 Scott G. Johnston
Scott G. Johnston Niloofar Karimian
Niloofar Karimian Edward D. Burton
Edward D. Burton- Southern Cross Geoscience, Southern Cross University, Lismore, NSW, Australia
Arsenic in oxic surface soils readily associates with Fe(III) oxide minerals such as ferrihydrite and goethite, predominantly as As(V). Fires are a common feature in many landscapes, creating high-temperature soil conditions which drive thermal transformation of these As(V)-bearing minerals. However, it is unknown whether fire-induced transformation of ferrihydrite and goethite can alter the mobility of As, or alter As(V) speciation (e.g., via pyrolysis induced electron-transfer generating the more mobile and toxic inorganic As(III) species). Here, we subject an organic-rich soil (∼15% organic C) mixed (4:1) with As(V)-bearing ferrihydrite and goethite (total As of 2.8–3.8 μmol g-1), to various temperatures (200–800∘C) and heating durations (5–120 min) and examine the consequences for As and Fe via X-ray absorption spectroscopy, X-ray diffraction, 57Fe Mössbauer spectroscopy and selective extracts. We show that heating transformed both ferrihydrite and goethite to mainly maghemite at temperatures >400∘C and tended to increase exchangeable surface-complexed As (AsEx) in ferrihydrite yet decrease AsEx in goethite. We demonstrate for the first time that ferrihydrite and goethite-bound As(V) can be rapidly reduced to As(III) during heating of organic-rich soil. Electrons were readily transferred to both Fe(III) and As(V), with reduction of As(V) to As(III) peaking at intermediate temperatures and time periods (maxima of ∼88% for ferrihydrite; ∼80% for goethite). Although As(III) formation was fast (within 5–10 min at temperatures >400∘C), it was followed by partial re-oxidation to As(V) at higher temperatures and longer time intervals. Additionally, combusted As-bearing ferrihydrite and goethite soil-mixtures display greatly enhanced (2–3 orders of magnitude) mobilization of inorganic As(III)aq species upon re-wetting with water. Mobilization of As(III)aq was positively correlated with solid-phase As(III) formation and was greater for goethite than ferrihydrite. These findings challenge the current prevailing view that As(V) reduction to As(III) in soil is mainly limited to waterlogged conditions and suggest that moderate-temperature fires of short duration in oxic soils, may generate substantial labile As(III) species and lead to a pulse of As(III)aq mobilization upon initial rainfall and re-wetting. Further investigation is recommended to explore the consequences for arsenic cycling in fire-prone natural landscapes and agricultural systems which involve controlled-burn practices.
Introduction
Ferrihydrite and goethite are common Fe(III)-minerals in soils (Schwertmann and Cornell, 2000; Cornell and Schwertmann, 2003). Ferrihydrite and goethite have a strong affinity for arsenic in soils and sediments (Brown et al., 1999; Voegelin et al., 2007; Asta et al., 2010; Johnston et al., 2011, 2015), sequestering arsenic via both adsorption and co-precipitation mechanisms (Waychunas et al., 1993, 2005; Dixit and Hering, 2003; Sherman and Randall, 2003; Root et al., 2007).
Arsenic is a toxic carcinogen of considerable global concern due to widespread contamination of groundwater, particularly in south-east Asia (Mukherjee et al., 2008; Ravenscroft et al., 2009; Fendorf et al., 2010a). In well-drained (oxic) surface soils, As occurs predominantly as oxyanions of As(V). In contrast, As(III) species are typically more abundant in reducing environments, such as waterlogged soils (Roberts et al., 2010). The relative abundance of As(V) and As(III) critically influences As mobility, toxicity and environmental behavior, with inorganic As(III) species generally considered to be more mobile and toxic than inorganic As(V) species (Smedley and Kinniburgh, 2002; Lee et al., 2005; Fendorf et al., 2010b).
Wildfires are an increasingly important phenomena throughout the world (Bladon et al., 2014). Fires can alter iron minerals in soils (Taylor and Schwertmann, 1974; Cornell and Schwertmann, 2003), transforming common Fe(III)-oxyhydroxides, such as ferrihydrite and goethite, to phases such as maghemite (γFe2O3) and hematite (αFe2O3) (Cudennec and Lecerf, 2005; Landers et al., 2009; Löhr et al., 2017). Pyrolysis of organic materials during the burning process influences Fe mineral genesis by promoting electron transfer to Fe(III), which generates Fe(II) and facilitates formation of maghemite (Taylor and Schwertmann, 1974; Grogan et al., 2003; Johnston et al., 2018).
Importantly, fires can also enhance trace metal/metalloid mobilization from burnt soils, altering soil properties and potentially degrading surface water quality in burnt catchments (Bladon et al., 2014; Abraham et al., 2017; Burton et al., 2019). An increasing body of field-based research provides examples of elevated trace metal/metalloid concentrations in soils and surface waters after wild-fires (e.g., Stein et al., 2012; Burke et al., 2013; Odigie and Flegal, 2014; Campos et al., 2015, 2016; Burton et al., 2016; Abraham et al., 2018). However, in general there remains a distinct lack of mechanistic studies exploring fire-induced geochemical changes to metal/metalloid speciation or to the mineralogy of their host-phase(s) and how this may be contributing to observations of enhanced trace metal/metalloid mobility (Cerrato et al., 2016).
Recent work by Johnston et al. (2016, 2018) revealed that fire-induced heating of schwertmannite, a meta-stable Fe(III)-oxyhydroxysulfate mineral, can enhance the mobility of co-precipitated As. This occurs by increasing the proportion of exchangeable/surface-complexed As species during Fe(III) mineral transformation (Johnston et al., 2016) and also by pyrolysis of soil organic matter causing transfer of electrons that reduce some As(V) to As(III) (Johnston et al., 2018). However, it is unknown whether a similar process occurs when As associated with ferrihydrite or goethite in soil is subject to fire-induced heating. It is important to resolve this uncertainty, as ferrihydrite and goethite are common Fe(III) mineral constituents in a wide range of natural soils - including some rice-based agricultural systems (Frommer et al., 2011) where straw and stubble is often burnt (Gadde et al., 2009) and where As in groundwater is known to be a major human health issue (Smedley and Kinniburgh, 2002).
This study aims to explore how heating and pyrolysis of organic-rich soil containing As(V)-bearing ferrihydrite and goethite may influence both As speciation and its subsequent mobility. Here, we mix As(V)-coprecipitated ferrihydrite and goethite with an organic-rich soil and subject these mixtures to heating (in air) at a range of temperatures (200–800°C) and time periods (5–120 min). We quantify changes in mineralogy and Fe and As speciation via X-ray absorption spectroscopy (XAS), X-ray diffraction (XRD), 57Fe Mössbauer spectroscopy and selective extractions. The findings are relevant to soils and landscapes subject to wildfires.
Materials and Methods
Mineral Synthesis
As(V)-coprecipitated ferrihydrite and goethite were synthesized according to Schwertmann and Cornell (1991). Sufficient Na2HAsO4⋅7H2O was dissolved in the initial solutions to generate an As(V) concentration in the resultant synthetic ferrihydrite and goethite of approximately 13–20 μmol g-1. Ferrihydrite and goethite suspensions were rinsed 5 times with deionized water and subsequently dried at 40°C before subsequent grinding to a fine powder. Mineralogy verified by XRD (Bruker D4 Endeavor).
An organic-rich and iron-rich surface soil (0–10 cm depth; ∼20% total organic carbon) was collected from a fire-prone, local seasonal wetland [Tuckean Nature Reserve, NSW, Australia – described by Burton et al. (2006)]. The near-total Fe and As content of the soil was 1570 μmol g-1 Fe and >0.1 μmol g-1 As. Soil was air-dried at 40°C and coarse particulates removed by sieving (500 μm). Ferrihydrite:soil and goethite:soil mixtures were prepared at ratios (w/w) of 4 parts soil and 1 part ferrihydrite or goethite. The primary reason for using a natural organic-rich soil from a fire-prone location was to provide a realistic and environmentally relevant matrix. The near-total Fe, As, S and organic C contents of the ferrihydrite:soil and goethite:soil mixtures prior to heating was 2160 and 2700 μmol g-1 Fe, 3.84 and 2.85 μmol g-1 As, 72.8 and 67.6 μmol g-1 S, and 16.1 and 15.4% organic carbon, respectively.
Heating and Thermal Transformation Experiments
The dried ferrihydrite:soil and goethite:soil mixtures were heated in ceramic crucibles (5 g mass of sample) for varying times (5, 10, 30, 60 and 120 min) at 200, 300, 400, 500, 600, 700, and 800°C in a high-temperature controlled muffle furnace (MTI Corporation, KSL-1700X) in air. These temperatures and times were selected to bracket the likely range of temperatures and exposures times associated with a ground-based wildfire front (Certini, 2005; Eggleton and Taylor, 2008).
Solid-Phase Extracts
Dried and ground material was stored in plastic vials at room temperature prior to being analyzed via separate selective extracts. Each sample was subject to a 1:5 water extract (1 g soil: 5 mL deionized water; 1 h orbital shaker at 125 rpm, in the dark) in order to examine aqueous As speciation. A freshly calibrated electrode and pH meter (HACH HQd) was used to measure pH immediately on the unfiltered water extract. Aqueous As speciation was determined by ion pairing chromatography (Wu and Sun, 2016) on filtered (0.45 μm; enclosed syringe filter) aliquots of water extracts that had been preserved with 10 μL of ultra-pure HCl and kept at 4°C until subsequent speciation analysis that was conducted within 4–8 h of water extraction. In brief, the As speciation of aliquots was determined via high performance liquid chromatography (HPLC; Perkin-Elmer Flexar) coupled with inductively coupled plasma mass spectrometry (ICP-MS; Perkin Elmer Nexion 350D), using a C18 (5 μm) column and mobile phase of 1 mM tetra-butyl ammonium hydroxide, 0.5 mM EDTA and 5% ethanol in de-ionized water (flow rate 1 mL min-1; 20 μL injection). Triplicate analysis on 12% of samples confirmed an analytical precision for As(III) within ±6% and for As(V) within ±10%.
A 1 M NaH2PO4 was used to extract exchangeable As (AsEx), and is an approach that targets weakly to strongly sorbed As (Keon et al., 2001). Near-total As (AsTot) and near-total Fe (FeTot) were extracted by microwave-assisted aqua regia digestion in PTFE containers (APHA, 2005). Filtered (0.45 μm; enclosed syringe filter) aliquots of all extracts were analyzed by ICP-MS (Perkin-Elmer ELAN-DRCe). Repeat extraction and analysis of 20% of samples indicated an analytical precision within ±8%. Reactive Fe(II) [Fe(II)HCl] was determined colorimetrically by the 1,10 phenanthroline method after extraction by 1 M HCl (Claff et al., 2010). Total reactive Fe (FeHCl) was also assessed after addition of hydroxyl-amine hydrochloride. Repeat extractions and analysis of 20% of Fe(II)HCl samples had analytical precision within ±12%. A LECO Trumac CNS analyzer was used to determine total organic carbon and total sulfur.
X-Ray Diffraction
Changes in mineralogy were examined by powder X-ray diffraction (XRD). Samples were scanned (5°–80° 2𝜃 with a 0.025° 2𝜃 step-size and a 3 s count-time) using a Bruker D4 Endeavor fitted with a Co X-ray source and Lynx-Eye detector. Diffraction patterns, along with integral peak intensity and full width at half maximum, were assessed using the DIFFRAC-plus evaluation software package (Bruker AXS, Karlsruhe, Germany).
X-Ray Absorption Spectroscopy
X-ray absorption spectroscopy was used to quantify As oxidation state in initial materials and all treatments. Arsenic K-edge X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS – selected samples only) spectra were collected at the 1.9T Wiggler XAS beamline at the Australian Synchrotron, Melbourne. A Si(111) monochromator controlled the X-ray energy which was calibrated against the first inflection point of the absorption edge of an As metal foil. Samples were sealed with Kapton tape in 1 mm thick Al sample holders prior to placement in a He-purged cryostat cooled to ∼4 K. Spectra (1–3 replicates) were collected by a 100 element array Ge solid-state detector (CANBERRA, France) in fluorescence mode. A fast-shutter system limited X-ray exposure and multiple scans of select samples revealed no apparent beam-induced change in As oxidation state.
Standard background subtraction and edge-height normalization was conducted by the AUTOBK algorithm in the Athena software program (Ravel and Newville, 2005). Normalized XANES spectra were merged and the speciation of As in selected samples quantitatively determined by least squares linear combination fitting (LCF) of As XANES spectra against diluted reference standards. Reference standards used in LCF include the initial synthetic As(V)-bearing ferrihydrite and As(V)-bearing goethite, sodium arsenate, as well as sodium arsenite, arsenite sorbed to hematite, arsenite sorbed to cellulose powder and arsenopyrite. No energy shift was allowed in the LCF and the sum of the fitted fractions was not constrained (LCF results are presented normalized to 100%). The fraction of As fitted by each individual As standard was summed for each valence state to determine total proportions of As(III) and As(V). Normalized EXAFS spectra (from selected ferrihydrite:soil samples only) were extracted using the AUTOBK algorithm (Rbkg = 2; k-weight = 3; spline k-range 2–11 Å-1) and Fourier-transformed over the k-range 1–8 Å-1 using a Kaiser-Bessel window (width = 2).
