray, A, et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. (2020) 34:6284–301. doi: 10.1096/fj.201903051R
66. Chen, J, Zhang, M, Aniagu, S, Jiang, Y, and Chen, T. PM2.5 induces cardiac defects via AHR-SIRT1-PGC-1α mediated mitochondrial damage. Environ Toxicol Pharmacol. (2024) 106:104393. doi: 10.1016/j.etap.2024.104393
67. Yu, SB, and Pekkurnaz, G. Mechanisms orchestrating mitochondrial dynamics for energy homeostasis. J Mol Biol. (2018) 430:3922–41. doi: 10.1016/j.jmb.2018.07.027
68. Youle, RJ, and van der Bliek, AM. Mitochondrial fission, fusion, and stress. Science. (2012) 337:1062–5. doi: 10.1126/science.1219855
69. Griparic, L, van der Wel, NN, Orozco, IJ, Peters, PJ, and van der Bliek, AM. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem. (2004) 279:18792–8. doi: 10.1074/jbc.M400920200
70. Tong, M, Zablocki, D, and Sadoshima, J. The role of Drp1 in mitophagy and cell death in the heart. J Mol Cell Cardiol. (2020) 142:138–45. doi: 10.1016/j.yjmcc.2020.04.015
71. Liu, W, Wang, Z, Kim, M-H, Gu, Y, Sim, H-J, Lee, J-C, et al. Exposure of young mice to atmospherically relevant PM2.5 has sex-dependent long-lasting impacts on the skeletal muscle system. Aging Dis. (2024). doi: 10.14336/AD.2024.1047
72. Fan, D, Pan, K, Guo, J, Liu, Z, Zhang, C, Zhang, J, et al. Exercise ameliorates fine particulate matter-induced metabolic damage through the SIRT1/AMPKα/PGC1-α/NRF1 signaling pathway. Environ Res. (2024) 245:117973. doi: 10.1016/j.envres.2023.117973
73. Wang, R, Han, X, Pang, H, Hu, Z, and Shi, C. Illuminating a time-response mechanism in mice liver after PM2.5 exposure using metabolomics analysis. Sci Total Environ. (2021) 767:144485. doi: 10.1016/j.scitotenv.2020.144485
74. Guo, Z, Hong, Z, Dong, W, Deng, C, Zhao, R, Xu, J, et al. PM2.5-induced oxidative stress and mitochondrial damage in the nasal mucosa of rats. Int J Environ Res Public Health. (2017) 14:134. doi: 10.3390/ijerph14020134
75. Li, R, Kou, X, Geng, H, Xie, J, Yang, Z, Zhang, Y, et al. Effect of ambient PM(2.5) on lung mitochondrial damage and fusion/fission gene expression in rats. Chem Res Toxicol. (2015) 28:408–18. doi: 10.1021/tx5003723
76. Liu, L, Li, Y, Chen, G, and Chen, Q. Crosstalk between mitochondrial biogenesis and mitophagy to maintain mitochondrial homeostasis. J Biomed Sci. (2023) 30:86. doi: 10.1186/s12929-023-00975-7
77. Popov, L-D. Mitochondrial biogenesis: An update. J Cell Mol Med. (2020) 24:4892–9. doi: 10.1111/jcmm.15194
78. Castellani, CA, Longchamps, RJ, Sun, J, Guallar, E, and Arking, DE. Thinking outside the nucleus: mitochondrial DNA copy number in health and disease. Mitochondrion. (2020) 53:214–23. doi: 10.1016/j.mito.2020.06.004
79. Seabright, AP, and Lai, Y-C. Regulatory roles of PINK1-Parkin and AMPK in ubiquitin-dependent skeletal muscle Mitophagy. Front Physiol. (2020) 11:608474. doi: 10.3389/fphys.2020.608474
80. Nguyen, TN, Padman, BS, and Lazarou, M. Deciphering the molecular signals of PINK1/Parkin Mitophagy. Trends Cell Biol. (2016) 26:733–44. doi: 10.1016/j.tcb.2016.05.008
81. Scarffe, LA, Stevens, DA, Dawson, VL, and Dawson, TM. Parkin and PINK1: much more than mitophagy. Trends Neurosci. (2014) 37:315–24. doi: 10.1016/j.tins.2014.03.004
