 Jaeeun Lee1
Jaeeun Lee1 Ji-Young Lee
Ji-Young Lee Hyunju Kang
Hyunju Kang- 1Department of Nutritional Sciences, University of Connecticut, Storrs, CT, United States
- 2Department of Food and Nutrition, Keimyung University, Daegu, Republic of Korea
With the increasing prevalence of alcohol-related diseases, expanding our understanding of the toxic effects of excessive alcohol consumption is critical for prevention and treatment of metabolic and inflammatory pathology. This review summarizes current knowledge on the metabolic dysfunction and inflammation caused by alcohol and their impact on the pathogenesis of alcohol-related liver disease (ALD), type 2 diabetes, cardiovascular disease, and obesity, and neurological damage. It highlights recent evidence that alcohol induces a cascade of reactive oxygen species (ROS)-mediated lipid peroxidation and nicotinamide adenine dinucleotide (NAD+) depletion, triggering mitochondrial dysfunction and metabolic imbalances in the liver, heart, pancreas, and brain. By integrating these mechanistic insights with emerging data on how disrupted lipid and glucose metabolism amplify immune dysregulation, the review underscores the interplay between metabolic and inflammatory pathways in exacerbating tissue injury across these organs. A deep understanding of these metabolic and inflammatory disruptions is therefore essential for developing novel therapeutic strategies, including metabolic and nutritional interventions, aimed at mitigating the health risks of excessive alcohol consumption.
1 Introduction
Excessive alcohol consumption is a major global health concern, responsible for an estimated 4.7% of all deaths in 2019, according to a new 2024 report from the World Health Organization (World Health Organization, 2024). It significantly contributes to the global burden of diseases, particularly liver disease, cardiovascular complications, and metabolic disorders. For the purposes of this review, “excessive alcohol consumption” refers to patterns of drinking that exceed public health guidelines, such as binge drinking (consuming four to five or more drinks on an occasion) or heavy weekly use (8–15 or more drinks per week for women and men, respectively), which can lead to health and safety risks (Crews et al., 2024; Koob, 2024). The toxicity of alcohol and its metabolites disrupts metabolic processes across multiple organs by inducing oxidative stress and depleting nicotinamide adenine dinucleotide (NAD+) (Tsermpini et al., 2022). Acetaldehyde, a toxic metabolite of alcohol, binds to proteins in mitochondria or microtubules, leading to structural damage and metabolic dysfunction. Furthermore, this harmful drinking pattern induces the overproduction of reactive oxygen species (ROS), culminating in oxidative stress and inflammation. This inflammatory state is a critical factor in the development of not only alcohol-related organ diseases and senescence-associated diseases but also in the pathophysiology of alcohol use disorder itself (Carvajal et al., 2023). Moreover, depletion of NAD+ levels due to alcohol oxidation can impair energy-generating pathways, including mitochondrial β-oxidation of fatty acids and the tricarboxylic acid (TCA) cycle, further exacerbating metabolic dysfunction (Bailey and Cunningham, 2002; Jeon and Carr, 2020).
Understanding the pathways sensitive to alcohol exposure is pivotal for the development of effective strategies to protect against alcohol-related conditions. This review summarizes the metabolic and inflammatory dysfunctions and diseases associated with excessive alcohol consumption. This exploration of alcohol-mediated dysfunctions can help identify new therapeutic approaches to protect against alcohol-related diseases.
2 Ethanol metabolism and its toxic effects
2.1 Ethanol oxidation
The primary pathway for alcohol metabolism is the oxidation of ethanol to acetaldehyde, catalyzed by cytosolic alcohol dehydrogenase (ADH), a process requiring NAD+ as a cofactor (Zakhari, 2006). Acetaldehyde, a highly toxic byproduct, is further oxidized to acetate by acetaldehyde dehydrogenase (ALDH), another NAD+-dependent reaction (Figure 1). This sequential ethanol oxidation by ADH and ALDH occurs primarily in the cytoplasm and mitochondria of hepatocytes, respectively. Acetate, the end product of ethanol oxidation, is released into the bloodstream and transported to various peripheral tissues, where it becomes acetyl coenzyme A (acetyl-CoA) by the action of acetyl-CoA synthetase (ACS) in the mitochondria. Acetyl-CoA serves as a key intermediate in several metabolic pathways, including the TCA cycle, fatty acid synthesis, and ketone body formation, depending on the energy demands and nutritional status of the cell (Shi and Tu, 2015).
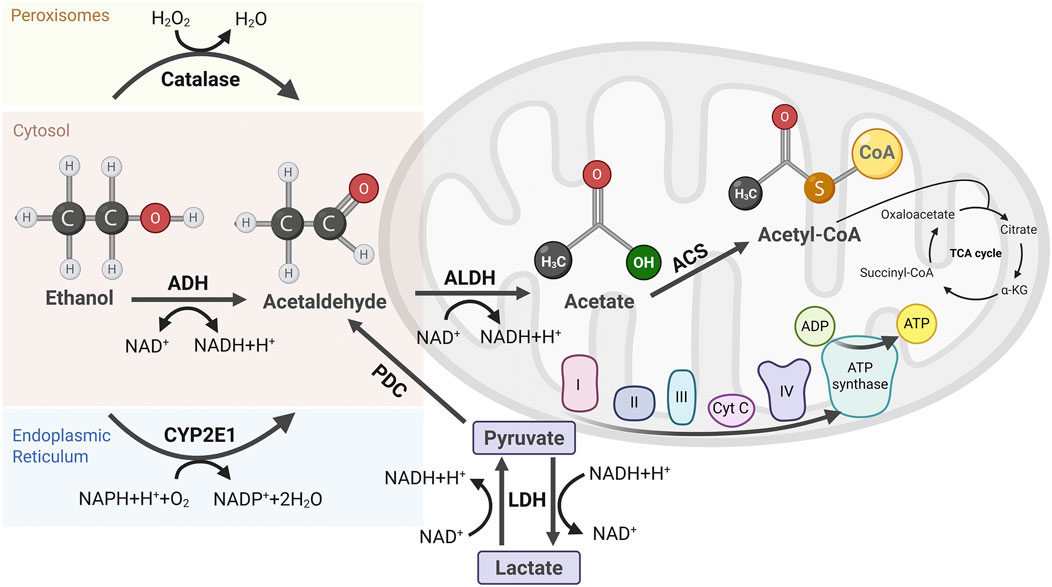
Figure 1. Pathways of Ethanol Metabolism and Its Byproducts. Ethanol metabolism occurs via three primary pathways. Alcohol dehydrogenase (ADH) in the cytosol oxidizes ethanol to acetaldehyde, producing NADH. Catalase in peroxisomes contributes minimally to ethanol oxidation. Cytochrome P450 2E1 (CYP2E1) in the endoplasmic reticulum (ER) generates acetaldehyde with concomitant ROS. Acetaldehyde is further oxidized to acetate by aldehyde dehydrogenase (ALDH) in mitochondria, accompanied by additional NADH generation. Acetate is then converted to acetyl-CoA by acetyl-CoA synthetase (ACS) locally or in other tissues, fueling the TCA cycle for energy production. Elevated NADH levels disrupt metabolic homeostasis, including the pyruvate-to-lactate conversion mediated by lactate dehydrogenase (LDH), altering redox balance and cellular metabolism.
During chronic alcohol consumption or elevated blood alcohol levels, cytochrome P450 2E1 (CYP2E1), located in the endoplasmic reticulum (ER), utilizes nicotinamide adenine dinucleotide phosphate (NADPH) and oxygen to oxidize ethanol and generate ROS during this process (Lu and Cederbaum, 2008). More recent in vivo studies in murine models have confirmed that chronic alcohol intake upregulates CYP2E1, exacerbating oxidative liver injury (Nath et al., 2011). ROS can damage mitochondrial DNA (mtDNA) by diffusing across mitochondrial membranes or through close contact between the ER and mitochondria, leading to oxidative modifications, such as base alterations (e.g., 8-oxo-2′-deoxyguanosine), single- and double-strand breaks, and cross-linking of mtDNA proteins (Koop, 2006).
Another minor ethanol oxidative pathway is present in peroxisomes, where ethanol is metabolized by catalase, especially in conditions of elevated hydrogen peroxide, often associated with chronic alcohol consumption (Zakhari, 2006). Also, non-oxidative ethanol metabolism occurs at the cellular level, although it is relatively minor. Fatty acid ethyl ester synthases catalyze the reaction between ethanol and fatty acids to form fatty acid ethyl esters (FAEEs), which can disrupt membrane integrity, induce inflammatory responses, and interfere with mitochondrial function (Lee et al., 2021). Although the accumulation of FAEEs exacerbates liver inflammation and fibrosis, key features of alcoholic steatosis (Park et al., 2023), their overall contribution to alcohol metabolism is considerably insignificant compared to the oxidative pathways (Figure 1).
Excessive alcohol consumption results in cellular and systemic toxicity driven by acetaldehyde and ROS, which disrupt cellular processes, leading to oxidative stress, inflammation, and mitochondrial dysfunction. This cascade compromises cellular integrity and contributes to the progression of alcohol-related diseases (ALDs) through widespread tissue and organ damage.