Mössbauer Spectroscopy
57Fe Mössbauer spectroscopy was used to quantify the valency of Fe, as well as assist in identifying changes in Fe mineralogy. Dried and powdered samples were ground to uniform size via an agate mortar and pestle, then weighed into self-supporting HDPE sample holders with a sample area of 1.76 cm2. Samples were prepared as to contain approximately 10 mg cm-2 of Fe. Spectra were collected at ambient temperature (290K to 292K) using an MR351A Mössbauer system with an MA-250 velocity transducer and the resultant data collected on a 1024 channel MCD (Fast ComTec GmbH). A 1.85 GBq 57Co source was mounted on the velocity transducer which was operated in constant-acceleration mode (set to ± 10 mm s-1) (Wissell Inc.). Transducer velocity was calibrated using an αFe foil at ambient temperature and fit to a sextet with a Bhf of 33.1T. Spectra were folded and fitted using the MossA software program (Prescher et al., 2012). Any mixed Fe(II)/Fe(III) sub-spectra were assigned a nominal valency of 2.5 (Dyar et al., 2006; Vandenberghe and De Grave, 2013).
Results
Total Organic C, Total Fe, Total As, and pH
Total organic C in the ferrihydrite:soil and goethite:soil mixtures tended to diminish at higher combustion temperatures and longer combustion durations (Tables 1, 2). In accord with this loss of organic C, the pH of water-extracts generally increased from around 4.5 in the initial materials to up to 7.5 in the combusted samples (Figure 1). The concentrations of AsTot and FeTot also tended to increase with losses in organic C at higher combustion temperatures and longer combustion times (Tables 1, 2). Although it is possible for some As to be volatilized during heating (Helsen et al., 2003), the relative behavior of As and Fe in these experiments (based on the ratio of AsTot to FeTot) constrains the loss of As to <12% (Tables 1, 2 and Supplementary Figure 1).
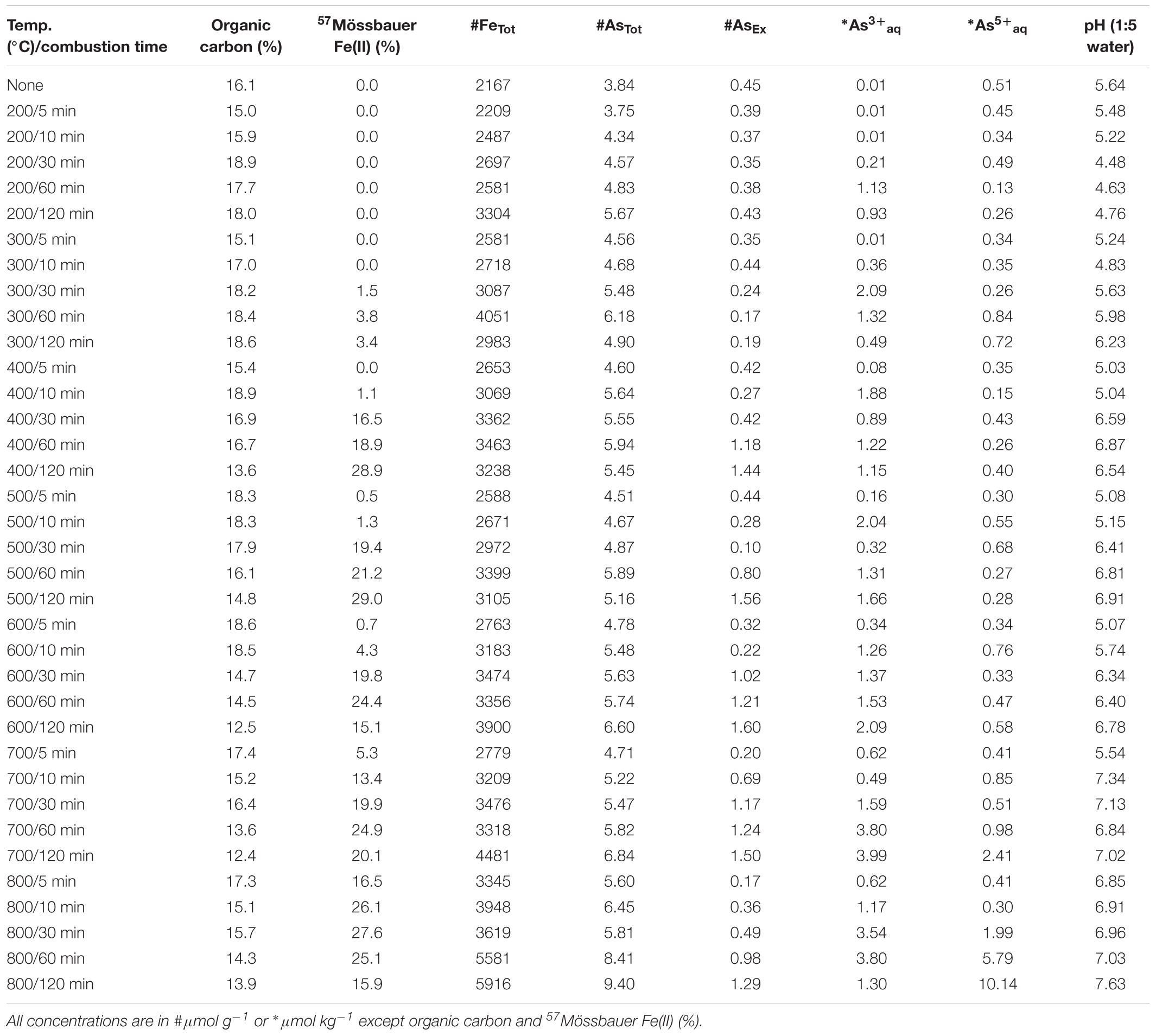
Table 1. Characteristics of soil:ferrihydrite mixtures.
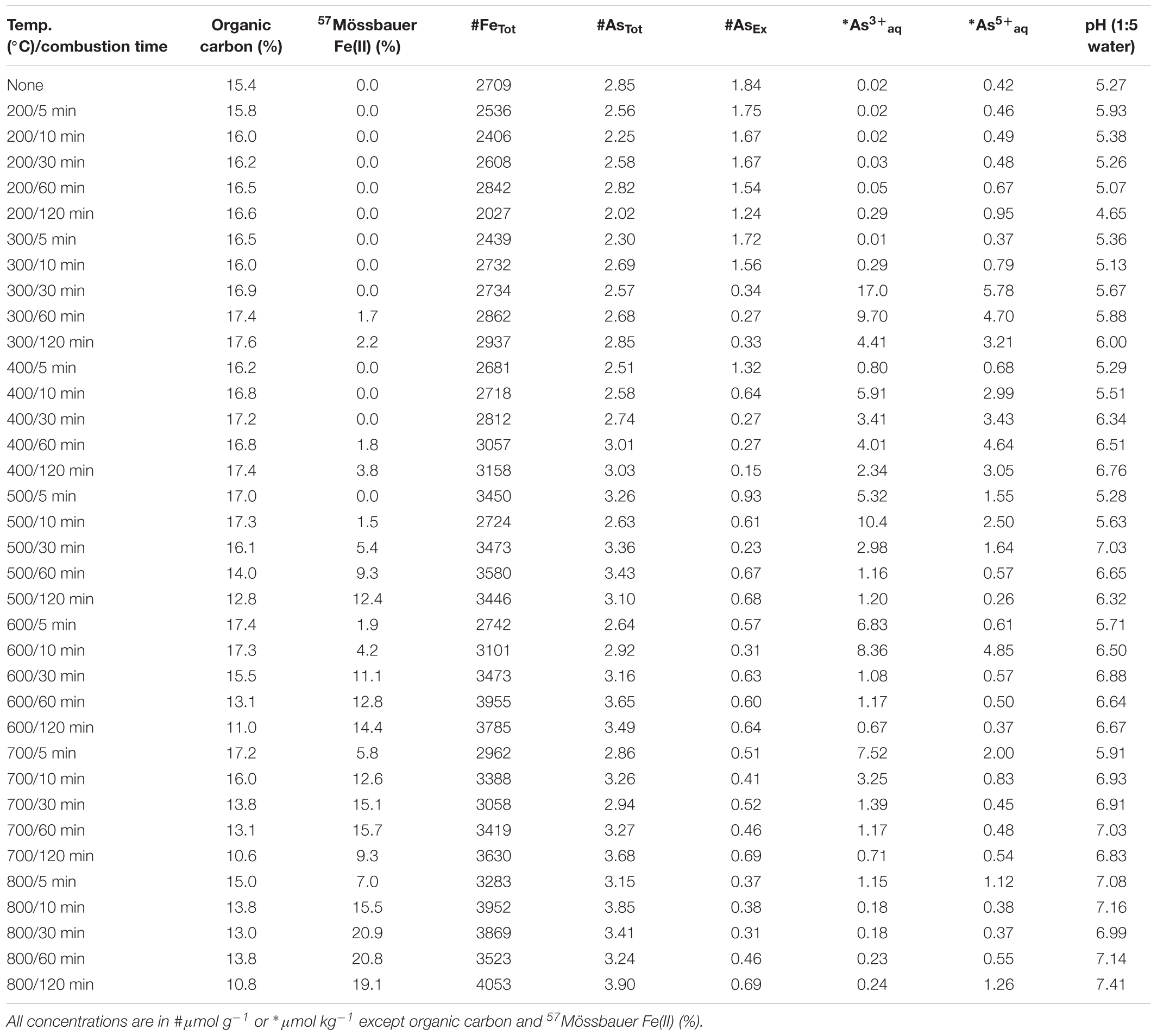
Table 2. Characteristics of soil:goethite mixtures.
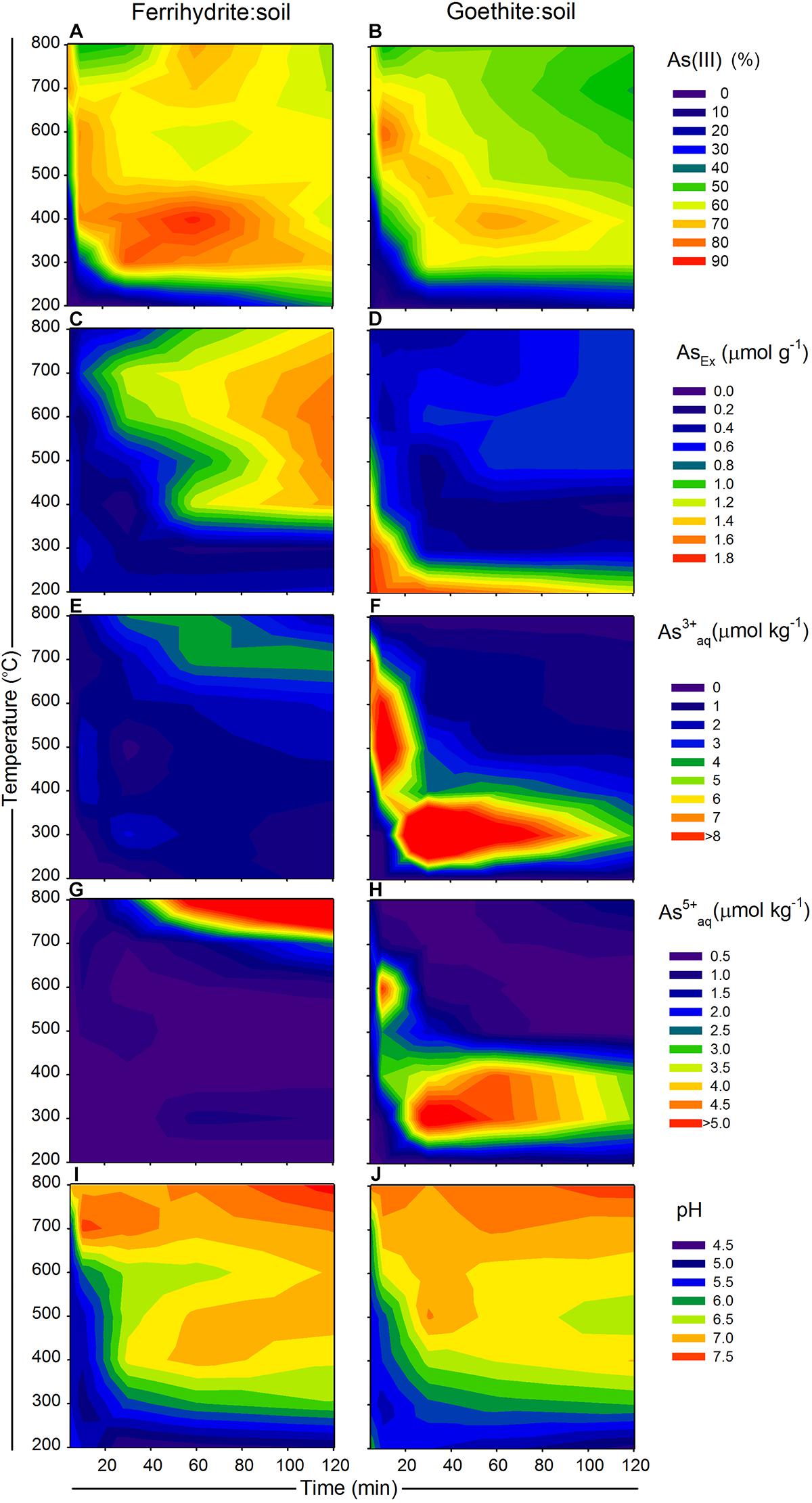
Figure 1. Changes in As species and availability of ferrihydrite:soil and goethite:soil mixtures in relation to combustion time (x-axis) and combustion temperature (y-axis), where (A,B) solid-phase As(III) [excluding the transitory S-coordinated As species formed at 700 and 800°C], (C,D) AsEx, (E,F) water-extractable As3+aq, (G,H) water-extractable As5+aq and (I,J) pH (1:5 in water). Solid-phase As(III) determined by linear combination fits of As K-edge XANES spectra. Contour XYZ plots constructed using Sigma Plot v. 12.0.
