 Jotheeswari Kothandaraman
Jotheeswari Kothandaraman David J. Heldebrant
David J. Heldebrant Johnny Saavedra Lopez
Johnny Saavedra Lopez Robert A. Dagle1
Robert A. Dagle1- 1Pacific Northwest National Laboratory, Richland, WA, United States
- 2The Gene and Linda Voiland School of Chemical Engineering and Bioengineering, Washington State University, Pullman, WA, United States
Amine-promoted hydrogenation of CO2 to methanol typically proceeds via a formamide intermediate when amines are used as additives or if the hydrogenation is performed in carbon capture solvents. The catalysts used for the hydrogenation of the formamide intermediate dictate the selectivity of the products formed: 1) Deoxygenative hydrogenation (C–O bond cleavage) resulting in N-methylation of amine and deactivation of the solvent, 2) Deaminative hydrogenation (C–N bond cleavage) resulting in formation of methanol and regeneration of the solvent. To date, catalytic reductions of CO2 with amine promoters suffer from poor selectively for methanol which we attribute to the limiting formamide intermediate, though to date, the conditions that favor C–N cleavage have yet to be fully understood. To better understand the reactivity of the formamide intermediates, a range of heterogenous catalysts were used to study the hydrogenation of formamide. Well-known gas phase CO2 hydrogenation catalysts catalyze the hydrogenation of formamide to N-methyl product via C–O bond cleavage. However, the selectivity can be readily shifted to selective C–N bond cleavage by addition of an additive with sufficient basicity for both homogenous and heterogeneous catalytic systems. The base additive shifts the selectivity by deprotonating a hemiaminal intermediate formed in situ during the formamide hydrogenation. This prevents dehydration process leading to N-methylated product, which is a key capture solvent deactivation pathway that hinders amine use in carbon capture, utilization, and storage (CCUS). The findings from this study provide a roadmap on how to improve the selectivity of known heterogenous catalysts, enabling catalytic reduction of captured CO2 to methanol.
1 Introduction
Anthropogenic CO2 emissions are considered as a main driver of global warming and climate change. To mitigate the rise in CO2 concentrations in the atmosphere, recycling of CO2 to fuels and materials may help provide economic incentives to jump-start the transition to a CO2-neutral, then ultimately a CO2-negative economy (Al-Mamoori et al., 2017; Bui et al., 2018; Gabrielli et al., 2020; Wei et al., 2022). We have recently posited that an integrated CO2 capture and conversion to materials (IC3M) approach, where the capture and conversion are performed in the same medium, such that the process can minimize or partially offset the energy intensive CO2 desorption, compression, and transportation stages (Heldebrant et al., 2022). In an IC3M approach, the captured CO2 can be directly converted to value-added chemicals which are then transported from the CO2 capture site instead of pipelines to move compressed supercritical CO2. Recent studies have focused on examining the viability of different media, catalysts, and experimental conditions for varied IC3M approaches (Heldebrant et al., 2022; Siegel et al., 2023).
CO2 capture solvents are most commonly studied than solids due to low capture costs, fast kinetics, enhanced heat and mass transfer, and ease of handling in a continuous flow process (Heldebrant and Kothandaraman, 2020; Heldebrant et al., 2022). Aqueous organic amines (such as 30wt% aqueous monoethanol amine) are the first-generation solvents that are commonly used for CO2 capture, with recent second-generation aqueous amines such as CANSOLV, piperazine and KS-1 have shown significant improvement in efficiency (Heldebrant and Kothandaraman, 2020). Recently, third generation solvents have been designed for water-lean conditions, lessening their energy demand. Irrespective of whether water-based or water-lean solvents are used, primary and/or secondary amines are the active moieties, reacting with CO2 to form carbamate and/or bicarbonate species. The catalytic reduction of carbamate and/or bicarbonate species to products, such as formates, methanol and cyclic carbonates, has been demonstrated (Li et al., 2013; McNamara and Hicks, 2014; Rezayee et al., 2015; Kothandaraman et al., 2016a; Kothandaraman et al., 2016b; Lao et al., 2016; Kar et al., 2018; Kar et al., 2019; Kothandaraman et al., 2019; Kothandaraman and Heldebrant, 2020b; Sen et al., 2020; Kothandaraman et al., 2021; Wei et al., 2021; Kothandaraman et al., 2022a; Heldebrant et al., 2022). Among these products, methanol is attractive as a fuel and feedstock for production of various value-added chemicals, representing a large market for CO2 reuse.
