Abstract
Smoking and particulate matter 2.5 (PM2.5) expose millions to cadmium (Cd), a toxic heavy metal linked to pro-inflammatory responses, oxidative stress, and disease pathogenesis. In the oral cavity, chronic Cd exposure contributes to the progression of periodontal diseases and oral cancers. However, the direct effect of Cd on oral tissues and the underlying mechanisms remains unclear. This study explored the impact of environmentally relevant concentrations of Cd on human gingival fibroblasts (HGFs) by evaluating cell viability, pro-inflammatory cytokine secretion (IL-6 and IL-8), COX-2 expression, and the activation of key signaling pathways: Akt, ERK1/2, and JNK. Cd exposure significantly reduced HGF viability, elevated IL-6 and IL-8 secretion, and upregulated COX-2 expression. These effects were attenuated by inhibitors targeting Akt, ERK1/2, and JNK pathways. By integrating cytokine profiling, COX-2 expression, and inhibitor-based pathway analysis, our study provides mechanistic insights into how low-level Cd exposure triggers early inflammatory responses in gingival fibroblasts. Our findings reveal that Cd exerts pro-inflammatory and cytotoxic effects on HGFs, which may play a role as one of the factors in the pathogenesis of smoking-related oral diseases. Targeting Akt, ERK1/2, and JNK signaling pathways could offer therapeutic strategies to attenuate Cd-induced oral pro-inflammatory responses and tissue damage.

Introduction
Smoking increases cadmium (Cd) levels, a toxic metal found in tobacco smoke at approximately 0.08 – 5.4 μg/g tobacco (Fresquez et al., 2013; Dinh et al., 2021; Richard et al., 2010). Salivary Cd is significantly higher in waterpipe and long-term smokers, reaching up to 352.72 μM which was also comparable to levels in plasma (Khabour et al., 2018; Bertuzzo, 2003). Cd levels in saliva and urine are proposed as indicators of smoking addiction, correlating with cigarette consumption (Talio et al., 2010). Salivary Cd levels were 0.03 μM in subjects smoking 3-5 cigarettes daily and 0.08-0.11 μM in those smoking 20-40 cigarettes daily, while non-smokers had levels around 0.006 μM. Cd is a known component of particulate matter 2.5 (PM2.5), linking airborne particulate to heavy metal contamination. A study showed that Tobacco smoke increases indoor PM2.5 and Cd levels in smoker households (Ruggieri et al., 2014). Moreover, prolonged exposure to these fine particles may damage oral tissues and promote dental caries (Zhu et al., 2024).
Interleukins (ILs) are key mediators of oral inflammation. IL-6 is associated with chronic inflammation, contributing to periodontal damage, periodontitis, Sjögren's syndrome, and oral squamous cell carcinoma (OSCC) (Nibali et al., 2012; Noh et al., 2013; Gasche et al., 2011). IL-8 is elevated in the gingival tissue of periodontitis patients, and both IL-6 and IL-8 are potential biomarkers for oral and oropharyngeal squamous cell carcinoma (Sahibzada et al., 2017; St John et al., 2004; Finoti et al., 2017). Smoking increases IL-6 and IL-8 levels in cells, animals, and humans, with smokers showing significantly higher levels in saliva compared to non-smokers (Hussein et al., 2020). Cd exposure has been shown to trigger IL-6 and IL-8 expression and secretion via MAPK and NF-κB pathways in neutrophils, macrophages, epithelial, and astrocytoma cells (Hossein-Khannazer et al., 2020; Olszowski et al., 2012; Phuagkhaopong et al., 2017), highlighting its role in inflammation.
COX-2 upregulation drives inflammation. In periodontitis, its increased expression correlates with alveolar bone loss in rat models (Li et al., 2021). Cd induces COX-2 in a dose-dependent manner in human gallbladder cells and activates the p38 MAPK pathway in brain endothelial cells (Sharma et al., 2020; Seok et al., 2006). These findings highlight the underlying mechanisms of Cd on COX-2 expression and its role in inflammatory responses.
While nicotine is widely recognized for its role in gum inflammation and periodontal disease, the contribution of Cd remains less defined despite the oral cavity being a primary site of Cd exposure through tobacco smoke and PM2.5. To address this gap, we investigated how Cd induces pro-inflammatory responses in human gingival fibroblasts (HGFs), focusing on cytokine secretion and activation of MAPK pathways. Unlike earlier studies that examined cytotoxicity, pro-inflammatory responses, or signaling in isolation (Issa et al., 2008; Yang et al., 2025; Chmielowska-Bąk and Deckert, 2012; Chen et al., 2022), our work integrates these aspects using environmentally relevant Cd concentrations. This approach provides clearer insight into the pro-inflammatory effects of Cd in the oral environment and their relevance to smoking-related and environmental health risks.
Materials and methods
Reagents
HGF cells obtained from ATCC CRL-2014, USA were used in this study. Culture media and supplements including Dulbecco’s Modified Eagle Medium (DMEM), fetal bovine serum (FBS), penicillin, streptomycin, and L-glutamine were purchased from Sigma-Aldrich (USA), Invitrogen (USA), and Gibco (USA). Cadmium chloride (CdCl2), MTT reagent were purchased from Sigma-Aldrich (USA), while dimethyl sulfoxide (DMSO) was obtained from LGC (UK). Specific pathway inhibitors: LY294002 (Akt), U0126 (ERK1/2), SB203580 (p38), and SP600125 (JNK) were purchased from Calbiochem (USA), Cell Signaling Technology (USA), Tocris (UK), and Sigma-Aldrich (USA), respectively. ELISA kits for IL-6 and IL-8 were from eBioscience (USA). Primary antibodies for signaling proteins and COX-2 were obtained from Cell Signaling Technology (USA) and Clarity ECL western blot substrate kits purchased from Bio-Rad (USA) were used as chemiluminescent reagents.
Cell culture
HGF cells at passages 3–10 were maintained in complete DMEM, with media refreshed every 2 days. Cells were incubated at 37°C in a humidified atmosphere with 5% CO2 until reaching 90% confluence.
A schematic overview of the experimental workflow is provided in Graphical Abstract for clarity.
Cell viability test
HGF cells (1x104 cells) were cultured in a 96-well plate for 24 h and treated with CdCl2 (0, 0.1, 1, 3, 6, 10, and 100 μM) for 24 h. CdCl2 was selected as the Cd source due to its stability and water solubility, providing a consistent release of Cd2+ ions which is suitable for in vitro toxicological studies. A 1000 mM stock solution was prepared in sterile water and stored at −20°C until use. Cell viability was assessed using the MTT assay (0.5 mg/ml for 2 h at 37°C) followed by DMSO to dissolve crystals. Absorbance was measured by a microplate reader (Biotek Instruments, CA, United States) at 570 nm (background 690 nm). DMEM without CdCl2 served as the control (100% viability). In addition, HGFs were co-treated with CdCl2 and inhibitors dissolved in DMSO (LY294002: Akt inhibitor, U0126: ERK1/2 inhibitor, SB203580: P38 inbibitor, and SP600125: JNK inhibitor) and cell viability was determined at 24 h.