Fe Mineralogy
The XRD patterns for the ferrihydrite:soil treatments indicate that ferrihydrite transformed to maghemite at temperatures >400°C and the magnitude of the transformation generally increased at longer combustion times (Figures 2, 3). In contrast, in the goethite:soil treatments, goethite transformed to a combination of both hematite and maghemite at heating temperatures of >300°C, with qualitative analysis of normalized XRD peak intensity indicating that hematite mainly occurred as a transitory intermediate phase prior to formation of maghemite (Figures 2, 4). Maghemite diffraction peaks sharpened progressively (as determined by full width at half maximum) as temperatures increased. This suggests evolution of increasing crystallinity and long-range structural order and/or increases in crystallite size due to sintering (Figures 3, 4).
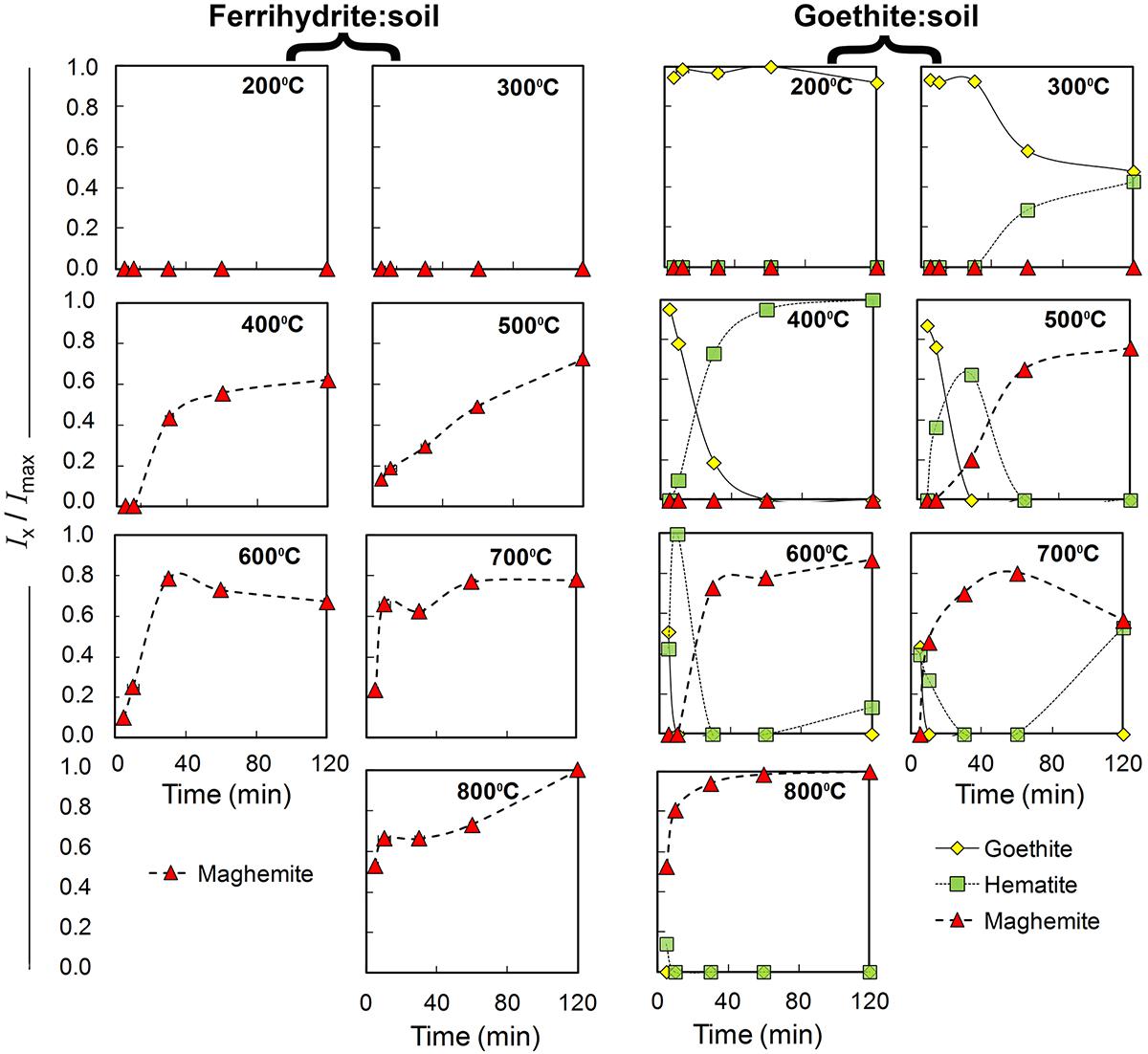
Figure 2. Qualitative estimates of changes in relative abundance of goethite, maghemite and hematite in ferrihydrite:soil and goethite:soil treatments in relation to combustion temperature and combustion duration. Based on normalized X-ray diffraction peak integral intensity (Goethite-111; Maghemite-220; and Hematite-113) derived from Figures 3, 4.
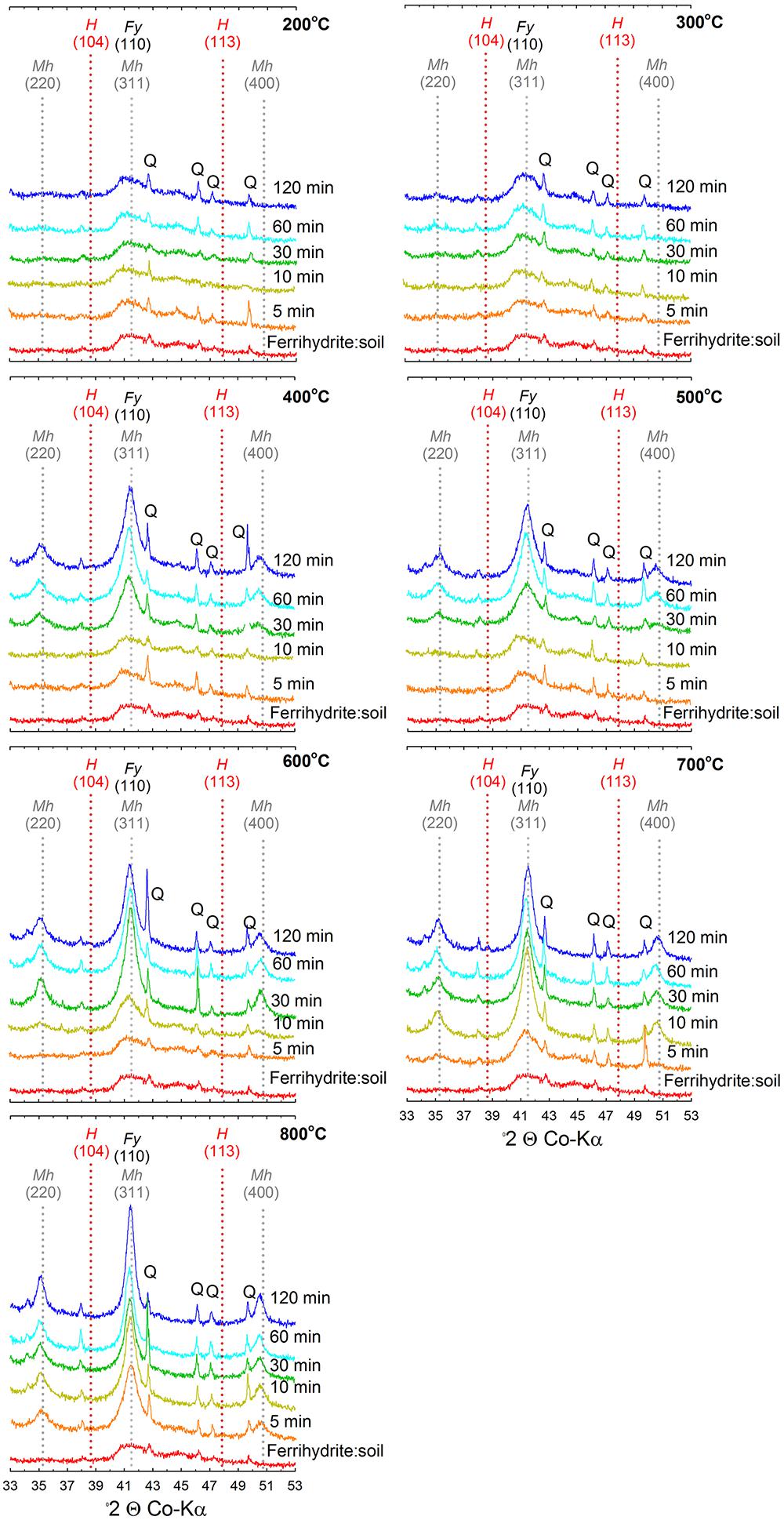
Figure 3. X-ray diffractograms of ferrihydrite:soil mixtures after heating at 200, 300, 400, 500, 600, 700, and 800°C for 5, 10, 30, 60, and 120 min. Fy, ferrihydrite; Mh, maghemite; H, hematite; Q, quartz.
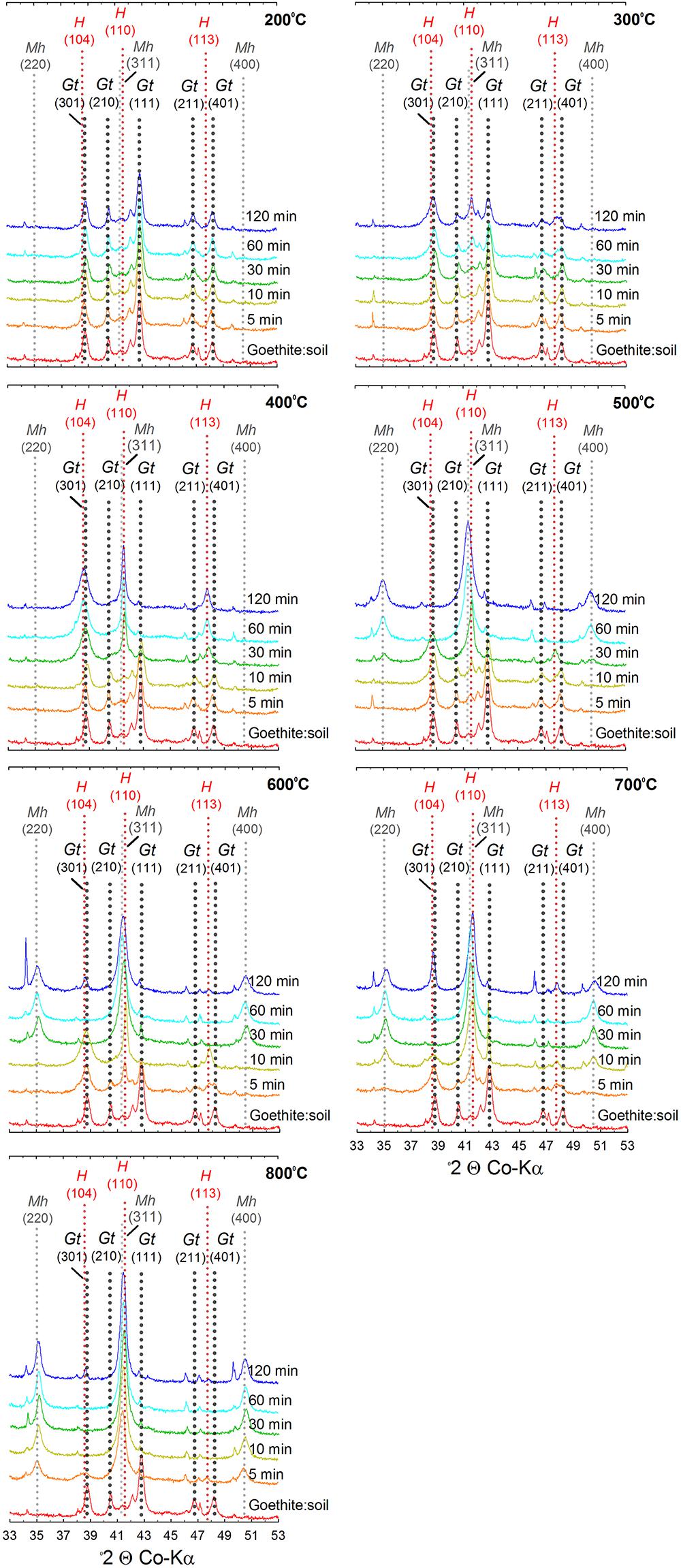
Figure 4. X-ray diffractograms of goethite:soil mixtures after heating at 200, 300, 400, 500, 600, 700, and 800°C for 5, 10, 30, 60, and 120 min. Gt, goethite; Mh, maghemite; H, hematite; Q, quartz.
Selected 57Fe Mössbauer spectra are displayed in Figures 5, 6 along with associated fit components, while all fitting parameters are summarized in Supplementary Tables 1, 2 and remaining fitted spectra are shown in Supplementary Figures 2–13. Changes in Fe mineralogy determined via 57Fe Mössbauer spectroscopy are broadly consistent with those indicated by XRD.