82. An, Z, Liu, G, Shen, L, Qi, Y, Hu, Q, Song, J, et al. Mitochondrial dysfunction induced by ambient fine particulate matter and potential mechanisms. Environ Res. (2024) 262:119930. doi: 10.1016/j.envres.2024.119930
83. Nsiah-Sefaa, A, and McKenzie, M. Combined defects in oxidative phosphorylation and fatty acid β-oxidation in mitochondrial disease. Biosci Rep. (2016) 36:e00313. doi: 10.1042/BSR20150295
84. Houten, SM, and Wanders, RJA. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J Inherit Metab Dis. (2010) 33:469–77. doi: 10.1007/s10545-010-9061-2
85. Houten, SM, Violante, S, Ventura, FV, and Wanders, RJA. The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annu Rev Physiol. (2016) 78:23–44. doi: 10.1146/annurev-physiol-021115-105045
86. Kim, JY, Hickner, RC, Cortright, RL, Dohm, GL, and Houmard, JA. Lipid oxidation is reduced in obese human skeletal muscle. Am J Physiol Endocrinol Metab. (2000) 279:E1039–44. doi: 10.1152/ajpendo.2000.279.5.E1039
87. Xie, W, Zhang, W, Ren, J, Li, W, Zhou, L, Cui, Y, et al. Metabonomics indicates inhibition of fatty acid synthesis, β-oxidation, and tricarboxylic acid cycle in Triclocarban-induced cardiac metabolic alterations in male mice. J Agric Food Chem. (2018) 66:1533–42. doi: 10.1021/acs.jafc.7b05220
88. Minnich, A, Tian, N, Byan, L, and Bilder, G. A potent PPARalpha agonist stimulates mitochondrial fatty acid beta-oxidation in liver and skeletal muscle. Am J Physiol Endocrinol Metab. (2001) 280:E270–9. doi: 10.1152/ajpendo.2001.280.2.E270
89. Adeva-Andany, MM, Carneiro-Freire, N, Seco-Filgueira, M, Fernández-Fernández, C, and Mouriño-Bayolo, D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion. (2019) 46:73–90. doi: 10.1016/j.mito.2018.02.009
90. Shi, L, and Tu, BP. Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr Opin Cell Biol. (2015) 33:125–31. doi: 10.1016/j.ceb.2015.02.003
91. Schäfer, E, Seelert, H, Reifschneider, NH, Krause, F, Dencher, NA, and Vonck, J. Architecture of active mammalian respiratory chain supercomplexes. J Biol Chem. (2006) 281:15370–5. doi: 10.1074/jbc.M513525200
92. Strauss, M, Hofhaus, G, Schröder, RR, and Kühlbrandt, W. Dimer ribbons of ATP synthase shape the inner mitochondrial membrane. EMBO J. (2008) 27:1154–60. doi: 10.1038/emboj.2008.35
93. Zhang, Y, Wang, J, Gong, X, Chen, L, Zhang, B, Wang, Q, et al. Ambient PM2.5 exposures and systemic biomarkers of lipid peroxidation and total antioxidant capacity in early pregnancy. Environ Pollut. (2020) 266:115301. doi: 10.1016/j.envpol.2020.115301
94. Xu, M-X, Ge, C-X, Qin, Y-T, Gu, T-T, Lou, D-S, Li, Q, et al. Prolonged PM2.5 exposure elevates risk of oxidative stress-driven nonalcoholic fatty liver disease by triggering increase of dyslipidemia. Free Radic Biol Med. (2019) 130:542–56. doi: 10.1016/j.freeradbiomed.2018.11.016
95. Zhang, J, Phillips, DIW, Wang, C, and Byrne, CD. Human skeletal muscle PPARalpha expression correlates with fat metabolism gene expression but not BMI or insulin sensitivity. Am J Physiol Endocrinol Metab. (2004) 286:E168–75. doi: 10.1152/ajpendo.00232.2003