Western blot analysis
HGF cells were cultured in 60-mm dishes and treated with 1 μM CdCl2 to measure Akt, ERK1/2, p38, and JNK with or without their specific inhibitors. 1 µM Cd was selected as it represented the highest concentration that did not cause cell death in our experiment. Protein lysates were collected and stored at −80°C. For western blotting, 20 μg protein lysates were separated using SDS-PAGE and transferred to a nitrocellulose membrane. The transferred membranes were blotted for phospho-Akt, phospho-Erk, phospho-p38, phospho-JNK, total-Akt, total-Erk, total-p38, total-JNK, β-actin, β-tubulin, or GAPDH. Additionally, COX-2 expression was measured after 1-hour CdCl2 treatment, with or without inhibitors. COX-2 antibody and GAPDH were detected. Antibodies were from Cell Signaling, USA, and chemiluminescent signals were visualized using the Amersham ImageQuant 800 Fluor visualizer (Cytiva, United States), with densitometry quantified by ImageJ.
Measurement of IL-6 and IL-8 by ELISA
HGF cells (1x104 cells) seeded in 96-well plates were co-treated with 1 μM CdCl2 and their specific inhibitors for 24 h. The supernatants were collected and centrifuged for subsequent ELISA analysis. The ELISA procedure was performed according to the manufacturer’s instructions. Briefly, 96-well plates were coated overnight with IL-6 or IL-8 capture antibodies. After washing and blocking the plates, standards or supernatants were added for 2 h, followed by detection antibodies for 1 h. HRP-labeled antibodies were incubated for 30 min. After washing, substrate was added, and the reaction stopped with 1 M sulfuric acid. Optical density was measured at 450 nm (background at 570 nm), and cytokine levels were interpolated from standard curves.
Statistical analysis
Statistical analyses were performed using Prism® version 10.3.1 (Prism Software Inc., United States). Data are presented as mean ± SEM. The IC50 of CdCl2 was determined using nonlinear regression. One-way ANOVA with Dunnett’s test was applied to evaluate cell viability (MTT assay) and IL-6 and IL-8 secretion (ELISA). For western blot analysis, one-way ANOVA with Tukey’s test was employed. A p-value of ≤ 0.05 was considered significant.
Results
Cd was cytotoxic to HGF cells
CdCl2 reduced HGF viability in a concentration-dependent manner, with significant cytotoxicity at ≥ 6 μM. The IC50 was 5.883 μM (Figure 1A). Since 1 μM CdCl2 was not cytotoxic, it was used in subsequent pro-inflammatory responses experiments. Co-treatment with inhibitors at 1 μM CdCl2 showed no significant effect on cell viability (Figure 1B). Although some values slightly exceeded 100%, they were not statistically significant compared to controls and likely reflect normal experimental variability.
FIGURE 1
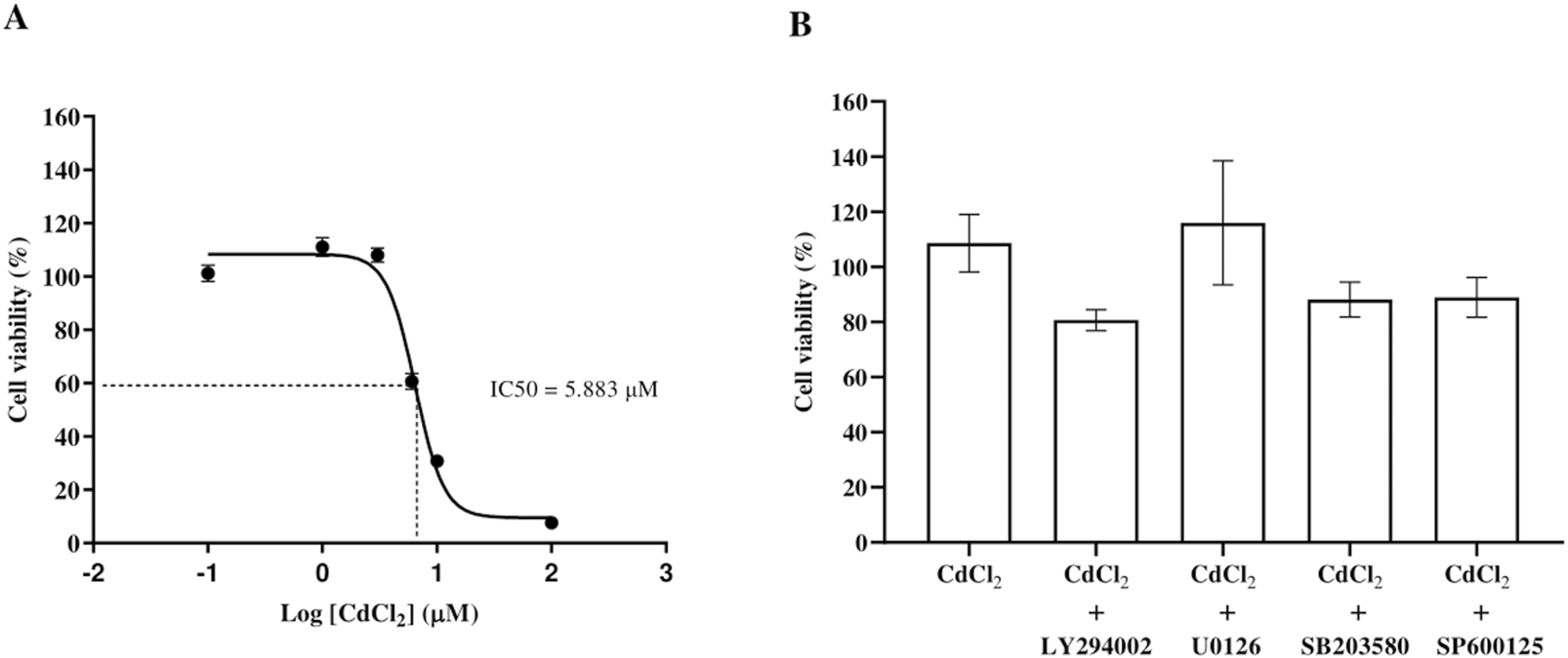
Dose-response curve of CdCl2-induced cytotoxicity in HGF and cell viability of HGFs following exposure to Cd and inhibitors. Cell viability was assessed after exposure to increasing concentrations of CdCl2 (0, 0.1, 1, 3, 6, 10, and 100 μM) for 24 h using MTT assays. The IC50 value, representing the concentration of CdCl2 required to reduce cell viability by 50%, was determined from the dose-response curve (A). The viability of HGFs was also assessed after treatment with 1 µM CdCl2 alone or in combination with specific pathway inhibitors: 10 µM LY294002 (Akt), 25 µM U0126 (ERK1/2), 25 µM SB203580 (p38), and 25 µM SP600125 (JNK) (B). HGFs cultured in control (DMEM) without treatment were the control group representing 100% cell viability. Data are shown as mean ± SEM (n = 3).