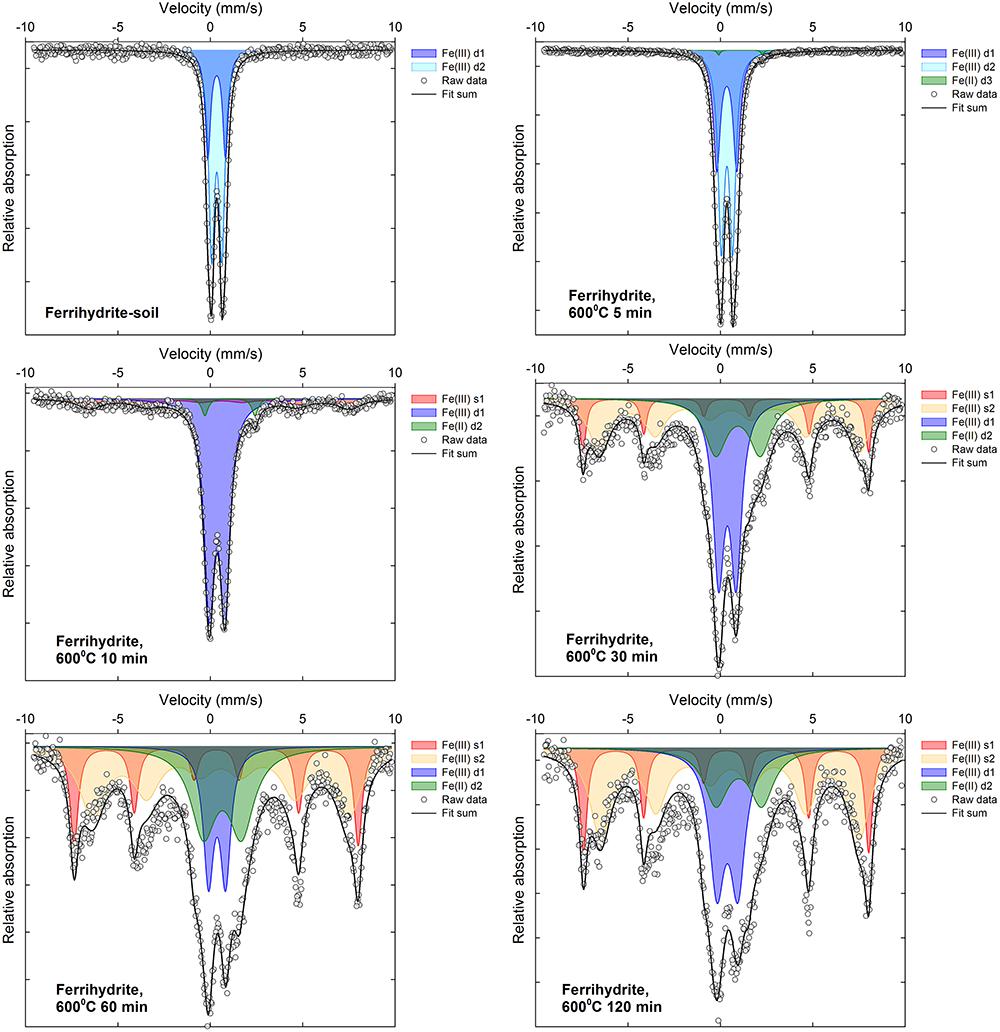
Figure 5. Fitted 57Fe Mössbauer spectra (292 K) for ferrihydrite:soil treatments, 600°C.
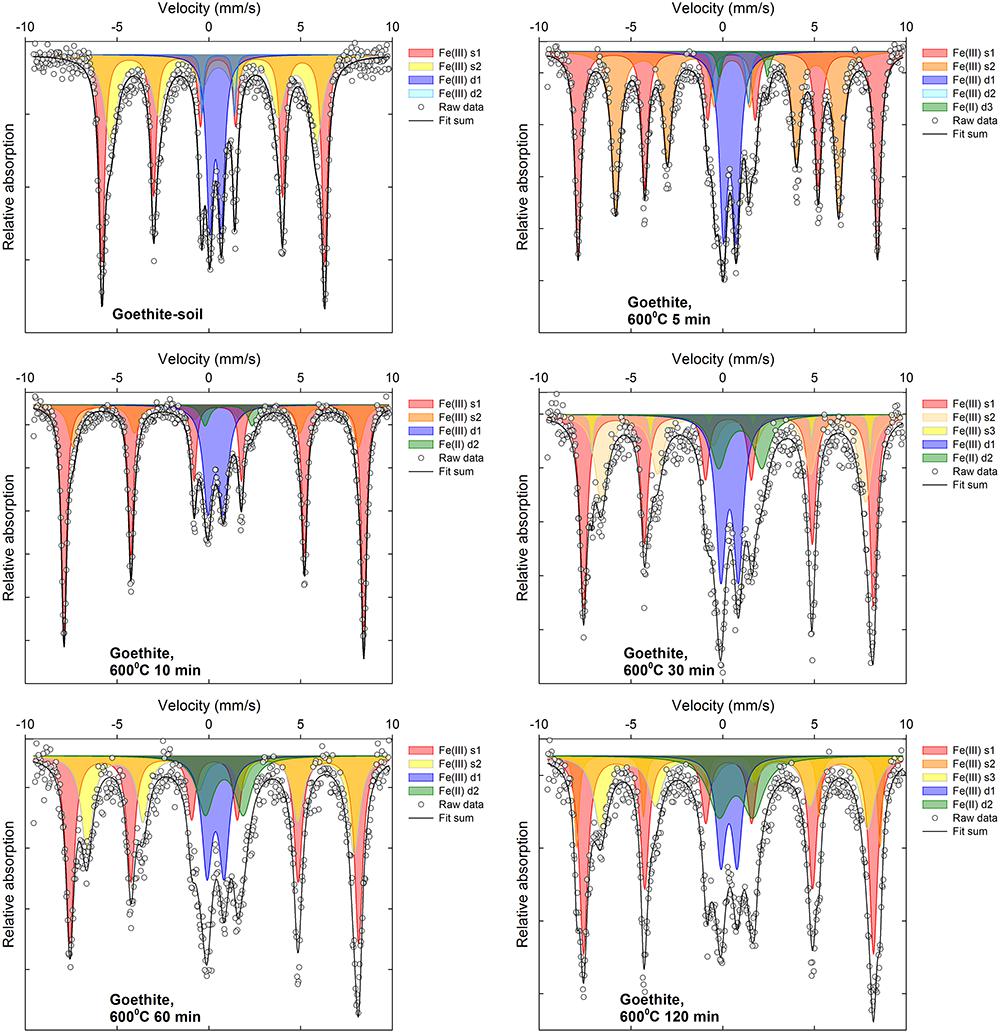
Figure 6. Fitted 57Fe Mössbauer spectra (292 K) for goethite:soil treatments, 600°C.
The initial (unheated) ferrihydrite:soil mixtures were characterized by two distinct Fe(III) doublets (d1 and d2, Supplementary Table 1), with an isomer shift (δFe∼0.35) and respective quadrupole splitting (Δ∼0.56 and 0.95) values that are consistent with ferrihydrite (Dyar et al., 2006; Vandenberghe and De Grave, 2013). Fe(II) doublet sub-spectra (δFe = ∼0.7–1.1; Δ = ∼2.0–2.8) appear in the ferrihydrite:soil samples subject to combustion temperatures ≥300°C (Figure 5, Supplementary Figures 2–7, and Supplementary Table 1). At heating temperatures from ≥400°C, the Mössbauer spectra for these samples exhibit development of Fe(III) sextets (s1, s2, and s3, Supplementary Table 1), which indicates increasing magnetic ordering, in accordance with the spectral behavior of maghemite at 292 K. Initially broad sextets consist of overlapping sub-spectra that are broadly consistent with those reported for maghemite (Dyar et al., 2006; Siddique et al., 2010; Vandenberghe and De Grave, 2013), with magnetic hyperfine field (Bhf) and isomer shift (δFe) parameters of ∼43–48 T and ∼0.27–0.59 mm s-1, respectively, while quadrupole shifts (2𝜀) remain close to zero.
Above 600–700°C and after 60 min combustion time for the ferrihydrite:soil treatments, sextet peaks sharpen and doublets account for a diminishing fraction of total spectral area. The sum of the magnetically ordered phases in fitted 57Fe Mössbauer spectra can be used to estimate the proportion of maghemite in the heated ferrihydrite:soil samples. This analysis suggests that the abundance of maghemite increases as both temperatures and combustion duration increase, corresponding to about 60–75% in samples heated to 700–800°C for ≥30 min. This 57Fe Mössbauer spectroscopy-derived maghemite abundance is significantly (r2 = 0.95; P < 0.05) positively correlated with the corresponding maghemite abundance estimated from the X-ray diffraction peak [maghemite (220)] integral intensity (Supplementary Figure 14).
For the goethite:soil mixtures, the initial (unheated) samples were characterized by two overlapping sextets (s1 and s2, Supplementary Table 2), with an isomer shift (δFe 0.37–0.38), quadrupole shift (2𝜀 -0.26-0.27) and respective magnetic hyperfine field values (Bhf of 34.7 and 37.6) that are consistent with those reported for goethite (Dyar et al., 2006; Vandenberghe and De Grave, 2013). Most of the remainder of the unheated goethite:soil sample 57Fe Mössbauer spectra can be fit by a doublet (d1, Supplementary Table 2), with an isomer shift (δFe 0.36) and quadrupole splitting (Δ 0.63) more consistent with that reported for poorly crystalline goethite (Vandenberghe and De Grave, 2013). Distinct Fe(II) doublet sub-spectra (δFe = ∼0.7–1.3; Δ = ∼2.0–2.6) appear in goethite:soil samples subject to combustion temperatures ≥400°C (Figure 6, Supplementary Figures 8–13, and Supplementary Table 2). Fe(III) sextets of heated goethite:soil treatments progressively develop at temperatures ≥300°C. In the 300 and 400°C treatments after ∼30 min of heating sextet sub-spectra emerge (e.g., s1 and s2, Supplementary Table 2), that display characteristics (δFe 0.35–0.38; 2𝜀 ∼-0.21; Bhf ∼40–51) consistent with those reported for hematite (Dyar et al., 2006; Vandenberghe and De Grave, 2013). Whereas in treatments ≥500°C, sextet sub-spectra initially develop hematite-like characteristics before progressively evolving two overlapping sextet sub-spectra (e.g., 800°C, 30 min, s1 and s2, Supplementary Table 2) similar to those reported for maghemite (δFe 0.30–0.63; 2𝜀 ∼0.0; Bhf ∼44–48) (Supplementary Figures 8–13 and Supplementary Table 2).
Solid-Phase Fe(II) Formation
Fe(II)HCl extracts and 57Fe Mössbauer-derived estimates of solid-phase Fe(II) are displayed in Figure 7. Similar to observations for As(III), maximum Fe(II)HCl occurred at high temperatures (>600°C) and short combustion time periods (5–30 min) as well as intermediate temperatures (400–500°C) at longer combustion time periods (30–60 min), in both ferrihydrite:soil and goethite:soil treatments (Figure 7). The Fe(II)HCl content of the goethite:soil treatments was considerably less than for ferrihydrite:soil, reflecting the more crystalline nature of goethite and its resistance to dissolution by dilute HCl.
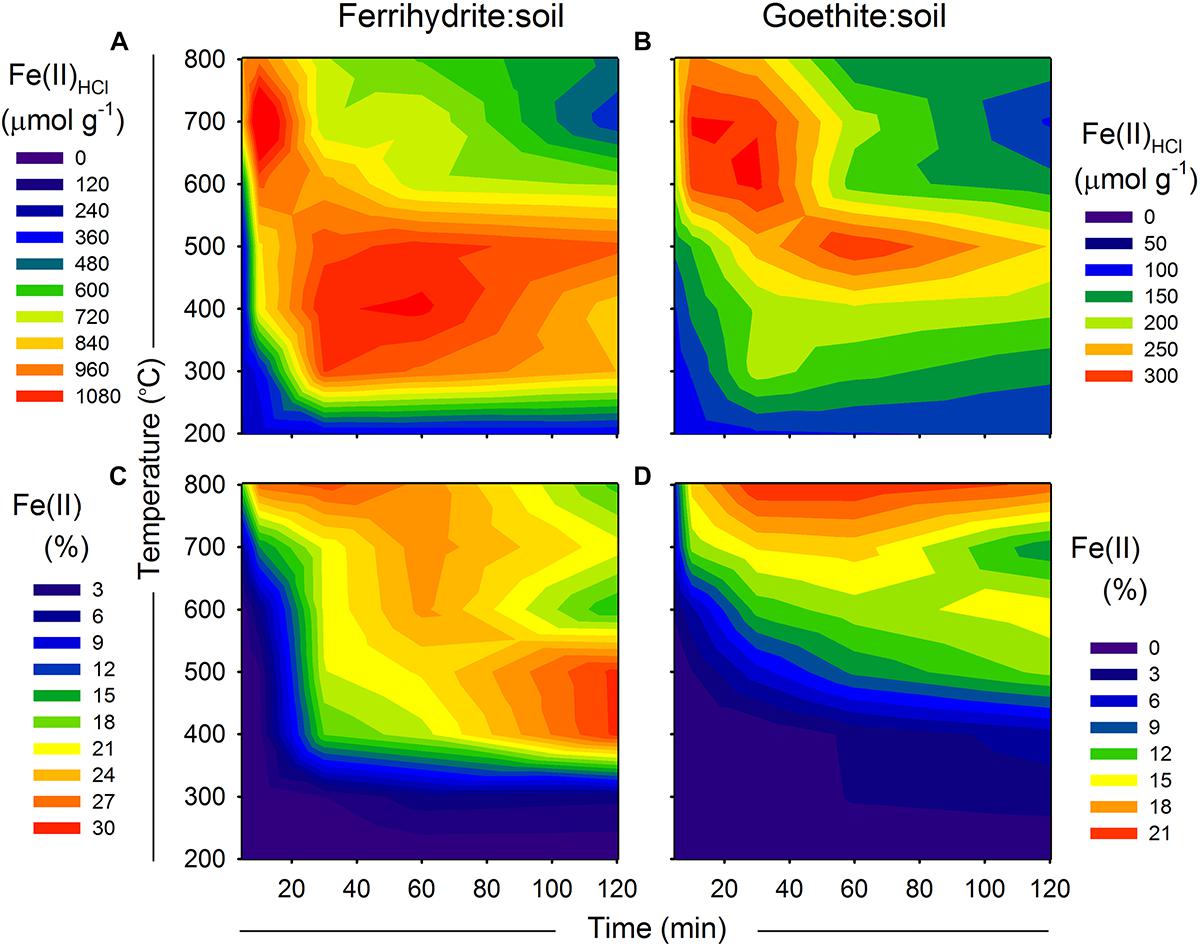
Figure 7. (A,B) Fe(II)HCl species and (C,D) 57Fe Mössbauer derived estimates of Fe(II) in relation to combustion time (x-axis) and temperature (y-axis) for ferrihydrite:soil and goethite:soil mixtures. Contour XYZ plots constructed using Sigma Plot v. 12.0.
