 Sudeshna Basu
Sudeshna Basu Alexander B. Verchovsky2
Alexander B. Verchovsky2 Anna Bogush
Anna Bogush- 1Department of Earth Sciences, University College London, London, United Kingdom
- 2Department of Planetary Sciences, Open University, Milton Keynes, United Kingdom
Stability and mobility of organic matter in shale is significant from the perspective of carbon cycle. Shale can only be an effective sink provided that the organic carbon present is stable and immobile from the host sites and, not released easily during geological processes such as low pressure-temperature burial diagenesis and higher pressure-temperature subduction. To examine this, three Jurassic shale samples of known mineralogy and total organic carbon content, with dominantly continental source of organic matter, belonging to the Haynesville-Bossier Formation were combusted by incremental heating from temperature of 200 to 1400°C. The samples were analyzed for their carbon and nitrogen release profiles, bulk δ13C composition and C/N atomic ratio, based on which, at least four organic carbon components are identified associated with different minerals such as clay, carbonate, and silicate. They have different stability depending on their host sites and occurrences relative to the mineral phases and consequently, released at different temperature during combustion. The components identified are denoted as, C-1 (organic carbon occurring as free accumulates at the edge or mouth of pore spaces), C-2 (associated with clay minerals, adsorbed or as organomineral nanocomposites; with carbonate minerals, biomineralized and/or occluded), C-3(a) (occurring with silicate minerals, biomineralized and/or occluded) and C-3(b) (graphitized carbon). They show an increasing stability and decreasing mobility from C-1 to C-3(b). Based on the stability of the different OC components, shale is clearly an efficient sink for the long term C cycle as, except for C-1 which forms a very small fraction of the total and is released at temperature of ∼200°C, OC can be efficiently locked in shale surviving conditions of burial diagenesis and, subduction at fore arc regions in absence of infiltrating fluids. Under low fluid flux, C-3(b) can be efficiently retained as a refractory phase in the mantle when subducted. It is evident that the association and interaction of the organic matter with the different minerals play an important role in its retention in the shale.
Introduction
Like many other elements, carbon cycles through different reservoirs on Earth including the atmosphere, mantle, hydrosphere (oceans and rivers), petrogenic reservoirs (mainly sedimentary rocks), soil and vegetal cover and biosphere (mainly as biomass). The carbon cycle has both slow and fast components referred to as the short and long term cycles respectively (Appendix 1) (Berner, 2003; Wallmann and Aloisi, 2012). The short term cycle includes photosynthesis, respiration and decomposition of biomass as well as air-ocean gas exchange. The carbon component in organic matter, referred to as the organic carbon (OC) can be incorporated in the plant biomass by photosynthesis. It subsequently cycles through the soil and oceanic sediments where, a fraction of it can be subject to decomposition. For the long term cycle, a fraction of the carbon subducted to the Earth’s mantle is returned to the surface via degassing as CO2 and CH4 from mid-oceanic ridges (MORs) and hotspots within plate settings, arcs and forearc regions in subduction settings (Chiodini et al., 2010; Kelemen and Manning, 2015; Aiuppa et al., 2019; Voyer et al., 2019). For mid-oceanic ridges the CO2 can be dissolved in the seawater as bicarbonate ions, although substantial release, to an extent diffuse, is also possible (Voyer et al., 2019). Contribution to degassing is restricted to shallow water areas where the ridge system is exposed above sea level. CO2 degassing from volcanic front is well-constrained as compared to fore arc or back arc regions, possibly related to lack of obvious high emission sources in the area between the trench and the degassing volcanic arc front (Voyer et al., 2019). Emplacement of Large Igneous Provinces and mountain building can be associated with metamorphic degassing. For better understanding of such open system processes, it is necessary to constraint the fluid composition and constrain the absolute timings and duration of the fluid flow (Evans, 2011). The time scale of interest, relative to the processes involved will determine if the carbon cycle is in a steady state.
Beside the mantle, another effective carbon sink can be the different rock types where it can be locked in mineralized form as in sedimentary rocks as carbonates (biogenic and authigenic), or in hydrothermal calcite vein formed by low temperature alteration of the upper oceanic crust following chemical weathering of silicate rocks (Alt et al., 1999; Coogan et al., 2016). A recent study has shown that CO2 maybe sequestered within the crust by calcite deposition or alternatively be incorporated into the biomass as inorganic carbon during microbial activity, consequently limiting the amount of C available for transfer to the deep mantle (Barry et al., 2019). OC is also contained in organic matter locked in ancient marine sediments represented by shale. The organic content in carbonaceous shale can be more than 20% by weight (Dayal, 2017). Given that sedimentary formations represent 66% of the rocks on the Earth’s surface of which 50% is shale (Blatt and Jones, 1975), it is the largest reservoir in terms of the total mass of OC on Earth. Consequently, its contribution to the global carbon budget and the role that it plays in the carbon cycle cannot be neglected. While rocks like limestone are primarily constituted of carbonate minerals, in shale, they coexist with OC. The carbonate content in shale is variable, deposited by primary precipitation, biochemical processes or recrystallization during diagenesis. OC degradation by microbial sulfate reduction can be related to carbonate accumulation in anoxic sediments when sedimentary organic matter is converted to bicarbonate ions, accounting for the coexistence of carbonate minerals with organic matter (Wallmann and Aloisi, 2012; Zeng et al., 2018). The hydrocarbon fluid in the petroleum and natural gas reservoirs, mostly biogenic although abiogenic origin cannot be ruled out, is also a considerable carbon sink (Etiope and Sherwood-Lollar, 2013). Immiscible hydrocarbon fluids can be present in the expected range of pressure and temperature in subduction zone, providing a mechanism for the transfer of slab carbon to the deep mantle (Huang et al., 2017; Li, 2017; Vitale Brovarone et al., 2017).
The subduction of the carbonate minerals is distinct from, but operates in tandem with the OC (Cook-Kollars et al., 2014). Based on net flux estimates, carbon can be considered to be in steady state between subduction and mantle outgassing (Jarrard, 2003). Alternatively, there may be an increase in carbon at the Earth’s surface over time or, a net flux to the mantle under the fore arcs controlled by the initial composition of the subducting material (Cook-Kollars et al., 2014; Kelemen and Manning, 2015; Clift, 2017). During fore arc metamorphism, subducted carbonate can be retained without any substantial decarbonation and loss of CO2 under peak pressure-temperature conditions of ∼3 GPa and 600°C, at depth of 120 km (Collins et al., 2015). Carbonates reduced to graphitized form, at very high to lower temperature of 430°C, can be transported to deeper regions resulting in long term (Gyrs) removal of reduced, light carbon from surficial reservoirs of the Earth (Galvez et al., 2013; Duncan and Dasgupta, 2017). Degassing of graphitic OC can be substantial up to the chlorite zone but restricted at higher metamorphic grades (Zhang et al., 2018). A highly stable OC component in the graphitized form can be retained as a refractory phase under mantle conditions during subduction (Duncan and Dasgupta, 2017). Subduction of carbonates to the deep mantle beyond 660 km (transition zone) maybe inhibited due to melting of carbonate components (Thomson et al., 2016). When fluids circulate along sheared and brecciated domains and plate interfaces, substantial decarbonation and carbonate dissolution can occur at depth of 80–120 km in subducting slabs (Ague and Nicolescu, 2014; Cook-Kollars et al., 2014; Collins et al., 2015; Schwarzenbach et al., 2018). The large volume of infiltrating H2O-rich fluid can be derived externally from underlying dehydrating mafic and ultramafic rocks.
We analyzed shale for carbon (and nitrogen) by stepwise combustion to decouple the different carbon components that are present. This study demonstrates that the stability of OC in shale is governed by their variable association with different mineral phases like carbonate, clay minerals and silicates related to how they are trapped/hosted with them. While free occurrence as accumulates, aggregated by clumping or binding renders them most unstable, a more stable and less mobile OC component occurs with silicate and carbonate minerals. It can occur as biominerals within the silicate walls of organisms such as diatoms or, within carbonate shells of corals or foraminifera. When occluded, the OC is protected or blocked within the mineral frame of silicate or carbonate matrix (Keil and Mayer, 2014). The stability can be variable when associated with clay and carbonate minerals, depending on whether they are adsorbed, occluded or biomineralized.
Objectives of the Study
Shale can be an efficient, long term geological reservoir locking up carbon both in its organic and inorganic forms. Carbon can be released from it during weathering and transported to soil and rivers and finally to the oceans as inherited or ‘old carbon’ (Di-Giovanni et al., 2002). The later can be substantial and, is in addition to contribution from soil originating from present or post-glacial vegetal cover referred to as the ‘new carbon’ which is less resistant to microbial degradation. It is critical to understand how much of OC in shale is lost prior to release and eventual transportation to rivers and oceans. Over time, this fraction of the OC can be again locked for millions of years as they are redeposited and reconsolidated into rocks. The availability of OC from shales to contribute toward soil and river flux is largely controlled by how easily it can be degraded by geological processes such as oxidative weathering or altered during burial diagenesis. This will also control the exposure of shale organic matter to microorganisms in the weathering profile (Petsch et al., 2005). The breakdown of the liberated OC is facilitated by microbial oxidation in the presence of atmospheric oxygen. Its susceptibility toward alteration under action of different reagents, has been seen to be related to its stability and/or mobility from different host sites in shale (Zhu et al., 2016). This has implications for sediment subduction as well. Depending on its stability, the OC can be effectively subducted to the deeper mantle, unless incorporated in advecting sedimentary piles or buoyant metasedimentary diapirs to be transported to the hotter mantle wedge (Tsuno and Dasgupta, 2011; Tsuno et al., 2012; Kelemen and Manning, 2015).