Cd activated Akt and MAPK pathways in HGFs
To study the pathways involved in Cd-induced effects on HGFs, we treated the cells with 1 μM CdCl2, alone or in combination with their specific pathway inhibitors. The treatment with 1 μM CdCl2 significantly increased the phosphorylation of Akt, ERK1/2, and JNK (Figure 2). Their specific inhibitors effectively blocked the activation of phosphorylated Akt, ERK1/2, and JNK (LY294002, U0126, SP600125, respectively). However, CdCl2 did not increase phosphorylated p38 levels, and treatment with SB203580 did not affect these levels.
FIGURE 2
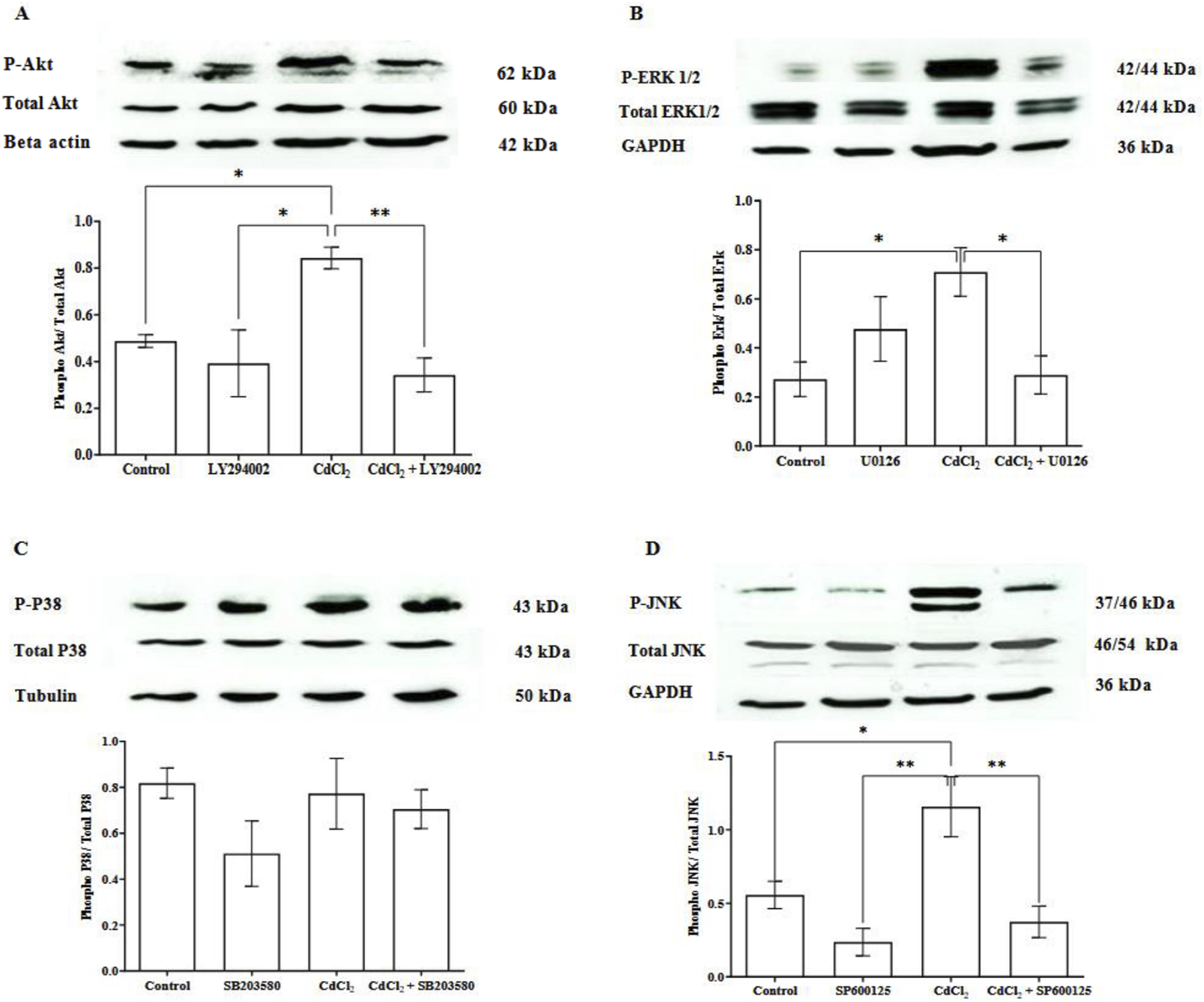
CdCl2-induced Akt, ERK 1/2, and JNK expression activation in cultured HGFs. HGF cells were incubated with 1 μM CdCl2 or co-treatment of 1 μM CdCl2 and each inhibitor: 10 µM LY294002 (Akt), 25 µM U0126 (ERK1/2), 25 µM SB203580 (p38), and 25 µM SP600125 (JNK). HGF cells were also treated with the inhibitor alone to determine its effect. Control refers to cells cultured in DMEM without CdCl2 treatment. Cells were incubated for 5 min for phosphorylated Akt measurement and 30 min for phosphorylated ERK1/2, p38, and JNK measurement. Levels of phospho-Akt, total Akt (A), phospho-ERK 1/2, ERK (B), phospho-p38 MAPK, p38 MAPK (C), and phospho-JNK, JNK (D) were investigated by Western blotting. Beta actin, GAPDH, and tubulin were used as housekeeping genes based on molecular weights of proteins of interest. Data are shown as mean ± SEM (n = 4). Statistical significance was determined using One-way ANOVA followed by Tukey’s multiple comparisons test represented by *P ≤ 0.05, **P ≤ 0.01.