Maximum values of Fe(II) of ∼29 and ∼21% were attained for the ferrihydrite:soil and goethite:soil treatments, respectively (Figure 7 and Tables 1,2). For ferrihydrite:soil treatments, Fe(II) determined via 57Fe Mössbauer spectroscopy (Figure 7) displays broadly similar overall trends in relation to combustion temperature and time as those derived from the Fe(II)HCl extractions. Whereas for goethite:soil treatments, trends in Fe(II) derived from 57Fe Mössbauer spectroscopy are quite distinct from those of Fe(II)HCl (Figure 7). 57Fe Mössbauer derived Fe(II) contents are generally higher than Fe(II)HCl and peaks occur at longer combustion times, particularly for goethite:soil treatments. Fe(II) determined via 57Fe Mössbauer spectroscopy is weakly positively correlated with that derived from the Fe(II)HCl extractions (Supplementary Figure 15). Differences between the two analytical approaches partly reflects how enhanced Fe mineral crystallinity or increased crystalline size induced by heating attenuates the effectiveness of the dilute HCl extractant, whereas 57Fe Mössbauer spectroscopy is not subject to this limitation.
Solid-Phase As(III) Formation
The summary LCF results from As K-edge XANES are presented in Figure 1, while individual spectra and select standards are shown in Figure 8 (see Supplementary Tables 3, 4 for fit results). At 200°C, there was very little change in As speciation in either ferrihydrite:soil or goethite:soil treatments (Figures 1, 8). Above 300°C, substantial amounts of As(V) was reduced to As(III), with the rate of As(III) formation being generally faster at higher temperatures (within 5 min at >500°C) for both ferrihydrite:soil or goethite:soil treatments. Maximum As(III) formation was attained at intermediate temperatures and intermediate time periods and was 88% for ferrihydrite:soil and 80% for goethite:soil (Figure 1 and Supplementary Tables 3, 4). In general, the ferrihydrite:soil treatments displayed greater As(III) formation than the corresponding goethite:soil treatments. Partial re-oxidation of As(III) to As(V) occurred in both ferrihydrite:soil and goethite:soil treatments at longer periods of combustion (Figures 1, 8). The proportion of As(III) formed via heating was significantly (P < 0.05) positively correlated to solid-phase Fe(II)HCl in both ferrihydrite:soil and goethite:soil treatments (Figure 9).
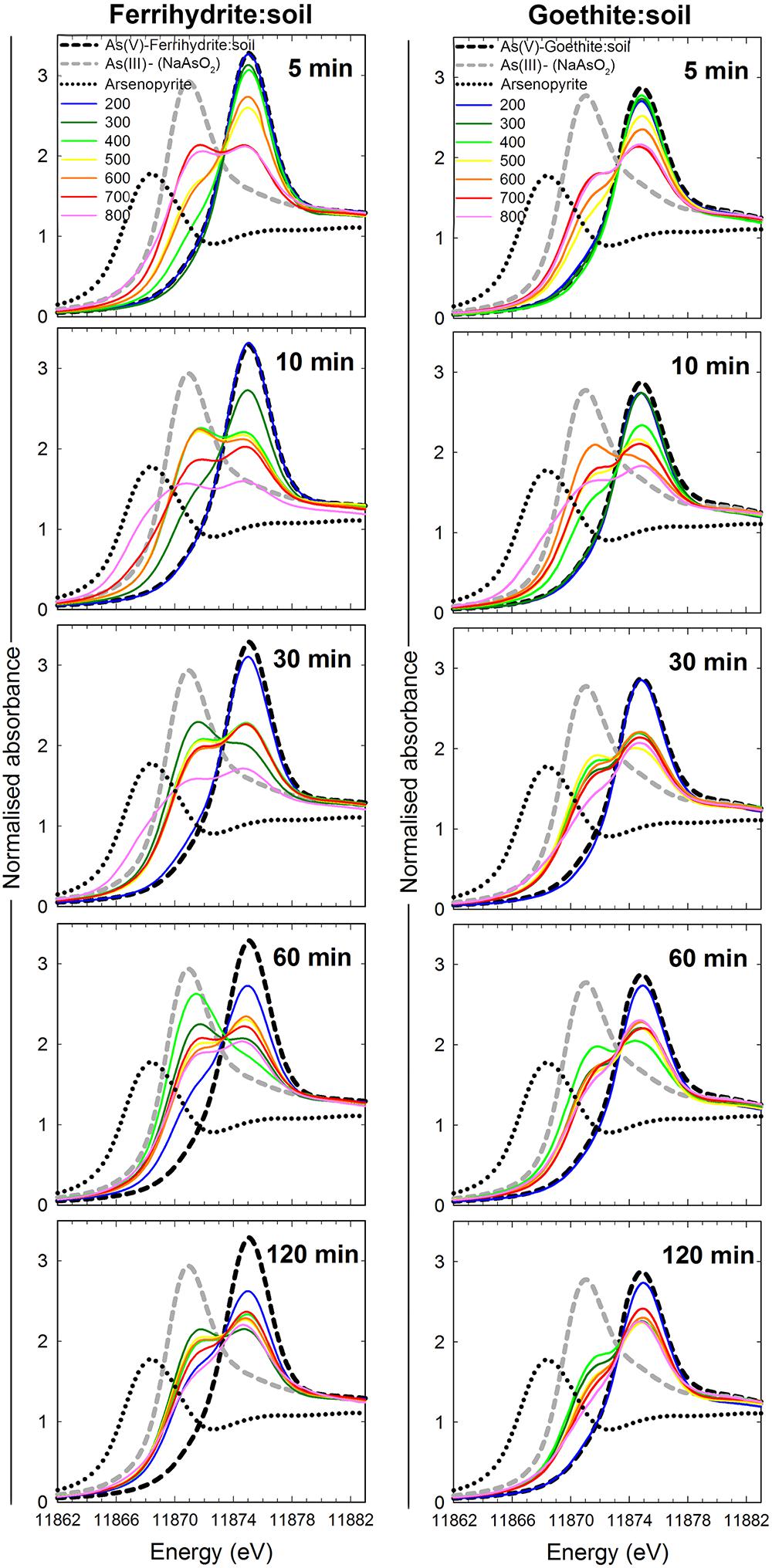
Figure 8. As K-edge XANES spectra of ferrihydrite:soil and goethite:soil mixtures after heating at 200, 300, 400, 500, 600, 700, and 800°C for 5, 10, 30, 60, and 120 min. Dashed lines are reference spectra for As(V)-ferrihydrite:soil, As(V)-goethite:soil, As(III), and Arsenopyrite.
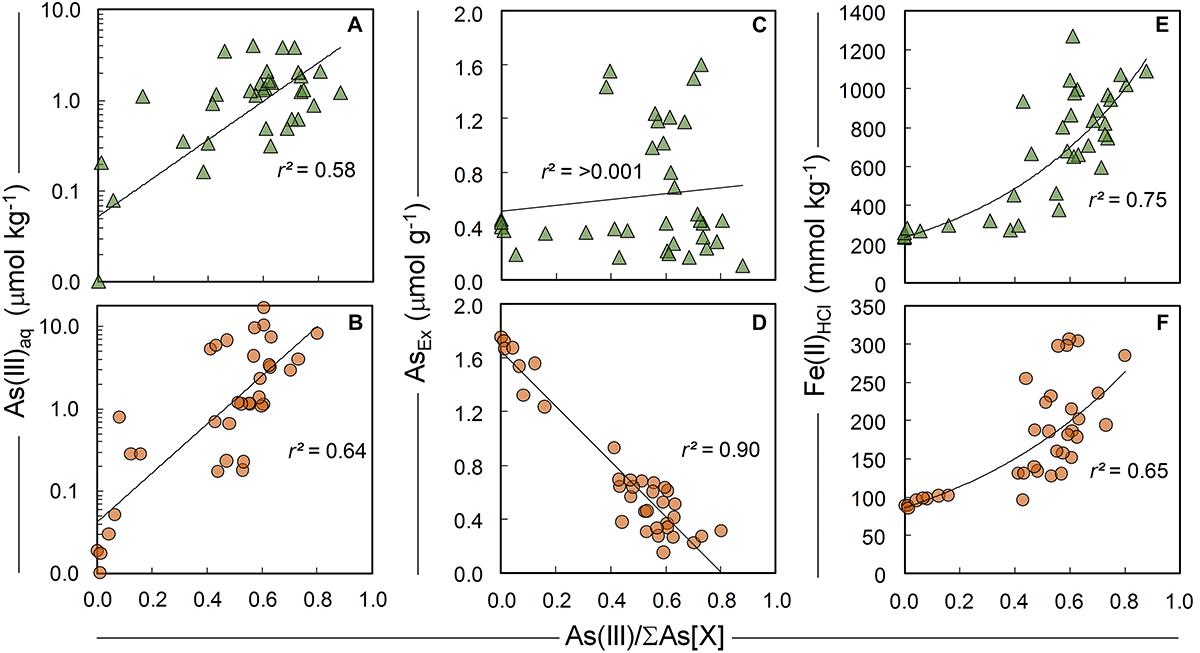
Figure 9. Bivariate relationships between the proportion of solid-phase As(III) species in ferrihydrite:soil (top row, triangles) and goethite:soil (bottom row, circles) treatments and (A,B) As(III)aq species in 1:5 water extracts and (C,D) AsEx and (D,E) concentration of Fe(II)HCl. As(III) determined by linear combination fits of As K-edge XANES spectra.
X-ray absorption near-edge structure spectra for the ferrihydrite:soil sample from 10 min in the 800°C treatment, display a distinct shoulder with a lower As K-edge energy than our As(III) standards (Figure 8). The XANES linear combination fit of this spectra assigned a 45% value to the arsenopyrite standard (Supplementary Table 3). Fourier transformed As-EXAFS spectra for select ferrihydrite:soil samples from the 800°C treatment reveal a backscattering peak consistent with first-shell As-O coordination in the unheated, 5 min and 60 min treatments, whereas the 10 min treatment displays a (transitory) backscattering peak that accords with that expected for a first-shell sulfur-coordinated As species (Supplementary Figure 16).
Exchangeable As
The AsEx content of the initial ferrihydrite:soil mixtures (0.45 μmol g-1) was around 12% of AsTot which indicates that the majority of As was not readily desorbed. Hence, most As was likely initially incorporated within the ferrihydrite structure and largely unable to participate in surface exchange reactions. The AsEx fraction of the ferrihydrite:soil mixtures increased considerably after heating >400°C, attaining a maximum of 1.6 μmol g-1 at 600°C, 120 min (Figure 1 and Table 1). In contrast, the AsEx content of the goethite:soil mixtures was initially quite high (1.84 μmol g-1), representing ∼64% of AsTot, indicating that the majority of the As was likely surface complexed (Table 2). The AsEx fraction in the goethite:soil mixtures decreased substantially at higher combustion temperatures and durations (Figure 1).
Water-Extractable As
Concentrations of water-extractable As(III) and As(V) species in relation to combustion temperature and time for ferrihydrite:soil and goethite:soil treatments are displayed in Figure 1. The extent of As(III)aq mobilization into the aqueous-phase in initial unheated treatments was very low (<0.02 μmol kg-1) and accounted for <5% of total Asaq. However, As mobilization in water increased by ∼20× for ferrihydrite:soil and ∼50× for goethite:soil as combustion temperatures increased, reaching a maximum of ∼22 μmol kg-1 in the goethite:soil treatments (Figure 1 and Table 2). Importantly, at temperatures ≥300°C, a substantial fraction of the Asaq was inorganic As(III)aq species, with inorganic As(III)aq species increasing up to a maximum of ∼470× for the ferrihydrite:soil and ∼840× for the goethite:soil treatments (Tables 1, 2).
Ferrihydrite and goethite displayed distinctly different patterns of Asaq mobilization. Ferrihydrite:soil treatments attained maximum Asaq mobilization at highest temperatures and longest combustion times, whereas in goethite:soil treatments the greatest mobilization of As(III)aq and As(V)aq occurred at intermediate temperatures and intermediate time periods (Figure 1). As(III)aq was significantly (P < 0.05) positively correlated with solid-phase As(III) determined via As K-edge XANES spectroscopy (Figure 9). As(III)aq was also significantly (P < 0.05) positively correlated with pH in ferrihydrite:soil treatments, but was poorly correlated in goethite:soil treatments (Supplementary Figure 17). In addition, total Asaq (∑ of As(V)aq + As(III)aq) was significantly (P < 0.05) positively correlated with the magnitude of Fe mineral transformation (maghemite formation) for the ferrihydrite:soil treatments. However, for the goethite:soil treatments Asaq was negatively correlated with maghemite yet significantly (P < 0.05) positively correlated with hematite abundance (Supplementary Figure 18).
Discussion
Transformation of Iron Minerals
Transformation of ferrihydrite to maghemite upon heating an organic-matter rich soil is consistent with previous investigations (e.g., Mazzetti and Thistlethwaite, 2002; Cornell and Schwertmann, 2003). Likewise, heating goethite alone can readily form hematite (Goss, 1987), while the progressive transformation of goethite to maghemite accords with several prior studies that demonstrate maghemite formation upon burning of organic-rich, goethite-bearing soil (Ketterings et al., 2000; Grogan et al., 2003).