2.2 Oxidative stress and cellular damage
A primary mechanism of alcohol-induced toxicity is the excessive generation of ROS during its metabolism. Ethanol is oxidized by ADH and CYP2E1 into acetaldehyde, which is subsequently converted into acetate by ALDH (Zakhari, 2006). This process generates large amounts of ROS, overwhelming the cell’s antioxidant defenses and triggering oxidative stress (Beckhauser et al., 2016). The accumulation of ROS causes oxidative damage of lipids, proteins, and DNA, resulting in cellular dysfunction and tissue injury (Juan et al., 2021).
The excessive production of ROS also depletes NAD+, disrupting the balance of the NAD+/NADH ratio, which is essential for cellular energy production and metabolic processes. Furthermore, in vitro cell culture studies have shown that ROS accumulation also activates enzymes such as NADPH oxidase-2 and cyclooxygenase-2, further exacerbating oxidative damage and cellular injury (Bedard and Krause, 2007; Onodera et al., 2015). Additionally, alcohol impairs cells’ antioxidant defenses, particularly by depleting the levels of glutathione, a key antioxidant that neutralizes ROS (Huang et al., 2009). This impairment exacerbates oxidative damage across major organ systems, such as the liver, brain, and cardiovascular system.
Another critical consequence of alcohol-induced oxidative stress is the repression of sirtuin 1 (SIRT1) activity, an NAD+-dependent deacetylase, which regulates inflammation and oxidative stress responses (Singh and Ubaid, 2020; Tsermpini et al., 2022). Indeed, an in vitro study using RAW 264.7 macrophages linked reduced SIRT1 activity by ethanol exposure to increased inflammation and metabolic dysfunction (Kang et al., 2021a). Taken together, these interconnected mechanisms, including ROS overproduction, NAD+ depletion, and SIRT1 repression, play a significant role in alcohol-related cellular and systemic damage.
The liver is the central organ responsible for most ethanol metabolism, primarily by ADH and CYP2E1, decreasing the NAD+/NADH ratio (Cederbaum, 2012). Elevated NADH levels inhibit fatty acid oxidation and promote lipogenesis by increasing levels of acetyl-CoA and malonyl-CoA (Cortés-Rojo et al., 2020). Malonyl-CoA further inhibits carnitine palmitoyltransferase 1, blocking the transport and oxidation of fatty acids in mitochondria, leading to lipid accumulation and steatosis (Dabravolski et al., 2021). Also, ROS generated by CYP2E1 further intensifies oxidative stress, lipid peroxidation, and inflammation (Ali et al., 2019), contributing to the progression of ALD from simple steatosis to alcoholic hepatitis and cirrhosis. Chronic alcohol consumption triggers progressive cycles of liver injury, repair, and scarring, ultimately leading to hepatic fibrosis, cirrhosis, and liver failure. The toxic byproducts of ethanol metabolism, such as acetaldehyde and ROS, damage liver cells and impair their ability to detoxify harmful substances, such as endotoxins and gut-derived bacteria (Osna et al., 2017). As liver cells are exposed to sustained damage, their detoxifying capacity decreases, exacerbating systemic toxicity (Osna et al., 2017).
2.3 Inflammation
Alcohol-induced oxidative stress triggers an inflammatory response in the liver, the primary site of alcohol metabolism and inflammation (Shukla et al., 2021). The breakdown of alcohol generates ROS and pro-inflammatory mediators, including cytokines and chemokines, which contribute to tissue damage and disease progression (Wheeler, 2003). In the liver, Kupffer cells (resident macrophages) play a central role in mediating alcohol-induced inflammation. Gut-derived lipopolysaccharides (LPS) translocate to the liver due to increased intestinal permeability, activating toll-like receptor 4 (TLR4) on Kupffer cells. This activation proceeds via the MyD88-dependent signaling pathway, which culminates in the activation of the transcription factor nuclear factor kappa B (NF-κB). Activated NF-κB then translocates to the nucleus and promotes the transcription of pro-inflammatory cytokines, including tumor necrosis factor (TNF) and interleukin-6 (IL-6) (Su et al., 2000). This chronic inflammatory state, driven by cytokines and ROS, promotes the progression of ALD, fibrosis, and cirrhosis. Alcohol also activates the NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome complex within these immune cells. This activation is triggered by endogenous signals stemming directly from alcohol-induced cellular stress, such as mitochondrial ROS overproduction and the release of damaged mitochondrial DNA (mtDNA) into the cytosol. The assembled NLRP3 inflammasome activates caspase-1, which in turn cleaves pro-interleukin-1β (pro-IL-1β) into its mature, highly inflammatory form, IL-1β, a critical mediator in ALD pathogenesis (Kelley et al., 2019).
Beyond the liver, ethanol metabolism significantly affects the gastrointestinal (GI) tract by disrupting the intestinal barrier and altering the gut microbiome. Although the liver is the primary site for ethanol metabolism, enzymes such as ADH and ALDH are also present in the intestinal mucosa (Jelski et al., 2014). Intestinal bacteria further contribute to ethanol metabolism, producing additional acetaldehyde, which damages intestinal cells by forming adducts with proteins and DNA. Both the small and large intestines are affected by these changes, leading to chronic inflammation, disruption of tissue homeostasis, and eventual organ dysfunction (Patel et al., 2015). Chronic ethanol consumption also results in gut dysbiosis, an imbalance between beneficial and pathogenic bacteria (Litwinowicz et al., 2020). This imbalance, coupled with excessive acetaldehyde production, exacerbates inflammation in the GI tract. As ethanol metabolism progresses, tight junction proteins such as claudins and occludins are downregulated, increasing the permeability of the intestinal mucosa, commonly known as a “leaky gut”. This increased permeability allows toxins and bacterial products, such as LPS, to enter the bloodstream, triggering systemic inflammation (Bishehsari et al., 2017). Recent in vivo animal studies have further established this mechanism also disrupts mucosal immune defenses, suppressing Paneth cells that produce antibacterial compounds, leading to bacterial overgrowth, which intensifies inflammatory responses in the intestine and liver (Zhong et al., 2020).
Alcohol further disrupts the gut barrier by reducing protective molecules like intestinal trefoil factor and increasing ROS production, which weakens the epithelial lining (Morris and Yeligar, 2018). Moreover, alcohol reduces the production of short-chain fatty acids (SCFAs) (Litwinowicz et al., 2020), including butyrate, which exacerbates gut permeability and systemic inflammation, linking alcohol consumption to diseases in other organs, particularly the liver (Fairfield and Schnabl, 2021). This allows harmful substances to infiltrate the bloodstream, amplifying systemic inflammation. Additionally, chronic alcohol exposure impairs vitamin metabolism, such as thiamine (vitamin B1), leading to deficiencies such as Wernicke-Korsakoff syndrome, which causes cognitive impairment (Martin et al., 2003). These interconnected mechanisms underscore the significant impact of alcohol on the gut-liver axis and its contribution to the progression of ALD and other alcohol-induced pathologies.
2.4 NAD+ metabolism and cellular energy disruption
NAD+ is required for ethanol oxidation by ADH and ALDH, but its continuous consumption during alcohol metabolism depletes cellular levels, disrupting redox balance and impairing other NAD+-dependent functions. NADH produced by ADH is transported into mitochondria through the α-glycerophosphate or malate-aspartate shuttles for re-oxidation (Lu et al., 2008; Cederbaum, 2012), a process that supports NAD+ regeneration (Xie et al., 2020). The efficiency of these transporters and the capacity of the mitochondrial respiratory chain to oxidize NADH determine the rate of ethanol oxidation and the cell’s ability to maintain redox homeostasis. We reported that alcohol depletes the cellular NAD+ pool and inhibits its synthesis. Alcohol inhibits the expression of nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme of the NAD+ salvage pathway, which was counteracted by an NAD+ precursor or a SIRT1 activator in ethanol-stimulated macrophages (Kang et al., 2021a; Kang et al., 2021b; Gwon et al., 2024). This inhibition, due to chronic alcohol consumption, exacerbates NAD+ deficiency in the liver and macrophages, further impairing metabolic functions, including fatty acid oxidation, glycolysis, and mitochondrial respiration, intensifying the metabolic imbalance and contributing to disease progression (Kang et al., 2022; Kang et al., 2020).
The reduction in NAD+ also hinders the activity of SIRT1, which relies on NAD+ as a cofactor (Zhang et al., 2009). As SIRT1 activity declines, the regulation of key metabolic processes, such as DNA repair, oxidative stress response, and inflammation control, is disrupted, further intensifying the cellular damage caused by alcohol. This dysfunction contributes to increased oxidative stress, greater DNA damage, and chronic inflammation associated with long-term alcohol exposure (Iside et al., 2020).
2.5 Mitochondrial dysfunction and dynamics
Mitochondria are crucial organelles responsible for energy production through oxidative phosphorylation. Alcohol metabolism severely impacts mitochondrial function by disrupting the NAD+/NADH balance and generating excessive ROS. Chronic alcohol consumption impairs mitochondrial oxidative phosphorylation, reducing ATP production and compromising the cell’s ability to generate energy efficiently (Hoek et al., 2002). Excessive ROS production in mitochondria damages mtDNA, proteins, and lipids, leading to impaired mitochondrial function. These oxidative modifications contribute to lipid peroxidation, protein dysfunction, and mtDNA mutations, further reducing mitochondrial energy capacity (Guo et al., 2013). Alcohol also disrupts mitochondrial dynamics, which regulate fission and fusion processes (Palma et al., 2020). Under normal conditions, these processes maintain mitochondrial health by removing damaged mitochondria through mitophagy and promoting content exchange to ensure functionality. However, alcohol promotes excessive mitochondrial fission and inhibits fusion, leading to mitochondrial fragmentation and impaired mitophagy (Palma et al., 2020; Siggins et al., 2023). Fragmented mitochondria are less efficient at energy production and more vulnerable to oxidative stress, which accelerates the decline in mitochondrial function (Liu et al., 2020).