It is evident that if the oxidation reactions proceed at a fast rate to enable degradation of the OC in shales to CO2 following exposure to the atmosphere by uplift and erosion, it cannot be sequestered in re-sedimented rocks to be isolated from the surficial carbon reservoirs for long periods of time. In subduction zones, with increasing burial, if OC is resistant to remobilization by CO2 formation, it cannot be released to the atmosphere by magmatic degassing but instead can be efficiently subducted in graphitized form by rhyolitic melt and sequestered in the deep mantle (Wallmann and Aloisi, 2012). In both these instances the fate of OC as to its susceptibility to oxidation, subsequently impacts the long term carbon cycle. This study aims to look into the effect of oxidation with increasing temperature on the mobility of carbon locked in shales, by combusting them with incremental heating. Depending on the host sites of different forms of carbon of various stability and their consequent mobility, their release during combustion will occur at different temperature. In the geological context, this has critical implication as stable and less mobile forms of carbon are more likely to be re-sedimented in marine rocks by escaping degradation or, if subducted, can be sequestered in the deep mantle.
Background: Decoupling Carbon Components in a Shale by Conventional Methods
Different types of OC have different degrees of resistance to oxidative degradation depending on their type (Wallmann and Aloisi, 2012) and their host sites in rocks (Zhu et al., 2016). But, isolating these components in terms of their variable stability and mobility can be quite challenging. Conventionally organic and inorganic carbon components (carbonates) in shale are decoupled by acid treatment, where the mineralized inorganic carbon is converted to CO2 during acidification. The OC is subsequently analyzed from the inorganic carbon free sample after injection into a TOC (total organic carbon) analyzer. The OC is then detected and measured after oxidizing it to CO2 and releasing it from the sample. The inorganic carbon can be quantified by the carbon analyzer by measuring the total carbon content and the non-carbonate carbon fraction following leaching with hydrochloric acid. Some loss of volatile and soluble components of OC is known to occur during acid treatment (Saikkonen and Rautiainen, 1990). In the process of separating the organic and inorganic components prior to analyses, information in terms of OC-carbonate association is lost. Also, no discrimination between the different OC components present in different sites is possible.
The OC occurs as both low molecular weight (free) and high molecular weight (kerogen) forms. Solvent extraction can solvate the free compounds thereby isolating them from the relatively immobile kerogen and mineral matrix (Wright et al., 2015) again failing to provide information pertaining to host site, stability and mobility. The heterogeneity of organic matter can be monitored by petrographic and spectroscopic methods. Use of organic petrography and scanning electron microscopy can be used to distinguish between different organic components in terms of their differences in porosity and, association with each other and mineral components (Yang et al., 2017). Raman imaging can provide structural information in terms of the degree of organization indicating graphitization of the organic matter (Henry et al., 2019), but none of these methods by themselves can relate the occurrences of the carbon components to their stability and mobility in the shale or provide any quantification of the components.
Thermal decomposition method like loss on ignition (LOI), that oxidizes organic matter at temperature of 500–550°C followed by decomposition of carbonate at 900–1000°C, can be used for decoupling and quantifying OC and inorganic carbon components in clay-poor shale, but can be affected by sample size and breakdown of clay minerals (Smith, 2003). On the other hand, a combination of methods have shown to be more informative. For example, multi-step pyrolysis at temperature of 350, 600, and 1000°C coupled with Fourier transform infrared (FTIR) spectrophotometry was successfully applied to reveal information on the different speciation of OC in terms of thermal maturity (Saikkonen and Rautiainen, 1990). In a different study, a combination of treatment with reagents after separation of clay-sized fractions, followed by pyrolysis and FTIR spectrophotometry revealed OC components having different stability/mobility levels, occurring at different sites in the shale (Zhu et al., 2016).
In this study, by simultaneously analyzing carbon and nitrogen in shale in small incremental steps for their concentration, isotopic (δ13C) and elemental composition (C/N), it has been possible to decouple the different carbon components based on their mobility/stability, without any acid treatment. Simultaneous analyses of carbon, and nitrogen enabled correlation with the carbonate and clay minerals. In addition, the organic δ13C (δ13Corganic) and N/C elemental ratio helped to constrain the source (marine/lacustrine/continental) of the organic matter in these shales. Measurement by simultaneous thermal analyses (STA), was used to support the observed mineral breakdown from the combustion experiments.
Methodology
Three Jurassic shales from two different cores of the Haynesville-Bossier formation were analyzed by multiple step combustion for carbon and nitrogen concentration, C/N atomic ratio and δ13C isotopic composition. Prior to the combustion, the mineralogy and the TOC content of two of the samples were determined on their powders by XRD (Bruker AXS D4 Endeavor X-ray diffractometer) and Flash Elemental Analyzer 1112 (Thermo) respectively. The δ13Corganic value was determined by Gas Ratio Isotope Ration Mass Spectrometry, following flash combustion, after cleaning the samples from any carbonate remains by acid digestion. The porosity and permeability were determined on the bulk shale after crushing.
Shales are highly friable and fissile. It is very difficult to get undamaged samples for study but here, we had access to relatively intact drill core samples. For step combustion, the samples were extracted from the central portion of the drill cores to minimize superficial effects of core retrieval and handling related contamination. They were then crushed to mm-sized fragments using agate mortar and pestle, easily achievable as they split along the fissile planes. Between 5 and 10 mg of samples were combusted from 200 to 1400°C (± 10°C), in incremental steps of 100–200°C each, using 2 torr of O2, introduced to the system from CuO under ultra-high vacuum conditions. The total number of steps varied between nine and 10 and the time of combustion was 30 min for each step. A fully automated mass spectrometric complex, Finesse, was used for carbon and nitrogen analyses (Verchovsky, 2017). The combustion system used was all metal with a CuO finger to clean up the nitrogen fraction before introducing the gas to the mass spectrometer. The CO2 was trapped using a cold finger.
Simultaneous thermal analysis using a NETZSCH STA 449 C was used to investigate mass changes [thermogravimetry (TG); differential TG (DTG)] and energy flows [differential scanning calorimetry (DSC)] as a function of temperature, to investigate the combustion experiments. STA analysis was conducted on two samples using ∼200 mg of bulk, crushed powder in an 85 μL alumina crucible (and an identical reference crucible) with an air purge gas flow rate of 100 mL/min, equilibration at 40°C for 10 min, followed by a heating rate of 10°C/min up to 1400°C.
Samples and Geological History
The samples belong to the Haynesville-Bossier Formation in northwestern Louisiana, eastern Texas, deposited during the Late Jurassic (156 to 145.5 Ma) in a marine environment. The organic rich shale is related to the tectonically formed East Texas Basin (ETB), about 125 to 150 km long and 50 km wide, considered a sub-basin of the larger basin of the Gulf of Mexico (Jackson and Laubach, 1988; Salvador, 1991). It was formed and developed during the Triassic through the Jurassic related to breakup, rifting and extension of Pangea with the opening of the Gulf of Mexico Province (Mainali, 2011). During the Jurassic, a possible elevated southern margin restricted circulation resulting in the deposition of anhydrite and salt, but eventually transited to open marine condition with progressive subsidence. In general, the Haynesville is significantly more calcareous and OC rich than the overlying Bossier, the later having higher clay content. The samples for this study belong to Bossier and retrieved from different depth of two cores which are in close proximity, separated by only ∼8 km between them. Because of the high temperature gradient of ∼ 60°C/km during the Jurassic, the Haynesville-Bossier shale was exposed to temperature of 150–200°C during the last ∼ 100 Myrs (Nunn, 2012).
Results
The mineralogy, porosity and permeability of two of the samples, each from a different core, are listed in Table 1 along with their TOC content. They have comparable TOC (1.2–1.5 wt%) and mainly consist of carbonate, clay, and quartz (>80%). In terms of content between carbonate and clay minerals there is a difference of < 15% between the samples. Sample S-2(1) has a higher carbonate content than S-2(3) (32 vs. 14%) but a lower clay content (34 vs. 46%). The carbonates are mostly calcite, with some dolomite present as well. Illite is the dominant clay mineral with some kaolinite, chlorite and mixed illite/smectite layer, although quantification is not possible. Other minerals present include K-feldspar, plagioclase, pyrite, and apatite. Both the samples have comparable porosity (9 to 11% of bulk volume) while the permeability of S-2(1) (∼10–4 md) is higher by an order of magnitude than S-2(3). The δ13Corganic (denoting source of organic matter) of the samples are also comparable (Table 2). High Tmax of > 400°C determined by rock eval pyrolysis on the samples indicates that they are thermally over matured.
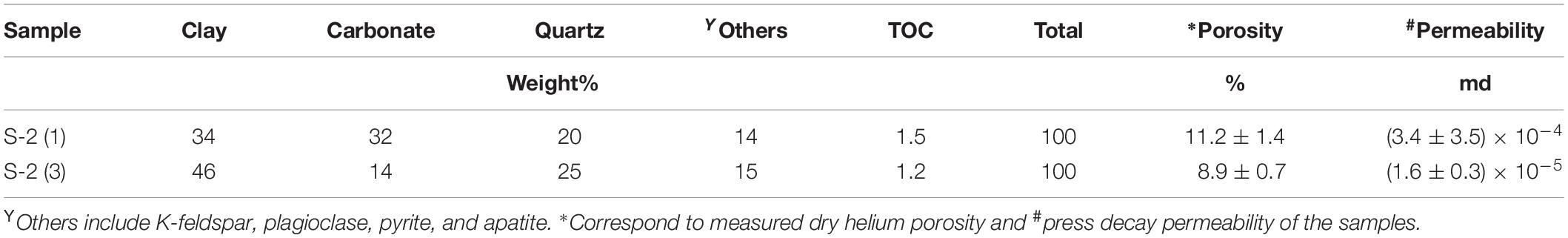
Table 1. XRD mineral compositions combined with TOC and normalized to 100%.