Unlike the traditional gas phase CO2 reduction to methanol (Porosoff et al., 2016; Huš et al., 2017; Pavlišič et al., 2020; Prasnikar et al., 2021; Kanuri et al., 2022), in the low-temperature amine-promoted catalytic reductions, ammonium carbamate/bicarbonate are reduced to methanol via a formamide intermediate (Scheme 1) (Rezayee et al., 2015; Kothandaraman et al., 2016a; Everett and Wass, 2017; Kar et al., 2018; Kothandaraman et al., 2022b). The hydrogenation of the formamide intermediate has been typically identified as a rate determining step (Kothandaraman and Heldebrant, 2020a; Rayder et al., 2020). One of the main challenges with integrated capture and conversion of CO2 to methanol in the presence of amine-based capture solvents is an N-methylation, which is formed from a C–O cleavage of N-methyl formamide rather than C–N cleavage of amide to methanol. N-methylation leads to deactivation of the capture solvent by converting the 2° amine to a 3° amine, thus preventing further carbamate formation (CO2 uptake) and thermal condensation of an ammonium formate salt to the formamide intermediate. Homogenous catalysts selective for hydrogenation of CO2 to methanol have been reported in the literature (Rezayee et al., 2015; Kothandaraman et al., 2016a; Everett and Wass, 2017; Kar et al., 2018). However, from a separation standpoint, heterogenous catalysts are preferred for continuous flow operations. Additionally, their superior durability is needed for process conditions and impurities common in post-combustion CO2 capture. While gas phase CO2 catalysis in presence of a solid heterogenous catalysts were widely investigated, our understanding on low-temperature condensed phase CO2 chemistry remains very limited.
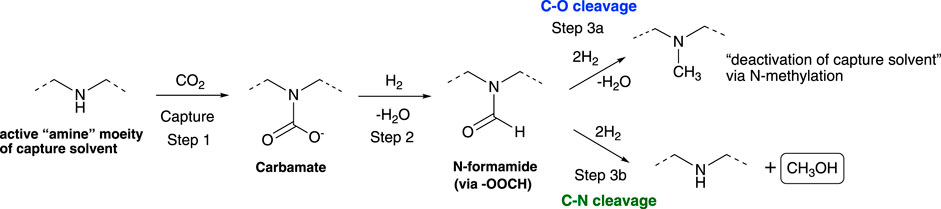
SCHEME 1. Hydrogenation of CO2 to methanol in the presence of a capture solvent.
To gain this knowledge we need to first study model systems to learn how to control the product selectivity in low-temperature CO2 hydrogenation chemistry. Herein, N-formyl piperidine was chosen as a model substrate to understand the C–O vs. C–N bond cleavage selectivity during the amide hydrogenation to methanol step (Step 3b, Scheme 1). In the literature, the selection of the catalyst is often reported to be important for C–N cleavage selectivity and the majority of them are organometallic catalysts (Werkmeister et al., 2014; Cabrero-Antonino et al., 2020). We found that the presence of an additive with sufficient basicity plays a crucial role in switching the selectivity from C–O cleavage to C–N cleavage. For the first time such trend has been identified across different heterogenous catalysts. We also show selectivity as high as 86% for C–N cleavage to methanol, even in the presence of a Cu-based catalyst. It is also important to note that the findings derived from this research are also relevant to the selective synthesis of amines and alcohols from hydrogenation of amides—amide functionalities are common in pharmaceuticals, natural products, and agrochemicals.
2 Materials and methods
Materials: The Cu/Zn/Al2O3 (64wt%) was obtained from Alfa Aesar. Copper Chromite and 1,3,5-trimethoxybenzene (TMB) were purchased from Sigma Aldrich. Al2O3 (γ-Al2O3) was purchased from Engelhard. Carbonylchlorohydrido[bis(2-(diphenylphosphinoethyl)amino]ruthenium (II) (also known as Ru-MACHO) was purchased from Strem chemicals. Unless otherwise mentioned, all other materials were obtained from commercial suppliers and used without further purification. All deuterated solvents were obtained from Cambridge Isotope Laboratories, Inc. All other catalysts (metal supported on metal oxides) were prepared by incipient wetness impregnation of nitrate metal precursors, followed by drying (8 h at 100°C) and calcination at 400°C (4 h under static air). TMB was used as an internal standard for NMR spectroscopy.
Standard experimental procedure for the hydrogenation of N-formyl piperidine: A Parr reactor (100 mL) equipped with a thermocouple, pressure transducer, turbine type impellor and reactor controller, was charged with a catalyst, base additive, and solvent, and sealed in a N2-atmosphere glovebox. The reactor was then pressurized with H2 (60 bar) and stirred at 170°C. The reactor was cooled to room temperature, and the gas in the reactor headspace was analyzed using a 2-channel Fusion MicroGC (Inficon). The remaining excess gas was slowly released after cooling the reactor to −78°C. TMB was added as an internal standard to the reaction mixture, and a small aliquot of the sample was analyzed by 1H NMR experiments.