Our recent findings from an in vitro study in Kupffer cells indicate that chronic ethanol exposure activates the mitochondrial unfolded protein response (UPRmt), an adaptive mechanism designed to manage the accumulation of misfolded proteins within mitochondria (Lee et al., 2025). Under normal conditions, UPRmt upregulates molecular chaperones and proteases to restore protein homeostasis (Pellegrino et al., 2013). However, prolonged ethanol exposure overwhelms this system, leading to further mitochondrial dysfunction. Notably, ethanol increases the nuclear translocation of activating transcription factor 5 and upregulates UPRmt-related genes in mouse Kupffer cells concomitantly with increased mitochondrial dysfunction.
3 Pathogenesis of alcohol-induced diseases
Excessive alcohol consumption disrupts metabolic processes far beyond the liver, leading to systemic dysfunctions that affect various organs. This disruption is primarily associated with lipid and glucose metabolism, resulting in metabolic imbalances that elevate the risk of conditions such as type 2 diabetes mellitus (T2DM), cardiovascular diseases (CVDs), and obesity. Alcohol’s impact on these pathways impairs insulin sensitivity, lipid metabolism, and energy balance, collectively driving the development and progression of these diseases. Figure 2 summarizes the systemic effects of chronic ethanol consumption, depicting its impact on multiple organ systems, including the brain, liver, pancreas, and musculoskeletal system, and its association with various diseases. Figure 3 presents the role of alcohol metabolism in metabolic and inflammatory disorders, detailing the disruption of lipid metabolism, intestinal barrier integrity, immune homeostasis, oxidative stress, and their downstream consequences leading to conditions such as obesity, T2DM, and ALD.
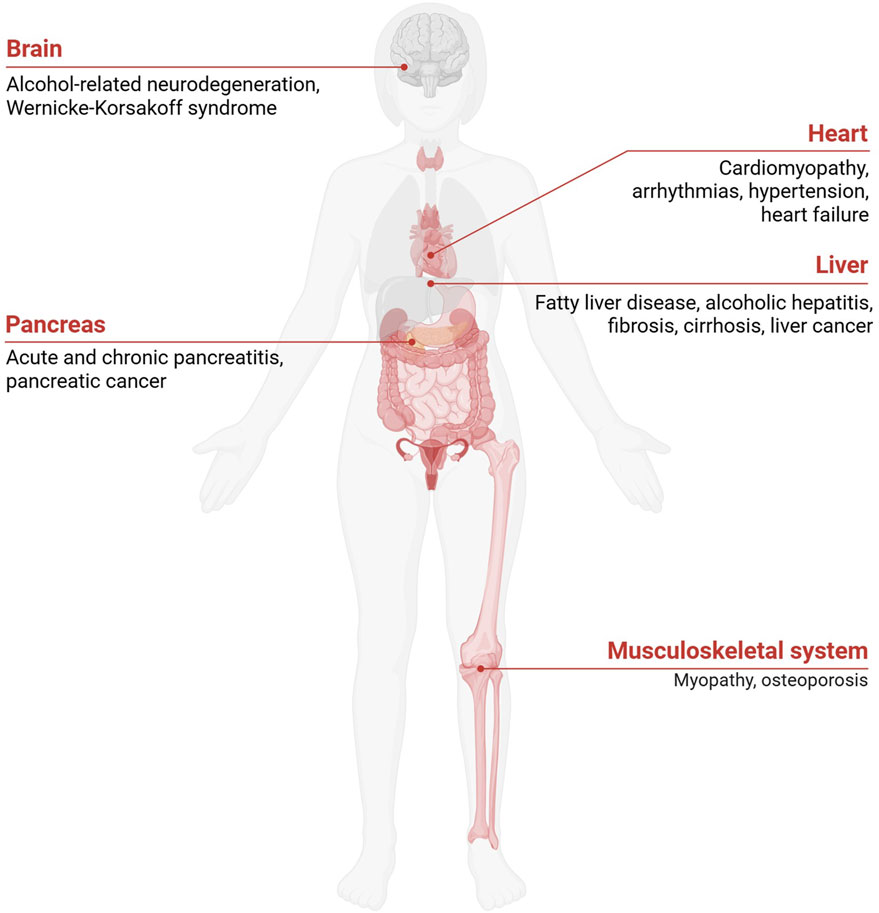
Figure 2. Pathological Consequences of Excessive Alcohol Consumption. Chronic alcohol consumption impacts multiple organ systems, including the brain, heart, liver, stomach, pancreas, kidney, musculoskeletal system, and reproductive system. Excessive alcohol consumption also promotes systemic inflammation and fat accumulation, exacerbating metabolic dysfunctions and increasing the risk of organ-specific diseases.
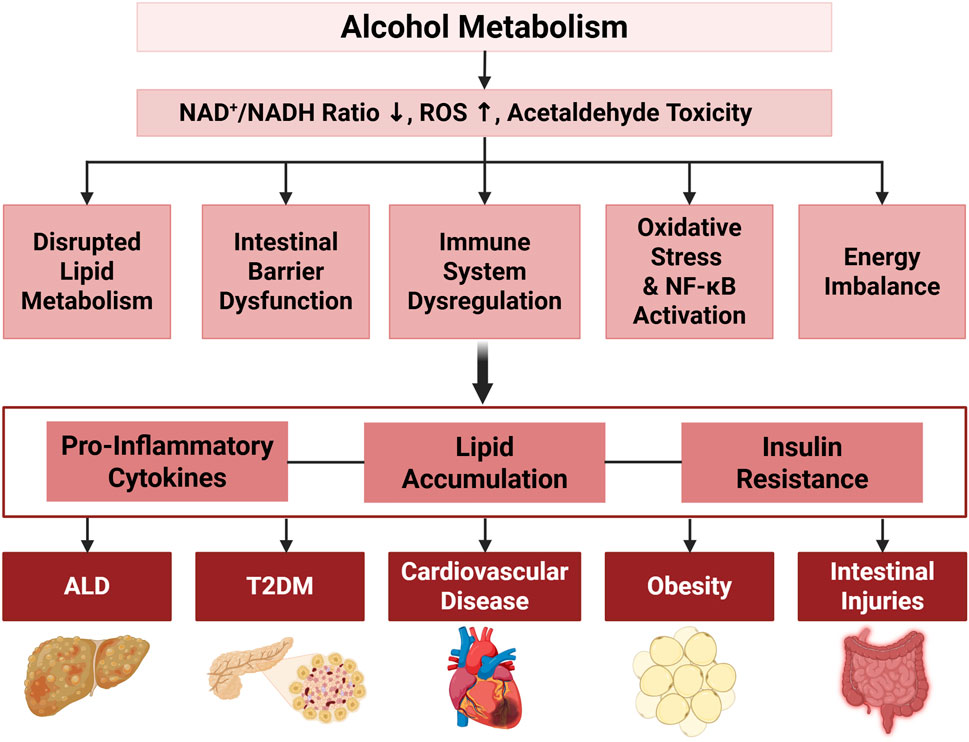
Figure 3. The Effects of Alcohol on Metabolic and Inflammatory Pathways Associated with Various Disorders. Alcohol metabolism disrupts cellular homeostasis by decreasing the NAD+/NADH ratio, increasing ROS, and inducing cellular toxicity, leading to disrupted lipid metabolism, intestinal microbiota and barrier function alterations, immune system dysregulation, oxidative stress with nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation, and energy imbalance through ADH1B activity. Key downstream effects include lipid accumulation and reduced β-oxidation, loss of beneficial gut bacteria with increased ROS, and elevated pro-inflammatory cytokines (e.g., Chemokine (C-C motif) ligand 2 (CCL2), IL-6, TNFα, IL-1β, IL-8). These processes contribute to obesity, lipid/glucose imbalances, and systemic inflammation, leading to the pathogenesis of ALD, T2DM, cardiovascular disease, obesity, and intestinal injuries.
3.1 Alcohol-related liver disease (ALD)
ALD is a major cause of alcohol-related mortality and encompasses a spectrum of liver conditions, ranging from alcoholic fatty liver (AFL) to cirrhosis and hepatocellular carcinoma (HCC) (Figure 2) (Prince et al., 2023). The progression of ALD is driven by chronic inflammation, oxidative stress, and metabolic dysfunction triggered by excessive alcohol consumption. The earliest stage of ALD is AFL, characterized by hepatic steatosis, or fat accumulation within hepatocytes (Yan et al., 2023). This stage can progress to alcoholic steatohepatitis, marked by liver inflammation, hepatocyte injury, and ballooning degeneration. Prolonged alcohol intake promotes persistent inflammation and fibrosis, eventually leading to cirrhosis, which significantly increases the risk of HCC (Fu et al., 2024).
