 Santiago Tassara1,2,3*
Santiago Tassara1,2,3* Martin Reich1,2
Martin Reich1,2 Brian A. Konecke4,5
Brian A. Konecke4,5 José María González-Jiménez6
José María González-Jiménez6 Adam C. Simon4Diego Morata1,2Fernando Barra1,2Adrian Fiege7
Adam C. Simon4Diego Morata1,2Fernando Barra1,2Adrian Fiege7 Manuel E. Schilling8Alexandre Corgne8
Manuel E. Schilling8Alexandre Corgne8- 1Department of Geology and Andean Geothermal Centre of Excellence (CEGA), FCFM, Universidad de Chile, Santiago, Chile
- 2Millennium Nucleus for Metal Tracing Along Subduction, FCFM, Universidad de Chile, Santiago, Chile
- 3Department of Geology and Geophysics, Yale University, New Haven, CT, United States
- 4Department of Earth and Environmental Sciences, University of Michigan, Ann Arbor, MI, United States
- 5Astromaterials Research and Exploration Science (ARES), NASA Johnson Space Center, Houston, TX, United States
- 6Departamento de Mineralogía y Petrología, Facultad de Ciencias, Universidad de Granada, Granada, Spain
- 7Department of Earth and Planetary Sciences, American Museum of Natural History, New York, NY, United States
- 8Facultad de Ciencias, Instituto de Ciencias de la Tierra, Universidad Austral de Chile, Valdivia, Chile
The oxidation state of the Earth’s mantle and its partial melting products exert a key control on the behavior and distribution of sulfur and chalcophile and siderophile elements between the mantle and crust, underpinning models of ore deposit formation. Whether the oxidized nature of magmas is inherited from the asthenospheric mantle source or acquired during ascent and differentiation is vigorously debated, limiting our understanding of the mechanisms of extraction of sulfur and metals from the mantle. Here, we focused on the redox-sensitive behavior of sulfur in apatite crystallized from quenched alkaline basaltic melts preserved within a peridotite xenolith from the El Deseado Massif auriferous province in southern Patagonia. We took advantage of this unique setting to elucidate the redox evolution of melts during their ascent through the subcontinental lithospheric mantle (SCLM) and grasp the inner workings of the Earth’s mantle during gold metallogenesis. Our data reveal that an initially reduced silicate melt (ΔFMQ −2.2 to −1.2) was oxidized to ΔFMQ between 0 and 1.2 during percolation and interaction with the surrounding peridotite wall-rock (ΔFMQ 0 to +0.8). This process triggered changes in sulfur speciation and solubility in the silicate melt, boosting the potential of the melt to scavenge ore metals such as gold. We suggest that large redox gradients resulting from the interaction between ascending melts and the surrounding mantle can potentially modify the oxidation state of primitive melts and enhance their metallogenic fertility. Among other factors including an enriched metal source and favorable geodynamic conditions, redox gradients in the mantle may exert a first-order control on the global-scale localization of crustal provinces endowed with gold deposits.
Introduction
The oxidation state of the Earth’s mantle is a fundamental parameter on models that attempt to explain the formation of metallogenic provinces on a lithospheric scale (Mungall, 2002; Sillitoe, 2008; Richards, 2015). The oxygen fugacity (fO2), as well as the contents of sulfur and chalcophile and siderophile elements (i.e., Au, Ag, Cu, Ni, Os, Ir, Ru, Rh, Pt, Pd, Re; hereafter “ore metals”) of primitive melts associated with the formation of giant ore deposits are frequently attributed to be either inherited directly from their asthenospheric mantle source (Carmichael, 1991; Kelley and Cottrell, 2009), or the result of variable degrees of differentiation at crustal levels (Lee et al., 2002, 2010; Jenner et al., 2010; Tang et al., 2018). In contrast, relatively little attention has been paid to melt–rock interaction processes occurring in the subcontinental lithospheric mantle (SCLM) that might lead to changes in the oxidation state and composition of ascending magmas after partial melting in the asthenosphere and before they reach crustal levels. For example, Chin et al. (2014) and Griffin et al. (2018) explored the effects of the SCLM on the composition of primary asthenosphere-derived melts during their ascent and concluded that metasomatism might be an important process that modifies the composition and fO2 of ascending melts and fluids. More recently, Tollan and Hermann (2019) also found that arc magmas oxidize during ascent and reaction with surrounding peridotite before reaching crustal levels. Therefore, melt–SCLM interaction processes might have a pivotal impact on the metal fertility of ascending magmas.
Ore metals and sulfur are stored primarily within accessory base metal sulfides and sulfide liquids in the mantle, and their release and transfer into the overriding crust is partially controlled by the fO2 of the magmatic system (Jugo, 2009; Lorand and Luguet, 2016). This parameter reflects the oxidation state of the system and is commonly expressed as logarithmic units relative to the fayalite–magnetite–quartz mineral buffer (ΔFMQ). Experimental studies have shown that sulfide (S2–) is the dominant sulfur species in silicate melts under reducing conditions (ΔFMQ < 0) (Jugo et al., 2005a, 2010). In contrast, at ΔFMQ > 2 the melt is dominated by sulfate (S6+) whereas a sharp transition from sulfide toward the more soluble and oxidized sulfate is observed around ΔFMQ + 1 (Jugo et al., 2010). Hence, the formation of oxidized magmas can efficiently promote the titration of sulfur and ore metals from the mantle, ultimately increasing the ore metal endowment of the overlying crust (Sillitoe, 2008; Richards, 2015). Furthermore, recent studies have provided evidence that restricted fertile blocks of the SCLM may act as fertile source regions from where ore metals were tapped by partial melting, fluxed melting, or by interaction with ascending melts (Griffin et al., 2013; Groves and Santosh, 2015; Groves et al., 2016; Tassara et al., 2017). However, the impact and extent of silicate melt–SCLM interaction on the composition and oxidation state of ascending melts that ultimately reach the crust to form ore deposits, and the mechanisms of ore metals extraction from the SCLM remain largely unexplored. Therefore, understanding the redox evolution of magmas during their ascent across the SCLM is crucial to unravel the mechanisms of metal enrichment of ascending magmas.
Here, we examine the effects of melt–mantle interaction on the fO2 and metal fertility by measuring directly the formal oxidation state(s) of sulfur in magmatic apatite contained within quenched silicate glass entrained in a peridotite xenolith from Patagonia, Argentina, which represents a silicate melt that ascended through the SCLM below the El Deseado Massif auriferous province. By combining micro-X-ray absorption near-edge structure (μ-XANES) spectroscopic analysis of sulfur in apatite with petrological and mineralogical data, we show that originally reduced silicate melts that infiltrated a relatively oxidized SCLM interacted with the surrounding peridotite wall-rock, triggering changes in sulfur speciation and solubility. We link this process to the formation of gold-rich magmas during ascent and interaction with the SCLM, providing new insights into how mantle processes may govern the localization of metallogenic provinces in the Earth’s crust.
Sample Background
This study focuses on a lherzolite xenolith, which represents a portion of the mantle beneath the world-class El Deseado Massif auriferous province (Figure 1). The Deseado Massif hosts several Au–Ag epithermal deposits associated with calc-alkaline rhyolites, basaltic andesites, and basalts from the late magmatic stages of the Chon Aike Large Igneous Province (CA-LIP) (Schalamuk et al., 1997). The CA-LIP formed during two major periods between 180 and 155 Ma (Féraud et al., 1999). The early Jurassic event was related to the thermal impact of the Karoo plume head (∼180 Ma), whereas the Middle to Late Jurassic events reflects the influence of active subduction on the western margin of Gondwana (∼155 Ma) (Pankhurst et al., 2000). Mantle xenoliths from the SCLM beneath the Deseado Massif were later extruded by extensive Neogene (∼3.5 Ma) back-arc plateau volcanism (Rivalenti et al., 2004).