96. Yoon, M. The role of PPARalpha in lipid metabolism and obesity: focusing on the effects of estrogen on PPARalpha actions. Pharmacol Res. (2009) 60:151–9. doi: 10.1016/j.phrs.2009.02.004
97. Maher, AC, Mohsen, A-W, Vockley, J, and Tarnopolsky, MA. Low expression of long-chain acyl-CoA dehydrogenase in human skeletal muscle. Mol Genet Metab. (2010) 100:163–7. doi: 10.1016/j.ymgme.2010.03.011
98. van Vlies, N, Tian, L, Overmars, H, Bootsma, AH, Kulik, W, Wanders, RJA, et al. Characterization of carnitine and fatty acid metabolism in the long-chain acyl-CoA dehydrogenase-deficient mouse. Biochem J. (2005) 387:185–93. doi: 10.1042/BJ20041489
99. Zhang, D, Liu, Z-X, Choi, CS, Tian, L, Kibbey, R, Dong, J, et al. Mitochondrial dysfunction due to long-chain acyl-CoA dehydrogenase deficiency causes hepatic steatosis and hepatic insulin resistance. Proc Natl Acad Sci USA. (2007) 104:17075–80. doi: 10.1073/pnas.0707060104
100. Ning, R, Shi, Y, Jiang, J, Liang, S, Xu, Q, Duan, J, et al. Mitochondrial dysfunction drives persistent vascular fibrosis in rats after short-term exposure of PM2.5. Sci Total Environ. (2020) 733:139135. doi: 10.1016/j.scitotenv.2020.139135
101. Saha, AK, and Ruderman, NB. Malonyl-CoA and AMP-activated protein kinase: an expanding partnership. Mol Cell Biochem. (2003) 253:65–70. doi: 10.1023/a:1026053302036
102. Jin, X, Xue, B, Ahmed, RZ, Ding, G, and Li, Z. Fine particles cause the abnormality of cardiac ATP levels via PPARɑ-mediated utilization of fatty acid and glucose using in vivo and in vitro models. Environ Pollut. (2019) 249:286–94. doi: 10.1016/j.envpol.2019.02.083
103. Cao, K, Xu, J, Cao, W, Wang, X, Lv, W, Zeng, M, et al. Assembly of mitochondrial succinate dehydrogenase in human health and disease. Free Radic Biol Med. (2023) 207:247–59. doi: 10.1016/j.freeradbiomed.2023.07.023
104. Gao, R, Ku, T, Ji, X, Zhang, Y, Li, G, and Sang, N. Abnormal energy metabolism and tau phosphorylation in the brains of middle-aged mice in response to atmospheric PM2.5 exposure. J Environ Sci (China). (2017) 62:145–53. doi: 10.1016/j.jes.2017.06.037
105. Meyer, JN, Leung, MCK, Rooney, JP, Sendoel, A, Hengartner, MO, Kisby, GE, et al. Mitochondria as a target of environmental toxicants. Toxicol Sci. (2013) 134:1–17. doi: 10.1093/toxsci/kft102
106. Vincent, AE, Ng, YS, White, K, Davey, T, Mannella, C, Falkous, G, et al. The spectrum of mitochondrial ultrastructural defects in mitochondrial myopathy. Sci Rep. (2016) 6:30610. doi: 10.1038/srep30610
107. Lanza, IR, and Nair, KS. Mitochondrial function as a determinant of life span. Pflugers Arch. (2010) 459:277–89. doi: 10.1007/s00424-009-0724-5
108. Lanza, IR, and Sreekumaran Nair, K. Regulation of skeletal muscle mitochondrial function: genes to proteins. Acta Physiol Oxf. (2010) 199:529–47. doi: 10.1111/j.1748-1716.2010.02124.x
109. Yu, T, Dohl, J, Elenberg, F, Chen, Y, and Deuster, P. Curcumin induces concentration-dependent alterations in mitochondrial function through ROS in C2C12 mouse myoblasts. J Cell Physiol. (2019) 234:6371–81. doi: 10.1002/jcp.27370
110. Brand, MD, and Nicholls, DG. Assessing mitochondrial dysfunction in cells. Biochem J. (2011) 435:297–312. doi: 10.1042/BJ20110162
111. Nicholls, DG. Mitochondrial membrane potential and aging. Aging Cell. (2004) 3:35–40. doi: 10.1111/j.1474-9728.2003.00079.x