Alcohol consumption triggers the activation of the NLRP3 inflammasome in Kupffer cells, which in turn activates caspase-1 and induces the release of IL-1β, exacerbating inflammation and liver damage (Brahadeeswaran et al., 2023). Additionally, alcohol-induced ROS production via CYP2E1 leads to mitochondrial dysfunction and oxidative stress, compromising liver regeneration and contributing to hepatocyte damage (Jin et al., 2013). Reactive nitrogen species (RNS), particularly peroxynitrite, further contribute to liver damage through nitrosative stress (Abdelmegeed and Song, 2014). Alcohol-induced disruptions in lipid metabolism also play a pivotal role in developing ALD by altering the NAD+/NADH ratio and impairing mitochondrial β-oxidation of fatty acids, which causes triglyceride accumulation in the liver (Figure 3) (Jeon and Carr, 2020). This metabolic imbalance is exacerbated by increased expression of sterol regulatory element-binding protein 1, a key regulator of lipid biosynthesis, and reduced activity of PPARα, a transcription factor essential for fatty acid oxidation (Hu et al., 2012). Moreover, alcohol-induced gut dysbiosis and increased intestinal permeability allow bacterial endotoxins to enter the liver, amplifying inflammation and hepatic damage (Bishehsari et al., 2017).
These intersecting mechanisms highlight the severe, multi-faceted nature of ALD. While alcohol moderation is often discussed as a preventive measure, substantial evidence indicates that toxic effects are present even at low to moderate levels of consumption (Cui and Koob, 2017; Eckardt et al., 1998). Furthermore, liver damage such as mitochondrial dysfunction can persist even after extended periods of abstinence, suggesting that recovery is neither immediate nor guaranteed (Salazar et al., 2025). Therefore, the most effective strategy to prevent the initiation and progression of ALD is the avoidance of alcohol. For individuals with existing damage, therapeutic approaches aimed at mitigating oxidative stress and cellular injury are being actively investigated (Wagner et al., 2024).
3.2 Type 2 diabetes mellitus (T2DM)
Excessive alcohol consumption is a known contributor to both the onset and progression of T2DM due to its detrimental impacts on insulin secretion and sensitivity. Alcohol impairs pancreatic β-cell function, reducing basal insulin secretion and increasing β-cell apoptosis (Steiner et al., 2015; Lin and Sun, 2010). This β-cell dysfunction, combined with alcohol-induced insulin resistance in the liver and skeletal muscle, disrupts glucose homeostasis, a hallmark of early-stage T2DM (Phielix and Mensink, 2008). Alcohol also interferes with glycogenolysis and gluconeogenesis, essential for maintaining blood glucose levels, which increases the risk of abnormal blood glucose regulation and hypoglycemia (Mauvais-Jarvis and Kahn, 2000; Kim and Kim, 2012). In the liver, alcohol activates hypoxia-inducible factor 1 (HIF-1), which upregulates glucose transporter 1, leading to increased glucose uptake and potential glucose toxicity (Morris and Yeligar, 2018; Nath et al., 2011).
Additionally, alcohol-induced dysregulation of key metabolic hormones, such as ghrelin and leptin, further contributes to the pathogenesis of T2DM. Ghrelin, a hormone that stimulates insulin secretion, is disrupted in T2DM patients who consume alcohol excessively, leading to increased hepatic glucose production and reduced peripheral glucose utilization (Kim and Kim, 2012). Leptin, which normally suppresses ghrelin and regulates glucose metabolism, is also dysregulated, exacerbating insulin resistance and glucose intolerance (Ju et al., 2011). Ethanol and its toxic metabolites accumulate in pancreatic tissues, causing cellular damage and inflammation, potentially leading to acute and chronic pancreatitis. The production of FAEEs during non-oxidative ethanol metabolism is particularly toxic to pancreatic cells, disrupting cellular membranes, promoting calcium overload, and inducing cell death, further contributing to alcoholic pancreatitis (Chowdhury and Gupta, 2006; Lee et al., 2021). Reduced insulin production diminishes the ability of the pancreas to maintain glucose homeostasis, worsening systemic effects such as hyperglycemia and insulin resistance, which exacerbate the metabolic complications of chronic alcohol consumption (Kim et al., 2010).
Taken together, these mechanisms indicate that excessive alcohol consumption accelerates both the development and severity of T2DM by impairing insulin function, altering glucose metabolism, and disrupting key metabolic hormones. These combined mechanisms underscore alcohol’s significant role in promoting T2DM progression and complications.
3.3 Cardiovascular disease (CVD)
The relationship between alcohol consumption and cardiovascular health is complex. Moderate alcohol intake has been associated with protective effects, including increased high-density lipoprotein (HDL) cholesterol levels, improved endothelial function, and reduced platelet aggregation that may lower the risk of coronary artery disease and atherosclerosis (Krenz and Korthuis, 2012; Perissinotto et al., 2010). However, these potential benefits are not attributed to all alcoholic beverages but are primarily linked to moderate red wine consumption, largely due to its high content of beneficial polyphenols (Castaldo et al., 2019). Furthermore, this association is often observed in the context of broader healthy lifestyle patterns, such as the Mediterranean diet, making it difficult to isolate the effects of red wine alone (Santos-Buelga et al., 2021). The evidence remains debated, and further research is required to fully elucidate the specific cardiovascular benefits of red wine and its components (Martínez-González and Hernández Hernández, 2024).
In contrast, excessive alcohol intake significantly elevates the risk of CVDs, such as alcohol-induced cardiomyopathy, characterized by reduced myocardial contractility, cardiac hypertrophy, and interstitial fibrosis (Figure 2) (Husain et al., 2014; Rehm et al., 2017). Chronic alcohol consumption disrupts lipid metabolism, raising low-density lipoprotein cholesterol and triglyceride levels, thereby contributing to atherosclerosis and hypertension (Husain et al., 2014). Additionally, excessive alcohol consumption elevates blood pressure and promotes vascular endothelial dysfunction, oxidative stress, and inflammation, all of which contribute to the development of CVDs (O'Keefe et al., 2018). The production of acetaldehyde and ROS in cardiac myocytes induces oxidative stress, mitochondrial dysfunction, and inflammation. These processes compromise structural and functional integrity of heart cells, impairing ATP production, disrupting calcium handling, and triggering apoptosis (El-Mas and Abdel-Rahman, 2019).
In addition, ethanol metabolism in the heart alters lipid metabolism, promoting dyslipidemia and fat accumulation within cardiac tissues by interfering with the regulation of lipid-modulating transcription factors, such as peroxisome proliferator-activated receptors (PPARs) (El-Mas and Abdel-Rahman, 2019). This lipid imbalance, along with the oxidative damage, accelerates the development of atherosclerosis and coronary artery disease. Chronic alcohol consumption also raises circulating catecholamine levels, leading to vasoconstriction and increased blood pressure, which heightens the risk of hypertension, stroke, and other cardiovascular complications (Hillbom et al., 2011). Cardiac dysrhythmias, such as atrial fibrillation, are also common in individuals who consume alcohol excessively, further compounding their cardiovascular risk (Husain et al., 2014).
Several genetic factors can influence individual susceptibility to alcohol-related cardiovascular damage. Variants of ADH and ALDH, such as ADH1B and ALDH2*2, alter the metabolism of alcohol and acetaldehyde, potentially increasing cardiovascular risks in certain populations (Chen et al., 2020). In summary, while moderate alcohol consumption may provide certain cardiovascular benefits, excessive intake overwhelmingly disrupts cardiovascular health, underscoring the critical balance needed to prevent alcohol-induced CVD.
3.4 Obesity and malnutrition
Alcohol’s high caloric content (7 kcal/g) can contribute to weight gain and obesity, especially in heavy drinkers. Beyond caloric intake, alcohol affects several metabolic pathways that promote fat accumulation, particularly in the liver and other peripheral tissues (Boyle et al., 2018; Ruhl and Everhart, 2005). Chronic alcohol consumption enhances lipolysis in adipose tissue, increasing circulating free fatty acids (FFAs), which are subsequently stored in the liver as triglycerides, contributing to alcoholic fatty liver disease (Li and Ding, 2017; Steiner and Lang, 2017). Ethanol metabolism significantly impacts adipose tissue by increasing lipolysis, altering hormone regulation, and promoting insulin resistance, leading to greater release of FFAs into the bloodstream (Mathur et al., 2023). Alcohol also disrupts the regulation of hormones involved in fat metabolism, such as adiponectin, leptin, and resistin (He et al., 2015). A primary target of this disruption is adiponectin, a hormone that promotes fatty acid oxidation, exerts anti-inflammatory effects, and improves insulin sensitivity (Xu et al., 2003). By suppressing the secretion and reducing plasma levels of adiponectin, alcohol triggers a cascade of metabolic dysfunctions, including impaired glucose tolerance, insulin resistance, and elevated circulating FFAs, which collectively exacerbate hepatic lipotoxicity and worsen obesity-related metabolic disorders (Steiner and Lang, 2017). Alcohol-induced upregulation of the microsomal ethanol-oxidizing system (MEOS), primarily involving CYP2E1, reduces fat utilization, favoring fat storage and further increasing the risk of obesity (Jiang et al., 2020; Suter et al., 2014).