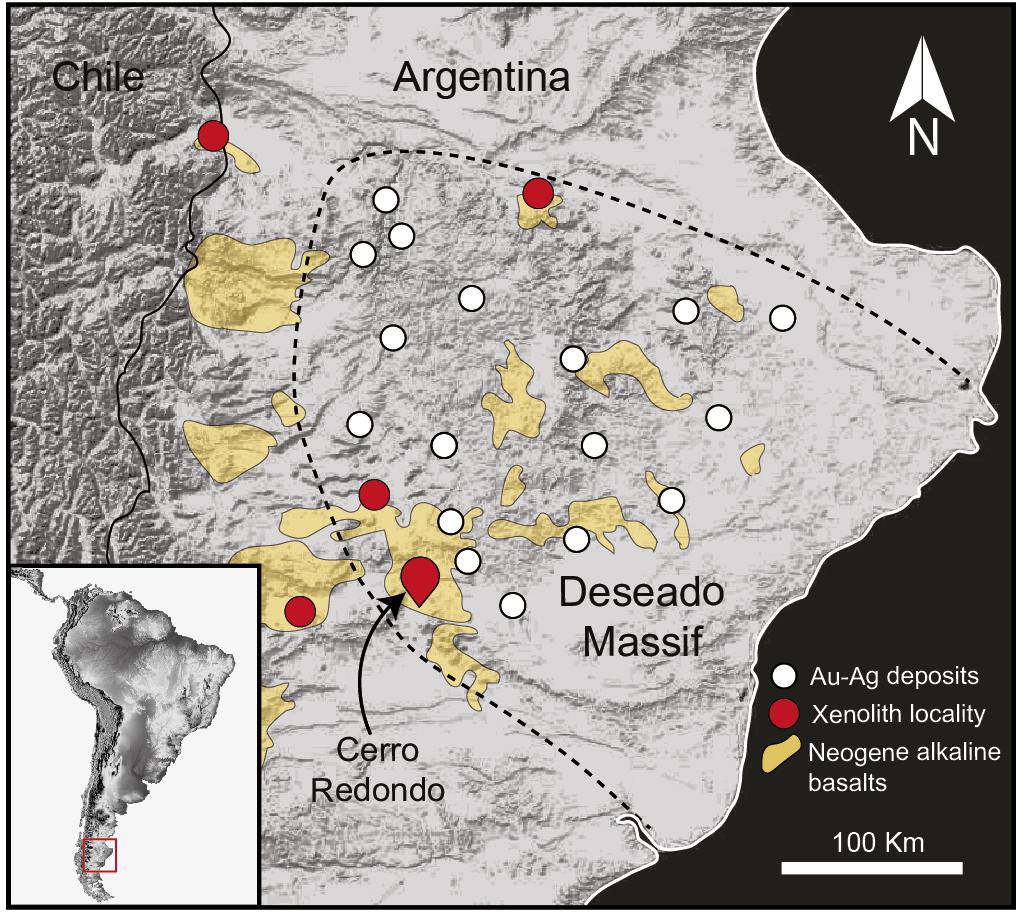
Figure 1. Simplified map of southern Patagonia. The dashed line delimits the Deseado Massif auriferous province. White circles indicate the location of low-sulfidation epithermal gold and silver deposits. Red circles indicate the location of other localities where mantle xenoliths have been reported. Modified after Tassara et al. (2017).
The studied peridotite is a relatively large (up to 25 cm) protogranular anhydrous lherzolite equilibrated in the spinel facies at temperatures of 1020–1150°C at 13.6 Kb (Tassara et al., 2017). The sample is characterized by the presence of quenched interstitial glass distributed along grain boundaries of the peridotite silicate matrix in the xenolith core forming an interconnected network of veinlets (Supplementary Figure 1 and Figure 2). These veinlets contain a mineral assemblage that includes armalcolite, apatite, ilmenite, K-feldspar, Au-bearing sulfides, and native gold particles embedded within a glassy matrix (Tassara et al., 2017; Figure 2). The interstitial silicate glass has strong mineralogical and trace element compositional differences with the host basalt, indicating that the origin of the studied glass veinlets is not related to the infiltration of the host basalt into the xenolith (Figure 3). Moreover, the interstitial glass and associated minerals were interpreted as remnants of a Au-bearing, Na-rich silica-undersaturated melt that infiltrated the peridotite. This melt extracted Ni–Cu sulfide liquids from the surrounding peridotite wall-rock and were physically entrained and transported within the silicate melt through the mantle (Bockrath et al., 2004; Ballhaus et al., 2006; Tassara et al., 2018).

Figure 2. Backscattered electron images of the interstitial glass veinlets in the studied lherzolite. Panels (A,B) show a general view of interstitial glass veinlets crosscutting olivine (Ol) grains with their partially crystallized mineral assemblage. Panels (C,D) show the reaction rims formed along clinopyroxene (Cpx) (C) and spinel (Sp) (D) when in contact with the glass veinlets. Panel (E) shows a detail of the mineral assemblages present in the interstitial glassy veinlets, including oxidized Ni–Cu sulfides, armalcolite (Arm), and apatite (Ap) with euhedral morphologies.
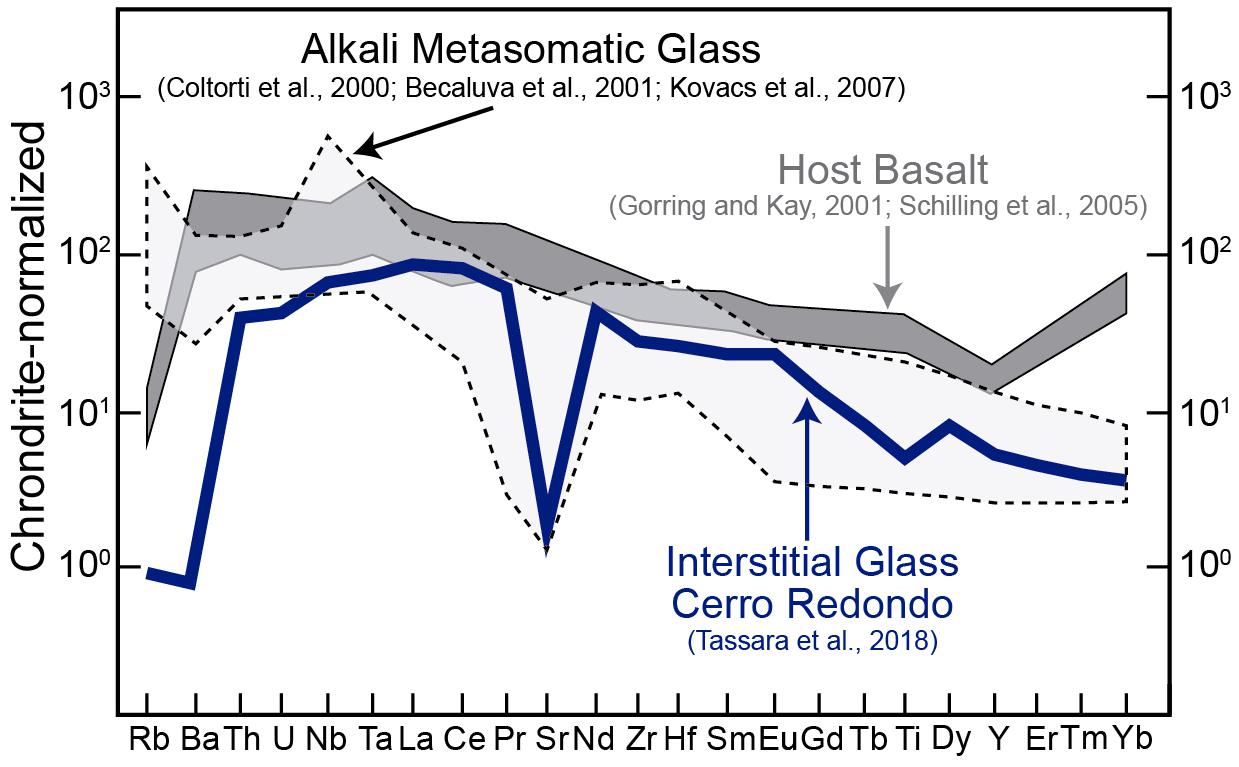
Figure 3. Chondrite-normalized trace element composition of interstitial silicate glasses in the Cerro Redondo peridotite xenolith. The trace element data for the interstitial glass is from Tassara et al. (2018). The dark shade area is the trace element data for the host basalt after from Gorring and Kay (2001) and Schilling et al. (2005). The light gray shaded field is the composition of alkaline metasomatic melts in the upper mantle after Coltorti et al. (2000), Beccaluva et al. (2001), and Kovacs et al. (2007). Figure modified after González-Jiménez et al. (2019).
Analytical Methods
Electron Probe Microanalysis
The chemical composition of apatite, silicates, and oxides was determined by using a CAMECA SX-100 electron microprobe at the University of Michigan in Ann Arbor. An acceleration voltage of 15 keV, a beam current of 10 nA, and a beam size of 2 μm were used for all analyses. Peak counting times of 20 s were used for all elements, except 5 s for F and 60 s for S, as this conditions are the best precautions to prevent electron beam damage of apatite (e.g., halogen migration; Konecke et al., 2019). During the analysis, both SiO2 and Al2O3 concentrations were monitored for contribution of the surrounding glass and mineral phases. Analyses indicating a contribution of the glass were discarded.