Cd induced IL-6 and IL-8 secretion from HGFs which were decreased by Akt and MAPK inhibitors
To demonstrate the pro-inflammatory effects of Cd on HGFs, we measured IL-6 and IL-8 levels following Cd exposure. Our results revealed that CdCl2 (1 μM) inceased IL-6 and IL-8 secretion after 24 hours of incubation (Figure 3). Based on our previous results, 1 μM CdCl2 did not activate p38 MAPK pathway, so we excluded p38 inhibitor in this experiment. The results showed that all tested inhibitors: 10 µM LY294002 (Akt), 25 µM U0126 (ERK1/2), and 25 µM SP600125 (JNK) decreased both IL-6 and IL-8 levels induced by CdCl2 in HGFs.
FIGURE 3
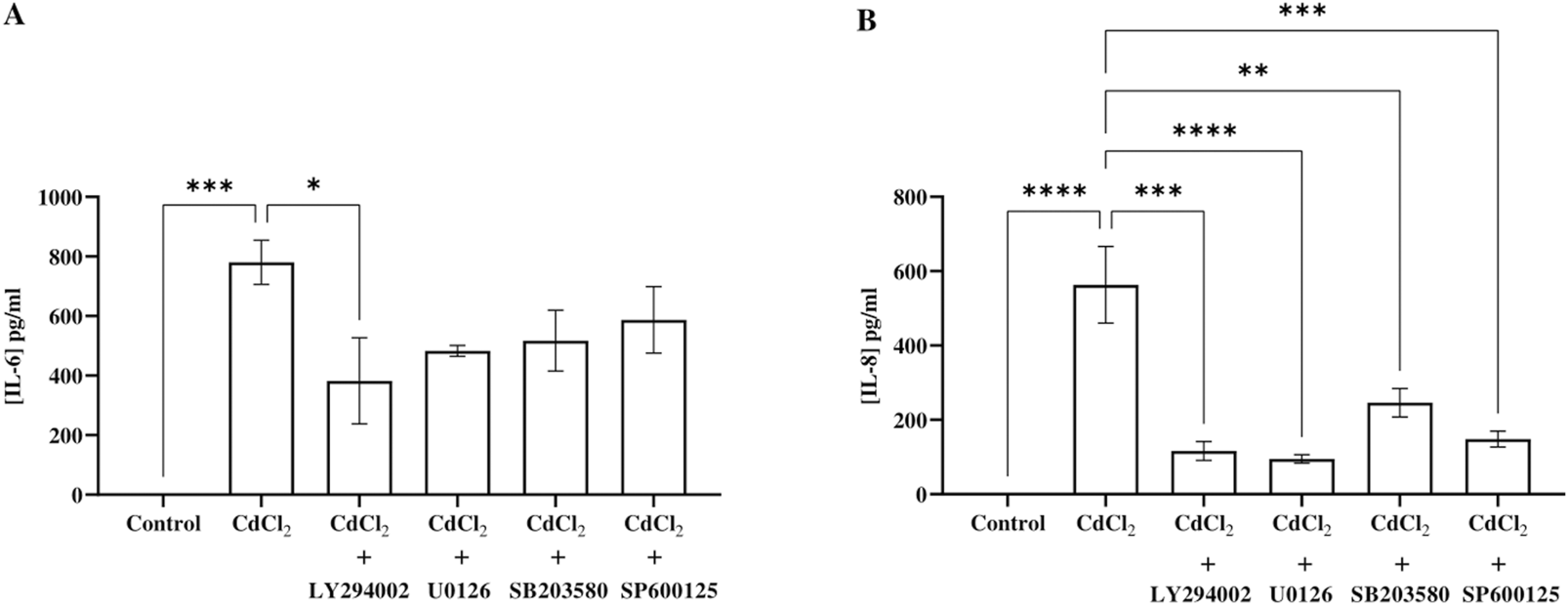
IL-6 and IL-8 secretion response to CdCl2 exposure and the effect of inhibitors. HGF cells were co-treated with 1 µM CdCl2 and specific inhibitors: 10 µM LY294002 (Akt), 25 µM U0126 (ERK1/2), and 25 µM SP600125 (JNK) prior to the measurement of IL-6 (A) and IL-8 levels (B) by ELISA. Control refers to cells cultured in DMEM without CdCl2 treatment. Data are shown as mean ± SEM. For all groups n = 3. Statistical significance was determined using One-way ANOVA followed by Dunnett’s multiple comparison test represented by *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Cd increased COX-2 expression in HGFs, which was inhibited by Akt and MAPK inhibitors
CdCl2 (1 μM) treatment induced COX-2 expression in HGFs 1 h after the exposure (Figure 4). This induction of COX-2 expression was significantly attenuated when the cells were co-treated with inhibitors (10 µM LY294002 for Akt, 25 µM U0126 for ERK1/2, and 25 µM SP600125 for JNK), indicating that these pathways contribute to COX-2 upregulation in response to Cd exposure.
FIGURE 4
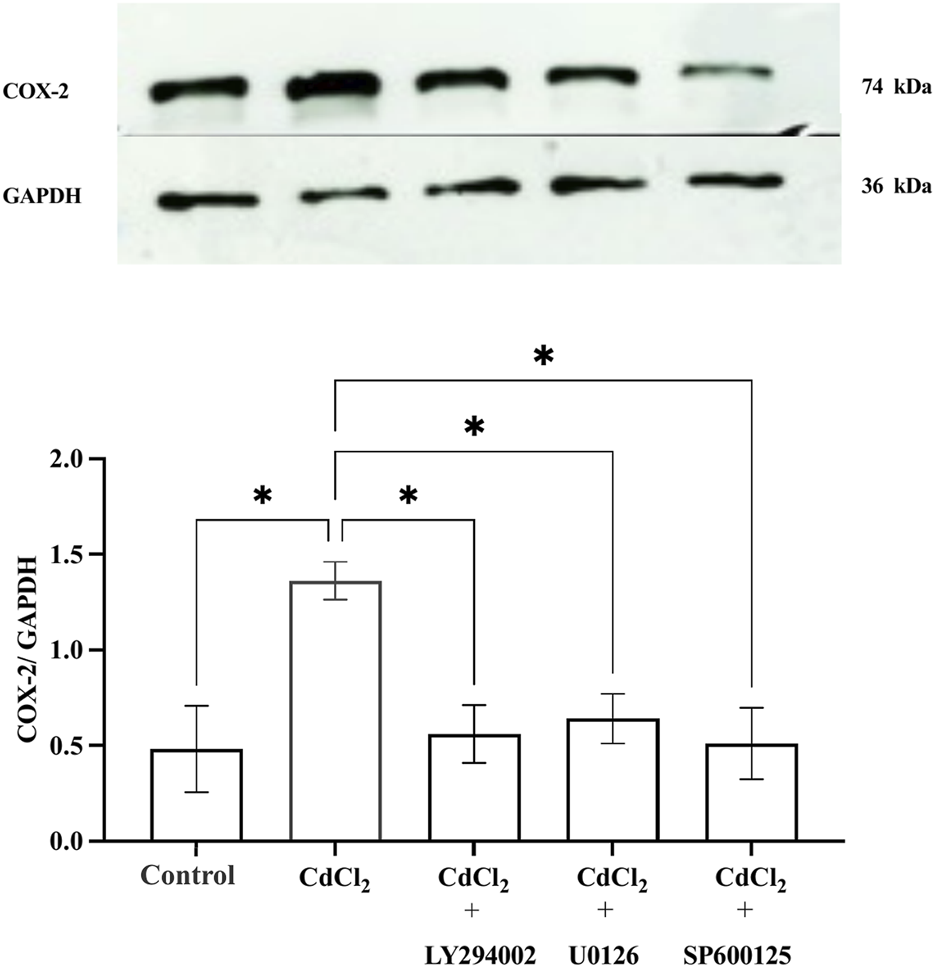
Effect of CdCl2 on COX-2 expression in HGFs. Western blot analysis of COX-2 expression in HGFs after 1-h incubation with 1 μM CdCl2, with and without specific pathway inhibitors: 10 µM LY294002 (Akt), 25 µM U0126 (ERK1/2), and 25 µM SP600125 (JNK) were determined. Control refers to cells cultured in DMEM without CdCl2 treatment. The upper bands represent COX-2, and the lower bands correspond to GAPDH which was used as housekeeping genes. Data are shown as mean ± SEM (n = 4). Statistical significance was determined using One-way ANOVA followed by Dunnett’s multiple comparison test represented by *P ≤ 0.05, **P ≤ 0.01.