X-ray diffractograms indicate that maghemite (as well as transitory hematite in the goethite:soil treatments) were the primary crystalline Fe-mineral phases that formed during heating at temperatures ≥300°C. 57Fe Mössbauer spectra evolve in a manner that is consistent with XRD results. For example, sextet parameters (i.e., low to negative quadrupole splitting and hyperfine fields mostly ∼50 T) are consistent with either maghemite or a mixture of maghemite and hematite, while the range of sub-spectra observed and the diversity of their respective fitting parameters most likely reflects a range of particle size characteristics (Vandenberghe et al., 2000; Murad and Cashion, 2004; Dyar et al., 2006).
57Fe Mössbauer data suggests that up to ∼29% of initial Fe(III) was reduced to Fe(II) species. Estimates of Fe(II) based on 1M HCl extracts and Mössbauer spectra show some agreement - despite their very different mechanisms. Unlike 1M HCl, 57Fe Mössbauer is largely free from common interferences associated with chemical extraction techniques (Dyar et al., 2006), combining these two approaches strengthens the overall findings regarding changes in Fe valency due to heating. However, it should be noted that quantitative interpretation of complex 57Fe Mössbauer spectra containing multiple, broad-peaks that overlap – like those observed here at intermediate temperatures - can still influenced by non-unique fits that are statistically equivalent (Vandenberghe et al., 2000).
Although there was abundant Fe(II), the mineralogical form(s) of this Fe(II) remains uncertain. A range of possibilities exist, including that Fe(II) species were sorbed to either char/ash materials or Fe(III) mineral surfaces, occurred as organic complexes, or formed very poorly crystalline nano-particles. It is not possible to conclude whether discrete Fe(II) mineral phase(s) may have formed based on existing data and further investigation is required to determine the likely form(s) of Fe(II) observed here.
Temperature and Time-Dependent Formation of As(III) and Fe(II)
This is the first study to demonstrate that ferrihydrite and goethite-bound As(V) can be reduced to As(III) as a result of heating an organic-rich soil. An important aspect is that As(III) formation was fast and occurred at moderate temperatures for both minerals (i.e., in 10 min at 400–500°C, >70% As(III) for ferrihydrite:soil and 40–60% As(III) for goethite:soil), although there was a tendency for As(III) to form faster at higher temperatures. In addition, As(III) partially re-oxidized to As(V) at longer time intervals and higher temperatures. The thermal and temporal patterns of As(V) reduction/re-oxidation for the ferrihydrite:soil and goethite:soil treatments are somewhat similar, with both displaying a distinct As(III) maxima at mid-temperatures and mid-heating durations.
Heating also caused up to 29% of Fe(III) to be reduced to Fe(II). Although thermal and temporal patterns of Fe(III) and As(V) reduction/re-oxidation were only superficially similar, Fe(II)HCl was significantly (P < 0.05) positively correlated with As(III) (Figure 9). The findings presented here for As and Fe broadly mirror those described by Johnston et al. (2018) for combustion of organic-rich As(V)-bearing schwertmannite soils.
Formation of As(III) and Fe(II) is a direct result of As(V)/Fe(III) interacting with reducing compounds derived from pyrolysis of organic matter. The breaking of covalent bonds in cellulose, hemicellulose and lignin during organic matter pyrolysis generates a wide range of low molecular weight products (Evans and Milne, 1987), including various reduced gaseous compounds (i.e., H2, CH4, CO, and H2S) (Liu et al., 2017), in addition to radicals with unpaired valence electrons. Atmospheres rich in carbon monoxide and H2 can readily transfer electrons to Fe(III)-oxides at temperatures above ∼300°C (Jozwiak et al., 2007).
The behavior of As in this study to some extent mirrors the findings of Helsen et al. (2004) who found reduced gaseous species generated during thermal decomposition of glucose encouraged the reduction of As(V) (as As2O5) to As(III). Our findings also accord with previous studies exploring combustion of Copper-Chrome-Arsenate (CCA) treated wood (Wasson et al., 2005) and Pteris vittata (Yan et al., 2008), in which pyrolysis was reported to drive partial reduction of As(V) to As(III) and cause partial volatilization of As(III) at temperatures >400°C (Helsen et al., 2003; Cuypers and Helsen, 2011). In addition, pyrolysis of organic matter at temperatures ≤500°C can cause reduction of other transition metals, including Cu and Ni (Richardson et al., 2010; Liu et al., 2012).
Perhaps one of the most important findings of this study is that most As(III) was formed at relatively moderate combustion temperatures (∼300–600°C) – for both ferrihydrite and goethite. Wildfires can readily generate such temperatures in topsoils (Certini, 2005; Blake et al., 2012). In contrast, at 200°C the reduction of As(V) [or Fe(III)] was both slow and limited. This observation likely reflects the fact that temperatures greater than ∼280–300°C are typically required to cause thermal decomposition of cellulose and the generation of abundant reducing gasses (Evans and Milne, 1987).
The initial reduction of As(V) was also rapid at higher temperatures (∼600–800°C), but was subsequently followed by partial re-oxidation of newly formed As(III) and Fe(II) species at longer combustion-time intervals. This observation accords with the heating in this study being performed in air – a feature which would lead to an oxidizing atmosphere being re-established at longer heating durations, after any reducing gasses formed during organic matter pyrolysis were eventually exhausted.
An alternative possible explanation for the apparent decrease in As(III) at longer heating durations and temperatures >500°C, could be some volatilization of As(III). For example, a study by Cuypers et al. (2009) observed some volatilization of As(III) during thermal treatment of As2O3 adsorbed to activated carbon, while Helsen et al. (2003) demonstrated some volatilization of As(III) during combustion of wood that was treated with CCA. Possible As(III) volatilization that might have occurred in these experiments can be estimated by assuming no Fe was lost as a result of heating, and examining changes in ratios of AsTot to FeTot for treatments in comparison to those of initial materials (see Supplementary Figure 1). This estimate provides an upper bound to possible As(III) volatilization and suggests that volatilization could only account for a mean loss of AsTot of ∼7% – i.e., too low to account for the 20–32% decrease in As(III) observed at longer time intervals of treatments with temperatures >600°C. This indicates that partial re-oxidation of newly formed As(III) was likely an important process, especially at longer durations of heating.
The (transitory) formation of a solid-phase As-species in the ferrihydrite:soil 10 min/800°C treatment with a distinctly lower K-edge XANES energy position than any of the As(III) reference standards, although somewhat unexpected, accords with similar observations made by Johnston et al. (2018) for heated schwertmannite:soil. Fourier transformed As K-edge EXAFS spectra for ferrihydrite:soil 10 min/800°C (Supplementary Figure 16) displays a distinct backscattering contribution consistent with first-shell As-S coordination (e.g., Root et al., 2009; Johnston et al., 2012; Langner et al., 2012). In contrast, unheated ferrihydrite:soil and treatments bracketing the 10 min time interval display a dominant backscattering contribution concordant with first-shell As(V)-O or As(III)-O (e.g., Waychunas et al., 1993; Manning et al., 1998). Pyrolysis of biomass at temperatures >600°C can decompose organic sulfur compounds and generate H2S (Liu et al., 2017), potentially forming inorganic sulfides in ash residues (Knudsen et al., 2004; Liu et al., 2017). The generation of a transitory As-sulfide species, possibly formed via reaction between As and H2S, may explain the temporary formation of first-shell As-S coordination as indicated by As K-edge EXAFS spectra. However, it is worth noting that neither 57Fe Mössbauer sub-spectra nor XRD patterns for the ferrihydrite:soil 10 min/800°C treatment display evidence of an FeS2 species. Hence, to definitively resolve this would require additional investigation.
Enhanced Mobilization of As(III) on Re-wetting
To our knowledge, this is also the first study to demonstrate that heating of soil which contains As(V)-bearing ferrihydrite and/or goethite can greatly increase the mobility of inorganic As(III)aq and As(V)aq species in water - in this case by up to three orders of magnitude for As(III). This enhanced mobility of Asaq, particularly As(III)aq species, likely reflects complex interactions between a combination of (a) changes in solid-phase As speciation, (b) changes in surface complexation of As during the thermally induced mineral transformation process, and (c) changes in the surface properties (i.e., charge, surface site density) of Fe mineral products and organic ash/char residues. Although heating caused additional Asaq mobilization from both the ferrihydrite:soil and goethite:soil treatments, they each displayed distinct and contrasting patterns of both AsEx formation and As(III)aq mobilization.
For the ferrihydrite:soil mixture, As(III)aq and total Asaq mobilization was greatest for higher temperature/longer duration heating treatments. This may partly be a consequence of increasing abundance of solid-phase As(III), for despite the fact that both As(III) and As(V) species can efficiently adsorb to a wide range of Fe-(hydr)oxides (Dixit and Hering, 2003; Morin et al., 2008; Burton et al., 2009; Bolanz et al., 2013), As(III) species are typically desorbed more readily (Herbel and Fendorf, 2006; Fendorf et al., 2010b). In addition, while ferrihydrite can be a very efficient host for both As(V) and As(III) (Dixit and Hering, 2003), maghemite is generally less efficient and its efficacy is highly contingent upon particle size (greater sorption efficiency for smaller particles) (Auffan et al., 2008). Hence, enhanced Asaq mobilization is also likely due to the thermally induced transformation of ferrihydrite to maghemite - for several reasons. Firstly, the structural re-ordering during maghemite formation was associated with increased surface-complexed As (as estimated by AsEx; Tables 1, 2) and secondly, the increasingly crystallinity and long-range order of neo-formed maghemite at higher temperature/heating durations (as evident from XRD) is likely to have been accompanied by diminished surface area/sorption site density, thereby facilitating easier desorption of As from the mineral surface. The significant positive correlation between total Asaq and maghemite abundance in ferrihydrite:soil treatments (Supplementary Figure 18) accords with these suggestions.
For goethite:soil treatments, maximum As(III)aq and As(V)aq mobilization in water occurred at distinctly lower temperatures (∼300–400°C), and also short (<30 min) heating durations at higher temperatures (>500°C). This pattern most closely mirrors that of solid-phase As(III) formation in goethite:soil treatments – a feature reflected by significant positive correlation (r2 = 0.64) between As(III)aq and solid-phase As(III) (Figure 9). In addition, Asaq mobilization in goethite:soil treatments also corresponds with the period where goethite is transforming to hematite and is thus also likely influenced by Fe mineral transformation processes, for similar reasons as outlined previously for the ferrihydrite:soil treatments.
Variations in temperature and heating duration caused complex and non-linear behavior of As(III)aq and As(V)aq. This likely reflects the varying extent and form(s) of As species incorporation/retention in char residues (Vithanage et al., 2017), contrasting effects of changes in pH (Dixit and Hering, 2003), competing interactions between solid-phase As(V) reduction/re-oxidation, as well as changes in the magnitude of AsEx formation.
As(III) species are not only more toxic than As(V), but they are also more mobile than As(V) in many groundwaters/surface waters (Takahashi et al., 2004; Burton et al., 2008; Fendorf et al., 2010a; Diwakar et al., 2015). Observations here of enhanced As(III)aq mobilization following heating have important implications for fire-prone landscapes and soils - especially given that ferrihydrite and goethite are ubiquitous components of many soils. The findings suggest that for an organic-rich soil containing As(V)-bound to ferrihydrite or goethite, fire alone may be enough to initiate As(III) mobilization into porewater or surface water upon initial soil re-wetting by rainfall.
Prior field-based studies on this topic are very limited. A United States Geological Survey study explored As speciation in soils and ash subjected to wildfire and found a predominance of As(V), as well as some As(III), readily mobilized in water leachate (Wolf et al., 2010). Although a number of studies have reported elevated As in surface waters after wildfires (Leak et al., 2003; Burke et al., 2013; Burton et al., 2016; Abraham et al., 2017), none of these studies present geochemical mechanistic explanations for why this might be the case - aside from invoking enhanced sediment erosion and likely desorption from higher pH ash/burned soil. The findings of this study provide a mechanism which may help explain past-observations of elevated As in surface waters after wildfires - yet simultaneously, the findings also highlight the need for future field-focused research.
Environmental Implications and Further Research
This study clearly shows that heating organic-rich soil containing As(V)-bearing ferrihydrite or goethite can lead to rapid and considerable formation of As(III). This enhances As(III)aq mobilization in water after subsequent re-wetting of soil. While the findings parallel those described by Johnston et al. (2018) for As(V)-bearing schwertmannite, the implications are far more significant given that both ferrihydrite and goethite are very common constituents in many soils. Importantly, for both minerals, electron transfer occurred quickly and at temperatures that have been found to occur in topsoil during wildfires and during prescribed burns in agricultural settings (Certini, 2005; Blake et al., 2012). This suggests that fire-induced As(III) formation and enhanced As mobility in topsoil may be an important factor influencing As cycling in many diverse settings. This newly identified fire-induced mechanism of As(III) formation expands upon the currently prevailing view that As(V) reduction to As(III) in soil systems is mainly limited to waterlogged conditions.
Predictions that wildfires will increase in size, frequency and intensity over large parts of the world due to climate change impacts lend these findings heightened environmental relevance – especially for water supply catchments (Bladon et al., 2014; Clarke et al., 2016). Although this study accords with (and helps to explain) prior field-observations of enhanced As mobilization in surface waters after wildfires, additional investigations are clearly warranted to examine how other factors, including fire intensity/duration, depth of heat penetration into soil, As concentrations, ferrihydrite/goethite content, may interact to influence As speciation and resultant surface water quality. Further, it is likely that heating samples in a temperature-controlled oven in air may lead to differences in pO2, pyrolysis products and therefore possible outcomes for As speciation, when compared to natural wildfire-induced combustion of soil. Wild fires can produce centimeter-scale vertical gradients in both soil temperature and pO2 (Certini, 2005) and although As(III) rapidly formed under the conditions of this study, there is a need to explore and validate the observations in variety of field settings.