Sulfur X-Ray Absorption Near Edge Structure Spectroscopy
In situ sulfur X-ray Absorption Near Edge Structure Spectroscopy (S-XANES) measurements at the S K-edge were conducted at the GSECARS 13-ID-E beamline, Advanced Photon Source (APS), Argonne National Laboratory, IL. The beamline can cover an energy range of 2.4–28 keV and uses a high-flux beam (≥4.5 × 1010 photons/s/100 mA/mm2) that produces a high spatial resolution micro-focused 2 × 2 μm (μ-XANES) beam by using Kirkpatrick–Baez (KB) focusing mirrors. The energy of the Si (111) channel-cut monochromator was calibrated to the 2481.8 (±0.2) eV white line of the spectrum for clear double-sided adhesive tape. Spectra were collected at ambient temperature and pressure and covered a range of energy from 2450 to 2550 eV, with a step size of 0.1–0.3 eV at the S K-edge (2464–2484 eV) and 1 eV for the pre- and post-edge regions (0.5–3 s scan durations per energy step). Step-scan durations of 1–3 s per energy step were used to achieve higher S X-ray counts required for high-quality spectra, especially in low S-bearing apatite. The analyses were performed directly on rock thick sections and the spectra was collected in fluorescence mode. X-ray fluorescence maps were performed in advance in order to locate the apatite crystals. Previous studies have confirmed that the S oxidation state in apatite remains constant when exposed to the synchrotron beam for over 1 h, demonstrating that beam damage is not an issue (Konecke et al., 2017, 2019).
Redox State Determination
The fO2 conditions of the oxide-silicate assemblage in the host peridotite were calculated using the methods described by Wood et al. (1990) for olivine–orthopyroxene–spinel equilibrium and at 13.6 Kb and 1020°C, yielding ΔFMQ values between 0 and +0.8 (Supplementary Tables 1, 2), consistent with redox constraints for most SCLM peridotites reported in the literature (Ballhaus, 1993; Richards, 2015).
The oxidation state of the infiltrating silicate melt was determined, on the other hand, by using two independent approaches based on the composition of armalcolite and apatite, respectively. Terrestrial occurrences of armalcolite [(Fe,Mg)Ti2O5] are exclusively associated with metasomatic melts in the upper mantle and high-Ti primitive lavas (Haggerty, 1987; Bowles, 1988; Contini et al., 1993; Grégoire et al., 2000). Compositional analysis of armalcolite shows that it contains a high concentration of Cr (1.73 and 2.16 wt.%) and is among the most MgO-enriched compositions reported in both lunar and terrestrial occurrences (Supplementary Table 3). Stoichiometric calculations indicate that armalcolite has near zero trivalent Fe and Ti concentrations (Supplementary Table 4). Similar compositions were reported for Cr-bearing armalcolite found in the Kerguelen island mantle xenoliths (Bowles, 1988; Haggerty, 1991; Grégoire et al., 2000; Figure 4). In an attempt to constrain the oxidation state of lunar basalts, Friel et al. (1977) performed experimental studies on the stability of synthetic armalcolite as a function of fO2 at 1200°C and 0.001 Kb. This experiment and observations in natural samples (Grégoire et al., 2000) demonstrated that Cr-rich and near-zero Fe3+ variety of armalcolite forms only under reducing conditions (fO2 between 10–12.5 and 10–13.5 Kb), i.e., ΔFMQ ≈ −2.2 to −1.20 (cf. Figure 4 in Friel et al., 1977). In addition, these authors showed that at even lower fO2 conditions (between 10–15.2 and 10–15.8 Kb), armalcolite breaks down to form Mg-rich armalcolite plus ilmenite. Based on experimental data, we constrained the oxidation conditions of the infiltrating silicate melt at the time of armalcolite crystallization in the mantle at ΔFMQ ≈−2.2 to −1.2, whereas the presence of ilmenite suggests ΔFMQ values closer to −2.2 (Friel et al., 1977). Importantly, it has been documented that whereas armalcolite is stable as a single phase at pressures up to 10 Kb, armalcolite + ilmenite + rutile assemblages form at pressures between 10 and 14 Kb. Thus, the fact that armalcolite + ilmenite is present in the studied veinlet indicates that either the armalcolite suffered slow cooling allowing it to partially transform to ilmenite (Lindsley et al., 1974), or that the crystallization of armalcolite suffered occurred at >10 Kb, where both phases are stable (Friel et al., 1977). Either case indicates that armalcolite did not simply crystallized at low pressure during rapid cooling of the sample but rather formed at depth and suffered at least some slow cooling before the xenolith was brought to the surface.
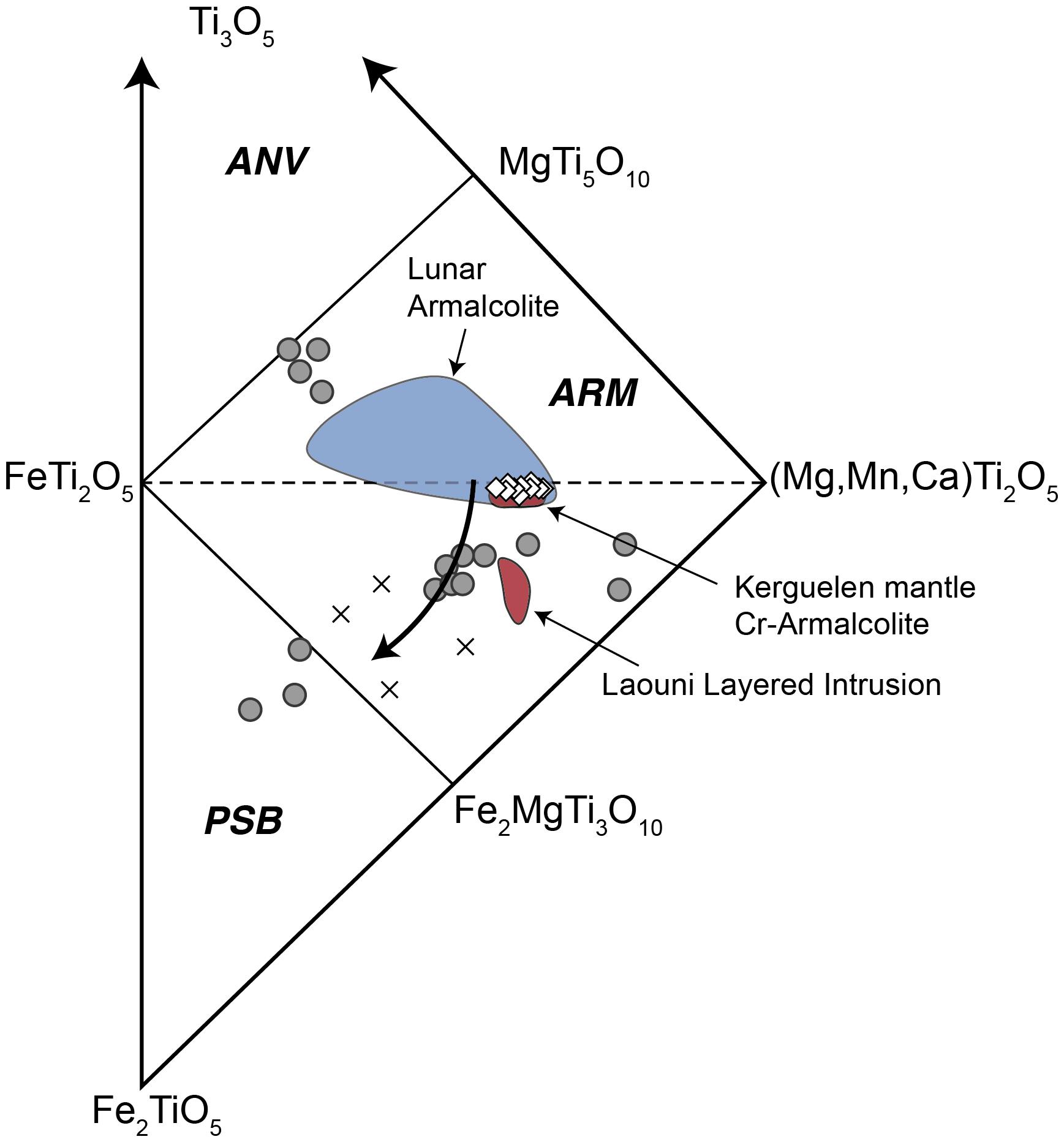
Figure 4. Pseudobrookite group diagram showing armalcolite end members. Modified from Grégoire et al. (2000). Pseudobrookite group diagram composition after Bowles (1988). ANV, anosovite; ARM, armalcolite; PSB, pseudobrookite. Open circles: armalcolite and pseudobrookite from Cancarix, Spain (Contini et al., 1993). Crosses: Armalcolite from Lebombo picrites (Cawthorn and Biggar, 1993). Polygons: Kimberlitic armalcolite from Jagersfontein (Haggerty, 1987). White diamonds: armalcolite from this study. The black arrow represents the compositional evolution of armalcolite at progressively higher fO2 after Medvedev (1996). End member calculation and classification according to Bowles (1988).