Conversely, significant weight loss and malnutrition are also frequently observed, particularly in individuals with severe alcohol use disorder. This paradox is often linked to alcohol-associated disordered eating patterns, where alcoholic calories substitute for nutritious food. This disruption primarily occurs at the brush border membrane of the small intestine, where alcohol alters the transport of nutrients like glucose, amino acids, vitamins, and minerals (Butts et al., 2023). Ethanol disrupts the structural integrity of intestinal cells, reduces villus height, and affects transporter proteins, leading to increased intestinal permeability and decreased absorption efficiency (Elamin et al., 2014). This compromised intestinal barrier not only leads to malabsorption but also exacerbates systemic inflammation by allowing bacterial endotoxins to translocate into the bloodstream, further driving metabolic dysfunction. Additionally, alcohol’s low nutritional value and substitution for food contribute to deficiencies, cellular damage, and metabolic imbalances (Kokavec, 2008). Chronic alcohol consumption can also modulate the hypothalamic-pituitary-adrenal axis, affecting hunger signals and reducing appetite, particularly for carbohydrate-rich foods (Inder et al., 1995; Kokavec, 2008). These combined effects impair digestion and promote a negative energy balance, exacerbating malnutrition and health complications (Bode and Bode, 1997).
Genetic factors can also play a role, as evidenced by the ADH1B gene variant (rs1229984), which has been linked to significant weight gain in individuals who consumed alcohol, further highlighting the genetic susceptibility to alcohol-induced obesity (Yokoyama et al., 2013). Taken together, these findings highlight alcohol’s paradoxical and damaging role in energy balance, contributing to obesity in some while causing severe malnutrition in others, often in the context of disordered eating. Therefore, limiting alcohol consumption is crucial for reducing both health risks and its associated metabolic complications.
3.5 Neurological damage
Ethanol metabolism significantly affects the brain as alcohol can cross the blood-brain barrier (Wu and Cederbaum, 2003) and be metabolized locally (Figure 2). Acetaldehyde exerts neurotoxic effects, such as impaired synaptic function and neuronal apoptosis, by forming adducts with proteins and DNA that disrupt cellular processes (Takeuchi and Saito, 2005; Nutt et al., 2021). Chronic ethanol exposure disrupts brain energy metabolism, leading to reduced glucose uptake and impaired mitochondrial function in neurons. These energy deficits, along with excessive ROS production, contribute to neurodegenerative conditions, including alcohol-related dementia and Wernicke-Korsakoff syndrome (León et al., 2021; Sanvisens et al., 2017).
Beyond direct toxicity from its metabolites, a central mechanism of alcohol-induced brain injury is the induction of a persistent neuroinflammatory state. While chronic alcohol use is known to activate microglia, it also triggers a reactive response in other glial cells like astrocytes. This widespread glial activation results in the release of a barrage of pro-inflammatory cytokines (e.g., TNF-α, IL-1β), chemokines, and additional ROS within the brain. This sustained neuroinflammation, driven by oxidative stress, damages neurons, disrupts synaptic plasticity, and is now understood to be a key driver of the cognitive deficits and emotional dysregulation characteristic of alcohol use disorder (Pervin and Stephen, 2021).
Moreover, alcohol disrupts neurotransmitter balance, with effects varying by brain region and neuronal cell type, by enhancing gamma-aminobutyric acid (GABA) activity and inhibiting glutamate signaling, leading to impairments in mood regulation, memory, and motor coordination (Oscar-Berman and Marinkovic, 2003). Over time, the combined insults of direct toxicity, oxidative stress, persistent neuroinflammation, and widespread neurotransmitter dysregulation impair synaptic plasticity, reduce cognitive function, and lead to significant neuronal loss and brain atrophy (León et al., 2021).
3.6 Alcoholic myopathy and muscle wasting
In addition to the organ systems discussed, ethanol exerts adverse effects on skeletal muscle by suppressing the mammalian target of rapamycin (mTOR) signaling, leading to reduced muscle protein synthesis (Levitt et al., 2022) and activating the ubiquitin-proteasome pathway for muscle protein degradation (Preedy et al., 2002). Additionally, its metabolites, e.g., acetaldehyde and acetate, further disrupt glucose and fatty acid metabolism (Steiner and Lang, 2015). In skeletal muscle, acetate derived from ethanol metabolism is converted into acetyl-CoA and enters the TCA cycle for energy production (Wilson and Matschinsky, 2020).
Chronic alcohol consumption reduces insulin sensitivity by impairing the activation of key proteins in insulin signaling, such as insulin receptor substrate-1 (IRS-1) and AKT, leading to reduced glucose uptake in muscle cells (Steiner et al., 2015). This disruption is further exacerbated by oxidative stress and pro-inflammatory cytokines, which interfere with normal insulin signal transduction and promote insulin resistance. The resulting decrease in glucose utilization leads to muscle wasting, a hallmark of alcoholic myopathy, characterized by muscle weakness, atrophy, and loss of muscle mass (Simon et al., 2017). Chronic alcohol exposure also inhibits protein synthesis by suppressing the mTOR pathway and increases protein degradation through the activation of the ubiquitin-proteasome system, involving muscle-specific E3 ubiquitin ligases, such as muscle ring finger 1 and atrogin-1, leading to muscle wasting (Simon et al., 2017). In addition, increased production of ROS in muscle, often driven by impaired mitochondrial function by chronic alcohol exposure, further damages muscle fibers and reduces ATP production, limiting muscle endurance and performance and causing structural damage, particularly in fast-twitch muscle fibers (Powers, 2014).
4 Summary and future directions
This review has highlighted the extensive detrimental effects of excessive alcohol consumption across multiple organ systems. These harmful effects stem from alcohol’s toxicity, its promotion of oxidative stress, and the substantial reduction in the NAD+/NADH ratio. Alcohol and its metabolites disturb the regulation of lipid and glucose metabolism, increasing the production of ROS and RNS, which further drives oxidative stress and inflammation, ultimately leading to cellular and organ damage. These interconnected pathways not only drive a range of chronic diseases, including liver disease, CVD, T2DM, and obesity, but also provide a clear roadmap for developing targeted therapeutic strategies.
While abstinence is paramount, emerging approaches aim to target the core mechanisms of injury. Promising pharmacological strategies include blocking inflammatory cell recruitment with chemokine receptor antagonists. Furthermore, nutritional and metabolic interventions counteract alcohol-induced damage. Supplementation with NAD+ precursors like nicotinamide riboside (NR) or nicotinamide mononucleotide (NMN) helps replenish the depleted NAD+ pool, while therapies targeting the gut-liver axis, such as probiotics or fecal microbiota transplantation work to repair intestinal barrier function and reduce systemic inflammation.
Ultimately, the shared mechanisms of oxidative stress, inflammatory responses, mitochondrial dysfunction, and metabolic disruption demonstrate alcohol’s systemic impact. This mechanistic overlap underscores the systemic risks posed by excessive alcohol consumption and emphasizes that reducing alcohol consumption is the safest strategy to mitigate the broad spectrum of alcohol-related health complications.
Author contributions
JL: Visualization, Writing – original draft. J-YL: Conceptualization, Writing – review and editing. HK: Conceptualization, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by the National Research Foundation of Korea (NRF) (No. RS-2025-16068795), funded by the Ministry of Science and ICT.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdelmegeed, M. A., and Song, B. J. (2014). Functional roles of protein nitration in acute and chronic liver diseases. Oxid. Med. Cell Longev. 2014, 149627. doi:10.1155/2014/149627
Ali, H., Assiri, M. A., Shearn, C. T., and Fritz, K. S. (2019). Lipid peroxidation derived reactive aldehydes in alcoholic liver disease. Curr. Opin. Toxicol. 13, 110–117. doi:10.1016/j.cotox.2018.10.003
Bailey, S. M., and Cunningham, C. C. (2002). Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic. Biol. Med. 32, 11–16. doi:10.1016/s0891-5849(01)00769-9
Beckhauser, T. F., Francis-Oliveira, J., and DE Pasquale, R. (2016). Reactive oxygen species: physiological and physiopathological effects on synaptic plasticity. J. Exp. Neurosci. 10, 23–48. doi:10.4137/JEN.S39887
Bedard, K., and Krause, K. H. (2007). The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313. doi:10.1152/physrev.00044.2005
Bishehsari, F., Magno, E., Swanson, G., Desai, V., Voigt, R. M., Forsyth, C. B., et al. (2017). Alcohol and gut-derived inflammation. Alcohol Res. 38, 163–171.
Bode, C., and Bode, J. C. (1997). Alcohol's role in gastrointestinal tract disorders. Alcohol Health Res. World 21, 76–83.