112. Nickel, A, Kohlhaas, M, and Maack, C. Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol. (2014) 73:26–33. doi: 10.1016/j.yjmcc.2014.03.011
113. Murphy, MP. How mitochondria produce reactive oxygen species. Biochem J. (2009) 417:1–13. doi: 10.1042/BJ20081386
114. Kushnareva, Y, Murphy, AN, and Andreyev, A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J. (2002) 368:545–53. doi: 10.1042/BJ20021121
115. Chen, Q, Vazquez, EJ, Moghaddas, S, Hoppel, CL, and Lesnefsky, EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. (2003) 278:36027–31. doi: 10.1074/jbc.M304854200
116. Brand, MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med. (2016) 100:14–31. doi: 10.1016/j.freeradbiomed.2016.04.001
117. Ribas, V, García-Ruiz, C, and Fernández-Checa, JC. Glutathione and mitochondria. Front Pharmacol. (2014) 5:151. doi: 10.3389/fphar.2014.00151
118. Handy, DE, Lubos, E, Yang, Y, Galbraith, JD, Kelly, N, Zhang, Y-Y, et al. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J Biol Chem. (2009) 284:11913–21. doi: 10.1074/jbc.M900392200
119. de Vries, HE, Witte, M, Hondius, D, Rozemuller, AJM, Drukarch, B, Hoozemans, J, et al. Nrf2-induced antioxidant protection: a promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic Biol Med. (2008) 45:1375–83. doi: 10.1016/j.freeradbiomed.2008.09.001
120. Sies, H, Berndt, C, and Jones, DP. Oxidative stress. Annu Rev Biochem. (2017) 86:715–48. doi: 10.1146/annurev-biochem-061516-045037
121. Bonnard, C, Durand, A, Peyrol, S, Chanseaume, E, Chauvin, M-A, Morio, B, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. (2008) 118:789–800. doi: 10.1172/JCI32601
122. Tsikas, D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: analytical and biological challenges. Anal Biochem. (2017) 524:13–30. doi: 10.1016/j.ab.2016.10.021
123. Zhang, Z, Su, H, Ahmed, RZ, Zheng, Y, and Jin, X. Critical biomarkers for myocardial damage by fine particulate matter: focused on PPARα-regulated energy metabolism. Environ Pollut. (2020) 264:114659. doi: 10.1016/j.envpol.2020.114659
124. Cheng, J, Wang, F, Yu, D-F, Wu, P-F, and Chen, J-G. The cytotoxic mechanism of malondialdehyde and protective effect of carnosine via protein cross-linking/mitochondrial dysfunction/reactive oxygen species/MAPK pathway in neurons. Eur J Pharmacol. (2011) 650:184–94. doi: 10.1016/j.ejphar.2010.09.033
125. Ribeiro, JP, Kalb, AC, Campos, PP, Cruz, A, Martinez, PE, Gioda, A, et al. Toxicological effects of particulate matter (PM2.5) on rats: bioaccumulation, antioxidant alterations, lipid damage, and ABC transporter activity. Chemosphere. (2016) 163:569–77. doi: 10.1016/j.chemosphere.2016.07.094
126. Park, J, Jang, J, So, B, Lee, K, Yeom, D, Zhang, Z, et al. Effects of particulate matter inhalation during exercise on oxidative stress and mitochondrial function in mouse skeletal muscle. Antioxidants (Basel). (2024) 13:113. doi: 10.3390/antiox13010113
127. Balaban, RS, Nemoto, S, and Finkel, T. Mitochondria, oxidants, and aging. Cell. (2005) 120:483–95. doi: 10.1016/j.cell.2005.02.001