Apatite [Ca5(PO4)3(F,Cl,OH)] also occurs embedded within the interstitial silicate glass, commonly forming isolated clusters of crystals of up to ∼8 μm in size (Figure 2). The apatite crystals are euhedral and show typical hexagonal and acicular shapes, indicating that it crystallized from the surrounding melt upon rapid quenching (Piccoli and Candela, 2002), possibly during the xenolith eruption (Tassara et al., 2017). Electron probe microanalysis (EPMA) revealed that apatite corresponds to the F-rich endmember (Supplementary Table 5), which suggests a late crystallization from the silicate melt associated with increasing differentiation (Nash, 1984). Sulfur concentrations in the analyzed apatite are between 40 and 170 μg/g with a few grains showing concentrations below detection limits (∼30 μg/g) (Supplementary Table 5). The sulfur μ-XANES spectra of the studied apatite grains revealed two dominant peaks at ∼2469.7 (sulfide) and ∼2481.8 eV (sulfate), indicating that both species were incorporated within the apatite structure (Figure 5). The sulfite (S4+) peak (∼2478 eV) very likely is present in all spectra but cannot be resolved due to the overlap of the broad sulfide peak. To alleviate this, the peak integration of the broad sulfide peak was used to determine the S6+/Stot area ratio calculation, identical to the fitting methods developed and implemented by Konecke et al. (2017, 2019). The integrated S6+/Stotal peak area ratios were determined following the methodology of Konecke et al. (2017) and range between 0.28 and 0.48 (Figure 5). In order to reduce crystal orientation effects on the relative integrated area of the different peaks of the μ-XANES spectra, several apatite spectra were collected, normalized (e.g., baseline removal and intensity normalization), and then merged (Konecke et al., 2017) to obtain an average spectrum representative of the bulk sulfur oxidation state of the studied apatite grains. Also, several apatite grains with different morphologies, including elongated (approximately parallel to the c-axis) and euhedral hexagonal apatite (approximately perpendicular to c-axis) were analyzed and no significant variation in the S6+/Stot ratios were observed. The averaged integrated S6+/Stotal peak area can be used to estimate the fO2 of the infiltrating silicate melt from which apatite crystallized. Experiments performed by Konecke et al. (2017, 2019) showed that apatite formed at ΔFMQ = 0 has a S6+/Stotal ratio of ∼0.14, whereas apatite that crystallized at ΔFMQ ≈ +1.2 is characterized by a S6+/Stotal ratio of ∼0.95. This implies that apatite undergoes a pronounced shift from sulfide- to sulfate-dominated species with changing oxidation conditions, similarly to silicate melts (Jugo et al., 2010). The integrated S6+/Stotal ratio of the individual studied apatite ranges from ∼0.28 to ∼0.48, and the integrated S6+/Stotal ratio resulting from merging all spectra is ∼0.35 (Figure 5). The latter values indicate that the oxidation state of the infiltrating silicate melt at the time of apatite crystallization was 0 < ΔFMQ < +1.2. Given that the S6+/Stotal ratio and log (fO2) follow a near linear trend within this range (Konecke et al., 2019), we infer that the final oxidation state of the silicate melt at apatite formation was between ΔFMQ +0.3 to +0.4 [±0.5, 2σ], which is within the fO2 estimated for the peridotite wall-rock. Considering that an increase in pressure shifts the stability field of sulfide toward more oxidizing conditions (Matjuschkin et al., 2016) and inferring that apatite follows a similar trend, we conclude that a ΔFMQ +0.3 to +0.4 represents a minimum fO2 value for the silicate melt.
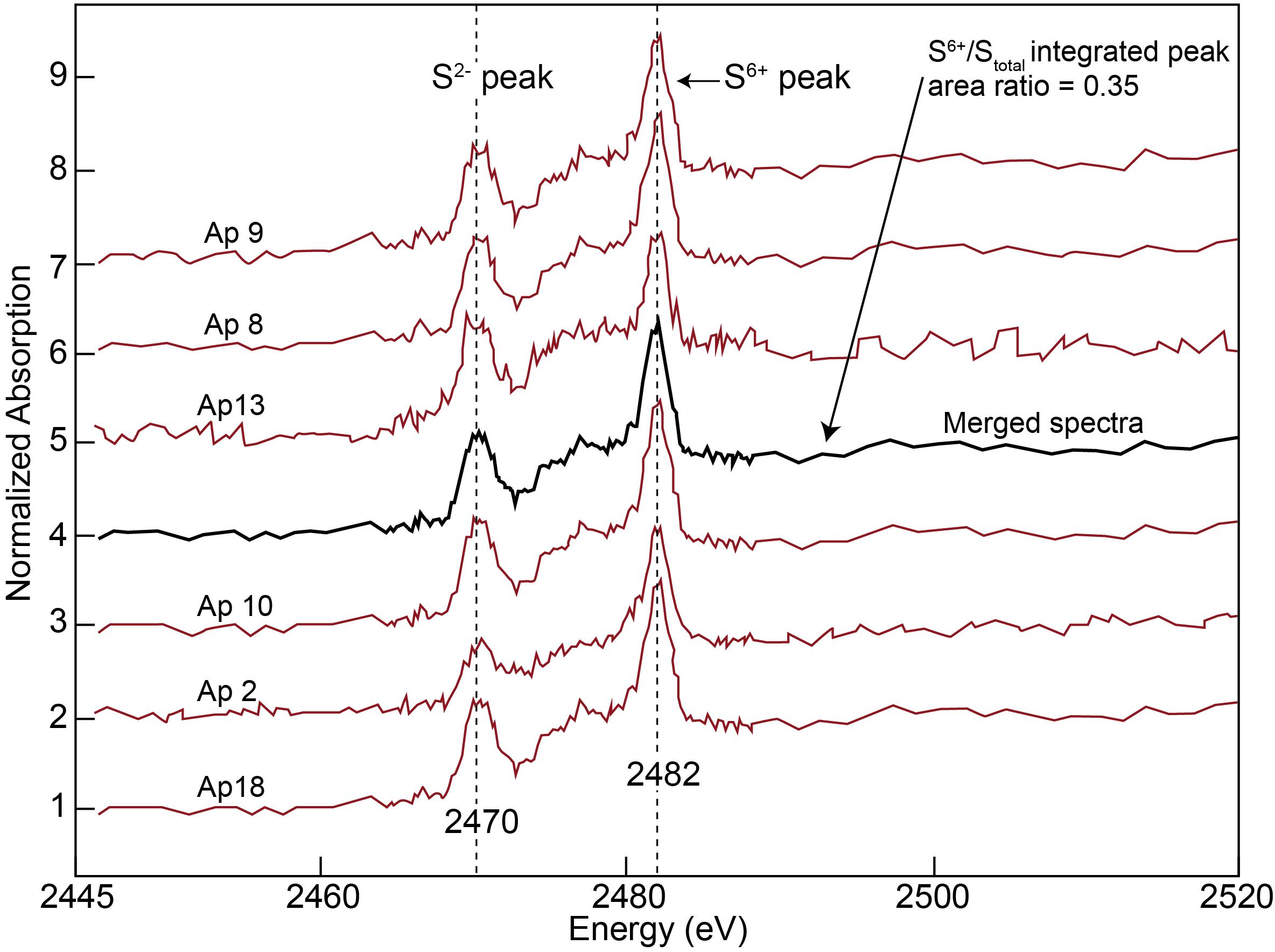
Figure 5. Sulfur μ-XANES spectra of the studied apatite. Averaged μ-XANES spectra showing the oxidation state of sulfur in apatite crystallized from a silicate melt that percolated through the subcontinental lithospheric mantle in southern Patagonia, beneath the Deseado Massif.
Discussion
The interstitial network of glass veinlets in the peridotite xenolith represents a Na-rich silica undersaturated melt that percolated through the SCLM and quenched during the rapid eruption of the xenolith (Tassara et al., 2018). Although the potential re-heating of the xenolith during entrapment in the host basalt could alter the original nature of the entrained veinlets, we observe no signs of perturbation in the integrity of the sample. Sharp contacts between the xenolith and host basalt, as well as the relatively large size of the xenolith indicate that ascent was very fast, precluding the possibility of ascending during sufficient time to transfer significant heat to the internal parts of the xenolith (O’Reilly and Griffin, 2010; and references therein). Based on our data, we propose that the mineral assemblage associated with the glassy vein (armalcolite and apatite) records two different oxidation states of the percolating silicate melt, and where armalcolite formed under highly reducing conditions (ΔFMQ −2.2 to −1.2), followed by the crystallization of apatite under more oxidizing conditions (ΔFMQ 0 to +1.2).