3 Results
First, the commercial methanol synthesis catalyst, Cu/ZnO/Al2O3, was studied to establish a baseline for the selective hydrogenation of N-formyl piperidine to methanol via C–N cleavage. Analogous to catalytic systems previously reported (Gredig et al., 1995; Gredig et al., 1996; 1997; Auer, 1999; Cui et al., 2014; Cabrero-Antonino et al., 2020), N-methyl piperidine formed with high conversion (93.2%) and selectivity (96%) via C–O cleavage of the N-formyl piperidine. In order to understand the reaction mechanism and conditions that favor the C–N cleavage over the C–O cleavage, the effect of reaction temperature, time and different additives on the selectivity and conversion was studied. As shown in entries 1–3, Table 1, the selectivity to methanol decreased with the increase in reaction time. The methanol yields for entries 1 (12 h), 2 (3 h) and 3 (1 h) were 4.6%, 9.8% and 9.6%, respectively. With the decrease in the reaction temperature from 170°C (entry 3, Table 1) to 150°C (entry 4, Table 1), the overall conversion decreased with some improvement in the methanol selectivity. These results show that the methanol acted as a N-methylating agent causing N-methylation of free amine with longer reaction time (Guillena et al., 2010). Thus, the methanol yield decreased while the N-methylation yield increased with increasing reaction time, as expected. To further confirm the role of methanol in N-methylation, piperidine was reacted with methanol under our typical reaction conditions (Cu/ZnO/Al2O3, under 60 bar H2 at 170°C for 12 h), a quantitative conversion of piperidine to N-methyl formamide was observed. Thus, longer reaction time is detrimental to the reaction selectivity due to the reactive nature of methanol.
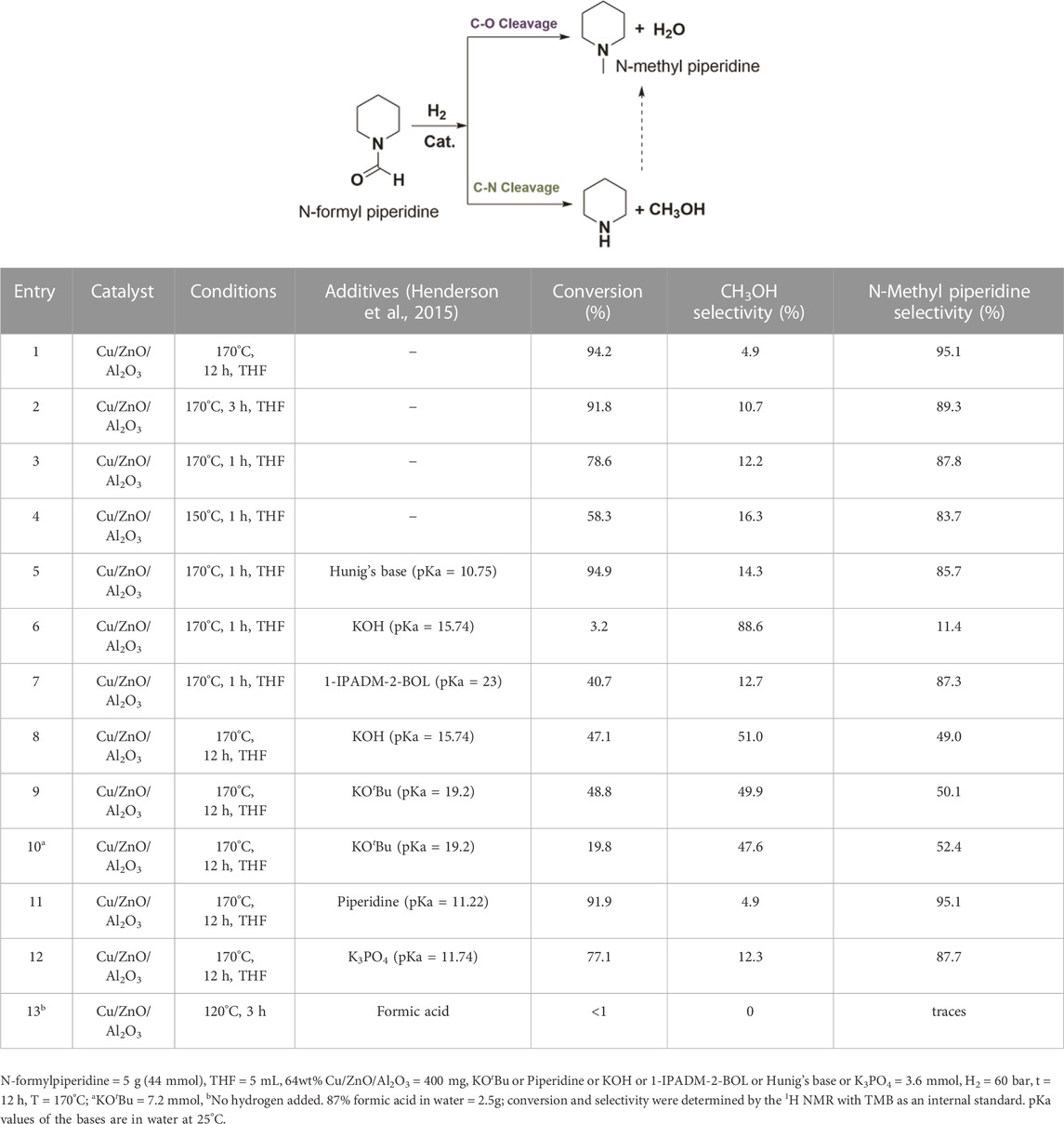
TABLE 1. Hydrogenation of N-formyl piperidine in the presence of Cu/ZnO/Al2O3 catalyst and different additives.