128. Fan, X, Dong, T, Yan, K, Ci, X, and Peng, L. PM2.5 increases susceptibility to acute exacerbation of COPD via NOX4/Nrf2 redox imbalance-mediated mitophagy. Redox Biol. (2023) 59:102587. doi: 10.1016/j.redox.2022.102587
129. Leclercq, B, Kluza, J, Antherieu, S, Sotty, J, Alleman, LY, Perdrix, E, et al. Air pollution-derived PM2.5 impairs mitochondrial function in healthy and chronic obstructive pulmonary diseased human bronchial epithelial cells. Environ Pollut. (2018) 243:1434–49. doi: 10.1016/j.envpol.2018.09.062
130. Wang, Y, Kong, L, Wu, T, and Tang, M. Urban particulate matter disturbs the equilibrium of mitochondrial dynamics and biogenesis in human vascular endothelial cells. Environ Pollut. (2020) 264:114639. doi: 10.1016/j.envpol.2020.114639
131. Leek, BT, Mudaliar, SR, Henry, R, Mathieu-Costello, O, and Richardson, RS. Effect of acute exercise on citrate synthase activity in untrained and trained human skeletal muscle. Am J Physiol Regul Integr Comp Physiol. (2001) 280:R441–7. doi: 10.1152/ajpregu.2001.280.2.R441
132. Zhao, R-Z, Jiang, S, Zhang, L, and Yu, Z-B. Mitochondrial electron transport chain, ROS generation and uncoupling (review). Int J Mol Med. (2019) 44:3–15. doi: 10.3892/ijmm.2019.4188
133. Letts, JA, and Sazanov, LA. Clarifying the supercomplex: the higher-order organization of the mitochondrial electron transport chain. Nat Struct Mol Biol. (2017) 24:800–8. doi: 10.1038/nsmb.3460
134. Wibom, R, Hagenfeldt, L, and von Döbeln, U. Measurement of ATP production and respiratory chain enzyme activities in mitochondria isolated from small muscle biopsy samples. Anal Biochem. (2002) 311:139–51. doi: 10.1016/s0003-2697(02)00424-4
135. Julienne, CM, Dumas, J-F, Goupille, C, Pinault, M, Berri, C, Collin, A, et al. Cancer cachexia is associated with a decrease in skeletal muscle mitochondrial oxidative capacities without alteration of ATP production efficiency. J Cachexia Sarcopenia Muscle. (2012) 3:265–75. doi: 10.1007/s13539-012-0071-9
136. Powers, SK, Kavazis, AN, and McClung, JM. Oxidative stress and disuse muscle atrophy. J Appl Physiol. (2007) 102:2389–97. doi: 10.1152/japplphysiol.01202.2006
137. Billet, S, Landkocz, Y, Martin, PJ, Verdin, A, Ledoux, F, Lepers, C, et al. Chemical characterization of fine and ultrafine PM, direct and indirect genotoxicity of PM and their organic extracts on pulmonary cells. J Environ Sci (China). (2018) 71:168–78. doi: 10.1016/j.jes.2018.04.022
138. Lıu, X, Deng, K, Chen, S, Zhang, Y, Yao, J, Weng, X, et al. 8-hydroxy-2′-deoxyguanosine as a biomarker of oxidative stress in acute exacerbation of chronic obstructive pulmonary disease. Turk J Med Sci. (2019) 49:93–100. doi: 10.3906/sag-1807-106
139. Gai, H-F, An, J-X, Qian, X-Y, Wei, Y-J, Williams, JP, and Gao, G-L. Ovarian damages produced by aerosolized fine particulate matter (PM2.5) pollution in mice: possible protective medications and mechanisms. Chin Med J. (2017) 130:1400–10. doi: 10.4103/0366-6999.207472
140. Cho, S-Y, and Roh, H-T. Impact of particulate matter exposure and aerobic exercise on circulating biomarkers of oxidative stress, antioxidant status, and inflammation in young and aged mice. Life (Basel). (2023) 13:1952. doi: 10.3390/life13101952