Discussion
This study demonstrates that Cd exerts harmful effects on normal HGFs through the activation of specific signaling pathways. Our findings show that Cd exposure activates the phosphorylated Akt, ERK1/2, and JNK signaling pathways, which mediate subsequent cellular responses. These include a significant decrease in cell viability, the induction of pro-inflammatory cytokines IL-6 and IL-8, and an increase in COX-2 expression. Therefore, our study provides a mechanistic link between Cd exposure, pro-inflammatory responses, and the development of smoking-related oral diseases.
The cytotoxic effects of Cd on HGF cells were evident, with a dose-dependent reduction in cell viability. The cytotoxic effects of Cd in our study align with previous reports demonstrating its dose- and time-dependent toxicity in normal cell types such as human lens epithelial cells and intestinal epithelial (IEC-18) cells in a dose-dependent manner (Duizer et al., 1999; Kishimoto et al., 1994). Cd also induced cytotoxicity in normal human keratinocytes (HaCaT cells) by reducing cell viability (LC50 of 11 µM), causing DNA damage and apoptosis (Li et al., 2022). In addition, Cd has the potential to transform normal cells into cancer cells. Chronic exposure to 10 μM Cd turned benign prostatic hyperplasia (BPH) cells to become malignant ones by increasing their ability to form colonies, invade, and migrate (Chandrasekaran et al., 2020). Cancer cells are more susceptible to Cd's cytotoxic effects than normal cells. A study reported that 20 nM Cd reduced the viability of A549 lung cancer cells, a much lower concentration than what affected normal HGFs in our study (Kim et al., 2020). Interestingly, clinical research found Cd accumulation in the oral keratinized mucosa (OKM) at concentrations of 0.76 µM in smokers and 1.28 µM in non-smokers (Satir et al., 2024). These levels are comparable to the 1 µM Cd concentration used in our study, making it a relevant and pathologically meaningful concentration for our study. We also determined the IC50 value of 5.883 μM, suggesting that even low Cd exposure levels can impair cellular functions in HGFs. Reported salivary Cd levels vary considerably depending on smoking behavior and method. The results ranged from 0.08–0.11 μM in heavy cigarette smokers to over 40 μM in long-term waterpipe users, and over 300 μM in long-term heavy tobacco users (Bertuzzo, 2003; Talio et al., 2010; Khabour et al., 2018). In our study, we mainly focused on the 1 μM concentration of Cd, which falls within the higher end of reported levels and is similar to what has been found in OKM. However, we did not explore the lowest concentration (0.1 μM) in detail, even though it is also closer to salivary Cd levels in smokers reported from some reports. Future studies should include this range to better reflect typical environmental exposure. Additionally, Cd may have a comparable or even greater cytotoxic potential at lower concentrations than nicotine, a major component of tobacco smoke, which has been shown to induce cytotoxic effects in fibroblast cells with IC50 values ranging from 6 µM to 25 mM (Holliday et al., 2019). Taken together, these findings suggest that both Cd and nicotine could synergistically contribute to cellular dysfunction and damage in the oral cavity.
Salivary Cd concentrations are significantly elevated in waterpipe and long-term smokers, with levels reported up to 352.72 μM in certain studies.
Understanding these mechanistic effects is critical because they can influence disease progression. Our study showed that Cd increased phosphorylation of Akt, ERK1/2, and JNK, with specific inhibitors blocking these pathways. However, we did not observe significant phosphorylation of p38. This finding contrasts with previous studies showing p38 activation in other cell types such as breast cancer, osteosarcoma, and bronchial epithelial cells, following Cd exposure (Casano et al., 2010; Hu et al., 2015; Cao et al., 2021). This discrepancy may reflect the cell-type-specific nature of p38 MAPK, which is more commonly activated by oxidative stress in immune or neuronal cells (Galán et al., 2000; Rockwell et al., 2004). Additionally, higher Cd concentrations (15–30 μM) were required for p38 activation in mouse hippocampal cells (HT4) (Wang et al., 2019), whereas we limited our analysis to 1 μM Cd, which was the highest concentration that did not induce cell death in our experiment to avoid cytotoxicity. At this lower concentration, Cd appears to preferentially activate Akt, ERK1/2 and JNK, which are more directly linked to inflammatory signaling in our study. This supports the notion that p38 activation is concentration-dependent and more relevant at higher Cd levels. Our findings suggest that Cd-induced pro-inflammatory responses in HGFs is primarily driven by phosphorylation of Akt, ERK1/2, and JNK, with p38 playing a minor role in this context.