Boyle, M., Masson, S., and Anstee, Q. M. (2018). The bidirectional impacts of alcohol consumption and the metabolic syndrome: cofactors for progressive fatty liver disease. J. Hepatol. 68, 251–267. doi:10.1016/j.jhep.2017.11.006
Brahadeeswaran, S., Dasgupta, T., Manickam, V., Saraswathi, V., and Tamizhselvi, R. (2023). NLRP3: a new therapeutic target in alcoholic liver disease. Front. Immunol. 14, 1215333. doi:10.3389/fimmu.2023.1215333
Butts, M., Sundaram, V. L., Murughiyan, U., Borthakur, A., and Singh, S. (2023). The influence of alcohol consumption on intestinal nutrient absorption: a comprehensive review. Nutrients 15, 1571. doi:10.3390/nu15071571
Carvajal, F., Sánchez-Gil, A., Cardona, D., Rincón-Cervera, M. A., and Lerma-Cabrera, J. M. (2023). The effect of very-long-chain n-3 polyunsaturated fatty acids in the central nervous system and their potential benefits for treating alcohol use disorder: reviewing pre-clinical and clinical data. Nutrients 15, 2993. doi:10.3390/nu15132993
Castaldo, L., Narváez, A., Izzo, L., Graziani, G., Gaspari, A., DI Minno, G., et al. (2019). Red wine consumption and cardiovascular health. Molecules 24, 3626. doi:10.3390/molecules24193626
Cederbaum, A. I. (2012). Alcohol metabolism. Clin. Liver Dis. 16, 667–685. doi:10.1016/j.cld.2012.08.002
Chen, C. H., Ferreira, J. C. B., Joshi, A. U., Stevens, M. C., Li, S. J., Hsu, J. H., et al. (2020). Novel and prevalent non-East Asian ALDH2 variants; Implications for global susceptibility to aldehydes' toxicity. EBioMedicine 55, 102753. doi:10.1016/j.ebiom.2020.102753
Chowdhury, P., and Gupta, P. (2006). Pathophysiology of alcoholic pancreatitis: an overview. World J. Gastroenterol. 12, 7421–7427. doi:10.3748/wjg.v12.i46.7421
Cortés-Rojo, C., Vargas-Vargas, M. A., Olmos-Orizaba, B. E., Rodríguez-Orozco, A. R., and Calderón-Cortés, E. (2020). Interplay between NADH oxidation by complex I, glutathione redox state and sirtuin-3, and its role in the development of insulin resistance. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1866, 165801. doi:10.1016/j.bbadis.2020.165801
Crews, F. T., Coleman, L. G., Macht, V. A., and Vetreno, R. P. (2024). Alcohol, HMGB1, and innate immune signaling in the brain. Alcohol Res. 44, 04. doi:10.35946/arcr.v44.1.04
Cui, C., and Koob, G. F. (2017). Titrating tipsy targets: the neurobiology of low-dose alcohol. Trends Pharmacol. Sci. 38, 556–568. doi:10.1016/j.tips.2017.03.002
Dabravolski, S. A., Bezsonov, E. E., Baig, M. S., Popkova, T. V., and Orekhov, A. N. (2021). Mitochondrial lipid homeostasis at the crossroads of liver and heart diseases. Int. J. Mol. Sci. 22, 6949. doi:10.3390/ijms22136949
Eckardt, M. J., File, S. E., Gessa, G. L., Grant, K. A., Guerri, C., Hoffman, P. L., et al. (1998). Effects of moderate alcohol consumption on the central nervous system. Alcohol Clin. Exp. Res. 22, 998–1040. doi:10.1111/j.1530-0277.1998.tb03695.x
EL-Mas, M. M., and Abdel-Rahman, A. A. (2019). Role of alcohol oxidative metabolism in its cardiovascular and autonomic effects. Adv. Exp. Med. Biol. 1193, 1–33. doi:10.1007/978-981-13-6260-6_1
Elamin, E., Masclee, A., Troost, F., Pieters, H. J., Keszthelyi, D., Aleksa, K., et al. (2014). Ethanol impairs intestinal barrier function in humans through mitogen activated protein kinase signaling: a combined in vivo and in vitro approach. PLoS One 9, e107421. doi:10.1371/journal.pone.0107421
Fairfield, B., and Schnabl, B. (2021). Gut dysbiosis as a driver in alcohol-induced liver injury. JHEP Rep. 3, 100220. doi:10.1016/j.jhepr.2020.100220
Fu, Y., Maccioni, L., Wang, X. W., Greten, T. F., and Gao, B. (2024). Alcohol-associated liver cancer. Hepatology 80, 1462–1479. doi:10.1097/HEP.0000000000000890
Guo, C., Sun, L., Chen, X., and Zhang, D. (2013). Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 8, 2003–2014. doi:10.3969/j.issn.1673-5374.2013.21.009
Gwon, Y., Moon, C. Y., Lee, E. H., Im, S. S., and Kang, H. Y. J. (2024). Anti-inflammatory effects of ginsenoside compound K in ethanol-stimulated macrophages by modulating sirtuin 1. J. Funct. Foods 117, 106218. doi:10.1016/j.jff.2024.106218
He, Z., Li, M., Zheng, D., Chen, Q., Liu, W., and Feng, L. (2015). Adipose tissue hypoxia and low-grade inflammation: a possible mechanism for ethanol-related glucose intolerance? Br. J. Nutr. 113, 1355–1364. doi:10.1017/S000711451500077X
Hillbom, M., Saloheimo, P., and Juvela, S. (2011). Alcohol consumption, blood pressure, and the risk of stroke. Curr. Hypertens. Rep. 13, 208–213. doi:10.1007/s11906-011-0194-y
Hoek, J. B., Cahill, A., and Pastorino, J. G. (2002). Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology 122, 2049–2063. doi:10.1053/gast.2002.33613
Hu, M., Wang, F., Li, X., Rogers, C. Q., Liang, X., Finck, B. N., et al. (2012). Regulation of hepatic lipin-1 by ethanol: role of AMP-activated protein kinase/sterol regulatory element-binding protein 1 signaling in mice. Hepatology 55, 437–446. doi:10.1002/hep.24708
Huang, M.-C., Chen, C.-H., Peng, F.-C., Tang, S.-H., and Chen, C.-C. (2009). Alterations in oxidative stress status during early alcohol withdrawal in alcoholic patients. J. Formos. Med. Assoc. 108, 560–569. doi:10.1016/S0929-6646(09)60374-0
Husain, K., Ansari, R. A., and Ferder, L. (2014). Alcohol-induced hypertension: mechanism and prevention. World J. Cardiol. 6, 245–252. doi:10.4330/wjc.v6.i5.245
Inder, W. J., Joyce, P. R., Ellis, M. J., Evans, M. J., Livesey, J. H., and Donald, R. A. (1995). The effects of alcoholism on the hypothalamic-pituitary-adrenal axis: interaction with endogenous opioid peptides. Clin. Endocrinol. (Oxf) 43, 283–290. doi:10.1111/j.1365-2265.1995.tb02033.x
Iside, C., Scafuro, M., Nebbioso, A., and Altucci, L. (2020). SIRT1 activation by natural phytochemicals: an overview. Front. Pharmacol. 11, 1225. doi:10.3389/fphar.2020.01225
Jelski, W., Laniewska-Dunaj, M., Strumnik, A., and Szmitkowski, M. (2014). The alcohol dehydrogenase isoenzyme alcohol dehydrogenase IV as a candidate marker of Helicobacter pylori infection. Arch. Med. Sci. 10, 951–955. doi:10.5114/aoms.2014.46215
Jeon, S., and Carr, R. (2020). Alcohol effects on hepatic lipid metabolism. J. Lipid Res. 61, 470–479. doi:10.1194/jlr.R119000547
Jiang, Y., Zhang, T., Kusumanchi, P., Han, S., Yang, Z., and Liangpunsakul, S. (2020). Alcohol metabolizing enzymes, microsomal ethanol oxidizing system, cytochrome P450 2E1, catalase, and aldehyde dehydrogenase in alcohol-associated liver disease. Biomedicines 8, 50. doi:10.3390/biomedicines8030050
Jin, M., Ande, A., Kumar, A., and Kumar, S. (2013). Regulation of cytochrome P450 2e1 expression by ethanol: role of oxidative stress-mediated pkc/jnk/sp1 pathway. Cell Death & Dis. 4, e554. doi:10.1038/cddis.2013.78
Ju, A., Cheon, Y. H., Lee, K. S., Lee, S. S., Lee, W. Y., Won, W. Y., et al. (2011). The change of plasma ghrelin and leptin levels by the development of type 2 diabetes mellitus in patients with alcohol dependence. Alcohol Clin. Exp. Res. 35, 905–911. doi:10.1111/j.1530-0277.2010.01420.x
Juan, C. A., Pérez DE LA Lastra, J. M., Plou, F. J., and Pérez-Lebeña, E. (2021). The Chemistry of reactive oxygen species (ROS) Revisited: Outlining their role in Biological Macromolecules (DNA, lipids and proteins) and induced pathologies. Int. J. Mol. Sci. 22, 4642. doi:10.3390/ijms22094642
Kang, H., Lee, Y., Bae, M., Park, Y. K., and Lee, J. Y. (2020). Astaxanthin inhibits alcohol-induced inflammation and oxidative stress in macrophages in a sirtuin 1-dependent manner. J. Nutr. Biochem. 85, 108477. doi:10.1016/j.jnutbio.2020.108477
Kang, H., Park, Y. K., and Lee, J. Y. (2021a). Inhibition of alcohol-induced inflammation and oxidative stress by astaxanthin is mediated by its opposite actions in the regulation of sirtuin 1 and histone deacetylase 4 in macrophages. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1866, 158838. doi:10.1016/j.bbalip.2020.158838
Kang, H., Park, Y. K., and Lee, J. Y. (2021b). Nicotinamide riboside, an NAD(+) precursor, attenuates inflammation and oxidative stress by activating sirtuin 1 in alcohol-stimulated macrophages. Lab. Invest 101, 1225–1237. doi:10.1038/s41374-021-00599-1
Kang, H., Kim, M. B., Park, Y. K., and Lee, J. Y. (2022). A mouse model of the regression of alcoholic hepatitis: Monitoring the regression of hepatic steatosis, inflammation, oxidative stress, and NAD(+) metabolism upon alcohol withdrawal. J. Nutr. Biochem. 99, 108852. doi:10.1016/j.jnutbio.2021.108852
Kelley, N., Jeltema, D., Duan, Y., and He, Y. (2019). The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 20, 3328. doi:10.3390/ijms20133328
Kim, S. J., and Kim, D. J. (2012). Alcoholism and diabetes mellitus. Diabetes Metab. J. 36, 108–115. doi:10.4093/dmj.2012.36.2.108
Kim, J. Y., Song, E. H., Lee, H. J., Oh, Y. K., Park, Y. S., Park, J.-W., et al. (2010). Chronic ethanol consumption-induced pancreatic {beta}-cell dysfunction and apoptosis through glucokinase nitration and its down-regulation. J. Biol. Chem. 285, 37251–37262. doi:10.1074/jbc.M110.142315
Kokavec, A. (2008). Is decreased appetite for food a physiological consequence of alcohol consumption? Appetite 51, 233–243. doi:10.1016/j.appet.2008.03.011
Koob, G. F. (2024). Alcohol use disorder treatment: problems and solutions. Annu. Rev. Pharmacol. Toxicol. 64, 255–275. doi:10.1146/annurev-pharmtox-031323-115847
Koop, D. R. (2006). Alcohol metabolism's damaging effects on the cell: a focus on reactive oxygen generation by the enzyme cytochrome P450 2E1. Alcohol Res. Health 29, 274–280.