To further understand and improve the C–N cleavage selectivity, we examined the proposed reaction mechanism for the hydrogenation of formamide (Cantillo, 2011; Volkov et al., 2016; Cabrero-Antonino et al., 2020). Based on the literature, the initial hydrogenation of formamide forms a hemiaminal intermediate, 3 (Scheme 2), which then can undergo C–N cleavage to form methanol and free amine via hydrogenation of a formaldehyde intermediate. Alternatively, the hemiaminal intermediate can undergo dehydration to form an imine intermediate via C–O cleavage. The subsequent hydrogenation of the imine intermediate forms the N-methylated amine. From the reaction mechanism, we hypothesized that the addition of catalytic amounts of base additives could improve the C–N cleavage selectivity by deprotonation of the proposed hemiaminal intermediate in Scheme 2. The deprotonation of hemiaminal can prevent the dehydration and subsequent formation of the imine intermediate.
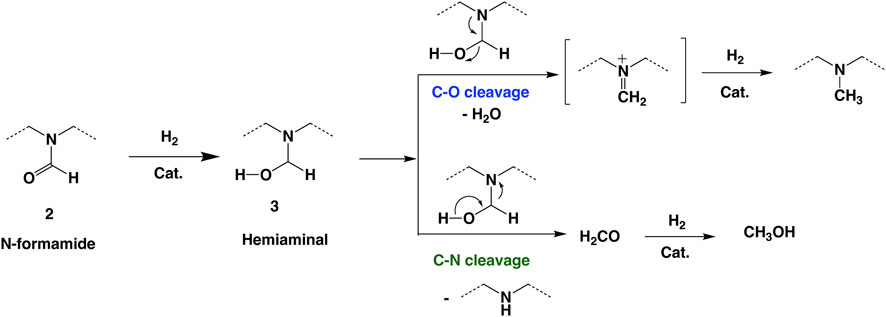
SCHEME 2. Proposed reaction mechanism for the hydrogenation for formamide.
To validate this mechanism, we first tested bases with varying pKa and the results are shown in entries 5–11, Table 1. With the addition of Hünig’s base (N,N-Diisopropylethylamine) (pKa = 10.75), there was some improvement in the methanol selectivity and conversion (entry 3 vs. 5, Table 1). The addition of KOH (pKa = 15.74) significantly improved the selectivity of methanol to 88.6% albeit with a low yield (3.2%) (entry 3 vs. 6, Table 1). The alkanolgaunidine based solvent additive, 1-IPADM-2-BOL (pKa = 23) (Zheng et al., 2016) significantly reduced the conversion with no change in the selectivity (entry 3 vs. 7, Table 1). This is because the amidinium based moieties are not stable under hydrogenation conditions in the presence of Cu/ZnO/Al2O3 catalyst. Longer reaction time improved the conversion to 47.1% (methanol yield = 24%) with a drop in selectivity to 51.0% (entry 8, Table 1), likely because the formed methanol became a methyl source to form N-methyl piperidine. The conversion and selectivity for the KOtBu (pKa = 19.2) added reaction performed similar to KOH (entry 9, Table 1). As the base additive content increased, the conversion decreased without significantly affecting selectivity because the excess base could block the active sites that are promoting hydrogenation (entry 10, Table 1). No significant change on the selectivity was observed with piperidine (pKa = 11.22; entry 1 vs. 11, Table 1), suggesting this additive is not strong enough to deprotonate the hemiaminal intermediate (3). There was some improved in the selectivity in the case of K3PO4 (pKa = 11.74; entry 1 vs. 12, Table 1) additive. Overall, base additives with pKa >15.7 are effective in deprotonating the hemiaminal species, promoting the formation of formaldehyde intermediate and consequently improving the selectivity to methanol (Note the pKa values listed in Table 1 are in water, and the actual pKa will likely differ in the THF solvent). In addition, in the case of entries 8 and 9, the methanol yield was more than the amount of base added, suggesting sub-stoichiometric amount of base is sufficient for improved C-N cleavage selectivity. This study provides a first example for the switch in the selectivity of C–O cleavage to C–N cleavage in amide hydrogenation by using a co-catalyst or additive without changing the catalyst in heterogeneous systems. The use of formic acid in place of hydrogen formed a significant amount of H2, CO and CO2 in the gas phase (by decomposition of formic acid) with no appreciable reaction with the N-formyl piperidine (entry 13, Table 1).