Melt Oxidation During Interaction With Surrounding Mantle
The reduced armalcolite-bearing silicate melt (ΔFMQ −2.2 to −1.2) infiltrated a relatively more oxidized peridotite (ΔFMQ 0 to +0.8), producing a large redox gradient, i.e., a ΔfO2 of near 3 log units between these two components (Figure 6A). The final redox conditions after oxidation of the reduced silicate melt and reduction of the relatively oxidized peridotite will depend on the mass balance between the more reduced and more oxidized components (Evans and Tomkins, 2011). Apatite crystallized from the surrounding silicate melt after the melt–peridotite interaction started, containing both sulfide and sulfate as evidenced by our μ-XANES data (Figure 5). The presence of sulfate in apatite requires the reaction between two components, i.e., a highly reduced melt with a S6+/Stotal ∼0 and low sulfur content (Jugo et al., 2010) that reacted with a more oxidized and sulfide-bearing peridotite (ΔFMQ 0 to +0.8).
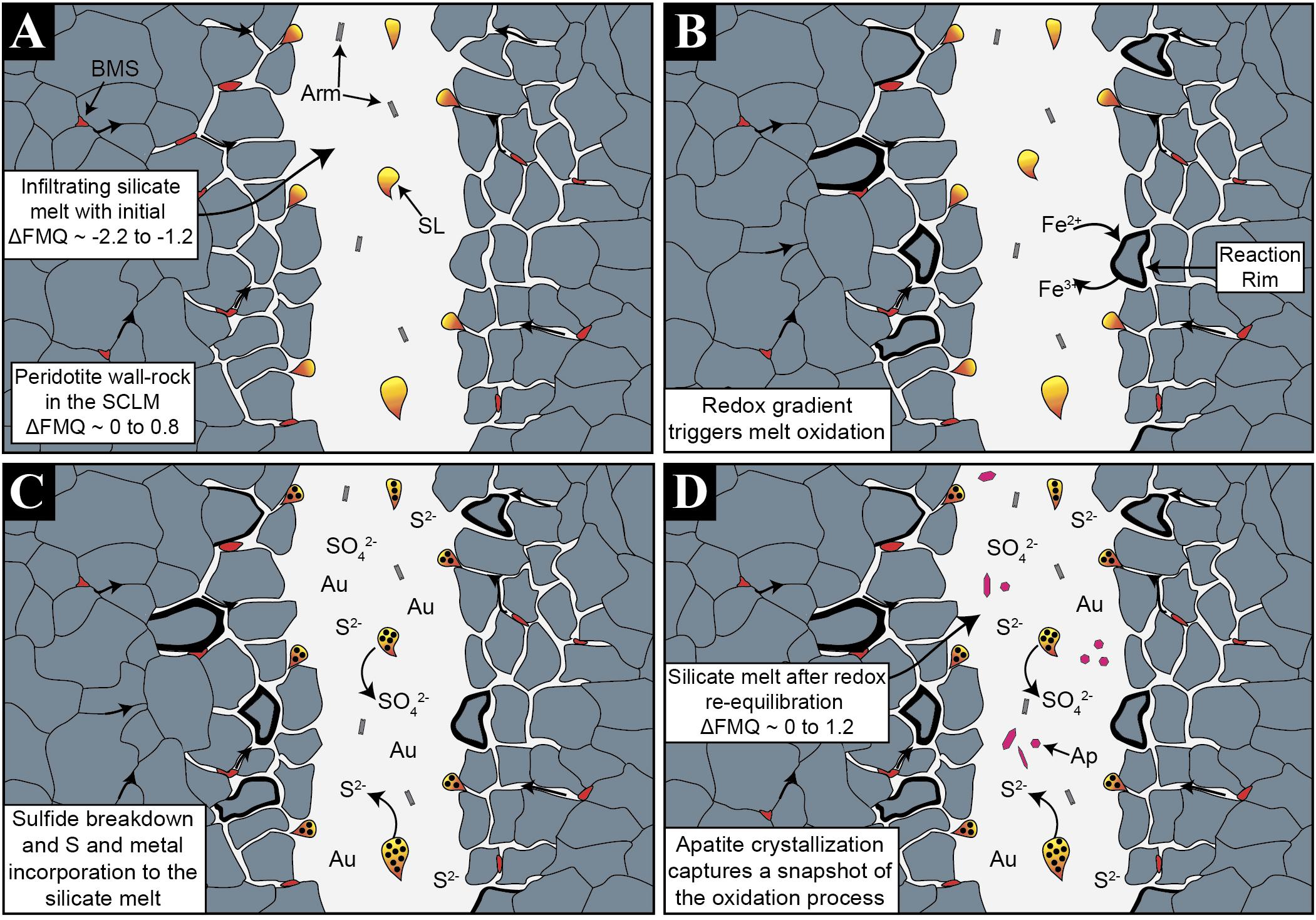
Figure 6. Mechanisms leading to oxidation of percolating melts and ore metals incorporation. (A) The reduced silicate melt enters the subcontinental lithospheric mantle, entraining immiscible sulfide liquids. A redox gradient is generated between the reduced melt and the more oxidized peridotite wall-rock. (B) The silicate melt starts to re-equilibrate with the surrounding peridotite wall-rock. (C) Increasing oxygen fugacity triggers changes in the sulfur solubility promoting metal sulfide breakdown and release of sulfide, sulfate, and Au into the percolating silicate melt. (D) Quenching upon rapid ascent induced apatite crystallization, which records a snapshot of the melt-SCLM interaction process.
In the SCLM, the oxidation state is governed by minerals that have a high content of Fe3+ such as spinel and clinopyroxene (Wood et al., 1990). These two phases are the first minerals to react with the melt when the redox conditions are disturbed, as has been reported during the mixing process between two compositionally distinct magmas (Fiege et al., 2017). Clinopyroxene and spinel from the studied peridotite show reaction rims when in contact with the reduced infiltrating silicate melt (Figures 2B,C) indicating that Fe2+ ↔ Fe3+ exchange between the peridotite wall-rock and the infiltrating silicate melt could have occurred, explaining the oxidation of the melt (Cooper et al., 1996; Figure 6B). Other than Fe, sulfur also has a large oxidation potential (Evans and Tomkins, 2011), and an increase in the oxidation state of the melt after melt–peridotite interaction would promote an increase in the sulfur solubility of the infiltrating silicate melt (Jugo et al., 2005b). Thus, at higher redox and sulfur solubility conditions (Jugo et al., 2005b), the sulfide liquids entrained in the percolating silicate melt will partially oxidize, breaking down and dissolving in the surrounding melt (Figure 6C). Decreasing pressure during ascent could potentially boost the breakdown of the sulfide phases, as the solubility of S in the melt will increase (Mavrogenes and O’Neill, 1999). However, if only this had been the mechanism of sulfide breakdown, no sulfate would be observed as the system would remain reduced (Matjuschkin et al., 2016). Therefore, an oxidation mechanism is still needed to account for the sulfate in the system. The apatite crystallized from the melt after the melt–peridotite interaction will incorporate both sulfide and sulfate in varying proportions, according to the S6+/Stotal ratio of the melt; therefore, apatite will record the oxidation state of the silicate melt during interaction with the surrounding peridotite wall-rock (Konecke et al., 2017; Figures 5, 6D).
Scavenging Sulfur and Ore Metals From the Lithospheric Mantle
The process described above explains the oxidation of silicate melts that ascend through the SCLM with the concomitant destabilization of ore metal-bearing sulfides (or sulfide melts) and incorporation of sulfur. Hence, sulfide breakdown will not only release sulfur but also its contained ore metals (Lorand and Luguet, 2016), such as Au (Figure 6D), which under certain circumstances could lead to metal enrichment in the percolating silicate melts. The solubility of Au in sulfur-bearing silicate melts is maximized when sulfur is dissolved as both sulfide and sulfate species (Cooper et al., 1996) and when the sulfide concentration is near the sulfide-saturated threshold (Botcharnikov et al., 2011; Li and Audétat, 2012; Zajacz et al., 2012; Jégo et al., 2016). Figure 7 shows that the estimated fO2 of the silicate melt after reacting with the peridotite wall-rock is between ΔFMQ 0 and +1.2. Sulfur μ-XANES in apatite indicates that the S6+/Stotal ratio is ∼0.35 and the presence of sulfide liquids within this silicate melt indicates that the melt was once sulfide saturated. These redox conditions are in good agreement with those that maximize Au solubility of sulfur-bearing basaltic melts, determined experimentally by Botcharnikov et al. (2011) at 1050°C and 2 Kb (Figure 7), meaning that the highest Au solubility was achieved during redox re-equilibration within the SCLM. In addition, the mineral assemblage associated with the studied melt contains native gold particles included within the sulfides as well as embedded within the quenched glass (Tassara et al., 2017). The native gold micro-particles in the sulfides have euhedral and hexagonal forms (Tassara et al., 2017) and likely crystallized from the sulfide liquids during desulfurization, after oxidation of the surrounding silicate melt (Tassara et al., 2018). Continued oxidation and desulfurization would result in the release of these Au micro-particles into the silicate melt. The Au micro-particles embedded within the glass have irregular shapes (Tassara et al., 2017), pointing to partial dissolution in the surrounding melt. This is in good agreement with Au solubility experiments (Botcharnikov et al., 2011; Jégo et al., 2016) and indicates that the studied silicate melt had an enhanced capability for transporting significant amounts of Au (Figure 7).