We also study how Cd induces pro-inflammatory responses in HGFs by measuring IL-6 and IL-8 levels. These two cytokines play important roles in chronic inflammation and are implicated in periodontal diseases and oral cancers. Cd exposure significantly elevated the secretion of pro-inflammatory cytokines IL-6 and IL-8 in our HGFs. The results are consistent with previous studies demonstrating Cd-induced cytokine upregulation in various cell types, including human astrocytes and breast cancer cells, highlighting its role in promoting pro-inflammatory responses (Phuagkhaopong et al., 2017; Bimonte et al., 2024). Cd also induced epidermal growth factor receptor (EGFR), one of the important factors for IL-1 and IL-6, in breast cancer cells and human lung adenocarcinoma cell lines (A549) (Wei et al., 2015; Kundu et al., 2011). The inflammatory effects of Cd become more concerning when considering environmental sources. As a component of PM2.5, Cd links air pollution to oral inflammation. Exposure to PM2.5 has been associated with higher salivary IL-6 levels in humans (He et al., 2022), which aligns with our findings of Cd-elevated IL-6 in HGFs. Since IL-6 plays a key role in inflammatory responses, tissue inflammation and structural damage in oral tissues after prolonged exposure to PM2.5 were observed in the previous study (Zhu et al., 2024). Additionally, the attenuation of IL-6 and IL-8 secretion by inhibitors targeting the Akt, ERK1/2, and JNK pathways in our study indicates that these pathways mediate the effects of Cd-induced IL-6 and IL-8 cytokine release in HGF cells.
In addition to cytokine release, Cd exposure also increased COX-2 expression, a key mediator of inflammation. In this study, we measured COX-2 expression after a 1-h incubation of CdCl2 in HGFS based on its short half-life (< 3.5 h) and previously observed an increase in COX-2 mRNA within 1-2 hours after tissue injury (Khan et al., 2007; Lukiw et al., 1997). Clinical and animal studies have shown that the peak of COX-2 expression was early at 1-2 h after carrageenin induction (Gilroy et al., 1999). Based on our results, CdCl2 did not induce p38 MAPK activation, we focused on Akt, ERK1/2, and JNK which are more relevant to Cd-induced pro-inflammatory responses in HGFs. In our study, Cd-induced COX-2 overexpression was reduced by inhibitors for Akt, ERK, and JNK pathways. Our findings align with previous studies showing Cd-induced COX-2 upregulation through Akt and ERK signaling in various cell types, including RAW264.7 cells and human gallbladder epithelial cells (Huang et al., 2014; Sharma et al., 2020). In animal study, the study in mice showed that Cd administration (5 mg/kg body weight) over 15 days elevated COX-2 and IL-6 levels in lung tissues (Kundu et al., 2009). In contrast, studies in HT4 cells at 15 µM Cd demonstrated that higher Cd concentrations induce COX-2 via the p38 pathway but not JNK (Rockwell et al., 2004). This supports the idea that higher Cd concentrations primarily activate the p38 MAPK pathway, while lower concentrations may preferentially activate the JNK pathway which was observed in our study. These findings emphasize the complexity of Cd-induced signaling pathways and Akt, ERK1/2, and JNK as primary drivers of pro-inflammatory responses in oral fibroblasts, while p38 plays a more prominent role under higher Cd-induced oxidative stress and cytotoxicity. Taken together, our findings highlight that Cd-induced IL-6, IL-8, and COX-2 expression in HGFs is not a non-specific stress effect but is regulated through Akt, ERK1/2, and JNK activation.
While previous studies have explored Cd’s cytotoxic effects (Issa et al., 2008), its correlation with periodontal (Yang et al., 2025), or signaling responses in other tissues (Chmielowska-Bąk and Deckert, 2012; Chen et al., 2022), these aspects were often examined separately. In contrast, our study combines cell viability, cytokine secretion, and COX-2 expression with mechanistic validation using pathway-specific inhibitors. This provides a mechanistic basis for the pro-inflammatory response observed in Cd-exposed oral tissues and distinguishes our study from prior works that did not examine intracellular signaling cascades in gingival fibroblasts. Moreover, we used Cd concentrations that reflect environmental or secondhand smoke exposure which differs from the higher levels used in earlier studies. This makes our findings more applicable to real-world oral health scenarios.
However, the present study has certain limitations. Inflammation is a complex biological process involving multiple cell types, vascular components, and molecular mediators, whereas our single-cell in vitro model focuses on intrinsic cellular immune responses, such as cytokine production and intracellular signalling. As a result, our model captures only intrinsic cell-autonomous responses and cannot fully replicate the interactions among different cell types that occur during inflammation. Future studies incorporating other cell types would further enhance the physiological relevance of the findings. Additionally, as this study used HGFs from a single donor, and genetic variability influences susceptibility to periodontitis, the response to Cd may differ across individuals. Including HGFs from diverse donors and other oral cell types in future work would help address this limitation. Lastly, in vivo studies are essential to validate our results and assess the long-term effects of Cd exposure on oral health.
In conclusion, this study highlights Cd's harmful effects on HGFs through activation of the Akt and MAPK (ERK1/2, JNK) signaling pathways. This leads to increased IL-6, IL-8, and COX-2 expression, contributing to oral pro-inflammatory responses. The findings support future research on targeting these pathways to reduce Cd-induced inflammation and the public health impact of smoking and environmental Cd exposure.
Statements
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
TP: Formal Analysis, Data curation, Investigation, Methodology, Project administration, Writing – original draft. SS: Methodology, Writing – review and editing. NS: Methodology, Writing – review and editing. NR: Writing – review and editing. PK: Writing – review and editing. PV: Writing – review and editing, Conceptualization, Formal Analysis, Funding acquisition, Supervision, Validation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research project is supported by Mahidol University (Basic Research Fund: fiscal year 2022 [BRF1-019/2565]) to PP and National Research Council of Thailand under grant number [350/2564] to PK.
Acknowledgments
We thank Pornpen Dararat, Rapeewan Settacomkul, and Pichsinee Woonfak for technical assistance.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/ftox.2025.1583865/full#supplementary-material
References
1
Bertuzzo C. Goodson J. M. Mager D. Pilatti G. L. Santos J. R. Silveira G. M. et al (2003). “Salivary cadmium levels associated with smoking,” in IADR/PER disease mechanisms - molecular genetics and oral manifestations. Goteborg, Sweden.