141. Bua, E, Johnson, J, Herbst, A, Delong, B, McKenzie, D, Salamat, S, et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. (2006) 79:469–80. doi: 10.1086/507132
142. Figueiredo, PA, Mota, MP, Appell, HJ, and Duarte, JA. The role of mitochondria in aging of skeletal muscle. Biogerontology. (2008) 9:67–84. doi: 10.1007/s10522-007-9121-7
143. Bou Saada, Y, Zakharova, V, Chernyak, B, Dib, C, Carnac, G, Dokudovskaya, S, et al. Control of DNA integrity in skeletal muscle under physiological and pathological conditions. Cell Mol Life Sci. (2017) 74:3439–49. doi: 10.1007/s00018-017-2530-0
144. Simonatto, M, Marullo, F, Chiacchiera, F, Musaró, A, Wang, JYJ, Latella, L, et al. DNA damage-activated ABL-MyoD signaling contributes to DNA repair in skeletal myoblasts. Cell Death Differ. (2013) 20:1664–74. doi: 10.1038/cdd.2013.118
145. Faleiro, I, Afonso, AI, Balsinha, A, Lucas, B, Martin, RM, Gomes, ER, et al. Adaptive changes in the DNA damage response during skeletal muscle cell differentiation. Front Cell Dev Biol. (2023) 11:1239138. doi: 10.3389/fcell.2023.1239138
146. Chen, C-H, Huang, L-Y, Lee, K-Y, Wu, C-D, Chiang, H-C, Chen, B-Y, et al. Effects of PM2.5 on skeletal muscle mass and body fat mass of the elderly in Taipei, Taiwan. Sci Rep. (2019) 9:11176. doi: 10.1038/s41598-019-47576-9
147. Zhang, F, Li, T, Chen, B, Li, N, Zhang, X, Zhu, S, et al. Air pollution weaken your muscle? Evidence from a cross-sectional study on sarcopenia in Central China. Ecotoxicol Environ Saf. (2023) 258:114962. doi: 10.1016/j.ecoenv.2023.114962
148. Lin, H, Guo, Y, Ruan, Z, Kowal, P, Di, Q, Zheng, Y, et al. Association of indoor and outdoor air pollution with hand-grip strength among adults in six low- and middle-income countries. J Gerontol A Biol Sci Med Sci. (2020) 75:340–7. doi: 10.1093/gerona/glz038
149. Flynn, JM, Meadows, E, Fiorotto, M, and Klein, WH. Myogenin regulates exercise capacity and skeletal muscle metabolism in the adult mouse. PLoS One. (2010) 5:e13535. doi: 10.1371/journal.pone.0013535
150. Zammit, PS. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin Cell Dev Biol. (2017) 72:19–32. doi: 10.1016/j.semcdb.2017.11.011
151. Yokoyama, S, Ohno, Y, Egawa, T, Ohashi, K, Ito, R, Ortuste Quiroga, HP, et al. MBNL1-associated mitochondrial dysfunction and apoptosis in C2C12 myotubes and mouse skeletal muscle. Int J Mol Sci. (2020) 21:6376. doi: 10.3390/ijms21176376
152. Bonaldo, P, and Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. (2013) 6:25–39. doi: 10.1242/dmm.010389
153. Yin, L, Li, N, Jia, W, Wang, N, Liang, M, Yang, X, et al. Skeletal muscle atrophy: from mechanisms to treatments. Pharmacol Res. (2021) 172:105807. doi: 10.1016/j.phrs.2021.105807
154. Zammit, PS, Relaix, F, Nagata, Y, Ruiz, AP, Collins, CA, Partridge, TA, et al. Pax7 and myogenic progression in skeletal muscle satellite cells. J Cell Sci. (2006) 119:1824–32. doi: 10.1242/jcs.02908
155. Richter, EA, and Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol Rev. (2013) 93:993–1017. doi: 10.1152/physrev.00038.2012
156. Kelley, DE, Mokan, M, Simoneau, JA, and Mandarino, LJ. Interaction between glucose and free fatty acid metabolism in human skeletal muscle. J Clin Invest. (1993) 92:91–8. doi: 10.1172/JCI116603