An additional potential environmental implication worthy of consideration relates to As cycling in rice-based agricultural systems subject to regular open-field burning. In some major rice growing regions of the world, the occurrence of arsenic in both groundwater and rice represents a major human health issue (Smedley and Kinniburgh, 2002; Islam et al., 2016). In many rice growing regions, large quantities of rice straw and stubble are commonly burnt in-field (Gadde et al., 2009), and rice plants/straw have been shown to accumulate As (Dittmar et al., 2010; Seyfferth et al., 2014; Islam et al., 2016). In rice paddy soils, both goethite and ferrihydrite are known to be key hosts for As (Seyfferth et al., 2010; Frommer et al., 2011). The findings of this study raise the possibility that burning of rice stubble may thermally alter As-bearing ferrihydrite or goethite in surface soil and thereby encourage the formation of As(III) species. Hence, rice-stubble burning may be an additional mechanism affecting the redox cycling/and mobility of As in frequently burnt rice-based agricultural systems and this possibility is worth examining.
Conclusion
This study demonstrates that heating an organic-rich soil containing common As(V)-bearing Fe minerals ferrihydrite and goethite, can form considerable As(III) and greatly enhance As(III)aq mobility upon re-wetting. This mechanism of As(III) formation and mobilization challenges the prevailing view that As(V) reduction to As(III) in soil systems is limited to aqueous reactions under waterlogged conditions. Furthermore, the fire-induced reduction of Fe(III) oxide-bound As(V) may have implications for As cycling and surface water/porewater As concentrations in fire-prone natural landscapes, as well as agricultural settings which involve the use of controlled fire regimes. However, the observations reported here would greatly benefit from further research into their applicability under natural conditions in field-based settings.
Data Availability
The raw data supporting the conclusions of this manuscript, but not presented above will be made available by the authors to any qualified researcher. All other data can be found in this manuscript.
Author Contributions
SJ designed the study, and analyzed and interpreted the data. SJ, NK, and EB conducted the XAS data collection and analysis. SJ wrote the manuscript with equal editorial input from NK and EB after the first draft.
Funding
Arsenic XAS was conducted at the Australian Synchrotron, Melbourne (Grant No. AS182/XAS/13322). Research expenses and salary support for Scott Johnston was provided by the Australian Research Council (Grant No. FT110100130), Great Lakes Council and Southern Cross University. HPLC-ICP-MS analysis that was possible due to Australian Research Council LIEF grant (LE150100007). This research was supported by the Environmental Analysis Laboratory (EAL) which is a Southern Cross University NATA accredited research support facility.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Peter Kappen for assistance and advice with collection of XAS data. We also thank Roz Hagan for her thorough and diligent work in the laboratory and for being a most fantastic all-round Technician. We also thank Don Brushett for HPLC-ICP-MS analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/feart.2019.00139/full#supplementary-material
References
Abraham, J., Dowling, K., and Florentine, S. (2017). Risk of post-fire metal mobilization into surface water resources: a review. Sci. Total Environ. 599–600, 1740–1755. doi: 10.1016/j.scitotenv.2017.05.096
Abraham, J., Dowling, K., and Florentine, S. (2018). Influence of controlled burning on the mobility and temporal variations of potentially toxic metals (PTMs) in the soils of a legacy gold mine site in Central Victoria, Australia. Geoderma 331, 1–14. doi: 10.1016/j.geoderma.2018.06.010
APHA (2005). Standard Methods for the Examination of Water and Wastewater. Baltimore, MD: American Public Health Association.
Asta, M. P., Ayora, C., Román-Ross, G., Cama, J., Acero, P., Gault, A. G., et al. (2010). Natural attenuation of arsenic in the Tinto Santa Rosa acid stream (Iberian Pyritic Belt, SW Spain): the role of iron precipitates. Chem. Geol. 271, 1–12. doi: 10.1016/j.chemgeo.2009.12.005
Auffan, M., Rose, J., Proux, O., Borschneck, D., Masion, A., Chaurand, P., et al. (2008). Enhanced adsorption of arsenic onto maghemites nanoparticles: as(III) as a probe of the surface structure and heterogeneity. Langmuir 24, 3215–3222. doi: 10.1021/la702998x
Bladon, K. D., Emelko, M. B., Silins, U., and Stone, M. (2014). Wildfire and the future of water supply. Environ. Sci. Technol. 48, 8936–8943. doi: 10.1021/es500130g
Blake, D., Lu, K., Horwitz, P., and Boyce, M. C. (2012). Fire suppression and burnt sediments: effects on the water chemistry of fire-affected wetlands. Int. J. Wildland Fire 21, 557–561.
Bolanz, R. M., Wierzbicka-Wieczorek, M., Èaplovièová, M., Uhlík, P., Göttlicher, J., Steininger, R., et al. (2013). Structural incorporation of As5+ into hematite. Environ. Sci. Technol. 47, 9140–9147. doi: 10.1021/es305182c
Brown, G. E., Foster, A. L., and Ostergren, J. D. (1999). Mineral surfaces and bioavailability of heavy metals: a molecular-scale perspective. Proc. Natl. Acad. Sci. U.S.A. 96, 3388–3395. doi: 10.1073/pnas.96.7.3388
Burke, M. P., Hogue, T. S., Kinoshita, A. M., Barco, J., Wessel, C., and Stein, E. D. (2013). Pre- and post-fire pollutant loads in an urban fringe watershed in Southern California. Environ. Monit. Assess. 185, 10131–10145. doi: 10.1007/s10661-013-3318-9
Burton, C. A., Hoefen, T. M., Plumlee, G. S., Baumberger, K. L., Backlin, A. R., Gallegos, E., et al. (2016). Trace elements in stormflow, ash, and burned soil following the 2009 station fire in Southern California. PLoS One 11:e0153372. doi: 10.1371/journal.pone.0153372
Burton, E. D., Bush, R. T., Johnston, S. G., Watling, K. M., Hocking, R. K., Sullivan, L. A., et al. (2009). Sorption of arsenic(V) and arsenic(III) to schwertmannite. Environ. Sci. Technol. 43, 9202–9207. doi: 10.1021/es902461x
Burton, E. D., Bush, R. T., and Sullivan, L. A. (2006). Sedimentary iron geochemistry in acidic waterways associated with coastal lowland acid sulfate soils. Geochim. Cosmochim. Acta 70, 5455–5468. doi: 10.1016/j.gca.2006.08.016
Burton, E. D., Bush, R. T., Sullivan, L. A., Johnston, S. G., and Hocking, R. K. (2008). Mobility of arsenic and selected metals during re-flooding of iron- and organic-rich acid-sulfate soil. Chem. Geol. 253, 64–73. doi: 10.1016/j.chemgeo.2008.04.006
Burton, E. D., Choppala, G., Karimian, N., and Johnston, S. G. (2019). A new pathway for hexavalent chromium formation in soil: fire-induced alteration of iron oxides. Environ. Pollut. 247, 618–625. doi: 10.1016/j.envpol.2019.01.094
Campos, I., Abrantes, N., Keizer, J. J., Vale, C., and Pereira, P. (2016). Major and trace elements in soils and ashes of eucalypt and pine forest plantations in Portugal following a wildfire. Sci. Total Environ. 572, 1363–1376. doi: 10.1016/j.scitotenv.2016.01.190
Campos, I., Vale, C., Abrantes, N., Keizer, J. J., and Pereira, P. (2015). Effects of wildfire on mercury mobilisation in eucalypt and pine forests. Catena 131, 149–159. doi: 10.1016/j.catena.2015.02.024
Cerrato, J. M., Blake, J. M., Hirani, C., Clark, A. L., Ali, A.-M. S., Artyushkova, K., et al. (2016). Wildfires and water chemistry: effect of metals associated with wood ash. Environ. Sci. Process. Impacts 18, 1078–1089. doi: 10.1039/c6em00123h
Certini, G. (2005). Effects of fire on properties of forest soils: a review. Oecologia 143, 1–10. doi: 10.1007/s00442-004-1788-8
Claff, S. R., Sullivan, L. A., Burton, E. D., and Bush, R. T. (2010). A sequential extraction procedure for acid sulfate soils: partitioning of iron. Geoderma 155, 224–230. doi: 10.1016/j.geoderma.2009.12.002
Clarke, H., Pitman, A. J., Kala, J., Carouge, C., Haverd, V., and Evans, J. P. (2016). An investigation of future fuel load and fire weather in Australia. Clim. Change 139, 591–605. doi: 10.1007/s10584-016-1808-9
Cornell, R. M., and Schwertmann, U. (2003). The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses. Weinheim: Wiley VHC.
Cudennec, Y., and Lecerf, A. (2005). Topotactic transformations of goethite and lepidocrocite into hematite and maghemite. Solid State Sci. 7, 520–529. doi: 10.1016/j.solidstatesciences.2005.02.002
Cuypers, F., De Dobbelaere, C., Hardy, A., Van Bael, M. K., and Helsen, L. (2009). Thermal behaviour of arsenic trioxide adsorbed on activated carbon. J. of Hazard. Mater. 166, 1238–1243. doi: 10.1016/j.jhazmat.2008.12.048
Cuypers, F., and Helsen, L. (2011). Pyrolysis of chromated copper arsenate (CCA) treated wood waste at elevated pressure: influence of particle size, heating rate, residence time, temperature and pressure. J. Anal. Appl. Pyrolysis 92, 111–122. doi: 10.1016/j.jaap.2011.05.002
Dittmar, J., Voegelin, A., Maurer, F., Roberts, L. C., Hug, S. J., Saha, G. C., et al. (2010). Arsenic in soil and irrigation water affects arsenic uptake by rice: complementary insights from field and pot studies. Environ. Sci. Technol. 44, 8842–8848. doi: 10.1021/es101962d
Diwakar, J., Johnston, S. G., Burton, E. D., and Shrestha, S. D. (2015). Arsenic mobilization in an alluvial aquifer of the Terai region, Nepal. J. Hydrol. Reg. Stud. 4, 59–79. doi: 10.1016/j.ejrh.2014.10.001
Dixit, S., and Hering, J. G. (2003). Comparison of arsenic(V) and arsenic(III) sorption onto iron oxide minerals: implications for arsenic mobility. Environ. Sci. Technol. 37, 4182–4189. doi: 10.1021/es030309t
Dyar, M. D., Agresti, D. G., Schaefer, M. W., Grant, C. A., and Sklute, E. C. (2006). Mössbauer spectroscopy of earth and planetary materials. Annu. Rev. Earth Planet. Sci. 34, 83–125.
Eggleton, R. A., and Taylor, G. (2008). Impact of fire on the Weipa Bauxite, northern Australia. Aust. J. Earth Sci. 55, S83–S86.
Evans, R. J., and Milne, T. A. (1987). Molecular characterization of the pyrolysis of biomass. Energy Fuels 1, 123–137. doi: 10.1021/ef00002a001
Fendorf, S., Michael, H. A., and van Green, A. (2010a). Spatial and temporal variations of groundwater arsenic in South and Southeast Asia. Science 328, 1123–1127. doi: 10.1126/science.1172974
Fendorf, S., Nico, P. S., Kocar, B. D., Masue, Y., and Tufano, K. J. (2010b). “Arsenic chemistry in soils and sediments,” in Developments in Soil Science, eds S. Balwant and G. Markus (Amsterdam: Elsevier), 357–378. doi: 10.1016/s0166-2481(10)34012-8
Frommer, J., Voegelin, A., Dittmar, J., Marcus, M. A., and Kretzschmar, R. (2011). Biogeochemical processes and arsenic enrichment around rice roots in paddy soil: results from micro-focused X-ray spectroscopy. Eur. J. Soil Sci. 62, 305–317. doi: 10.1111/j.1365-2389.2010.01328.x
Gadde, B., Bonnet, S., Menke, C., and Garivait, S. (2009). Air pollutant emissions from rice straw open field burning in India, Thailand and the Philippines. Environ. Pollut. 157, 1554–1558. doi: 10.1016/j.envpol.2009.01.004
Goss, C. J. (1987). The kinetics and reaction mechanism of the goethite to hematite transformation. Mineral. Mag. 51, 437–451. doi: 10.1021/es4043275
Grogan, K. L., Gilkes, R. J., and Lottermoser, B. G. (2003). Maghemite formation in burnt plant litter at East Trinity, North Queensland, Australia. Clays Clay Miner. 51, 390–396. doi: 10.1346/ccmn.2003.0510404
Helsen, L., Van den Bulck, E., Van Bael, M. K., and Mullens, J. (2003). Arsenic release during pyrolysis of CCA treated wood waste: current state of knowledge. J. Anal. Appl. Pyrolysis 68–69, 613–633. doi: 10.1016/s0165-2370(03)00055-x
Helsen, L., Van den Bulck, E., Van Bael, M. K., Vanhoyland, G., and Mullens, J. (2004). Thermal behaviour of arsenic oxides (As2O5 and As2O3) and the influence of reducing agents (glucose and activated carbon). Thermochim. Acta 414, 145–153. doi: 10.1016/j.tca.2003.12.016
Herbel, M., and Fendorf, S. (2006). Biogeochemical processes controlling the speciation and transport of arsenic within iron coated sands. Chem. Geol. 228, 16–32. doi: 10.1016/j.chemgeo.2005.11.016
Islam, S., Rahman, M. M., Islam, M. R., and Naidu, R. (2016). Arsenic accumulation in rice: consequences of rice genotypes and management practices to reduce human health risk. Environ. Int. 96, 139–155. doi: 10.1016/j.envint.2016.09.006
Johnston, S. G., Bennett, W. W., Burton, E. D., Hockmann, K., Dawson, N., and Karimian, N. (2018). Rapid arsenic(V)-reduction by fire in schwertmannite-rich soil enhances arsenic mobilisation. Geochim. Cosmochim. Acta 227, 1–18. doi: 10.1016/j.gca.2018.01.031
Johnston, S. G., Burton, E. D., Keene, A. F., Planer-Friedrich, B., Voegelin, A., Blackford, M. G., et al. (2012). Arsenic mobilization and iron transformations during sulfidization of As(V)-bearing jarosite. Chem. Geol. 334, 9–24. doi: 10.1016/j.chemgeo.2012.09.045
Johnston, S. G., Burton, E. D., and Moon, E. M. (2016). Arsenic mobilization is enhanced by thermal transformation of schwertmannite. Environ. Sci. Technol. 50, 8010–8019. doi: 10.1021/acs.est.6b02618
Johnston, S. G., Diwakar, J., and Burton, E. D. (2015). Arsenic solid-phase speciation in an alluvial aquifer system adjacent to the Himalayan forehills, Nepal. Chem. Geol. 419, 55–66. doi: 10.1016/j.chemgeo.2015.10.035
Johnston, S. G., Keene, A. F., Burton, E. D., Bush, R. T., and Sullivan, L. A. (2011). Iron and arsenic cycling in intertidal surface sediments during wetland remediation. Environ. Sci. Technol. 45, 2179–2185. doi: 10.1021/es103403n
Jozwiak, W. K., Kaczmarek, E., Maniecki, T. P., Ignaczak, W., and Maniukiewicz, W. (2007). Reduction behavior of iron oxides in hydrogen and carbon monoxide atmospheres. Appl. Catal. A Gen. 326, 17–27. doi: 10.1016/j.apcata.2007.03.021
Keon, N. E., Swartz, C. H., Brabander, D. J., Harvey, C., and Hemond, H. F. (2001). Validation of an arsenic sequential extraction method for evaluating mobility in sediments. Environ. Sci. Technol. 35, 2778–2784. doi: 10.1021/es001511o
Ketterings, Q. M., Bigham, J. M., and Laperche, V. (2000). Changes in soil mineralogy and texture caused by slash-and-burn fires in Sumatra, Indonesia. Soil Sci. Soc. Am. J. 64, 1108–1117.