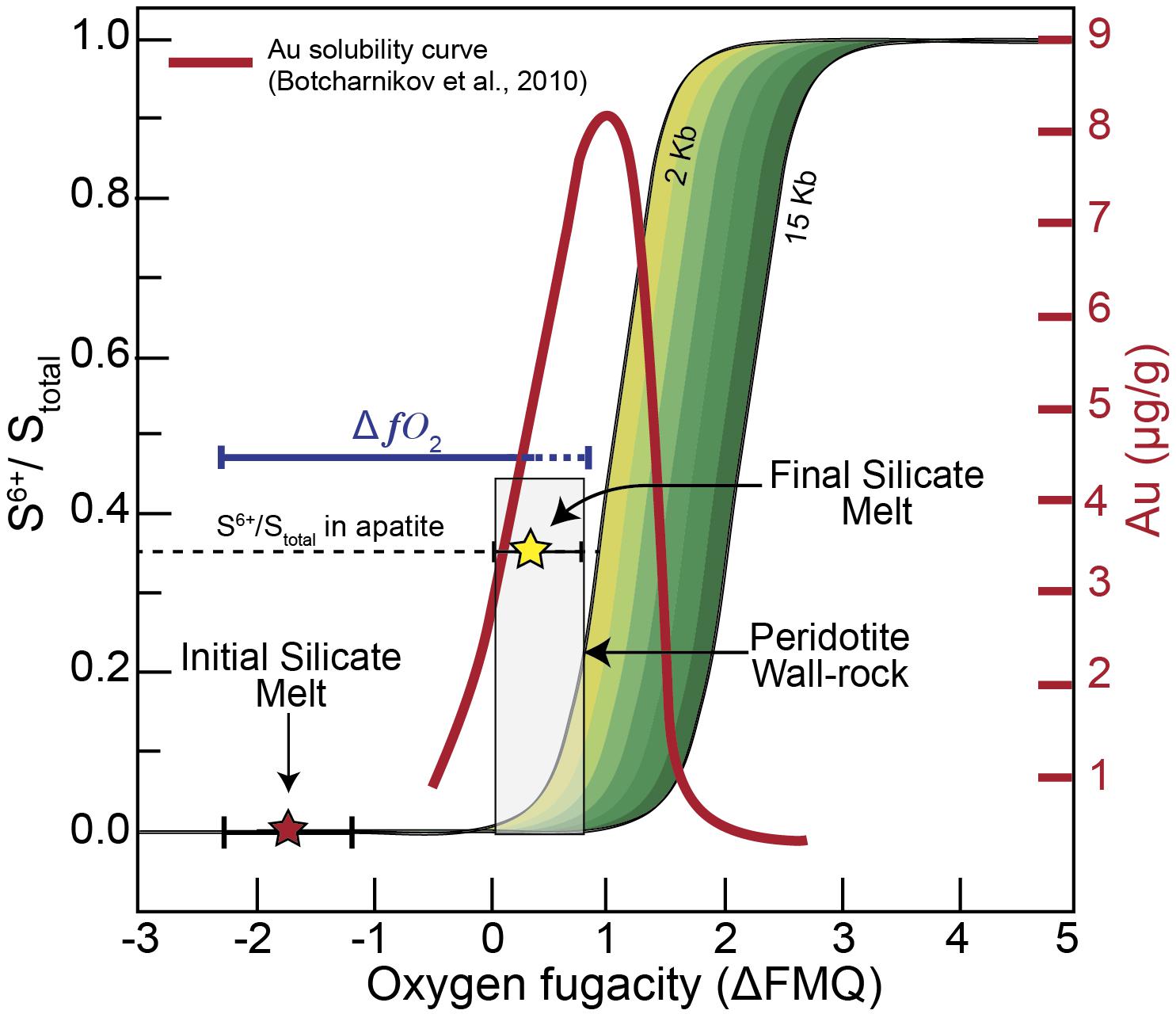
Figure 7. Redox evolution of the silicate melt and the effect of redox gradients on Au solubility. Plot of S6+/Stotal vs. oxygen fugacity, expressed log units relative to the fayalite–magnetite–quartz mineral buffer (ΔFMQ). The diagram shows the initial and final redox conditions of the originally reduced silicate melt that interacts with the more oxidized peridotite wall-rock (1020°C and 13.6 Kb). The empirical approach for the determined fO2 conditions of the initial and final silicate melt is from experiments performed at 1200°C and 0.001 to 14 Kb (armalcolite), and 1000°C 3 Kb (apatite). The green shaded area illustrates the changes in S6+/Stot in silicate melts with pressure after Jugo et al. (2010) and Matjuschkin et al. (2016). The red line corresponds to Au solubility in S-bearing basaltic glasses at 1050°C and 2 Kb (Botcharnikov et al., 2011). The redox gradient generated between the melt and the peridotite wall-rock, defined as ΔfO2, promotes oxidation of the infiltrating melt leading to optimal Au solubility conditions.
In summary, mantle-derived and metal-rich sulfides entrained within reduced percolating silicate melts (e.g., Bockrath et al., 2004; Ballhaus et al., 2006; Tassara et al., 2018) may release significant amounts of sulfur and Au due to the transient increase in fO2 caused by melt–peridotite interaction during melt ascent across the SCLM (Figure 6), acting as an efficient mechanism for sourcing metals from the deep lithosphere, and resulting in Au enrichment of ascending silicate melts by almost one order of magnitude.
Concluding Remarks
The role of melt–peridotite interactions is being increasingly recognized in the literature as a key factor controlling the fO2 of silicate melts percolating through the mantle in various tectonic settings (e.g., Tollan and Hermann, 2019). Our data from the El Deseado Massif auriferous province in southern Patagonia are in agreement with this point of view. Moreover, we suggest that changes in the redox state of ascending magmas during ascent throughout the SCLM can boost the potential for efficient extraction of Au and other metals from selected mantle regions, increasing their ore-fertility. Sillitoe (2008) emphasized that the recurrent generation of major gold deposits and belts in North and South America remains uncertain. Among the factors explaining the occurrence of metallogenic provinces dominated by one or more ore metals, widely contemplated possibilities include heterogeneously distributed metal preconcentration, favorable redox conditions, or other parameters somewhere above the subducted slab, between the mantle wedge and upper crust (Sillitoe, 2008). We argue that the formation of restricted blocks of crust highly endowed with gold deposits requires, in addition to hydrothermal processes in the upper crust, the efficient extraction of Au from refertilized and oxidized domains of SCLM (Rielli et al., 2017). Evidence presented here suggests that redox gradients between ascending magmas and the surrounding mantle can trigger the destabilization of mantle sulfides and sulfide melts, resulting in the release of their Au cargo into the silicate melt fraction. Our model proposes an efficient mechanism for the transfer of Au and associated metals from enriched portions of the SCLM to ascending magmas, helping to improve our understanding of metallogenic processes that operate on a lithospheric scale.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
All the authors contributed substantially to the manuscript as a team including sampling, data acquisition and interpretation, construction of the model, and writing, and analyzed and discussed the data and revised the manuscript, which was written by ST and MR. Specifically, MR, DM, FB, and JG-J conceived and designed the study. ST and BK carried out the EPMA data acquisition, under the supervision of AS. ST and BK carried out the μ-XANES data acquisition, under the supervision of AF. MS and AC provided the samples. ST and MR were involved in the preparation of figures.