157. Ke, R, Xu, Q, Li, C, Luo, L, and Huang, D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol Int. (2018) 42:384–92. doi: 10.1002/cbin.10915
158. Doenst, T, Nguyen, TD, and Abel, ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. (2013) 113:709–24. doi: 10.1161/CIRCRESAHA.113.300376
159. Tsao, TS, Li, J, Chang, KS, Stenbit, AE, Galuska, D, Anderson, JE, et al. Metabolic adaptations in skeletal muscle overexpressing GLUT4: effects on muscle and physical activity. FASEB J. (2001) 15:958–69. doi: 10.1096/fj.00-0381
160. Tan, VP, and Miyamoto, S. HK2/hexokinase-II integrates glycolysis and autophagy to confer cellular protection. Autophagy. (2015) 11:963–4. doi: 10.1080/15548627.2015.1042195
161. Liu, C, Xu, X, Bai, Y, Wang, T-Y, Rao, X, Wang, A, et al. Air pollution-mediated susceptibility to inflammation and insulin resistance: influence of CCR2 pathways in mice. Environ Health Perspect. (2014) 122:17–26. doi: 10.1289/ehp.1306841
162. Govers, R. Cellular regulation of glucose uptake by glucose transporter GLUT4. Adv Clin Chem. (2014) 66:173–240. doi: 10.1016/b978-0-12-801401-1.00006-2
163. Doehner, W, Gathercole, D, Cicoira, M, Krack, A, Coats, AJS, Camici, PG, et al. Reduced glucose transporter GLUT4 in skeletal muscle predicts insulin resistance in non-diabetic chronic heart failure patients independently of body composition. Int J Cardiol. (2010) 138:19–24. doi: 10.1016/j.ijcard.2008.07.004
164. Kiens, B. Skeletal muscle lipid metabolism in exercise and insulin resistance. Physiol Rev. (2006) 86:205–43. doi: 10.1152/physrev.00023.2004
165. Yu, J, Zheng, J, Liu, XF, Feng, ZL, Zhang, XP, Cao, LL, et al. Exercise improved lipid metabolism and insulin sensitivity in rats fed a high-fat diet by regulating glucose transporter 4 (GLUT4) and musclin expression. Braz J Med Biol Res. (2016) 49:e5129. doi: 10.1590/1414-431X20165129
166. Qin, F, Cui, S, Dong, Y, Xu, M, Wang, Z, Qu, C, et al. Aerobic exercise ameliorates particulate matter-induced lung injury in aging rats. Environ Pollut. (2021) 280:116889. doi: 10.1016/j.envpol.2021.116889
167. So, B, Park, J, Jang, J, Lim, W, Imdad, S, and Kang, C. Effect of aerobic exercise on oxidative stress and inflammatory response during particulate matter exposure in mouse lungs. Front Physiol. (2021) 12:773539. doi: 10.3389/fphys.2021.773539
168. Qin, F, Xu, M-X, Wang, Z-W, Han, Z-N, Dong, Y-N, and Zhao, J-X. Effect of aerobic exercise and different levels of fine particulate matter (PM2.5) on pulmonary response in Wistar rats. Life Sci. (2020) 254:117355. doi: 10.1016/j.lfs.2020.117355
169. Marmett, B, Nunes, RB, de Souza, KS, Lago, PD, and Rhoden, CR. Aerobic training reduces oxidative stress in skeletal muscle of rats exposed to air pollution and supplemented with chromium picolinate. Redox Rep. (2018) 23:146–52. doi: 10.1080/13510002.2018.1475993
170. Zhang, Z, Hoek, G, Chang, L-Y, Chan, T-C, Guo, C, Chuang, YC, et al. Particulate matter air pollution, physical activity and systemic inflammation in Taiwanese adults. Int J Hyg Environ Health. (2018) 221:41–7. doi: 10.1016/j.ijheh.2017.10.001