Knudsen, J. N., Jensen, P. A., Lin, W., Frandsen, F. J., and Dam-Johansen, K. (2004). Sulfur transformations during thermal conversion of herbaceous biomass. Energy Fuels 18, 810–819. doi: 10.1021/ef034085b
Landers, M., Gilkes, R. J., and Wells, M. A. (2009). Rapid dehydroxylation of nickeliferous goethite in lateritic nickel ore: x-ray diffraction and TEM investigation. Clays Clay Miner. 57, 751–770. doi: 10.1346/ccmn.2009.0570608
Langner, P., Mikutta, C., and Kretzschmar, R. (2012). Arsenic sequestration by organic sulphur in peat. Nat. Geosci. 5, 66–73. doi: 10.1038/ngeo1329
Leak, M., Passuello, R., and Tyler, B. (2003). I’ve seen fire, I’ve seen rain. I’ve seen muddy waters that I thought would never clean again. Waterworks 6, 38–44.
Lee, M.-K., Saunders James, A., Wilkin Richard, T., and Mohammad, S. (2005). “Geochemical modeling of arsenic speciation and mobilization: implications for bioremediation,” in Advances in Arsenic Research, Vol. 915, eds P. A. O’Day, D. Vlassopoulos, X. Meng, and L. G. Benning (Washington, DC: American Chemical Society), 398–413. doi: 10.1021/bk-2005-0915.ch029
Liu, W.-J., Li, W.-W., Jiang, H., and Yu, H.-Q. (2017). Fates of chemical elements in biomass during its pyrolysis. Chem. Rev. 117, 6367–6398. doi: 10.1021/acs.chemrev.6b00647
Liu, W.-J., Tian, K., Jiang, H., Zhang, X.-S., Ding, H.-S., and Yu, H.-Q. (2012). Selectively improving the bio-oil quality by catalytic fast pyrolysis of heavy-metal-polluted biomass: take copper (Cu) as an example. Environ. Sci. Technol. 46, 7849–7856. doi: 10.1021/es204681y
Löhr, S. C., Murphy, D. T., Nothdurft, L. D., Bolhar, R., Piazolo, S., and Siegel, C. (2017). Maghemite soil nodules reveal the impact of fire on mineralogical and geochemical differentiation at the Earth’s surface. Geochim. Cosmochim. Acta 200, 25–41. doi: 10.1016/j.gca.2016.12.011
Manning, B. A., Fendorf, S. E., and Goldberg, S. (1998). Surface structures and stability of arsenic(III) on goethite: spectroscopic evidence for inner-sphere complexes. Environ. Sci. Technol. 32, 2383–2388. doi: 10.1021/es9802201
Mazzetti, L., and Thistlethwaite, P. J. (2002). Raman spectra and thermal transformations of ferrihydrite and schwertmannite. J. Raman Spectrosc. 33, 104–111. doi: 10.1002/jrs.830
Morin, G., Ona-Nguema, G., Wang, Y., Menguy, N., Juillot, F., Proux, O., et al. (2008). Extended X-ray absorption fine structure analysis of arsenite and arsenate adsorption on maghemite. Environ. Sci. Technol. 42, 2361–2366. doi: 10.1021/es072057s
Mukherjee, A., Bhattacharya, P., Savage, K., Foster, A., and Bundschuh, J. (2008). Distribution of geogenic arsenic in hydrologic systems: controls and challenges. J. Contam. Hydrol. 99, 1–7. doi: 10.1016/j.jconhyd.2008.04.002
Murad, E., and Cashion, J. (eds). (2004). “Iron oxides,” in Mössbauer Spectroscopy of Environmental Materials and Their Industrial Utilization, (Boston, MA: Springer), 159–188.
Odigie, K. O., and Flegal, A. R. (2014). Trace metal inventories and lead isotopic composition chronicle a forest fire’s remobilization of industrial contaminants deposited in the Angeles national forest. PLoS One 9:e107835. doi: 10.1371/journal.pone.0107835
Prescher, C., McCammon, C., and Dubrovinsky, L. (2012). MossA: a program for analyzing energy-domain Mössbauer spectra from conventional and synchrotron sources. J. Appl. Crystallogr. 45, 329–331. doi: 10.1107/s0021889812004979
Ravel, B., and Newville, M. (2005). ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Rad. 12, 537–541. doi: 10.1107/S0909049505012719
Ravenscroft, P., Brammer, H., and Richards, K. (2009). Arsenic Pollution: A Global Synthesis. West Sussex: Wiley-Blackwell.
Richardson, Y., Blin, J., Volle, G., Motuzas, J., and Julbe, A. (2010). In situ generation of Ni metal nanoparticles as catalyst for H2-rich syngas production from biomass gasification. Appl. Catal. A Gen. 382, 220–230. doi: 10.1016/j.apcata.2010.04.047
Roberts, L. C., Hug, S. J., Dittmar, J., Voegelin, A., Kretzschmar, R., Wehrli, B., et al. (2010). Arsenic release from paddy soils during monsoon flooding. Nat. Geosci. 3, 53–59. doi: 10.1038/ngeo723
Root, R. A., Dixit, S., Campbell, K. M., Jew, A. D., Hering, J. G., and O’Day, P. A. (2007). Arsenic sequestration by sorption processes in high-iron sediments. Geochim. Cosmochim. Acta 71, 5782–5803. doi: 10.1016/j.gca.2007.04.038
Root, R. A., Vlassopoulos, D., Rivera, N. A., Rafferty, M. T., Andrews, C., and O’Day, P. A. (2009). Speciation and natural attenuation of arsenic and iron in a tidally influenced shallow aquifer. Geochim. Cosmochim. Acta 73, 5528–5553. doi: 10.1016/j.gca.2009.06.025
Schwertmann, U., and Cornell, R. M. (1991). Iron Oxides in the Laboratory: Preparation and Characterization. Hoboken, NJ: John Wiley & Sons.
Schwertmann, U., and Cornell, R. M. (2000). Iron Oxides in the Laboratory: Preparation and Characterization. Weinheim: Wiley VHC.
Seyfferth, A. L., McCurdy, S., Schaefer, M. V., and Fendorf, S. (2014). Arsenic concentrations in paddy soil and rice and health implications for major rice-growing regions of Cambodia. Environ. Sci. Technol. 48, 4699–4706. doi: 10.1021/es405016t
Seyfferth, A. L., Webb, S. M., Andrews, J. C., and Fendorf, S. (2010). Arsenic localization, speciation, and co-occurrence with iron on rice (Oryza sativa L.) roots having variable Fe coatings. Environ. Sci. Technol. 44, 8108–8113. doi: 10.1021/es101139z
Sherman, D. M., and Randall, S. R. (2003). Surface complexation of arsenic(V) to iron(III) (hydr)oxides: structural mechanism from ab initio molecular geometries and EXAFS spectroscopy. Geochim. Cosmochim. Acta 67, 4223–4230. doi: 10.1016/s0016-7037(03)00237-0
Siddique, M., Ahmed, E., and Butt, N. M. (2010). Particle size effect on Mössbauer parameters in γ-Fe2O3 nanoparticles. Physica B Condens. Matter 405, 3964–3967. doi: 10.1016/j.physb.2010.06.039
Smedley, P. L., and Kinniburgh, D. G. (2002). A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochem. 17, 517–568. doi: 10.1016/s0883-2927(02)00018-5
Stein, E. D., Brown, J. S., Hogue, T. S., Burke, M. P., and Kinoshita, A. (2012). Stormwater contaminant loading following southern California wildfires. Environ. Toxicol. Chem. 31, 2625–2638. doi: 10.1002/etc.1994
Takahashi, Y., Minamikawa, R., Hattori, K. H., Kurishima, K., Kihou, N., and Yuita, K. (2004). Arsenic behavior in paddy fields during the cycle of flooded and non-flooded periods. Environ. Sci. Technol. 38, 1038–1044. doi: 10.1021/es034383n
Taylor, R. M., and Schwertmann, U. (1974). Maghemite in soils and its origin: 1 properties and observations on soil maghemites. Clay Miner. 10, 289–298. doi: 10.1180/claymin.1974.010.4.07
Vandenberghe, R. E., Barrero, C. A., da Costa, G. M., Van San, E., and De Grave, E. (2000). Mössbauer characterization of iron oxides and (oxy)hydroxides: the present state of the art. Hyperfine Interact. 126, 247–259.
Vandenberghe, R. E., and De Grave, E. (2013). “Application of Mössbauer spectroscopy in earth sciences,” in Mössbauer Spectroscopy: Tutorial Book, eds Y. Yoshida and G. Langouche (Berlin: Springer), 91–185. doi: 10.1007/978-3-642-32220-4_3
Vithanage, M., Herath, I., Joseph, S., Bundschuh, J., Bolan, N., Ok, Y. S., et al. (2017). Interaction of arsenic with biochar in soil and water: a critical review. Carbon 113, 219–230. doi: 10.1016/j.carbon.2016.11.032
Voegelin, A., Weber, F.-A., and Kretzschmar, R. (2007). Distribution and speciation of arsenic around roots in a contaminated riparian floodplain soil: micro-XRF element mapping and EXAFS spectroscopy. Geochim. Cosmochim. Acta 71, 5804–5820. doi: 10.1016/j.gca.2007.05.030
Wasson, S. J., Linak, W. P., Gullett, B. K., King, C. J., Touati, A., Huggins, F. E., et al. (2005). Emissions of chromium, copper, arsenic, and PCDDs/Fs from open burning of CCA-treated wood. Environ. Sci. Technol. 39, 8865–8876. doi: 10.1021/es050891g
Waychunas, G. A., Kim, C. S., and Banfield, J. F. (2005). Nanoparticulate iron oxide minerals in soils and sediments: unique properties and contaminant scavenging mechanisms. J. Nanopart. Res. 7, 409–433. doi: 10.1007/s11051-005-6931-x
Waychunas, G. A., Rea, B. A., Fuller, C. C., and Davis, J. A. (1993). Surface chemistry of ferrihydrite: part 1. EXAFS studies of the geometry of coprecipitated and adsorbed arsenate. Geochim. Cosmochim. Acta 57, 2251–2269. doi: 10.1016/0016-7037(93)90567-g
Wolf, R. E., Hoefen, T. M., Hageman, P. L., Morman, S. A., and Plumlee, G. S. (2010). Speciation of Arsenic, Selenium, and Chromium in Wildfire Impacted Soils and Ashes, Open-File Report 2010–1242. Reston, VA: U.S. Geological Survey, 29.
Wu, D., and Sun, S. (2016). Speciation analysis of As, Sb and Se. Trends Environ. Anal. Chem. 11, 9–22. doi: 10.1016/j.teac.2016.05.001
Keywords: arsenic, fire, water quality, XANES, ferrihydrite, goethite, maghemite, soil
Citation: Johnston SG, Karimian N and Burton ED (2019) Fire Promotes Arsenic Mobilization and Rapid Arsenic(III) Formation in Soil via Thermal Alteration of Arsenic-Bearing Iron Oxides. Front. Earth Sci. 7:139. doi: 10.3389/feart.2019.00139
Received: 31 March 2019; Accepted: 15 May 2019;
Published: 29 May 2019.
Edited by:
Jon Hawkings, Florida State University, United StatesReviewed by:
Robert A. Root, The University of Arizona, United StatesPetr Drahota, Charles University, Czechia
Copyright © 2019 Johnston, Karimian and Burton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Scott G. Johnston, c2NvdHQuam9obnN0b25Ac2N1LmVkdS5hdQ==