Table 2. Bulk composition of samples in terms of carbon (C) and nitrogen (N) concentration, δ13C (bulk and organic), and atomic C/N ratios.
In Tables 2, 3, the carbon and nitrogen concentration and isotopic ratios along with elemental C/N values are listed for bulk values and individual combustion steps of each sample respectively. There are some differences in the carbon content when compared to XRD measurements (Table 1) that can be related to shale heterogeneity, also as the two methods involved analyses of different weight of samples. But, the release profiles of all the three samples for step combustion are comparable. The peak nitrogen release occurs at 500–600°C, at a slightly lower temperature as compared to the peak carbon release at 600–700°C. The total carbon content of the samples is between 2 and 3 wt.% while the nitrogen concentration varies between 1000 and 1400 ppm. Their δ13Corganic (measured independently) are ∼−27‰ and comparable. Given marine carbonates have δ13C (up to + 4‰) with a considerable range when affected by diagenesis (Murata et al., 1969), the δ13C of −20 to −16‰ for the three bulk shales from this study, indicates mixing between OC and inorganic carbon, the later contributed by the carbonates. The peak release of carbon is coincident with elevated δ13C suggestive of breakdown of the carbonate minerals. The peak release of nitrogen for S-2(3) and S-3(3) are coincident with low C/N ratios suggestive of contribution from clay minerals and/or associated organic matter. The nitrogen release profile of S-2(1) is more prolonged than the other two samples. The C/N of the samples are in the range of 15–30. Instrumental blank corrections for both carbon and nitrogen were below significance.
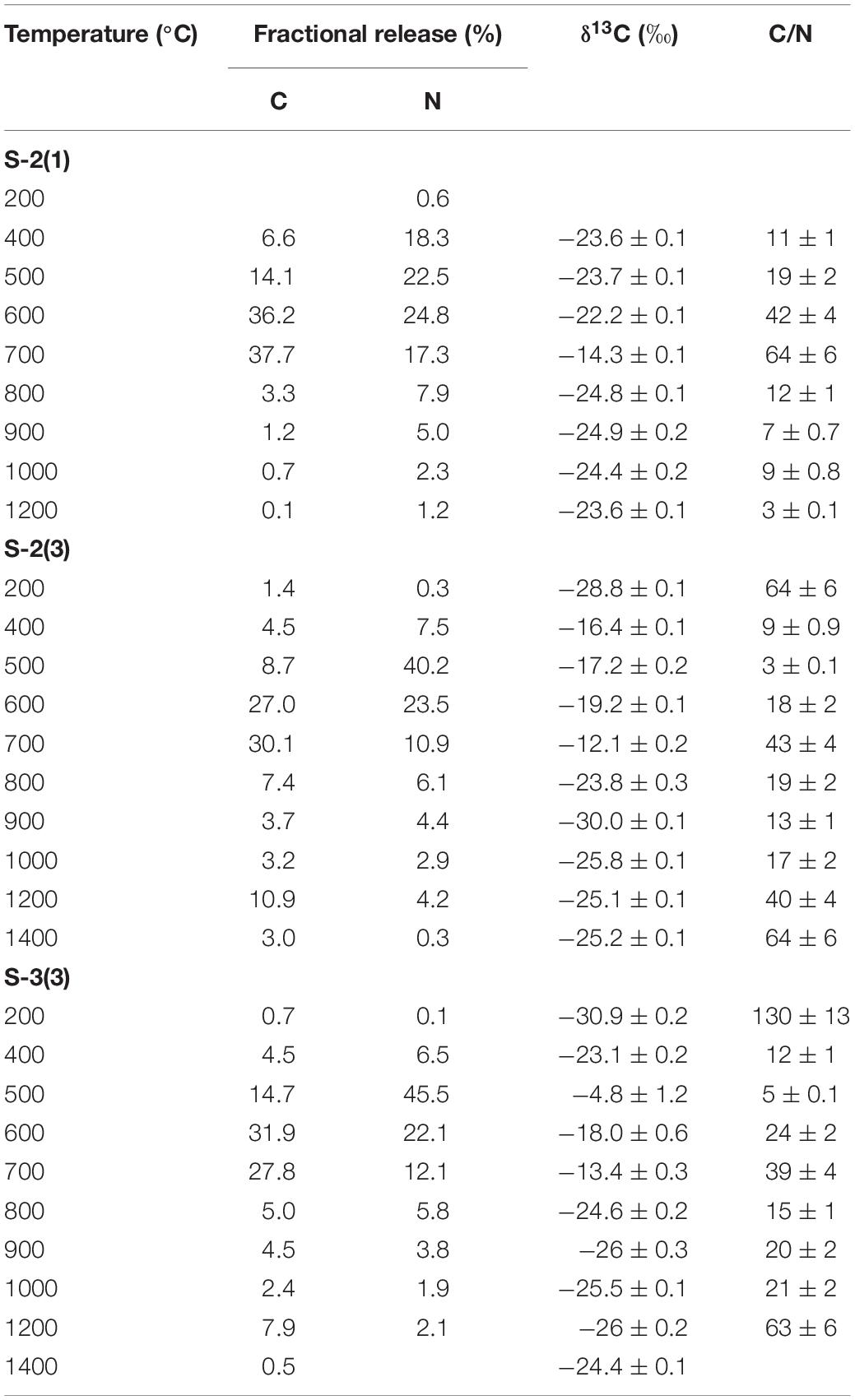
Table 3. Individual step release of carbon (C) and nitrogen (N) from shale during combustion with corresponding δ13C and C/N ratios.
For STA, five characteristic regions for shales in the studied interval from 50 to 1400°C could be identified (Appendix 2). At 50 to 150°C, there is mainly loss of free water/moisture physisorbed on the surfaces of particles, and clay-bound fluids resulting in a slight weight loss [0.69 wt.% for S-2(1) and 0.79 wt.% for S-2(3)] and a slight visible endothermic process. At the second temperature region of 300–650°C, there is a mass loss [6.12 wt.% for S-2(1); 3.16 wt.% for S-2(3)] attributed to decomposition of organic matter (300–600°C), an exothermic process shown on DSC curve, with decomposition of clay, e.g., degradation of kaolinite/illite (500–550°C) and chlorite (580–625°C). The complex peak in this temperature range [mainly for S-2(3)] with a shoulder at the lower temperature corresponding to the oxidation reaction of light hydrocarbons, while the main peak corresponds to the oxidation of heavy hydrocarbons and fixed carbon (Sun et al., 2015). Also, presence of any pyrite, might influence the TGA curve in this temperature region due to oxidation of that mineral at around 500°C (Zhang et al., 2014). The decomposition of carbonates was observed in the DTG curve from ∼625–850°C (dolomite 625–720°C and calcite 780–840°C) with mass losses of 4.1 wt.% for S-2(1) and 3.5 wt.% for S-2(3). At 850–1150°C, the mass losses of 1.5 wt.% for S-2(1) and 2.6 wt.% for S-2(3) can be attributed to decomposition and transformation of more stable mineral phases such as quartz, silicate and iron-bearing phases (Hajpal and Torok, 2004). At the highest temperature of 1150–1400°C, the mass loss of 0.7 wt.% for S-2(1) and 0.6 wt.% for S-2(3) with a broad endothermic region can indicate decomposition/transformation and or melting of the residual sample.
Discussion
Source of OM
The organic matter may be present in different forms from multiple sources (e.g., marine and continental), that may have different thermal maturity and stability. Marine organic matter is constituted of phytoplankton debris or detritus with proteins/amino acids, carbohydrates, and lipids as the main chemical components (Burdige, 2007). On the other hand, continental organic matter is composed of terrestrial biomass including plant residue and soil organic matter. Since continental derived organic matter consists of already altered and degraded remains of the living terrestrial biomass (e.g., soil humus), it is less susceptible to further degradation and alteration as compared to marine organic matter. The organic matter for the three samples in this study is predominantly derived from continental source with any marine contribution being minor, as seen previously from C/N and δ13C in shale retrieved from various depth of the studied cores (Basu et al., 2018). This is corroborated by the measured δ13Corganic ratio of ∼ −27‰ in all the three samples (Table 2), in agreement with expected δ13C of −27‰ for land plant organic matter, as to ∼ −20‰ in marine algal organic matter (Meyers, 2014).
Multiple OC Components Based on Step Combustion Release Profile
The peak release of nitrogen (∼70% of the total) in S-2(3) and S-3(3) corresponds to 500–600°C indicating the breakdown of organic matter and some clay minerals in this temperature range, also suggested from STA experiments. For S-2(1), the nitrogen release is much more prolonged from 400 to 700°C. While all other attributes such as TOC and nitrogen content are comparable, S-2(1) has a notably 18% higher carbonate content. The prolonged release can be related to physical protection of the organic matter by the carbonate minerals in S-2(1). But in all the three samples, peak release of carbon (>55%) occurred between 600 and 700°C with elevated δ13C, corresponding to the breakdown of carbonate minerals, as also seen from STA experiments. Alternatively, the prolonged release can be an effect of fabric/texture of the sample, which is beyond the scope of this study. It is possible, that in S-2(1), the mineral matrix has specific arrangement of its constituents (e.g., alternation of organic and clay nanolayers) (Salmon et al., 2000), aiding in physical protection of the organic matter and preventing its breakdown. This can only be confirmed from detailed study of the nano-scale textural arrangement of the shales.