Following this observation, we also tested other heterogenous catalysts to understand the effect of base additives (Table 2). Similar to Cu/ZnO/Al2O3 catalyst, the other Cu catalysts, copper chromite and Cu/Al2O3, also showed suppression in N-methylation upon the addition of a base. Even with the Ag/Al2O3, the first heterogenous catalyst reported in the literature for selective C–N cleavage, we noticed in the absence of a base additive, the C–O cleavage was predominant, and a base is necessary for the C–N cleavage selectivity (Xie et al., 2018; Cabrero-Antonino et al., 2020; Long et al., 2023). The following catalysts were also screened for the C–N cleavage selectivity: Pd/Al2O3 and Pd/ZnO/Al2O3. In both the cases, the addition of KOtBu, switched the selectivity towards C–N cleavage. Au/Al2O3 was inactive for hydrogenation of N-formyl piperidine. Similar to previous reports (Kothandaraman et al., 2021; Koch et al., 2023), Ru/Al2O3 catalyst selectively formed methane after hydrogenation with no detectable amounts of methanol or N-methyl piperidine (Entry 15, Table 2).
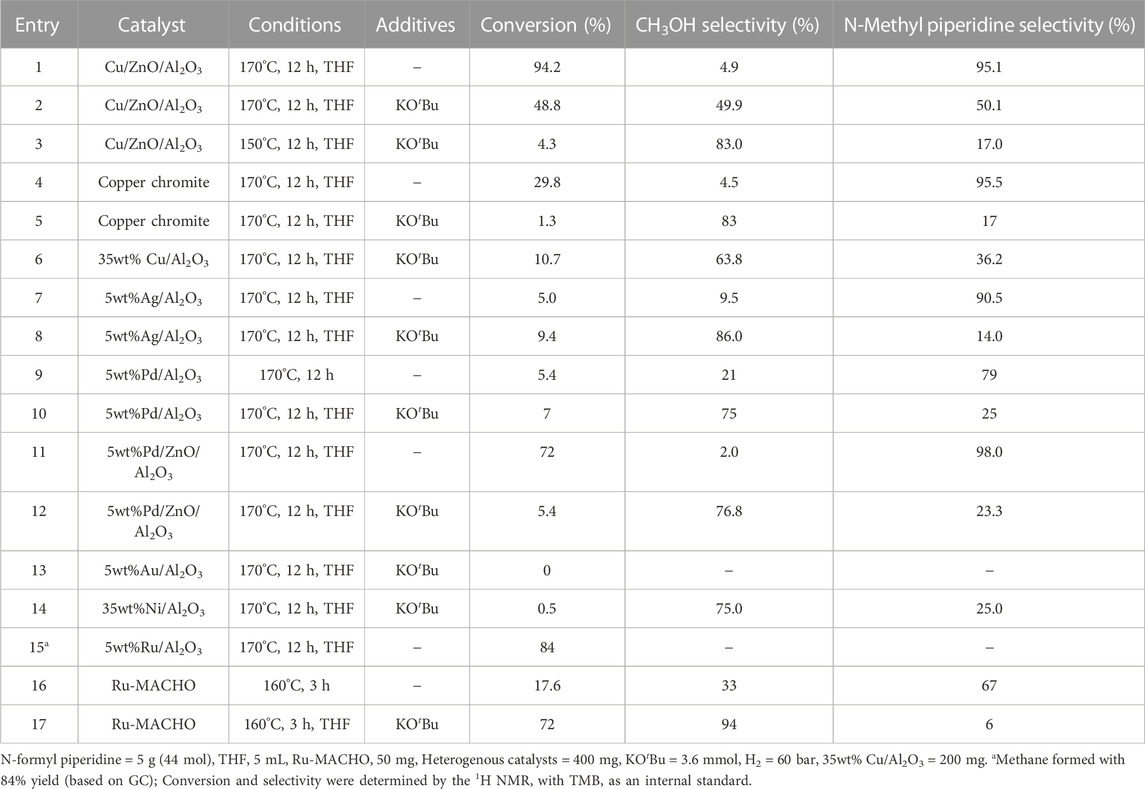
TABLE 2. Hydrogenation of N-formyl piperidine in the presence of different catalysts.
Next, to understand if a similar switch in the selectivity can be observed in the presence of a homogenous catalyst, we tested the well-known low temperature methanol synthesis catalyst (from CO2 hydrogenation), Ru-MACHO, for the hydrogenation of N-formyl piperidine (Kothandaraman et al., 2016a; Kar et al., 2018). Hydrogenation of CO2 to methanol in the presence of Ru-MACHO has been widely demonstrated to proceed via a formamide intermediate. Entry 16, Table 2, to our surprise, in the absence of base addition and solvent, Ru-MACHO formed N-methyl piperidine with good selectivity. Notably, similar to heterogeneous catalysts, we observed a switch in the selectivity with Ru-MACHO catalyst with the addition of a base.