Funding
This study was funded by the Iniciativa Científica Milenio through Millennium Nucleus for Metal Tracing along Subduction Grant NC130065. Additional funding for analytical work was provided by the FONDAP project 15090013 “Centro de Excelencia en Geotermia de Los Andes, CEGA.” ST acknowledges CONICYT for support through a Ph.D. scholarship #21170857. Grants RTI2018-099157-A-100 and RYC-2015-1796 provided funding for sample preparation and EPMA analyses. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The DID-UACh project #S-201505 financed the fieldwork.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Tony Lanzirotti and Matt Newville for assistance with μ-XANES analyses at the GeoSoilEnviroCars (Sector 13), Advanced Photon Source (APS), Argonne National Laboratory.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/feart.2020.00029/full#supplementary-material
References
Ballhaus, C. (1993). Redox states of lithospheric and asthenospheric upper mantle. Contribut. Mineral. Petrol. 114, 331–348. doi: 10.1007/bf01046536
Ballhaus, C., Bockrath, C., Wohlgemuth-Ueberwasser, C., Laurenz, V., and Berndt, J. (2006). Fractionation of the noble metals by physical processes. Contribut. Mineral. Petrol. 152, 667–684. doi: 10.1007/s00410-006-0126-z
Beccaluva, L., Bianchini, G., Coltorti, M., Perkins, W. T., Siena, F., Vaccaro, C., et al. (2001). Multistage evolution of the European lithospheric mantle) new evidence from Sardinian peridotite xenoliths. Contribut. Mineral. Petrol. 142, 284–297. doi: 10.1007/s004100100288
Bockrath, C., Ballhaus, C., and Holzheid, A. (2004). Fractionation of the platinum-group elements during mantle melting. Science 305, 1951–1953. doi: 10.1126/science.1100160
Botcharnikov, R. E., Linnen, R. L., Wilke, M., Holtz, F., Jugo, P. J., and Berndt, J. (2011). High gold concentrations in sulfide-bearing magma under oxidizing conditions. Nat. Geosci. 4, 112–115. doi: 10.1038/ngeo1042
Bowles, J. F. W. (1988). Definition and range of naturally occurring minerals with the pseudobrookite structure. Am. Mineral. 73, 1377–1383.
Carmichael, I. S. E. (1991). The redox states of basic and silicic magmas: a reflection of their source regions? Contribut. Mineral. Petrol. 106, 129–141. doi: 10.1007/bf00306429
Cawthorn, R. G., and Biggar, G. M. (1993). Crystallization of titaniferous chromite, magnesian ilmenite and armalcolite in tholeiitic suites in the Karoo Igneous Province. Contribut. Mineral. Petrol. 114, 221–235. doi: 10.1007/bf00307757
Chin, E. J., Lee, C.-T. A., and Barnes, J. D. (2014). Thickening, refertilization, and the deep lithosphere filter in continental arcs: constraints from major and trace elements and oxygen isotopes. Earth Planet. Sci. Lett. 397, 184–200. doi: 10.1016/j.epsl.2014.04.022
Coltorti, M., Becaluva, L., Bonadiman, C., Salvini, L., and Siena, F. (2000). Glasses in mantle xenoliths as geochemical indicators of metasomatic agents. Earth Planet. Sci. Lett. 183, 303–320. doi: 10.1016/s0012-821x(00)00274-0
Contini, S., Venturelli, G., and Toscani, L. (1993). Cr-Zr armalcolite-bearing lamproites of Cancarix, SE Spain. Mineral. Magaz. 57, 203–216. doi: 10.1180/minmag.1993.057.387.02
Cooper, R. F., Fanselow, J. B., and Poker, D. B. (1996). The mechanism of oxidation of a basaltic glass: chemical diffusion of network modifying cations. Geochim. Cosmochim. Acta 60, 3253–3265. doi: 10.1016/0016-7037(96)00160-3
Evans, K. A., and Tomkins, A. G. (2011). The relationship between subduction zone redox budget and arc magma fertility. Earth Planet. Sci. Lett. 308, 401–409. doi: 10.1016/j.epsl.2011.06.009
Féraud, G., Alric, V., Fornari, M., Bertrand, H., and Haller, M. (1999). 40Ar/39Ar dating of the Jurassic volcanic province of patagonia: migrating magmatism related to Gondwana break-up and subduction. Earth Planet. Sci. Lett. 172, 83–96. doi: 10.1016/s0012-821x(99)00190-9
Fiege, A., Ruprecht, P., and Simon, A. C. (2017). A magma mixing redox trap that moderates mass transfer of sulfur and metals. Geochem. Perspect. Lett. 3, 190–199. doi: 10.7185/geochemlet.1722
Friel, J. J., Harker, R. I., and Ulmer, G. C. (1977). Armalcolite stability as a function of pressure and oxygen fugacity. Geochim. Cosmochim. Acta 41, 403–410. doi: 10.1016/0016-7037(77)90268-x
González-Jiménez, J. M., Roqué-Rosell, J., Jiménez-Franco, A., Tassara, S., Nieto, F., Gervilla, F., et al. (2019). Magmatic platinum nanoparticles in metasomatic silicate glasses and sulfides from Patagonian mantle xenoliths. Contribut. Mineral. Petrol. 174, 47.
Gorring, M. L., and Kay, S. M. (2001). Mantle processes and sources of Neogene slab window magmas from southern Patagonia Argentina. J. Petrol. 42, 1067–1094. doi: 10.1093/petrology/42.6.1067
Grégoire, M., Lorand, J.-P., O’Reilly, S. Y., and Cottin, J. Y. (2000). Armalcolite-bearing, Ti-rich metasomatic assemblages in harzburgitic xenoliths from Kerguelen Islands: implications for the oceanic mantle budget of high-field strength elements. Geochim. Cosmochim. Acta 64, 673–694. doi: 10.1016/s0016-7037(99)00345-2
Griffin, W. L., Begg, G. C., and O’Reilly, S. Y. (2013). Continental-root control on the genesis of magmatic ore deposits. Nat. Geosci. 6, 905–910. doi: 10.1038/nature13532
Griffin, W. L., Huang, J.-X., Thomassot, E., Gain, S. E. M., Toledo, V., and O’Reilly, S. Y. (2018). Super-reducing conditions in ancient and modern volcanic systems: sources and behaviour of carbon-rich fluids in the lithospheric mantle. Mineral. Petrol. 112, 1–14.
Groves, D. I., Goldfarb, R. J., and Santosh, M. (2016). The conjunction of factors that lead to formation of giant gold provinces and deposits in non-arc settings. Geosci. Front. 7, 303–314. doi: 10.1016/j.gsf.2015.07.001
Groves, D. I., and Santosh, M. (2015). Province-scale commonalities of some world-class gold deposits: implications for mineral exploration. Geosci. Front. 6, 389–399. doi: 10.1016/j.gsf.2014.12.007
Haggerty, S. E. (1987). “Metasomatic mineral titanates in upper mantle xenoliths,” in Mantle Xenoliths, ed. P. H. Nixon, (Hoboken, NJ: John Wiley and Sons), 671–690.
Haggerty, S. E. (1991). “Oxide mineralogy in the upper mantle,” in Oxide minerals: Petrologic and Magnetic Significance, ed. D. H. Lindsley, (Chantilly, VA: Mineralogical Society of America), 355–416. doi: 10.1515/9781501508684-013
Jégo, S., Nakamura, M., Kimura, J.-I., Iizuka, Y., Chang, Q., and Zellmer, G. F. (2016). Is gold solubility subject to pressure variations in ascending arc magmas? Geochim. Cosmochim. Acta 188, 224–243. doi: 10.1016/j.gca.2016.05.034
Jenner, F. E., O’Neill, H., Arculus, R. J., and Mavrogenes, J. A. (2010). The magnetite crisis in the evolution of arc-related magmas and the initial concentration of Au, Ag, and Cu. J. Petrol. 51, 2445–2464. doi: 10.1093/petrology/egq063
Jugo, P. J. (2009). Sulfur content at sulfide saturation in oxidized magmas. Geology 37, 415–418. doi: 10.1130/g25527a.1
Jugo, P. J., Luth, R. W., and Richards, J. P. (2005a). An experimental study of the sulfur content in basaltic melts saturated with immiscible sulfide or sulfate liquids at 1300 °C and 1.0 GPa. J. Petrol. 46, 783–798. doi: 10.1093/petrology/egh097
Jugo, P. J., Wilke, M., and Botcharnikov, R. E. (2005b). Sulfur K-edge XANES analysis of natural and synthetic basaltic glasses: implications for S speciation and S content as a function of oxygen fugacity. Geochim. Cosmochim. Acta 74, 5926–5938. doi: 10.1016/j.gca.2010.07.022
Jugo, P. J., Wilke, M., and Botcharnikov, R. E. (2010). Sulfur K-edge XANES analysis of natural and synthetic basaltic glasses: implications for S speciation and S content as a function of oxygen fugacity. Geochim. Cosmochim. Acta 74, 5926–5938. doi: 10.1016/j.gca.2010.07.022
Kelley, K., and Cottrell, E. (2009). Water and the oxidation state of subduction zone magmas. Science 325, 605–607. doi: 10.1126/science.1174156
Konecke, B. A., Fiege, A., Simon, A. C., Linsler, S., and Holtz, F. (2019). An experimental calibration of a sulfur-in-apatite oxybarometer for mafic systems. Geochim. Cosmochim. Acta 265, 242–258. doi: 10.1016/j.gca.2019.08.044
Konecke, B. A., Fiege, A., Simon, A. C., Parat, F., and Stechern, A. (2017). Co-variability of S6+, S4+, and S2- in apatite as a function of oxidation state: implications for a new oxybarometer. Am. Mineral. 102, 548–557. doi: 10.2138/am-2017-5907
Kovacs, I., Hidas, K., Hermann, J., Sharygin, V., Szabó, C., and Ntaflos, T. (2007). Fluid induced melting in mantle xenoliths and some implications for the continental lithospheric mantle from Minusinsk region (Khakasia, southern Siberia). Geol. Carpathica 58, 211–228.