Based on the carbon and nitrogen release profiles and the associated δ13C and N/C ratios, up to five different components can be identified associated with different release temperature as described in Table 4.
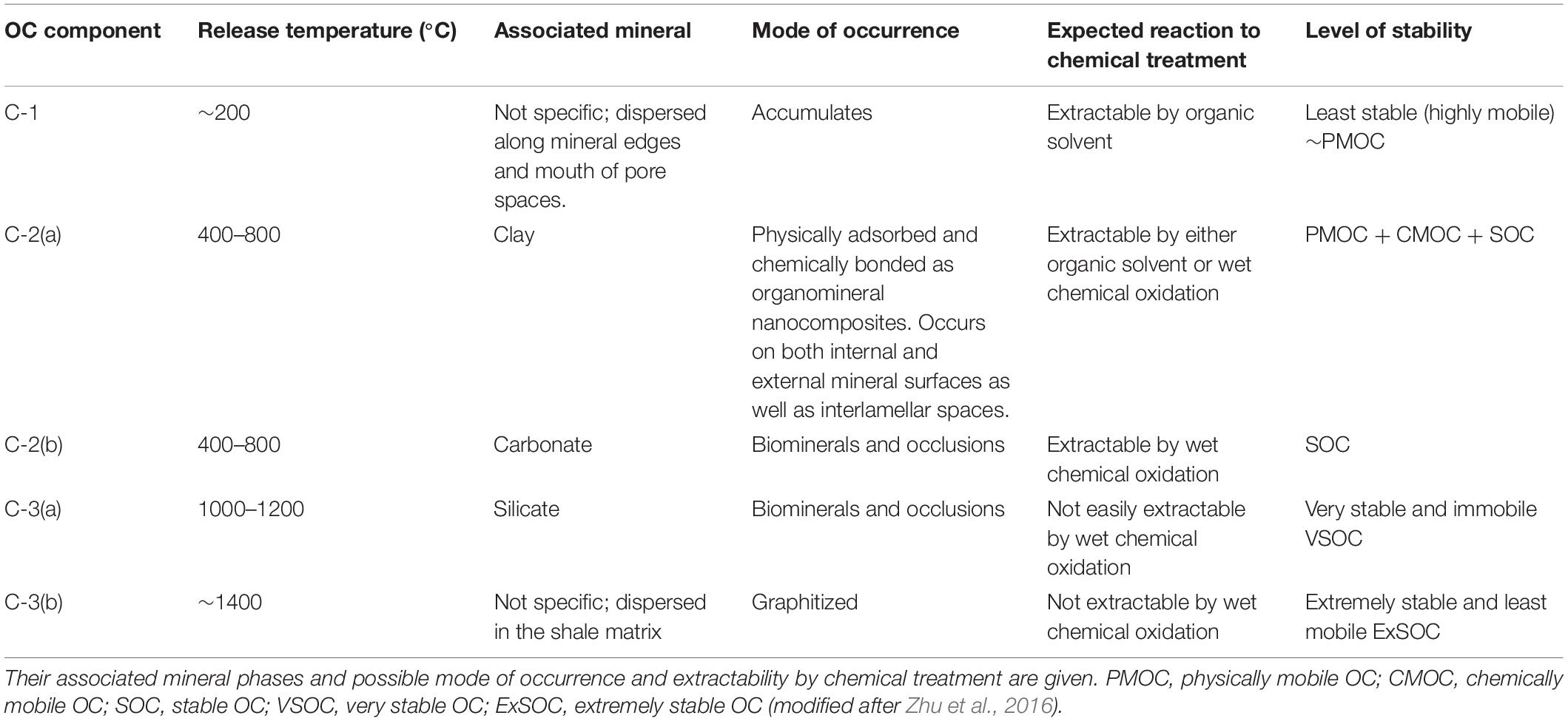
Table 4. OC components present in a shale as identified based on their variable release temperature during step heating.
<400°C
A very small fraction of total carbon (<1.5% of the total) is released at low temperature of 200°C for samples S-2(3) and S-3(3), also accompanied by very low nitrogen content (<0.5%) and with depleted δ13C of −30 to −28‰. In agreement with observations from STA experiments, this component can be attributed to free phases that can accumulate at the mouth of pore spaces or along mineral edges, with least preservation protection. The high C/N and extremely depleted δ13C is suggestive of adsorbed thermogenic, methane in these two samples (Golding et al., 2013). The possibility of this adsorbed component to be the result of depressurization during core retrieval cannot be completely ruled out. In S-2(1), a comparable, carbon component at an early temperature step is lacking although a small fractional nitrogen release is observed. This nitrogen dominated component is formed during decomposition of organic matter, with degraded matter being preferentially adsorbed on charged clay mineral surfaces as exchangeable nitrogen (Scholten, 1991). Interaction of organic matter by physisorption with silicate mineral matrix has been identified during chemical extraction of mineral matrices in shale using acid/ether extracts (Jeong and Kobylinski, 1983).
Such a low temperature component of the organic matter, will be unstable and mobile, and easily lost from the shale during geological processes. This readily mobile component is denoted as C-1 (Figures 1–3) and contributes toward a very small fraction of the total OC present.
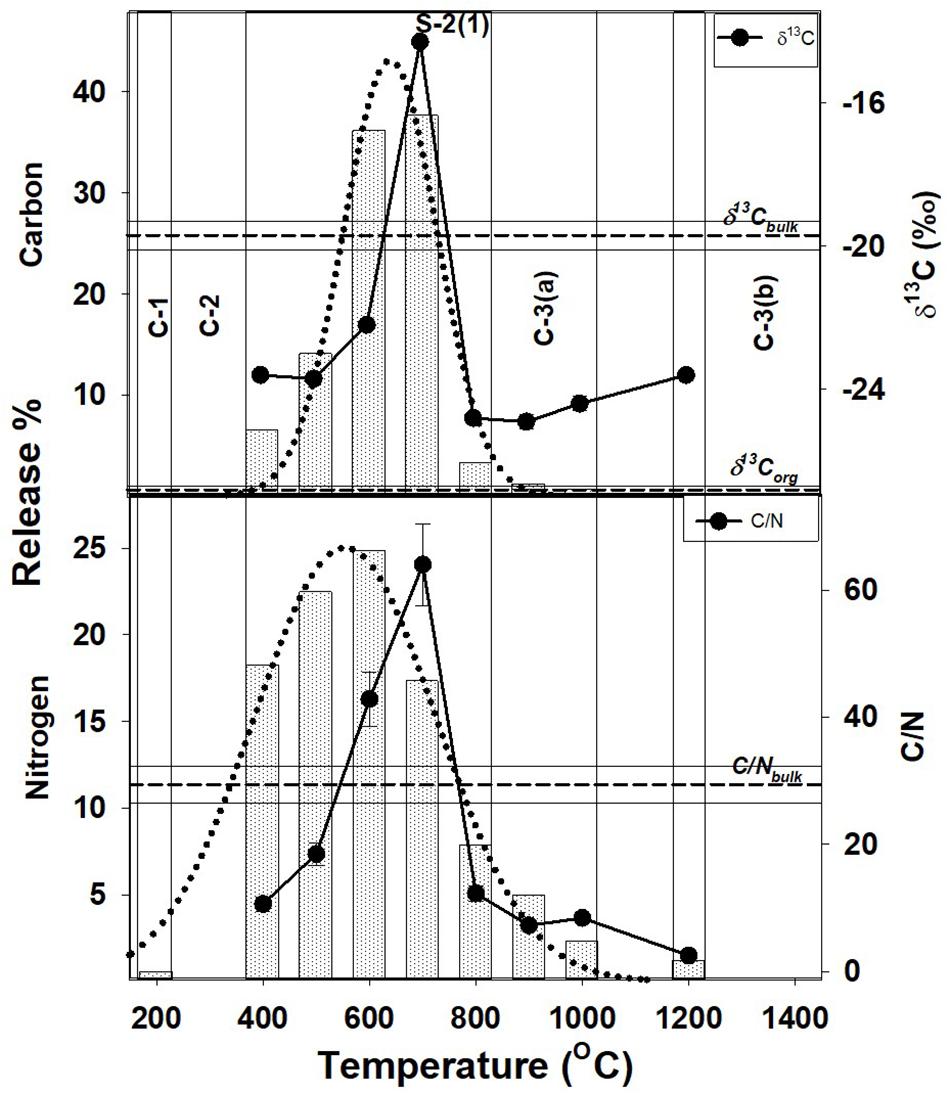
Figure 1. Step combustion temperature vs. carbon and nitrogen release percent for S-2(1). The δ13C and C/N for each temperature step are shown along with the bulk C/N and δ13C (C/Nbulk, δ13Cbulk), and, organic δ13C (δ13Corganic) ratios. The different OC components, C-1, C-2, C-3(a) and C-3-(b), released at different temperature due to differences in their stability resulting from their variable mode of association with the minerals present, are also shown.