Krenz, M., and Korthuis, R. J. (2012). Moderate ethanol ingestion and cardiovascular protection: from epidemiologic associations to cellular mechanisms. J. Mol. Cell Cardiol. 52, 93–104. doi:10.1016/j.yjmcc.2011.10.011
Lee, J., Lim, J. W., and Kim, H. (2021). Lycopene inhibits oxidative stress-mediated inflammatory responses in ethanol/Palmitoleic acid-stimulated pancreatic Acinar AR42J cells. Int. J. Mol. Sci. 22, 2101. doi:10.3390/ijms22042101
Lee, J., Woo, H., Kang, H., Park, Y. K., and Lee, J. Y. (2025). Nicotinamide riboside targets mitochondrial unfolded protein response to reduce alcohol-induced damage in Kupffer cells. J. Pathol. 265, 110–122. doi:10.1002/path.6372
León, B. E., Kang, S., Franca-Solomon, G., Shang, P., and Choi, D. S. (2021). Alcohol-induced neuroinflammatory response and mitochondrial dysfunction on aging and Alzheimer's disease. Front. Behav. Neurosci. 15, 778456. doi:10.3389/fnbeh.2021.778456
Levitt, D. E., Luk, H. Y., and Vingren, J. L. (2022). Alcohol, resistance Exercise, and mTOR pathway signaling: an evidence-Based Narrative review. Biomolecules 13, 2. doi:10.3390/biom13010002
Li, Y., and Ding, W.-X. (2017). Adipose tissue autophagy and homeostasis in alcohol-induced liver injury. Liver Res. 1, 54–62. doi:10.1016/j.livres.2017.03.004
Lin, Y., and Sun, Z. (2010). Current views on type 2 diabetes. J. Endocrinol. 204, 1–11. doi:10.1677/joe-09-0260
Litwinowicz, K., Choroszy, M., and Waszczuk, E. (2020). Changes in the composition of the human intestinal microbiome in alcohol use disorder: a systematic review. Am. J. Drug Alcohol Abuse 46, 4–12. doi:10.1080/00952990.2019.1669629
Liu, Y. J., Mcintyre, R. L., Janssens, G. E., and Houtkooper, R. H. (2020). Mitochondrial fission and fusion: a dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 186, 111212. doi:10.1016/j.mad.2020.111212
Lu, Y., and Cederbaum, A. I. (2008). CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 44, 723–738. doi:10.1016/j.freeradbiomed.2007.11.004
Lu, M., Zhou, L., Stanley, W. C., Cabrera, M. E., Saidel, G. M., and Yu, X. (2008). Role of the malate-aspartate shuttle on the metabolic response to myocardial ischemia. J. Theor. Biol. 254, 466–475. doi:10.1016/j.jtbi.2008.05.033
Martin, P. R., Singleton, C. K., and Hiller-Sturmhöfel, S. (2003). The role of thiamine deficiency in alcoholic brain disease. Alcohol Res. Health 27, 134–142.
Martínez-González, M. Á., and Hernández Hernández, A. (2024). Effect of the Mediterranean diet in cardiovascular prevention. Rev. Esp. Cardiol. Engl. Ed. 77, 574–582. doi:10.1016/j.rec.2024.01.006
Mathur, M., Yeh, Y. T., Arya, R. K., Jiang, L., Pornour, M., Chen, W., et al. (2023). Adipose lipolysis is important for ethanol to induce fatty liver in the National Institute on Alcohol Abuse and Alcoholism murine model of chronic and binge ethanol feeding. Hepatology 77, 1688–1701. doi:10.1002/hep.32675
Mauvais-Jarvis, F., and Kahn, C. R. (2000). Understanding the pathogenesis and treatment of insulin resistance and type 2 diabetes mellitus: what can we learn from transgenic and knockout mice? Diabetes Metab. 26, 433–448.
Morris, N. L., and Yeligar, S. M. (2018). Role of HIF-1α in alcohol-mediated multiple organ dysfunction. Biomolecules 8, 170. doi:10.3390/biom8040170
Nath, B., Levin, I., Csak, T., Petrasek, J., Mueller, C., Kodys, K., et al. (2011). Hepatocyte-specific hypoxia-inducible factor-1α is a determinant of lipid accumulation and liver injury in alcohol-induced steatosis in mice. Hepatology 53, 1526–1537. doi:10.1002/hep.24256
Nutt, D., Hayes, A., Fonville, L., Zafar, R., Palmer, E. O. C., Paterson, L., et al. (2021). Alcohol and the brain. Nutrients 13, 3938. doi:10.3390/nu13113938
O'Keefe, E. L., Dinicolantonio, J. J., O'Keefe, J. H., and Lavie, C. J. (2018). Alcohol and CV health: Jekyll and Hyde J-Curves. Prog. Cardiovasc Dis. 61, 68–75. doi:10.1016/j.pcad.2018.02.001
Onodera, Y., Teramura, T., Takehara, T., Shigi, K., and Fukuda, K. (2015). Reactive oxygen species induce Cox-2 expression via TAK1 activation in synovial fibroblast cells. FEBS Open Bio 5, 492–501. doi:10.1016/j.fob.2015.06.001
Oscar-Berman, M., and Marinkovic, K. (2003). Alcoholism and the brain: an overview. Alcohol Res. Health 27, 125–133.
Osna, N. A., Donohue, T. M., and Kharbanda, K. K. (2017). Alcoholic liver disease: pathogenesis and current Management. Alcohol Res. 38, 147–161.
Palma, E., Riva, A., Moreno, C., Odena, G., Mudan, S., Manyakin, N., et al. (2020). Perturbations in mitochondrial dynamics are closely involved in the progression of alcoholic liver disease. Alcohol Clin. Exp. Res. 44, 856–865. doi:10.1111/acer.14299
Park, S. H., Seo, W., Xu, M. J., Mackowiak, B., Lin, Y., He, Y., et al. (2023). Ethanol and its Nonoxidative metabolites promote acute liver injury by inducing ER stress, Adipocyte death, and lipolysis. Cell Mol. Gastroenterol. Hepatol. 15, 281–306. doi:10.1016/j.jcmgh.2022.10.002
Patel, S., Behara, R., Swanson, G. R., Forsyth, C. B., Voigt, R. M., and Keshavarzian, A. (2015). Alcohol and the intestine. Biomolecules 5, 2573–2588. doi:10.3390/biom5042573
Pellegrino, M. W., Nargund, A. M., and Haynes, C. M. (2013). Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 1833, 410–416. doi:10.1016/j.bbamcr.2012.02.019
Perissinotto, E., Buja, A., Maggi, S., Enzi, G., Manzato, E., Scafato, E., et al. (2010). Alcohol consumption and cardiovascular risk factors in older lifelong wine drinkers: the Italian Longitudinal Study on Aging. Nutr. Metab. Cardiovasc Dis. 20, 647–655. doi:10.1016/j.numecd.2009.05.014
Pervin, Z., and Stephen, J. M. (2021). Effect of alcohol on the central nervous system to develop neurological disorder: pathophysiological and lifestyle modulation can be potential therapeutic options for alcohol-induced neurotoxication. AIMS Neurosci. 8, 390–413. doi:10.3934/Neuroscience.2021021
Phielix, E., and Mensink, M. (2008). Type 2 diabetes mellitus and skeletal muscle metabolic function. Physiol. Behav. 94, 252–258. doi:10.1016/j.physbeh.2008.01.020
Powers, S. K. (2014). Can antioxidants protect against disuse muscle atrophy? Sports Med. 44 (Suppl. 2), S155–S165. doi:10.1007/s40279-014-0255-x
Preedy, V. R., Adachi, J., Asano, M., Koll, M., Mantle, D., Niemela, O., et al. (2002). Free radicals in alcoholic myopathy: indices of damage and preventive studies. Free Radic. Biol. Med. 32, 683–687. doi:10.1016/s0891-5849(01)00794-8
Prince, D. S., Nash, E., and Liu, K. (2023). Alcohol-associated liver disease: Evolving Concepts and treatments. Drugs 83, 1459–1474. doi:10.1007/s40265-023-01939-9
Rehm, J., Hasan, O. S. M., Imtiaz, S., and Neufeld, M. (2017). Quantifying the contribution of alcohol to cardiomyopathy: a systematic review. Alcohol 61, 9–15. doi:10.1016/j.alcohol.2017.01.011
Ruhl, C. E., and Everhart, J. E. (2005). Joint effects of body weight and alcohol on elevated serum alanine aminotransferase in the United States population. Clin. Gastroenterol. Hepatol. 3, 1260–1268. doi:10.1016/s1542-3565(05)00743-3
Salazar, C., Barreto, M., Adriasola-Carrasco, A. A., Carvajal, F., Lerma-Cabrera, J. M., and Ruiz, L. M. (2025). Lactobacillus rhamnosus GG modulates mitochondrial function and antioxidant responses in an ethanol-exposed in vivo model: evidence of HIGD2A-dependent OXPHOS remodeling in the liver. Antioxidants (Basel) 14, 627. doi:10.3390/antiox14060627
Santos-Buelga, C., González-Manzano, S., and González-Paramás, A. M. (2021). Wine, polyphenols, and Mediterranean diets. What else is there to say? Molecules 26, 5537. doi:10.3390/molecules26185537
Sanvisens, A., Zuluaga, P., Fuster, D., Rivas, I., Tor, J., Marcos, M., et al. (2017). Long-term mortality of patients with an alcohol-related Wernicke–korsakoff syndrome. Alcohol Alcohol. 52, 466–471. doi:10.1093/alcalc/agx013
Shi, L., and Tu, B. P. (2015). Acetyl-CoA and the regulation of metabolism: mechanisms and consequences. Curr. Opin. Cell Biol. 33, 125–131. doi:10.1016/j.ceb.2015.02.003
Shukla, P. K., Meena, A. S., Dalal, K., Canelas, C., Samak, G., Pierre, J. F., et al. (2021). Chronic stress and corticosterone exacerbate alcohol-induced tissue injury in the gut-liver-brain axis. Sci. Rep. 11, 826. doi:10.1038/s41598-020-80637-y
Siggins, R. W., Mcternan, P. M., Simon, L., Souza-Smith, F. M., and Molina, P. E. (2023). Mitochondrial dysfunction: at the nexus between alcohol-associated immunometabolic dysregulation and tissue injury. Int. J. Mol. Sci. 24, 8650. doi:10.3390/ijms24108650
Simon, L., Jolley, S. E., and Molina, P. E. (2017). Alcoholic myopathy: pathophysiologic mechanisms and clinical Implications. Alcohol Res. 38, 207–217.