171. Dent, JR, Stocks, B, Campelj, DG, and Philp, A. Transient changes to metabolic homeostasis initiate mitochondrial adaptation to endurance exercise. Semin Cell Dev Biol. (2023) 143:3–16. doi: 10.1016/j.semcdb.2022.03.022
172. Zheng, L, Rao, Z, Wu, J, Ma, X, Jiang, Z, and Xiao, W. Resistance exercise improves glycolipid metabolism and mitochondrial biogenesis in skeletal muscle of T2DM mice via miR-30d-5p/SIRT1/PGC-1α axis. Int J Mol Sci. (2024) 25:12416. doi: 10.3390/ijms252212416
173. Burd, NA, Mitchell, CJ, Churchward-Venne, TA, and Phillips, SM. Bigger weights may not beget bigger muscles: evidence from acute muscle protein synthetic responses after resistance exercise. Appl Physiol Nutr Metab. (2012) 37:551–4. doi: 10.1139/h2012-022
174. Thirupathi, A, Wang, M, Lin, JK, Fekete, G, István, B, Baker, JS, et al. Effect of different exercise modalities on oxidative stress: a systematic review. Biomed Res Int. (2021) 2021:1947928. doi: 10.1155/2021/1947928
175. Hashimoto, T, and Brooks, GA. Mitochondrial lactate oxidation complex and an adaptive role for lactate production. Med Sci Sports Exerc. (2008) 40:486–94. doi: 10.1249/MSS.0b013e31815fcb04
176. Qaisar, R, Bhaskaran, S, and Van Remmen, H. Muscle fiber type diversification during exercise and regeneration. Free Radic Biol Med. (2016) 98:56–67. doi: 10.1016/j.freeradbiomed.2016.03.025
177. Pisoschi, AM, and Pop, A. The role of antioxidants in the chemistry of oxidative stress: a review. Eur J Med Chem. (2015) 97:55–74. doi: 10.1016/j.ejmech.2015.04.040
178. Ning, R, Li, Y, Du, Z, Li, T, Sun, Q, Lin, L, et al. The mitochondria-targeted antioxidant MitoQ attenuated PM2.5-induced vascular fibrosis via regulating mitophagy. Redox Biol. (2021) 46:102113. doi: 10.1016/j.redox.2021.102113
179. Jong, CJ, Sandal, P, and Schaffer, SW. The role of taurine in mitochondria health: more than just an antioxidant. Molecules. (2021) 26:4913. doi: 10.3390/molecules26164913
180. Bo, L, Jiang, S, Xie, Y, Kan, H, Song, W, and Zhao, J. Effect of vitamin E and omega-3 fatty acids on protecting ambient PM2.5-induced inflammatory response and oxidative stress in vascular endothelial cells. PLoS One. (2016) 11:e0152216. doi: 10.1371/journal.pone.0152216
181. Xu, X, Liu, C, Xu, Z, Tzan, K, Zhong, M, Wang, A, et al. Long-term exposure to ambient fine particulate pollution induces insulin resistance and mitochondrial alteration in adipose tissue. Toxicol Sci. (2011) 124:88–98. doi: 10.1093/toxsci/kfr211
Keywords: particulate matter, skeletal muscle, mitochondrial function, exercise, fine dust
Citation: Ding Y, Wan Q and Liu W (2025) Effects of atmospherically relevant PM2.5 on skeletal muscle mitochondria: a review of damage mechanisms and potential of exercise interventions. Front. Public Health. 13:1615363. doi: 10.3389/fpubh.2025.1615363
Edited by:
Tong Wang, Duke University, United StatesReviewed by:
Xue Wu, University of California, San Francisco, United StatesLin Liu, Tianjin Medical University Eye Hospital, China
Yilin Han, Eli Lilly, United States
Kexin Zhang, University of Massachusetts Medical School, United States
Copyright © 2025 Ding, Wan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenduo Liu, bHdkMTIwNEBqYm51LmFjLmty
†These authors have contributed equally to this work