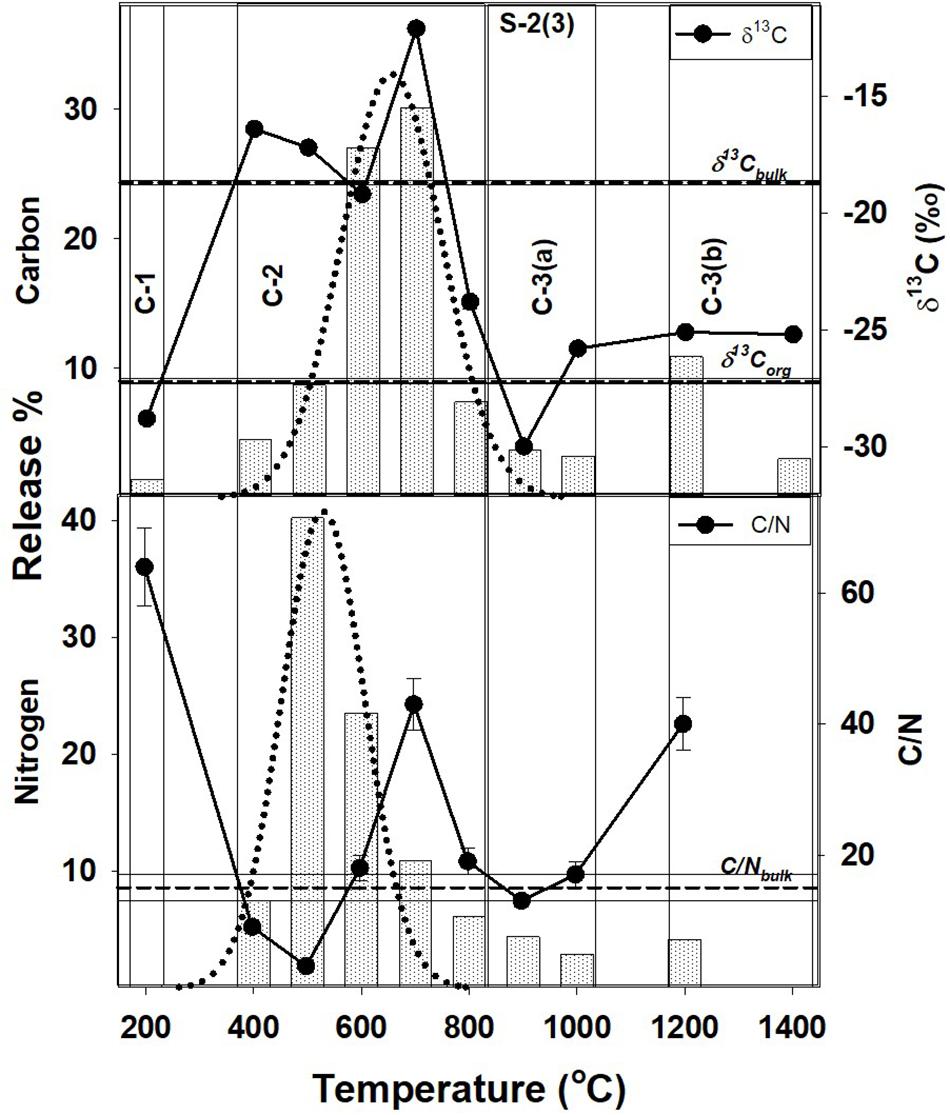
Figure 2. Step combustion temperature vs. carbon and nitrogen release percent for S-2(3). The isotopic and elemental ratios for each temperature step are shown along with the bulk values and the corresponding OC components [C-1, C-2, C-3(a), and C-3(b)]. For further details, refer to text and Figure 1.
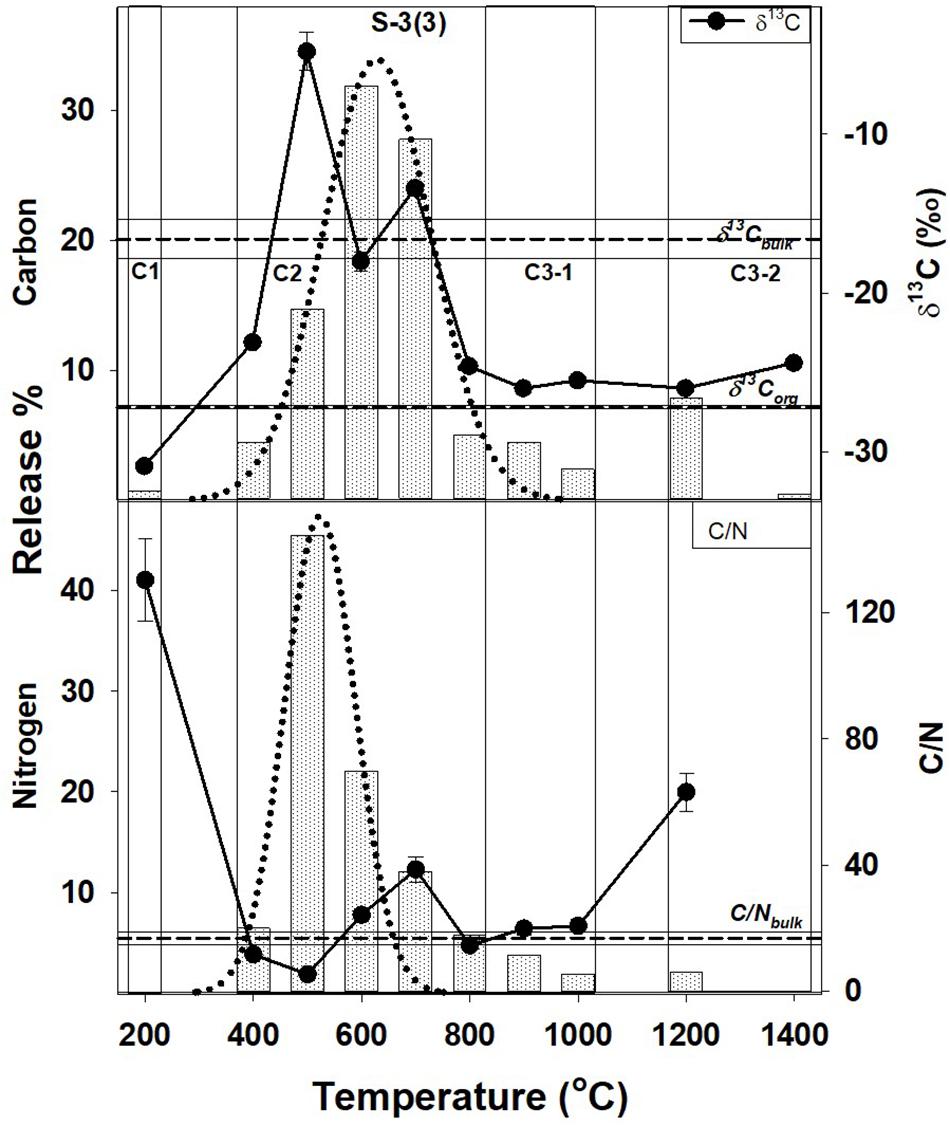
Figure 3. Step combustion temperature vs. carbon and nitrogen release percent for S-3(3). The isotopic and elemental ratios for each temperature step are shown along with the bulk values and the corresponding OC components [C-1, C-2, C-3(a), and C-3(b)]. For further details, refer to text and Figure 1.
400–800°C
This temperature range corresponds to the breakdown of clay and carbonate minerals and release of associated organic matter that may be adsorbed on the mineral surfaces, along with heavy hydrocarbons that may be present. It constitutes the bulk of total carbon released, contributed from the breakdown of carbonate minerals such as calcite and dolomite. Based on TOC and bulk carbon concentration, this temperature range also accounts for > 60% of the TOC in the shales (assuming all carbon that is released at < 400°C and > 800°C to be OC). The peak release of nitrogen indicating breakdown of clay minerals (500–600°C) occurs at a slightly lower temperature as compared to carbon corresponding to the breakdown of the carbonate minerals (600–700°C). It implies that clay-associated OC [denoted as C-2(a); Figures 1–3], can be relatively less stable and more mobile than the carbonate associated OC [denoted as C-2(b); Figures 1–3]. While C-2 can be physically/chemically adsorbed on clay surfaces, they are more likely to be biomineralized with the carbonates. But given the overlap as to the breakdown of the clay and carbonate phases, any differences between relative stability of these two components will be minor.
The instability of clay minerals in shale is related to their hydrophyllic and charged nature, particularly for expandable clay such as smectite where it can result in swelling. However, whether such swelling related to osmosis by the action of an external fluid can occur significantly in a compacted, impermeable shale is debatable, although internal microfractures can be efficient fluid conduits (Santarelli and Carminati, 1995; Wilson and Wilson, 2014). In addition, some water may be present in intra-aggregate pore spaces in smectitic clay since deposition and through burial, compression and lithification, but this is a bound state and not free (Touret et al., 1990). Based on modeling, it has been observed that interlamellar water in smectites is stable up to at least 180°C corresponding to a depth of 6 km without affecting the mineral stability (Odriozola and Guevara-Rodríguez, 2004). This is in agreement with observation from the step combustion experiment of this study, where clay mineral instability is only initiated at temperature > 200°C. A recent study has shown that the OC enrichment is strongly correlated to the high surface area clay minerals where, it occurs as intercalation with phyllosilicate minerals as organomineral nanocomposites that persists through a range of burial depth up to the oil window (Kennedy et al., 2014). With illitization, the OC and clays organize themselves into nano-scale aggregates without any net loss, although there might be significant decrease in the total mineral surface area. Together with results from this study, it is clear that the occurrence of high surface area detrital clay minerals helps in the preservation of OC as nanocomposites and aggregates not only during burial but also at higher temperature during subduction.