Singh, V., and Ubaid, S. (2020). Role of Silent Information regulator 1 (SIRT1) in regulating oxidative stress and inflammation. Inflammation 43, 1589–1598. doi:10.1007/s10753-020-01242-9
Steiner, J. L., and Lang, C. H. (2015). Dysregulation of skeletal muscle protein metabolism by alcohol. Am. J. Physiol. Endocrinol. Metab. 308, E699–E712. doi:10.1152/ajpendo.00006.2015
Steiner, J. L., and Lang, C. H. (2017). Alcohol, adipose tissue and lipid dysregulation. Biomolecules 7, 16. doi:10.3390/biom7010016
Steiner, J. L., Crowell, K. T., and Lang, C. H. (2015). Impact of alcohol on Glycemic control and insulin action. Biomolecules 5, 2223–2246. doi:10.3390/biom5042223
Su, G. L., Klein, R. D., Aminlari, A., Zhang, H. Y., Steinstraesser, L., Alarcon, W. H., et al. (2000). Kupffer cell activation by lipopolysaccharide in rats: role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology 31, 932–936. doi:10.1053/he.2000.5634
Suter, M. A., Ma, J., Vuguin, P. M., Hartil, K., Fiallo, A., Harris, R. A., et al. (2014). In utero exposure to a maternal high-fat diet alters the epigenetic histone code in a murine model. Am. J. Obstet. Gynecol. 210, 463.e1–463.e11. doi:10.1016/j.ajog.2014.01.045
Takeuchi, M., and Saito, T. (2005). Cytotoxicity of acetaldehyde-derived advanced glycation end-products (AA-AGE) in alcoholic-induced neuronal degeneration. Alcohol Clin. Exp. Res. 29, 220S–224S. doi:10.1097/01.alc.0000190657.97988.c7
Tsermpini, E. E., Plemenitaš Ilješ, A., and Dolžan, V. (2022). Alcohol-induced oxidative stress and the role of antioxidants in alcohol Use disorder: a systematic review. Antioxidants (Basel) 11, 1374. doi:10.3390/antiox11071374
Wagner, J., Park, L. M., Mukhopadhyay, P., Matyas, C., Trojnar, E., Damadzic, R., et al. (2024). PCSK9 inhibition attenuates alcohol-associated neuronal oxidative stress and cellular injury. Brain Behav. Immun. 119, 494–506. doi:10.1016/j.bbi.2024.04.022
Wheeler, M. D. (2003). Endotoxin and Kupffer cell activation in alcoholic liver disease. Alcohol Res. Health 27, 300–306.
Wilson, D. F., and Matschinsky, F. M. (2020). Ethanol metabolism: the good, the bad, and the ugly. Med. Hypotheses 140, 109638. doi:10.1016/j.mehy.2020.109638
World Health Organization (2024). Global status report on alcohol and health and treatment of substance use disorders. Geneva: World Health Organization.
Wu, D., and Cederbaum, A. I. (2003). Alcohol, oxidative stress, and free radical damage. Alcohol Res. Health 27, 277–284.
Xie, N., Zhang, L., Gao, W., Huang, C., Huber, P. E., Zhou, X., et al. (2020). NAD(+) metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target Ther. 5, 227. doi:10.1038/s41392-020-00311-7
Xu, A., Wang, Y., Keshaw, H., Xu, L. Y., Lam, K. S., and Cooper, G. J. (2003). The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Invest 112, 91–100. doi:10.1172/JCI17797
Yan, C., Hu, W., Tu, J., Li, J., Liang, Q., and Han, S. (2023). Pathogenic mechanisms and regulatory factors involved in alcoholic liver disease. J. Transl. Med. 21, 300. doi:10.1186/s12967-023-04166-8
Yokoyama, A., Yokoyama, T., Matsui, T., Mizukami, T., Matsushita, S., Higuchi, S., et al. (2013). Alcohol dehydrogenase-1B genotype (rs1229984) is a strong determinant of the relationship between body weight and alcohol intake in Japanese alcoholic men. Alcohol Clin. Exp. Res. 37, 1123–1132. doi:10.1111/acer.12069
Zakhari, S. (2006). Overview: how is alcohol metabolized by the body? Alcohol Res. Health 29, 245–254.
Zhang, T., Berrocal, J. G., Frizzell, K. M., Gamble, M. J., Dumond, M. E., Krishnakumar, R., et al. (2009). Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene Promoters. J. Biol. Chem. 284, 20408–20417. doi:10.1074/jbc.M109.016469
Zhong, W., Wei, X., Hao, L., Lin, T. D., Yue, R., Sun, X., et al. (2020). Paneth cell dysfunction Mediates alcohol-related steatohepatitis through promoting bacterial translocation in mice: role of Zinc deficiency. Hepatology 71, 1575–1591. doi:10.1002/hep.30945
Glossary
ACS acetyl-CoA synthetase
ADH alcohol dehydrogenase
AFL alcoholic fatty liver
ALD alcohol-related liver disease
ALDH aldehyde dehydrogenase
acetyl-CoA acetyl coenzyme A
CCL2 chemokine (C-C motif) ligand 2
CYP2E1 cytochrome P450 2E1
FAEE fatty acid ethyl esters
FFA free fatty acid
GI gastrointestinal
IL-1β interleukin-1β
IL-6 interleukin-6
LDH lactate dehydrogenase
LPS lipopolysaccharide
mtDNA mitochondrial DNA
NAMPT nicotinamide phosphoribosyltransferase
NAD+ nicotinamide adenine dinucleotide
NADH a reduced form of nicotinamide adenine dinucleotide
NADPH nicotinamide adenine dinucleotide phosphate
NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells
NLRP3 NOD-like receptor family pyrin domain containing 3
PPAR peroxisome proliferator-activated receptor
RNS reactive nitrogen species
ROS reactive oxygen species
SCFA short-chain fatty acid
SIRT1 sirtuin 1
TCA tricarboxylic acid
T2DM type 2 diabetes mellitus
TLR4 toll-like receptor 4
TNF tumor necrosis factor
UPRmt mitochondrial unfolded protein response.
Keywords: alcohol, metabolic dysfunction, inflammation, alcohol-related disease, oxidative stress
Citation: Lee J, Lee J-Y and Kang H (2025) Excessive alcohol consumption: a driver of metabolic dysfunction and inflammation. Front. Toxicol. 7:1670769. doi: 10.3389/ftox.2025.1670769
Received: 22 July 2025; Accepted: 16 September 2025;
Published: 29 September 2025.
Edited by:
Parames C. Sil, Bose Institute, IndiaReviewed by:
Jose Manuel Lerma-Cabrera, University of Almeria, SpainRobert Wade Siggins, Louisiana State University, United States
Copyright © 2025 Lee, Lee and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyunju Kang, aHl1bmp1a2FuZ0BrbXUuYWMua3I=