2
Bimonte V. M. Catanzaro G. Po A. Trocchianesi S. Besharat Z. M. Spinello Z. et al (2024). The endocrine disruptor cadmium modulates the androgen-estrogen receptors ratio and induces inflammatory cytokines in luminal (A) cell models of breast cancer. Endocrine83 (3), 798–809. 10.1007/s12020-023-03594-2
3
Cao X. Fu M. Bi R. Zheng X. Fu B. Tian S. et al (2021). Cadmium induced BEAS-2B cells apoptosis and mitochondria damage via MAPK signaling pathway. Chemosphere263, 128346. 10.1016/j.chemosphere.2020.128346
4
Casano C. Agnello M. Sirchia R. Luparello C. (2010). Cadmium effects on p38/MAPK isoforms in MDA-MB231 breast cancer cells. Biometals23, 83–92. 10.1007/s10534-009-9268-6
5
Chandrasekaran B. Dahiya N. R. Tyagi A. Kolluru V. Saran U. Baby B. V. et al (2020). Chronic exposure to cadmium induces a malignant transformation of benign prostate epithelial cells. Oncogenesis9, 23. 10.1038/s41389-020-0202-7
6
Chen X. Bi M. Yang J. Cai J. Zhang H. Zhu Y. et al (2022). Cadmium exposure triggers oxidative stress, necroptosis, Th1/Th2 imbalance and promotes inflammation through the TNF-α/NF-κB pathway in swine small intestine. J. Hazard Mater421, 126704. 10.1016/j.jhazmat.2021.126704
7
Chmielowska-Bąk J. Deckert J. (2012). A common response to common danger? Comparison of animal and plant signaling pathways involved in cadmium sensing. J. Cell Commun. Signal6, 191–204. 10.1007/s12079-012-0173-3
8
Dinh Q. P. Novirsa R. Jeong H. Nugraha W. C. Addai-Arhin S. Viet P. H. et al (2021). Mercury, cadmium, and lead in cigarettes from international markets: concentrations, distributions and absorption ability of filters. J. Toxicol. Sci.46, 401–411. 10.2131/jts.46.401
9
Duizer E. Gilde A. J. Versantvoort C. H. Groten J. P. (1999). Effects of cadmium chloride on the paracellular barrier function of intestinal epithelial cell lines. Toxicol. Appl. Pharmacol.155, 117–126. 10.1006/taap.1998.8589
10
Finoti L. S. Nepomuceno R. Pigossi S. C. Corbi S. C. Secolin R. Scarel-Caminaga R. M. (2017). Association between interleukin-8 levels and chronic periodontal disease: a PRISMA-compliant systematic review and meta-analysis. Med. Baltim.96, e6932. 10.1097/MD.0000000000006932
11
Fresquez M. R. Pappas R. S. Watson C. H. (2013). Establishment of toxic metal reference range in tobacco from US cigarettes. J. Anal. Toxicol.37, 298–304. 10.1093/jat/bkt021
12
Galán A. García-Bermejo M. L. Troyano A. Vilaboa N. E. DE Blas E. Kazanietz M. G. et al (2000). Stimulation of p38 mitogen-activated protein kinase is an early regulatory event for the cadmium-induced apoptosis in human promonocytic cells. J. Biol. Chem.275, 11418–11424. 10.1074/jbc.275.15.11418
13
Gasche J. A. Hoffmann J. Boland C. R. Goel A. (2011). Interleukin-6 promotes tumorigenesis by altering DNA methylation in oral cancer cells. Int. J. Cancer129, 1053–1063. 10.1002/ijc.25764
14
Gilroy D. W. Colville-Nash P. R. Willis D. Chivers J. Paul-Clark M. J. Willoughby D. A. (1999). Inducible cyclooxygenase may have anti-inflammatory properties. Nature medicine5(6), 698–701. 10.1038/9550
15
He L. Norris C. Cui X. Li Z. Barkjohn K. K. Teng Y. et al (2022). Oral cavity response to air pollutant exposure and association with pulmonary inflammation and symptoms in asthmatic children. Environmental research, 206, 112275. 10.1016/j.envres.2021.112275
16
Holliday R. S. Campbell J. Preshaw P. M. (2019). Effect of nicotine on human gingival, periodontal ligament and oral epithelial cells. A systematic review of the literature. J. Dent.86, 81–88. 10.1016/j.jdent.2019.05.030
17
Hossein-Khannazer N. Azizi G. Eslami S. Alhassan Mohammed H. Fayyaz F. Hosseinzadeh R. et al (2020). The effects of cadmium exposure in the induction of inflammation. Immunopharmacol. Immunotoxicol.42, 1–8. 10.1080/08923973.2019.1697284
18
Hu K. H. Li W. X. Sun M. Y. Zhang S. B. Fan C. X. Wu Q. et al (2015). Cadmium induced apoptosis in MG63 cells by increasing ROS, activation of p38 MAPK and inhibition of ERK 1/2 pathways. Cell Physiol. Biochem.36, 642–654. 10.1159/000430127
19
Huang Y. Y. Xia M. Z. Wang H. Liu X. J. Hu Y. F. Chen Y. H. et al (2014). Cadmium selectively induces MIP-2 and COX-2 through PTEN-mediated Akt activation in RAW264.7 cells. Toxicological sciences: an official journal of the Society of Toxicology, 138(2), 310–321. 10.1093/toxsci/kfu013
20
Hussein B. J. Atallah H. N. Al-Dahhan N. A. A. (2020). Salivary levels of interleukin-6, interleukin-8 and tumor necrosis factor-alpha in smokers aged 35-46 Years with dental caries disease. Medico-Legal Update. 10.37506/mlu.v20i4.2040
21
Issa Y. Brunton P. Waters C. M. Watts D. C. (2008). Cytotoxicity of metal ions to human oligodendroglial cells and human gingival fibroblasts assessed by mitochondrial dehydrogenase activity. Dent. Mater.24, 281–287. 10.1016/j.dental.2007.09.010
22
Khabour O. F. Alzoubi K. H. Al-Sheyab N. A. Azab M. A. Massadeh A. M. Alomary A. A. et al (2018). Plasma and saliva levels of three metals in waterpipe smokers: a case control study. Inhal. Toxicol.30, 224–228. 10.1080/08958378.2018.1500663
23
Khan A. A. Iadarola M. Yang H. Y. Dionne R. A. (2007). Expression of COX-1 and COX-2 in a clinical model of acute inflammation. The journal of pain8(4), 349–354. 10.1016/j.jpain.2006.10.004
24
Kim A. Park S. Sung J. H. (2020). Cell viability and immune response to low concentrations of nickel and cadmium: an in vitro model. Int. J. Environ. Res. Public Health17, 9218. 10.3390/ijerph17249218
25
Kishimoto T. Oguri T. Ohno M. Matsubara K. Yamamoto K. Tada M. (1994). Effect of cadmium (CdCl2) on cell proliferation and production of EDRF (endothelium-derived relaxing factor) by cultured human umbilical arterial endothelial cells. Archives Toxicol.68, 555–559. 10.1007/s002040050113