Carbonate minerals are not susceptible to breakdown during interaction and reaction with the formation water that is trapped in the pore spaces. The formation water is expected to be saline and already saturated with the minerals in the host rock (Kaszuba et al., 2013). So although the role of carbonate minerals for stabilizing OC is less significant as compared to the clay minerals, it is still possible to trap OC within the stable structure of the carbonate minerals as formanifera, corals and cocolithospores augmenting its stability (Ingalls et al., 2003, 2004). Interaction of organic matter with the carbonate matrix may also be in the form of chemical bonding (Jeong and Kobylinski, 1983). Interaction of clay minerals and carbonate with OC till their breakdown at 400–800°C (clay) and 500–800°C (carbonate), are important in the preservation of carbon in shale (Figures 1–3). This is in agreement with carbon retention in sediments at temperature of up to 600°C in intact subducting slabs in absence of infiltrating fluids (Cook-Kollars et al., 2014; Collins et al., 2015).
>800°C
The OC at higher temperature steps is contemporaneously released with decomposition and transformation of stable mineral phases such as quartz, K-feldspar and other silicates, as indicated by STA analyses. This component is marked as C-3(a) and is distinct from C-3(b) (Figures 1–3), the later released at temperature exceeding 1000°C. Like calcite, silicates can also be important for stabilizing OC in biominerals (Carter and Mitterer, 1978; Maita et al., 1982) accounting for the release of C-3(a) at temperature corresponding to the breakdown of the silicate minerals. Organic matter can also be contained and protected in pore spaces surrounded by silicate minerals (Ma et al., 2017), as an occlusion in a mineral matrix.
C-3(b), observed in S-2(3), and S-3(3) represents graphitized carbon marked by high C/N and slightly enriched δ13C (as compared to δ13Corganic). An enrichment of δ13C during graphitization of carbonaceous matter occurs as 12C-13C bonds in C-complexes are broken with increasing thermal stress (Fomina et al., 2019). Although C-3(b) is a very small fraction of the total OC present in shale, its survival during combustion at temperature > 1000°C, imply that it can be preserved even in the hottest subduction zones. Transfer of carbon to the deep Earth locked as graphite at temperature of up to 1200°C with negligible fluid flux, correspond to depths of ∼80–250 km depending on the geothermal gradient (Stern, 2002; Galvez et al., 2013; Zhang et al., 2018). However, given the low density of graphite (2.3 gm/cc) as compared to that of mantle peridotite (3.1–3.4 gm/cc), it can seggregate as a refractory phase in the shallow mantle wedge with restricted deep subduction, while the associated metasediments and carbonates from the slab can form CO2-rich melt (Kelemen and Manning, 2015). Mantle graphite is indeed known from much shallower depths than diamonds, and also can be related to subduction (Kennedy and Kennedy, 1976; Schulze et al., 1997).
There is a small possibility that this high temperature component is related to highly stable mineral phases like Fe-Ti-Al oxides formed by recrystallization and transformation during the combustion experiments in this study. Such a phase formed can trap OC as occluded or coated phases. Formation of such a mineral phase during these experiments is unlikely as it is not consistently observed in all the three samples [not observed for sample S-2(1) at 1400°C].
The different components released at different temperature during combustion of shungites, a unique rock bearing 1 to 100% amorphous carbon, has been related to the variable oxidation temperature of different organic matter type having distinct C/N and, carbon and nitrogen isotopic composition (Verchovsky et al., 2018). In our study, given the consistent δ13Corganic of – 27‰ in all the three samples indicating a continental source, and their δ13Cbulk values explainable by mixing between OC and carbonate components, such a possibility is unlikely. This is supported by C/N and δ13C mixing relationship observed in a previous study (Basu et al., 2018). It is possible that in the shungites, the observed differential release pattern of nitrogen (with accompanying carbon) is a consequence of how the components are sited in their different forms. For example, they can be present as exchangeable or fixed nitrogen or, hosted in the core of the carbonaceous matrix encapsulated from the influence of any hot, infiltrating fluids (Scholten, 1991; Boudou et al., 2008).
OC Components and Their Host Sites
Organic matter in shale can be present as adsorbed component, both on internal and external surfaces of minerals or, accumulated freely at the mouth or edge of the pore spaces and, the interlayer spaces of the clay minerals (Zhu et al., 2016). The associated carbon, occurring as freely accumulated OC along with physically adsorbed OC on external clay surfaces, can be easily extracted by organic solvent and referred to as the physically mobile OC (PmOC). Chemically bonded OC (CmOC) present in the internal surfaces of the minerals can only be removed by wet chemical oxidation by breaking the chemical bonds. But the most stable OC (SOC), occurs in the interlayer spacing of clay minerals, black carbon and occluded in carbonates and is not affected by wet chemical oxidation. Based on treatment with chemical reagents, OC components have the following sequence of mobility, PmOC > CmOC > SOC, which corresponds to a reverse trend in their stability (Zhu et al., 2016). The OC component can be sorbed to the surfaces interacting variably, sometimes via multiple points that enhances its overall stability (Keil and Mayer, 2014).
In this study, based on step combustion experiments and temperature release up to five components can be identified. In an order of increasing release temperature reflecting increasing stability and decreasing mobility from their host sites in a shale, they are C-1 < C-2(a) < C-2(b) < C-3(a) < C-3(b) (Table 4). C-1 occurs as a free component while C-2(a) is associated with clay minerals and both are equivalent to PmOC. C-2(a) corresponds to the physically adsorbed component on the surfaces of clay minerals. C-2(a) also includes the more retentive, chemically adsorbed component present in the internal surfaces of the clay minerals or, in their interlamellar spaces and comparable to CmOC and SOC respectively. Consequently, C-2(a) is released over a range of temperature in our experiments from 400 to 800°C. Closely associated with C-2(a), must be the OC occurring as biomineral or occluded phases associated with the carbonate minerals and denoted as C-2(b), which correspond to SOC. The C-3(a) occurs as biomineral or as occluded phase with silicate minerals, not distinguished by chemical reagents. Based on this study, we denote this component as very stable OC (VSOC). The graphitized OC also identified as SOC by Zhu et al. (2016), is categorized in this study as extremely stable OC (ExSOC) and is the most stable of all OC components in the shale.
Conclusion
Stepwise combustion of three shales from the Jurassic Haynesville-Bossier Formation suggests that the OC present in them occurs as different components. As the organic matter from the three samples are identical in terms of δ13Corganic, TOC content and thermal maturity, any selective degradation related to its type, should be minimum, although cannot be ruled out completely. Rather, the different components are released at different temperature steps due to differences in their stability, as related to their association with various mineral phases such as clay minerals, carbonate, and silicates. This highlights the significance of organic-mineral interaction in the preservation of OC components. The OC is hosted differently with these mineral phases sometimes sorbed as small molecules to the external surface or chemically sorbed to the internal surfaces of the clay minerals. It may be biomineralized or occluded by carbonate and silicate while a very small fraction can occur as free accumulates or in the graphitized form. In absence of any externally infiltrating fluid in a closed system, mineral associated OC is well-preserved till the breakdown of the shale constituent minerals such as clay and carbonate. This corresponds to peak release of 500–600°C for the phyllosilicate clay minerals and 600–700°C for the carbonate, indicating depth of ∼80 to 100 km, depending on the thermal regime of the subduction zone (Syracuse et al., 2010).
The OC components present in a shale in order of their increasing stability (and decreasing mobility), released at different temperature during the combustion of shale are:
C-1 – Released at 200°C, occurs as free accumulates at the mouth of the pore spaces or edge of minerals in a shale. It is likely to be extracted using organic solvent and, is prone to be lost from the shale during diagenesis. It constitutes a very small fraction of the total OC.
C-2(a) – Released predominantly at 400–700°C, it is the OC component that is associated with clay minerals. It can be physically adsorbed on the external mineral surfaces and can be extracted using organic solvent. A more stable form can be chemically adsorbed in the internal surfaces of the clay minerals forming intercalations as organomineral nanocomposites, and can only be released by breaking the chemical bonds by wet chemical oxidation. C-2(a) can occur in the interlamellar spaces making them highly stable that would be non-extractable by chemical oxidation. A large fraction of the total OC in shale occurs as C-2(a).
C-2(b) – Released predominantly at 500–800°C, it refers to the OC component that is associated with the carbonate minerals, either occluded or in the biomineralized form. It should be extractable by wet chemical oxidation.
C-3(a) – Released at 800–1000°C, this OC component is associated with the silicate minerals in the biomineralized form or as occlusions. It occurs as a very stable form and is unlikely to be extractable by wet chemical oxidation as easily as C-2(a) or C-2(b).
C-3(b) – Released at 1200–1400°C, this corresponds to the graphitized OC that should be non-reactive to wet chemical oxidation. It is a small fraction of the total OC but is highly stable in the shale.
Except C-1, all other components are retained till high temperature in the shale suggesting that they can survive during geological processes such as burial diagenesis and subduction. C-3(b), if subducted, can remain as a refractory phases in the shallow mantle without being oxidized.
Future studies should consider how shale fabric (relationship between the different minerals and the resulting pore spaces within the volume of the shale), texture (size distribution of constituent particles in terms of clay, silt and sand sized particles) and structure (bedding planes, fissility, sedimentary laminations) influence the interaction between minerals and organic matter affecting the stability/mobility of the OC components present.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
SB wrote and edited the manuscript, and conducted the combustion experiments, and XRD and TOC measurements. AV provided instrumental guidance, performed the combustion experiments, and edited the manuscript. AB performed the STA experiments and edited the manuscript. AJ edited the manuscript and provided input for the overall work, collaborating with SB. A-LJ performed the organic carbon isotopic measurements and edited the manuscript.