Lee, C.-T. A., Luffi, P., Chin, E. J., Bouchet, R., Dasgupta, R., Morton, D. M., et al. (2002). Copper systematics in arc magmas and implications for crust-mantle differentiation. Science 336, 64–68. doi: 10.1126/science.1217313
Lee, C.-T. A., Luffi, P., Le Roux, V., Dasgupta, R., Albarède, F., and Leeman, W. P. (2010). The redox state of arc mantle using Zn/Fe systematics. Nature 468, 681–685. doi: 10.1038/nature09617
Li, Y., and Audétat, A. (2012). Partitioning of V, Mn, Co, Ni, Cu, Zn, As, Mo, Ag, Sn, Sb, W, Au, Pb, and Bi between sulfide phases and hydrous basanite melt at upper mantle conditions. Earth Planet. Sci. Lett. 355-356, 327–340. doi: 10.1016/j.epsl.2012.08.008
Lindsley, D. H., Kesson, S. E., Hartzman, M. J., and Cushman, M. K. (1974). “The stability of armalcolite: experimental studies in the system MgO-Fe-Ti-O,” in Proceedings of the 11 Lunar and Planetary Science Conference, (New York, NY: Pergamon Press Inc), 521–534.
Lorand, J.-P., and Luguet, A. (2016). “Chalcophile and siderophile elements in mantle rocks: trace elements controlled by trace minerals,” in Highly Siderophile and Strongly Chalcophile Elements in High-Temperature Geochemistry and Cosmochemistry. Reviews in Mineralogy and Geochemistry 81, eds J. Harvey and J. M. D. Day, (Chantilly, VA: The Mineralogical Society of America).
Medvedev, A. Y. (1996). Synthetic armalcolite and pseudobrookite. Mineral. Mag. 60, 347–353. doi: 10.1180/minmag.1996.060.399.09
Matjuschkin, C., Blundy, J. D., and Brooker, R. A. (2016). The effect of pressure on sulfur speciation in mid-to deep-crustal arc magmas and implications for the formation of porphyry copper deposits. Contribut. Mineral. Petrol. 171:66.
Mavrogenes, J. A., and O’Neill, H. S. C. (1999). The relative effects of pressure, temperature and oxygen fugacity on the solubility of sulfide in mafic magmas. Geochim. Cosmochim. Acta 63, 1173–1180. doi: 10.1016/s0016-7037(98)00289-0
Mungall, J. E. (2002). Roasting the mantle: slab melting and the genesis of major Au and Au-rich Cu deposits. Geology 30, 915–918.
Nash, W. P. (1984). “Phosphate minerals in terrestrial igneous and metamorphic rocks,” in Phosphate Minerals, eds J. O. Nriagu and P. B. Moore, (Berlin: Springer-Verlag), 442.
O’Reilly, S. Y., and Griffin, W. L. (2010). “Rates of magma ascent: constraints from mantle-derived xenoliths,” in Timescales of Magmatic Processes: From Core To Atmosphere, eds A. Dosseto, S. Turner, and J. Van Orman, (Oxford: John Wiley & Sons).
Pankhurst, R. J., Riley, T. R., Fanning, C. M., and Kelley, S. P. (2000). Episodic silicic volcanism in Patagonia and the Antarctic Peninsula: chronology of magmatism associated with the break-up of Gondwana. J. Petrol. 41, 605–625. doi: 10.1093/petrology/41.5.605
Piccoli, P. M., and Candela, P. A. (2002). “Apatite in igneous systems,” in Phosphates: Geochemical, Geobiological, and Materials Importance, eds M. J. Kohn and J. M. Hughes, (Chantilly, VA: Mineralogical Society of America), 255–292. doi: 10.2138/rmg.2002.48.6
Richards, J. P. (2015). The oxidation state, and sulfur and Cu contents of arc magmas: implications for metallogeny. Lithos 233, 27–45. doi: 10.1016/j.lithos.2014.12.011
Rielli, A., Tomkins, A. G., Nebel, O., Brugger, J., Etschmann, B., Zhong, R., et al. (2017). Evidence of sub-arc mantle oxidation by sulphur and carbon. Geochem. Perspect. Lett. 3, 124–132. doi: 10.7185/geochemlet.1713
Rivalenti, G., Mazzucchelli, M., Laurora, A., Ciuffi, S. I. A., Zanetti, A., Vannucci, R., et al. (2004). The backarc mantle lithosphere in patagonia, South America. J. South Am. Earth Sci. 17, 121–152. doi: 10.1016/j.jsames.2004.05.009
Schalamuk, I. B., Zubia, M., Genini, A., and Fernandez, R. R. (1997). Jurassic epithermal Au-Ag deposits of patagonia, Argentina. Ore Geol. Rev. 12, 173–176.
Schilling, M. E., Conceição, R. V., Mallmann, G., Koester, E., Kawashita, K., Hervé, F., et al. (2005). Spinel-facies mantle xenoliths from Cerro Redondo, Argentine Patagonia: Petrographic, geochemical, and isotopic evidence of interaction between xenoliths and host basalt. Lithos 82, 485–502. doi: 10.1016/j.lithos.2004.09.028
Sillitoe, R. H. (2008). Major gold deposits and belts of the North and South American cordillera: distribution, tectonomagmatic settings, and metallogenic considerations. Econ. Geol. 103, 663–687. doi: 10.2113/gsecongeo.103.4.663
Tang, M., Erdman, M., Eldridge, G., and Lee, C.-T. A. (2018). The redox “filter” beneath magmatic orogens and the formation of continental crust. Sci. Adv. 4:eaar4444. doi: 10.1126/sciadv.aar4444
Tassara, S., González-Jiménez, J. M., Reich, M., Saunders, E., Luguet, A., Morata, D., et al. (2018). Highly siderophile elements mobility in the subcontinental lithospheric mantle beneath southern Patagonia. Lithos 314-315, 579–596. doi: 10.1016/j.lithos.2018.06.022
Tassara, S., González-Jiménez, J. M., Reich, M., Schilling, M. E., Morata, D., Begg, G. C., et al. (2017). Plume-subduction interaction forms large auriferous provinces. Nat. Commun. 8:843. doi: 10.1038/s41467-017-00821-z
Tollan, P., and Hermann, J. (2019). Arc magmas oxidized by water dissociation and hydrogen incorporation in orthopyroxene. Nat. Geosci. 12, 667–671. doi: 10.1038/s41561-019-0411-x
Wood, B. J., Bryndzia, L. T., and Johnson, K. E. (1990). Mantle oxidation state and its relationship to tectonic environment and fluid speciation. Science 248, 337–345. doi: 10.1126/science.248.4953.337
Keywords: melt–mantle interaction, oxidation state, apatite, XANES, gold, ore deposits
Citation: Tassara S, Reich M, Konecke BA, González-Jiménez JM, Simon AC, Morata D, Barra F, Fiege A, Schilling ME and Corgne A (2020) Unraveling the Effects of Melt–Mantle Interactions on the Gold Fertility of Magmas. Front. Earth Sci. 8:29. doi: 10.3389/feart.2020.00029
Received: 15 October 2019; Accepted: 27 January 2020;
Published: 11 February 2020.
Edited by:
Anastassia Borisova, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Elena F Bazarkina, UPR2940 Institut Neel (NEEL), FranceAnne-Sophie Bouvier, Université de Lausanne, Switzerland
Zoltan Zajacz, Université de Genève, Switzerland
Copyright © 2020 Tassara, Reich, Konecke, González-Jiménez, Simon, Morata, Barra, Fiege, Schilling and Corgne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Santiago Tassara, c2FudGlhZ28udGFzc2FyYUB5YWxlLmVkdQ==