4 Conclusion
In summary, we studied the effect of reaction temperature, time and different additives on the C-O and C-N cleavage selectivity during catalytic hydrogenation of formamide. We show that in the absence of any additives, hydrogenation of N-formyl piperidine formed N-methyl piperidine by C–O bond cleavage with high selectivity (>90%). On the other hand, in the presence of a base additive, the selectivity switched to methanol formation by C–N bond cleavage. We also showed that the (in situ formed) product methanol can also act as a N-methylating agent upon longer residence time. Such reactivity trend has been identified for the first time across a range of heterogeneous catalysts and the homogeneous catalyst, Ru-MACHO. The strong base additive can deprotonate the hemiaminal intermediate, preventing the subsequent dehydration step to form N-methyl piperidine. The results from this study show the crucial role of base additives in determining the product selectivity. The ease of which the selectivity can be shifted from C–O cleavage to C–N cleavage provides a simple and effective means to suppress amine deactivation by N-methylation. This study provides new fundamental understandings on how to better design integrated capture and conversion systems to upcycle CO2 into useful products like methanol.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This material is based upon work supported by the DOE’s Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division under FWP 75428. The authors would also like to acknowledge the U.S. Department of Energy’s (DOE’s) Office of Fossil Energy and Carbon Management (FECM) and SoCalGas for co-funding this work under FWP 80562 and TCF-19-17862. The Pacific Northwest National Laboratory is operated by Battelle for the DOE.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2023.1158499/full#supplementary-material
References
Al-Mamoori, A., Krishnamurthy, A., Rownaghi, A. A., and Rezaei, F. (2017). Carbon capture and utilization update. Energy Technol. 5, 834–849. doi:10.1002/ente.201600747
Auer, S. (1999). Synthesis of methylamines from CO2, H2 and NH3 over Cu–Mg–Al mixed oxides. J. Mol. Catal. A Chem. 141, 193–203. doi:10.1016/s1381-1169(98)00263-5
Bui, M., Adjiman, C. S., Bardow, A., Anthony, E. J., Boston, A., Brown, S., et al. (2018). Carbon capture and storage (CCS): the way forward. Energy & Environ. Sci. 11, 1062–1176. doi:10.1039/c7ee02342a
Cabrero-Antonino, J. R., Adam, R., Papa, V., and Beller, M. (2020). Homogeneous and heterogeneous catalytic reduction of amides and related compounds using molecular hydrogen. Nat. Commun. 11, 3893. doi:10.1038/s41467-020-17588-5
Cantillo, D. (2011). Mechanistic Insights on the ruthenium-catalyzed hydrogenation of amides – C–N vs. C–O cleavage. Eur. J. Inorg. Chem. 2011, 3008–3013. doi:10.1002/ejic.201100443
Cui, X. J., Dai, X. C., Zhang, Y., Deng, Y. Q., and Shi, F. (2014). Methylation of amines, nitrobenzenes and aromatic nitriles with carbon dioxide and molecular hydrogen. Chem. Sci. 5, 649–655. doi:10.1039/c3sc52676c
Everett, M., and Wass, D. F. (2017). Highly productive CO2 hydrogenation to methanol - a tandem catalytic approach via amide intermediates. Chem. Commun. 53, 9502–9504. doi:10.1039/c7cc04613h
Gabrielli, P., Gazzani, M., and Mazzotti, M. (2020). The role of carbon capture and utilization, carbon capture and storage, and biomass to enable a net-zero-CO2 emissions chemical industry. Ind. Eng. Chem. Res. 59, 7033–7045. doi:10.1021/acs.iecr.9b06579
Gredig, S. V., Koeppel, R. A., and Baiker, A. (1995). Synthesis of methylamines from carbon dioxide and ammonia. J. Chem. Soc. Chem. Commun., 73–74. doi:10.1039/c39950000073
Gredig, S. V., Koeppel, R., and Baiker, A. (1996). Comparative study of synthesis of methylamines from carbon oxides and ammonia over Cu/A12O3. Catal. Today 29, 339–342. doi:10.1016/0920-5861(95)00301-0
Gredig, S. V., Koeppel, R., and Baiker, A. (1997). Synthesis of methylamines from CO2, H2 and NH3. Catalytic behaviour of various metal-alumina catalysts. Appl. Catal. A General 162, 249–260. doi:10.1016/s0926-860x(97)00107-5
Guillena, G. D, J. R., and Yus, M. (2010). Hydrogen autotransfer in the N-alkylation of amines and related compounds using alcohols and amines as electrophiles. Chem. Rev. 110, 1611–1641. doi:10.1021/cr9002159
Heldebrant, D. J., Kothandaraman, J., Dowell, N. M., and Brickett, L. (2022). Next steps for solvent-based CO2 capture; integration of capture, conversion, and mineralisation. Chem. Sci. 13, 6445–6456. doi:10.1039/d2sc00220e
Heldebrant, D. J., and Kothandaraman, J. (2020). “Chapter 3 solvent-based absorption,” in Carbon capture and storage. Editors Mai Bui, and N. M. Dowell (London, United Kingdom: The Royal Society of Chemistry), 36–68.