Funding
Funding for this work was provided to SB by an industry consortium constituted by BG Group (now Shell), who also provided the specimen used under award 159877.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/feart.2019.00297/full#supplementary-material
References
Ague, J. J., and Nicolescu, S. (2014). Slab melting as a barrier to deep carbon subduction. Nat. Geosci. 7, 355–360. doi: 10.1038/NGEO2143
Aiuppa, A., Fischer, T. P., Plank, T., and Bani, P. (2019). CO2 flux emissions from the Earth’s most actively degassing volcanoes, 2005-2015. Sci. Rep. 9:5442. doi: 10.1038/s41598-019-41901-y
Alt, J. C., Teagle, D. A. H., and Damon, A. H. (1999). The uptake of carbon during alteration of ocean crust. Geochim. Cosmochim. Acta 63, 1527–1535. doi: 10.1016/S0016-7037(99)00123-4
Barry, P. H., de Moor, J. M., Giovannelli, D., Schrenk, M., Hummer, D. R., Lopez, T., et al. (2019). Forearc carbon sink reduces long-term volatile recycling into the mantle. Nature 568, 487–492. doi: 10.1038/s41586-019-1131-5
Basu, S., Ahmed, J., Jones, A. P., and Verchovsky, A. B. (2018). Characterisation of carbón components and their isotopic composition in gas shales. Energy Proc. 146, 47–52. doi: 10.1016/j.egypro.2018.07.007
Berner, R. A. (2003). The long-term carbon cycle, fossil fuels and atmospheric composition. Nature 426, 323–326. doi: 10.1038/nature02131
Blatt, H., and Jones, R. (1975). Proportions of exposed igneous, metamorphic, and sedimentary rocks. GSA Bull. 86, 1085–1088.
Boudou, J.-P., Schimmelmann, A., Ader, M., Mastalerz, M., Sebilo, M., and Gengembre, L. (2008). Organic nitrogen chemistry during low-grade metamorphism. Geochim. Cosmochim. Acta 72, 1199–1221. doi: 10.1016/j.gca.2007.12.004
Burdige, D. J. (2007). Preservation of organic matter in marine sediments: controls, mechanisms, and an imbalance in sediment organic carbon budgets? Chem. Rev. 107, 467–485. doi: 10.1021/cr050347q
Carter, P. W., and Mitterer, R. M. (1978). Amino acid composition of organic matter associated with carbonate and non-carbonate sediments. Geochim. Cosmochim Acta 42, 1231–1238. doi: 10.1016/0016-7037(78)90116-3
Chiodini, G., Granieri, D., Avino, R., Caliro, S., Costa, A., Minopoli, G., et al. (2010). Non-volcanic CO2 earth degassing: case of mefite d’Ansanto (southern Apennines), Italy. Geophys. Res. Lett. 37:L11303. doi: 10.1029/2010GL042858
Clift, P. D. (2017). A revised budget for Cenozoic sedimentary carbon subduction. Rev. Geophys. 55, 97–125. doi: 10.1002/2016RG000531
Collins, N. C., Bebout, G. E., Angiboust, S., Agard, P., Scambelluri, M., Crispini, L., et al. (2015). Subduction zone metamorphic pathway for deep carbon cycling: II. Evidence from HP/UHP metabasaltic rocks and ophicarbonates. Chem. Geol. 412, 132–150. doi: 10.1016/j.chemgeo.2015.06.012
Coogan, L. A., Parrish, R. R., and Roberts, N. M. W. (2016). Early hydrothermal carbon uptake by the upper oceanic crust: insight from in situ U-Pb dating. Geology 44, 147–150. doi: 10.1130/G37212.1
Cook-Kollars, J., Bebout, G. E., Collins, N. C., Angiboust, S., and Agard, P. (2014). Subduction zone metamorphic pathway for deep carbon cycling: I. Evidence from HP/UHP metasedimentary rocks, Italian Alps. Chem. Geol. 386, 31–48. doi: 10.1016/j.chemgeo.2014.07.013
Dayal, A. M. (2017). “Deposition and Diagenesis,” in Shale Gas: Exploration and Environmental and Economic Impacts, eds A. M. Dayal, and D. Mani, (Amsterdam: Elsevier Inc.), 13–23. doi: 10.1016/B978-0-12-809573-7.00002-0
Di-Giovanni, C., Disnar, J.-R., and Macaire, J.-J. (2002). Estimation of the annual organic carbon yield related to carbonated rocks chemical weathering: implications for the global organic carbon cycle understanding. Glob. Planet. Change 32, 195–210. doi: 10.1016/S0921-8181(01)00141-2
Duncan, M. S., and Dasgupta, R. (2017). Rise of Earth’s atmospheric oxygen controlled by efficient subduction of organic carbon. Nat. Geosci. 10, 387–392. doi: 10.1038/NGEO2939
Etiope, G., and Sherwood-Lollar, B. (2013). Abiotic methane on Earth. Rev. Geophys. 51, 276–299. doi: 10.1002/rog.20011
Evans, K. (2011). Metamorphic carbon fluxes: how much and how fast? Geology 39, 95–96. doi: 10.1130/focus012011.1
Fomina, E., Kozlov, E., Lokhov, K., Lokhova, O., and Bocharov, V. (2019). Carbon sources and the graphitization of carbonaceous matter in precambrian rocks of the keivy terrane (Kola Peninsula, Russia). Minerals 9:94. doi: 10.3390/min9020094
Galvez, M. E., Beyssac, O., Martinez, I., Benzerara, K., Chaduteau, C., Malvoisin, B., et al. (2013). Graphite formation by carbonate reduction during subduction. Nat. Geosci. 6, 473–477. doi: 10.1038/NGEO1827
Golding, S. D., Boreham, C. J., and Esterle, J. (2013). Stable isotope geochemistry of coal bed and shale gas and related production waters: a review. Int. J. Coal Geol. 120, 24–40. doi: 10.1016/j.coal.2013.09.001
Hajpal, M., and Torok, A. (2004). Mineralogical and colour changes of quartz sandstones by heat. Environ. Geol. 46, 311–322. doi: 10.1007/s00254-004-1034-z
Henry, D. G., Jarvis, I., Gillmore, G., and Stephenson, M. (2019). Raman spectroscopy as a tool to determine the thermal maturity of organic matter: application to sedimentary, metamorphic and structural geology. Earth Sci. Rev. (in press). doi: 10.1016/j.earscirev.2019.102936
Huang, F., Daniel, I., Cardon, H., Montagnac, G., and Sverjensky, D. A. (2017). Immiscible hydrocarbon fluids in the deep carbon cycle. Nat. Commun. 8:15798. doi: 10.1038/ncomms15798
Ingalls, A. E., Aller, R. C., Lee, C., and Wakeham, S. G. (2004). Organic matter diagenesis in shallow water carbonate sediments. Geochim. Cosmochim. Acta 68, 4363–4379. doi: 10.1016/j.gca.2004.01.002
Ingalls, A. E., Lee, C., and Druffel, E. R. M. (2003). Preservation of organic matter in mound-forming coral skeletons. Geochim. Cosmochim Acta 67, 2827–2841. doi: 10.1016/S0016-7037(03)00079-6
Jackson, M. L. W., and Laubach, S. E. (1988). Cretaceous and tertiary compressional tectonics as the cause of the Sabine Arch, East Texas and Northwest Louisiana. Gulf Coast Assoc. Geol. Soc. Trans. 38, 245–256.
Jarrard, R. D. (2003). Subduction fluxes of water, carbon dioxide, chlorine and potassium. Geochem. Geophys. Geosyst. 4:8905. doi: 10.1029/2002GC000392
Jeong, K. M., and Kobylinski, T. P. (1983). Organic-Mineral matter interactions in green river oil shale. Geochem. Chem. Oil Shales 230, 493–512. doi: 10.1021/bk-1983-0230.ch028
Kaszuba, J., Yardley, B., and Andreani, M. (2013). Experimental perspectives of mineral dissolution and precipitation due to carbon dioxide-water-rock interactions. Rev. Miner. Geochem. 77, 153–188. doi: 10.2138/rmg.2013.77.5
Keil, R. G., and Mayer, L. M. (2014). Mineral matrices and organic matter. Treatise Geochem. 2014, 337–359. doi: 10.1016/B978-0-08-095975-7.01024-X
Kelemen, P. B., and Manning, C. E. (2015). Reevaluating carbon fluxes in subduction zones, what goes down, mostly comes up. Proc. Natl. Acad. Sci. U.S.A. 112, E3997–E4006. doi: 10.1073/pnas.1507889112
Kennedy, C. S., and Kennedy, G. C. (1976). The equilibrium boundary between graphite and diamond. J. Geophys. Res. 81, 2467–2470. doi: 10.1029/JB081i014p02467
Kennedy, M. J., Löhr, S. C., Fraser, S. A., and Baruch, E. T. (2014). Direct evidence for organic carbon preservation as clay-organic nanocomposites in a Devonian black shale; from deposition to diagenesis. Earth Planet. Sci. Lett. 388, 59–70. doi: 10.1016/j.epsl.2013.11.044
Li, Y. (2017). Immiscible C-H-O fluids formed at subduction zone conditions. Geochem. Perspect. Lett. 3, 12–21. doi: 10.7185/geochemlet.1702
Ma, L., Taylor, K. G., Dowey, P. J., Courtois, L., Gholinia, A., and Lee, P. D. (2017). Multi-scale 3D characterisation of porosity and organic matter in shales with variable TOC content and thermal maturity: examples from the lublin and baltic basins, Poland and Lithuania. Int. J. Coal Geol. 180, 100–112. doi: 10.1016/j.coal.2017.08.002
Mainali, P. (2011). Chemostratigraphy and the Paleoceanography of the Bossier-Haynesville Formation, East Texas Basin, TX and LA, USA. Master’s thesis, University of Texas, Arlington.