26
Kundu S. Sengupta S. Bhattacharyya A. (2011). EGFR upregulates inflammatory and proliferative responses in human lung adenocarcinoma cell line (A549), induced by lower dose of cadmium chloride. Inhalation toxicology, 23(6), 339–348. 10.3109/08958378.2011.572931
27
Kundu S. Sengupta S. Chatterjee S. Mitra S. Bhattacharyya A. (2009). Cadmium induces lung inflammation independent of lung cell proliferation: a molecular approach. Journal of inflammation (London, England), 619. 10.1186/1476-9255-6-19
28
Li J. Y. Cui D. L. Xie Y. M. Su J. Z. Zhang M. Y. Niu Y. Y. et al (2022). Mechanisms of Cd-induced cytotoxicity in normal human skin keratinocytes: implication for human health. Int. J. Mol. Sci.23, 11767. 10.3390/ijms231911767
29
Li X. Liang X. Li S. Qi X. Du N. Yang D. (2021). Effect of environmental tobacco smoke on COX-2 and SHP-2 expression in a periodontitis rat model. Oral Dis.27, 338–347. 10.1111/odi.13538
30
Lukiw W. J. Bazan N. G. (1997). Cyclooxygenase 2 RNA message abundance, stability, and hypervariability in sporadic Alzheimer neocortex. J. Neu. Res.50(6), 937–945. 10.1002/(SICI)1097-4547(19971215)50:6<937:AID-JNR4>3.0.CO;2-E
31
Nibali L. Fedele S. D’Aiuto F. Donos N. (2012). Interleukin-6 in oral diseases: a review. Oral Dis.18, 236–243. 10.1111/j.1601-0825.2011.01867.x
32
Noh M. K. Jung M. Kim S. H. Lee S. R. Park K. H. Kim D. H. et al (2013). Assessment of IL-6, IL-8 and TNF-α levels in the gingival tissue of patients with periodontitis. Exp. Ther. Med.6, 847–851. 10.3892/etm.2013.1222
33
Olszowski T. Baranowska-Bosiacka I. Gutowska I. Chlubek D. (2012). Pro-inflammatory properties of cadmium. Acta Biochim. Pol.59, 475–482. 10.18388/abp.2012_2080
34
Phuagkhaopong S. Ospondpant D. Kasemsuk T. Sibmooh N. Soodvilai S. Power C. et al (2017). Cadmium-induced IL-6 and IL-8 expression and release from astrocytes are mediated by MAPK and NF-κB pathways. Neurotoxicology60, 82–91. 10.1016/j.neuro.2017.03.001
35
Richard J. O. Qiang L. Stephens W. E. David H. Tara E.-M. Cummings K. M. et al (2010). Cigarettes sold in China: design, emissions and metals. Tob. Control19, i47–i53. 10.1136/tc.2009.030163
36
Rockwell P. Martinez J. Papa L. Gomes E. (2004). Redox regulates COX-2 upregulation and cell death in the neuronal response to cadmium. Cell Signal16, 343–353. 10.1016/j.cellsig.2003.08.006
37
Ruggieri S. Drago G. Perrino C. Canepari S. Balzan M. Cuttitta G. et al (2014). Higher indoor PM2.5 concentration of cadmium (Cd) and thallium (Tl) is related to domestic smoking.
38
Sahibzada H. A. Khurshid Z. Khan R. S. Naseem M. Siddique K. M. Mali M. et al (2017). Salivary IL-8, IL-6 and TNF-α as potential diagnostic biomarkers for oral cancer. Diagn. (Basel)7, 21. 10.3390/diagnostics7020021
39
Satir S. Kaya D. I. Ozsoy S. C. (2024). Effect of tobacco use on cadmium accumulation in the oral keratinized mucosa. BMC Oral Health24, 257. 10.1186/s12903-024-04001-6
40
Seok S. M. Park D. H. Kim Y. C. Moon C. H. Jung Y. S. Baik E. J. et al (2006). COX-2 is associated with cadmium-induced ICAM-1 expression in cerebrovascular endothelial cells. Toxicol. Lett.165, 212–220. 10.1016/j.toxlet.2006.04.007
41
Sharma P. Caldwell T. S. Rivera M. N. Gullapalli R. R. (2020). Cadmium exposure activates Akt/ERK Signaling and pro-inflammatory COX-2 expression in human gallbladder epithelial cells via a ROS dependent mechanism. Toxicol Vitro67, 104912. 10.1016/j.tiv.2020.104912
42
St John M. A. Li Y. Zhou X. Denny P. Ho C. M. Montemagno C. et al (2004). Interleukin 6 and interleukin 8 as potential biomarkers for oral cavity and oropharyngeal squamous cell carcinoma. Arch. Otolaryngol. Head. Neck Surg.130, 929–935. 10.1001/archotol.130.8.929
43
Talio M. C. Luconi M. O. Masi A. N. Fernández L. P. (2010). Cadmium monitoring in saliva and urine as indicator of smoking addiction. Sci. Total Environ.408, 3125–3132. 10.1016/j.scitotenv.2010.03.052
44
Wang H. Abel G. M. Storm D. R. Xia Z. (2019). Cadmium exposure impairs adult hippocampal neurogenesis. Toxicol. Sci.171, 501–514. 10.1093/toxsci/kfz152
45
Wei Z. Song X. Shaikh Z. A. et al (2015). Cadmium promotes the proliferation of triple-negative breast cancer cells through EGFR-mediated cell cycle regulation. Toxicology and applied pharmacology, 289(1), 98–108. 10.1016/j.taap.2015.09.006
46
Yang S. Li J. Wu Y. (2025). The association of lead and cadmium exposure with periodontitis: a systematic review and meta-analysis. BMC Oral Health25, 935. 10.1186/s12903-025-06195-9
47
Zhu L. Tang M. Cai Y. Wang P. (2024). Association between exposure to environmental pollutants and increased oral health risks, a comprehensive review. Front. Public Health12, 1482991. 10.3389/fpubh.2024.1482991
Summary
Keywords
cadmium, fibroblasts, cyclooxygenase, interleukin-6, interleukin-8
Citation
Parakaw T, Srihirun S, Sibmooh N, Ruangsawasdi N, Khemawoot P and Vivithanaporn P (2025) Cadmium decreases human gingival fibroblast viability and induces pro-inflammatory response associated with Akt and MAPK pathway activation. Front. Toxicol. 7:1583865. doi: 10.3389/ftox.2025.1583865
Received
26 February 2025
Accepted
08 July 2025
Published
23 July 2025
Volume
7 - 2025
Edited by
Ariane Zamoner, Federal University of Santa Catarina, Brazil
Reviewed by
Maicon Roberto Kviecinski, Federal University of Santa Catarina, Brazil
Katiuska Marins, Unidade Central de Educação Faem Faculdade (UCEFF), Brazil
Marcelo Farina, Universidade Federal de Santa Catarina, Brazil
Updates
Copyright
© 2025 Parakaw, Srihirun, Sibmooh, Ruangsawasdi, Khemawoot and Vivithanaporn.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pornpun Vivithanaporn, pornpun.viv@mahidol.ac.th
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.