Henderson, R. K., Hill, A. P., Redman, A. M., and Sneddon, H. F. (2015). Development of GSK's acid and base selection guides. Green Chem. 17, 945–949. doi:10.1039/c4gc01481b
Huš, M., Kopač, D., Štefančič, N. S., Jurković, D. L., Dasireddy, V. D. B. C., and Likozar, B. (2017). Unravelling the mechanisms of CO2 hydrogenation to methanol on Cu-based catalysts using first-principles multiscale modelling and experiments. Catal. Sci. Technol. 7, 5900–5913. doi:10.1039/c7cy01659j
Kanuri, S., Roy, S., Chakraborty, C., Datta, S. P., Singh, S. A., and Dinda, S. (2022). An insight of CO2 hydrogenation to methanol synthesis: thermodynamics, catalysts, operating parameters, and reaction mechanism. Int. J. Energy Res. 46, 5503–5522. doi:10.1002/er.7562
Kar, S., Goeppert, A., and Prakash, G. K. S. (2019). Integrated CO2 capture and conversion to formate and methanol: connecting two threads. Acc. Chem. Res. 52, 2892–2903. doi:10.1021/acs.accounts.9b00324
Kar, S., Kothandaraman, J., Goeppert, A., and Prakash, G. K. S. (2018). Advances in catalytic homogeneous hydrogenation of carbon dioxide to methanol. J. CO2. Util. 23, 212–218. doi:10.1016/j.jcou.2017.10.023
Koch, C. J., Alagaratnam, A., Goeppert, A., and Prakash, G. K. S. (2023). Phosphate assisted integrated carbon dioxide capture and conversion to methane. Sustain. Energy Fuels 7, 2824–2829. doi:10.1039/d3se00390f
Kothandaraman, J., Goeppert, A., Czaun, M., Olah, G. A., and Prakash, G. K. S. (2016b). CO2 capture by amines in aqueous media and its subsequent conversion to formate with reusable ruthenium and iron catalysts. Green Chem. 18, 5831–5838. doi:10.1039/c6gc01165a
Kothandaraman, J., Goeppert, A., Czaun, M., Olah, G. A., and Prakash, G. K. (2016a). Conversion of CO2 from air into methanol using a polyamine and a homogeneous ruthenium catalyst. J. Am. Chem. Soc. 138, 778–781. doi:10.1021/jacs.5b12354
Kothandaraman, J., and Heldebrant, D. J. (2020a). Catalytic coproduction of methanol and glycol in one pot from epoxide, CO2, and H2. RSC Adv. 10, 42557–42563. doi:10.1039/d0ra09459e
Kothandaraman, J., and Heldebrant, D. J. (2020b). Towards environmentally benign capture and conversion: heterogeneous metal catalyzed CO2 hydrogenation in CO2 capture solvents. Green Chem. 22, 828–834. doi:10.1039/c9gc03449h
Kothandaraman, J., Lopez, J. S., Jiang, Y., Walter, E. D., Burton, S. D., Dagle, R. A., et al. (2022a). Integrated capture and conversion of CO2 to methanol in a post-combustion capture solvent: heterogeneous catalysts for selective C-N bond cleavage. Adv. Energy Mater. 12, 2202369. doi:10.1002/aenm.202202369
Kothandaraman, J., Lopez, J. S., Jiang, Y., Walter, E. D., Burton, S. D., Dagle, R. A., et al. (2022b). Integrated capture and conversion of CO2 to methanol in a post-combustion capture solvent: heterogeneous catalysts for selective C-N bond cleavage. Adv. Energy Mater. 12, 2202369. doi:10.1002/aenm.202202369
Kothandaraman, J., Saavedra Lopez, J., Jiang, Y., Walter, E. D., Burton, S. D., Dagle, R. A., et al. (2021). Integrated capture and conversion of CO2 to methane using a water-lean, post-combustion CO2 capture solvent. ChemSusChem 14, 4812–4819. doi:10.1002/cssc.202101590
Kothandaraman, J., Zhang, J., Glezakou, V.-A., Mock, M. T., and Heldebrant, D. J. (2019). Chemical transformations of captured CO2 into cyclic and polymeric carbonates. J. CO2 Utili. 32, 196–201. doi:10.1016/j.jcou.2019.04.020
Lao, D. B., Galan, B. R., Linehan, J. C., and Heldebrant, D. J. (2016). The steps of activating a prospective CO2 hydrogenation catalyst with combined CO2 capture and reduction. Green Chem. 18, 4871–4874. doi:10.1039/c6gc01800a
Li, Y. N., He, L. N., Liu, A. H., Lang, X. D., Yang, Z. Z., Yu, B., et al. (2013). In situ hydrogenation of captured CO2 to formate with polyethyleneimine and Rh/monophosphine system. Green Chem. 15, 2825–2829. doi:10.1039/c3gc41265b
Long, Y., He, J., Zhang, H., Chen, Y., Liu, K., Fu, J., et al. (2023). Highly selective monomethylation of amines with CO2/H2 via Ag/Al2O3 as catalyst. Chem. Eur. J.