Maita, Y., Montani, S., and Ishii, J. (1982). Early diagenesis of amino acids in Okhotsk Sea sediments. Deep Sea Res. Part A Oceanogr. Res. Pap. 29, 485–498. doi: 10.1016/0198-0149(82)90072-3
Meyers, P. A. (2014). Why are the δ13Corg values in Phanerozoic black shales more negative than in modern marine organic matter? Geochem. Geophys. Geosyst. 15, 3085–3106. doi: 10.1002/2014GC005305
Murata, K. J., Friedman, I., and Madsen, B. M. (1969). Isotopic composition of diagenetic carbonates in marine Miocene formations of California and Oregon. U.S. Geol. Survey Prof. Pap. 614-B, 1–24. doi: 10.3133/pp614B
Nunn, J. A. (2012). Burial and thermal history of the Haynesville Shale: implications for overpressure, gas generation, and natural hydrofracture. GCAGS J. 1, 81–96.
Odriozola, G., and Guevara-Rodríguez, F. de, J. (2004). Na-montmorillonite hydrates under basin conditions: hybrid monte carlo and molecular dynamics simulations. Langmuir 20, 2010–2016. doi: 10.1021/la035784j
Petsch, S. T., Edwards, K. J., and Eglinton, T. I. (2005). Microbial transformations of organic matter in black shales and implications for global biogeochemical cycles. Palaeogeogr. Palaeoclimatol. Palaeoecol. 219, 157–170. doi: 10.1016/j.palaeo.2004.10.019
Saikkonen, R. J., and Rautiainen, I. A. (1990). Determination of total and non-carbonate carbon in rock samples by a method using infrared absorption. Bull. Geol. Soc. Finland 62, 149–156. doi: 10.17741/bgsf/62.2.005
Salmon, V., Derenne, S., Lallier-Vergès, E., Largeau, C., and Beaudoin, B. (2000). Protection of organic matter by mineral matrix in a Cenomanian black shale. Organ. Geochem. 31, 463–474. doi: 10.1016/S0146-6380(00)00013-19
Salvador, A. (1991). “Origin and development of the Gulf of Mexico basin,” in The Gulf of Mexico Basin. The Geology of North America, ed. A. Salvador, (Boulder, CO: Geological Society of America), doi: 10.1130/DNAG-GNA-J.389
Santarelli, F. J., and Carminati, S. (1995). Do shales swell? A critical review of available evidence. Soc. Pet. Eng. Pap. SPE 29421, 741–756. doi: 10.2118/29421-MS
Scholten, S. O. (1991). The Distribution of Nitrogen Isotopes in Sediments. Dissertation, Faculteit Aardwetenschappen, Utrecht University Repository, Utrecht.
Schulze, D. J., Valley, J. W., Viljoen, K. S., Stiefenhofer, J., and Spicuzza, M. (1997). Carbon isotope composition of graphite in mantle eclogites. J. Geol. 105, 379–386. doi: 10.1086/515933
Schwarzenbach, E. M., Caddick, M. J., Petroff, M., Gill, B. C., Cooperdock, E. H. G., and Barnes, J. D. (2018). Sulphur and carbon cycling in the subduction zone mélange. Sci. Rep. 8:15517. doi: 10.1038/s41598-018-33610-9
Smith, J. G. (2003). Aspects of the Loss-on-ignition (loi) technique in the context of clay-rich, glaciolacustrine sediments. Geografiska Ann. Ser. A Phys. Geogr. 85, 91–97. doi: 10.1111/1468-0459.00191
Sun, Y.-H., Bai, F.-T., Lü, X.-S., Li, Q., Liu, Y.-M., Guo, M.-Y., et al. (2015). A novel energy-efficient pyrolysis process: self-pyrolysis of oil shale triggered by topochemical heat in a horizontal fixed bed. Sci. Rep. 5:8290. doi: 10.1038/srep08290
Syracuse, E. M., van Keken, P. E., and Abers, G. A. (2010). The global range of subduction zone thermal models. Phys. Earth Planet. Inter. 183, 73–90. doi: 10.1016/j.pepi.2010.02.004
Thomson, A. R., Walter, M. J., Kohn, S. C., and Brooker, R. A. (2016). Slab melting as a barrier to deep carbon subduction. Nature 529, 76–79. doi: 10.1038/nature16174
Touret, O., Pons, C. H., Tessier, D., and Tardy, Y. (1990). Etude de la repartition de l’eau dans des argiles saturees Mg2+ aux fortes teneurs en eau. Clay Miner. 25, 217–233. doi: 10.1180/claymin.1990.025.2.07
Tsuno, K., and Dasgupta, R. (2011). Melting phase relation of nominally anhydrous, carbonated pelitic-eclogite at 2.5–3.0 GPa and deep cycling of sedimentary carbon. Contrib. Mineral. Petrol. 161, 743–763. doi: 10.1007/s00410-010-0560-9
Tsuno, K., Dasgupta, R., Danielson, L., and Righter, K. (2012). Flux of carbonate melt from deeply subducted pelitic sediments: geophysical and geochemical implications for the source of Central American volcanic arc. Geophys. Res. Lett. 39:L16307. doi: 10.1029/2012GL052606
Verchovsky, A. B. (2017). Origin of isotopically light nitrogen in meteorites. Geochem. Int. 55, 957–970. doi: 10.1134/S0016702917110106
Verchovsky, A. B., Gol’tsyn, N. A., Prasolov, E. M., and Lokhov, K. I. (2018). Nitrogen isotopic composition of shungite from the onega structure, Russia, and the origin of the organic matter. Geochem. Int. 56, 1341–1353. doi: 10.1134/S0016702918130086
Vitale Brovarone, A., Isabelle, M., Elmaleh, A., Compagnoni, R., Chaduteau, C., Ferraris, C., et al. (2017). Massive production of abiotic methane during subduction evidenced in metamorphosed ophicarbonates from the Italian Alps. Nat. Commun. 8:14134. doi: 10.1038/ncomms14134
Voyer, M. L., Hauri, E. H., Cottrell, E., Kelley, K. A., Salters, V. J. M., Langmuir, C. H., et al. (2019). Carbon fluxes and primary magma CO2 contents along the global mid-ocean ridge system. Geochem. Geophys. Geosyst. 20, 1387–1424. doi: 10.1029/2018GC007630
Wallmann, K., and Aloisi, G. (2012). “The global carbon cycle: geological processes,” in Fundamentals of Geobiology, eds A. H. Knoll, D. E. Canfield, and K. O. Konhauser, (Hoboken, NJ: Blackwell Publishing Ltd.), 20–34.
Wilson, M. J., and Wilson, L. (2014). Clay mineralogy and shale instability: an alternative conceptual analysis. Clay Miner. 49, 127–145. doi: 10.1180/claymin.2014.049.2.01
Wright, M. C., Court, R. W., Kafantaris, F.-C. A., Spathopoulos, F., and Sephton, M. A. (2015). A new rapid method for shale oil and shale gas assessment. Fuel 153, 231–239. doi: 10.1016/j.fuel.2015.02.089
Yang, J., Hatcherian, J., Hackley, P. C., and Pomerantz, A. E. (2017). Nanoscale geochemical and geomechanical characterization of organic matter in shale. Nat. Commun. 8:2179. doi: 10.1038/s41467-017-02254-0
Zeng, X., Cai, J., Dong, Z., Bian, L., and Li, Y. (2018). Relationship between mineral and organic matter in shales: the case of shahejie formation, Dongying Sag, China. Minerals 8:222. doi: 10.3390/min8060222
Zhang, S., Ague, J. J., and Vitale Brovarone, A. (2018). Degassing of organic carbon during regional metamorphism of pelites, Wepawaug Schist, Connecticut, USA. Chem. Geol. 490, 30–44. doi: 10.1016/j.chemgeo.2018.05.003
Zhang, Y., Ge, X., Nakano, J., Liu, L., Wang, X., and Zhang, Z. (2014). Pyrite transformation and sulfur dioxide release during calcination of coal gangue. RSC Adv. 4, 42506–42513. doi: 10.1039/C4RA06954D
Keywords: carbon cycle, carbon, nitrogen, clay minerals, carbonate, silicate
Citation: Basu S, Verchovsky AB, Bogush A, Jones AP and Jourdan A-L (2019) Stability of Organic Carbon Components in Shale: Implications for Carbon Cycle. Front. Earth Sci. 7:297. doi: 10.3389/feart.2019.00297
Received: 16 June 2019; Accepted: 29 October 2019;
Published: 19 November 2019.
Edited by:
Sami Mikhail, University of St Andrews, United KingdomReviewed by:
Vadim Reutsky, V.S. Sobolev Institute of Geology and Mineralogy (RAS), RussiaAlberto Vitale Brovarone, UMR 7590 Institut de Minéralogie, de Physique des Matériaux et de Cosmochimie (IMPMC), France
Copyright © 2019 Basu, Verchovsky, Bogush, Jones and Jourdan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sudeshna Basu, U3VkZXNobmEuYmFzdUB1Y2wuYWMudWs=