Mcnamara, N. D., and Hicks, J. C. (2014). CO2 capture and conversion with a multifunctional polyethyleneimine-tethered iminophosphine iridium catalyst/adsorbent. ChemSusChem 7, 1114–1124. doi:10.1002/cssc.201301231
Pavlišič, A., Huš, M., Prašnikar, A., and Likozar, B. (2020). Multiscale modelling of CO2 reduction to methanol over industrial Cu/ZnO/Al2O3 heterogeneous catalyst: linking ab initio surface reaction kinetics with reactor fluid dynamics. J. Clean. Prod. 275, 122958. doi:10.1016/j.jclepro.2020.122958
Porosoff, M. D., Yan, B., and Chen, J. G. (2016). Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: challenges and opportunities. Energy Environ. Sci. 9, 62–73. doi:10.1039/c5ee02657a
Prasnikar, A., Jurkovic, D. L., and Likozar, B. (2021). Reaction path analysis of CO2 reduction to methanol through multisite microkinetic modelling over Cu/ZnO/Al2O3 catalysts. Appl. Catal. B Environ. 292, 120190. doi:10.1016/j.apcatb.2021.120190
Rayder, T. M., Adillon, E. H., Byers, J. A., and Tsung, C.-K. (2020). A bioinspired multicomponent catalytic system for converting carbon dioxide into methanol autocatalytically. Chem 6, 1742–1754. doi:10.1016/j.chempr.2020.04.008
Rezayee, N. M., Huff, C. A., and Sanford, M. S. (2015). Tandem amine and ruthenium-catalyzed hydrogenation of CO2 to methanol. J. Am. Chem. Soc. 137, 1028–1031. doi:10.1021/ja511329m
Sen, R., Goeppert, A., Kar, S., and Prakash, G. K. S. (2020). Hydroxide based integrated CO(2) capture from air and conversion to methanol. J. Am. Chem. Soc. 142, 4544–4549. doi:10.1021/jacs.9b12711
Siegel, R. E., Pattanayak, S., and Berben, L. A. (2023). Reactive capture of CO2: opportunities and challenges. ACS Catal. 13, 766–784. doi:10.1021/acscatal.2c05019
Volkov, A., Tinnis, F., Slagbrand, T., Trillo, P., and Adolfsson, H. (2016). Chemoselective reduction of carboxamides. Chem. Soc. Rev. 45, 6685–6697. doi:10.1039/c6cs00244g
Wei, D., Junge, H., and Beller, M. (2021). An amino acid based system for CO2 capture and catalytic utilization to produce formates. Chem. Sci. 12, 6020–6024. doi:10.1039/d1sc00467k
Wei, D., Sang, R., Moazezbarabadi, A., Junge, H., and Beller, M. (2022). Homogeneous carbon capture and catalytic hydrogenation: toward a chemical hydrogen battery system. JACS Au 2, 1020–1031. doi:10.1021/jacsau.1c00489
Werkmeister, S., Junge, K., and Beller, M. (2014). Catalytic hydrogenation of carboxylic acid esters, amides, and nitriles with homogeneous catalysts. Org. Process Res. Dev. 18, 289–302. doi:10.1021/op4003278
Xie, Y., Hu, P., Bendikov, T., and Milstein, D. (2018). Heterogeneously catalyzed selective hydrogenation of amides to alcohols and amines. Catal. Sci. Technol. 8, 2784–2788. doi:10.1039/c8cy00112j
Zheng, F., Heldebrant, D. J., Mathias, P. M., Koech, P., Bhakta, M., Freeman, C. J., et al. (2016). Bench-scaletesting and process performance projections of CO2Capture by CO2–binding organic liquids (CO2BOLs) with and without polarity-swing-assisted regeneration. Energy fuels 30, 1192–1203. doi:10.1021/acs.energyfuels.5b02437
Keywords: methanol synthesis, hydrogenation, C-N cleavage, N-methylation, catalysis
Citation: Kothandaraman J, Heldebrant DJ, Lopez JS and Dagle RA (2023) Mechanistic insights to drive catalytic hydrogenation of formamide intermediates to methanol via deaminative hydrogenation. Front. Energy Res. 11:1158499. doi: 10.3389/fenrg.2023.1158499
Received: 04 February 2023; Accepted: 02 August 2023;
Published: 25 September 2023.
Edited by:
Melis Seher Duyar, University of Surrey, United KingdomCopyright © 2023 Kothandaraman, Heldebrant, Lopez and Dagle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jotheeswari Kothandaraman, am90aGVlc3dhcmkua290aGFuZGFyYW1hbkBwbm5sLmdvdg==