 Malgorzata N. Drwal1
Malgorzata N. Drwal1 Vishal B. Siramshetty1
Vishal B. Siramshetty1 Priyanka Banerjee1,2
Priyanka Banerjee1,2 Andrean Goede1
Andrean Goede1 Robert Preissner1,3
Robert Preissner1,3 Mathias Dunkel1*
Mathias Dunkel1*- 1Structural Bioinformatics Group, Institute for Physiology, Charité – University Medicine Berlin, Berlin, Germany
- 2Graduate School of Computational Systems Biology, Humboldt-Universität zu Berlin, Berlin, Germany
- 3BB3R – Berlin Brandenburg 3R Graduate School, Freie Universität Berlin, Berlin, Germany
To assess the toxicity of new chemicals and drugs, regulatory agencies require in vivo testing for many toxic endpoints, resulting in millions of animal experiments conducted each year. However, following the Replace, Reduce, Refine (3R) principle, the development and optimization of alternative methods, in particular in silico methods, has been put into focus in the recent years. It is generally acknowledged that the more complex a toxic endpoint, the more difficult it is to model. Therefore, computational toxicology is shifting from modeling general and complex endpoints to the investigation and modeling of pathways of toxicity and the underlying molecular effects. The U.S. Toxicology in the twenty-first century (Tox21) initiative has screened a large library of compounds, including approximately 10K environmental chemicals and drugs, for different mechanisms responsible for eliciting toxic effects, and made the results publicly available. Through the Tox21 Data Challenge, the consortium has established a platform for computational toxicologists to develop and validate their predictive models. Here, we present a fast and successful method for the prediction of different outcomes of the nuclear receptor and stress response pathway screening from the Tox21 Data Challenge 2014. The method is based on the combination of molecular similarity calculations and a naïve Bayes machine learning algorithm and has been implemented as a KNIME pipeline. Molecules are represented as binary vectors consisting of a concatenation of common two-dimensional molecular fingerprint types with topological compound properties. The prediction method has been optimized individually for each modeled target and evaluated in a cross-validation as well as with the independent Tox21 validation set. Our results show that the method can achieve good prediction accuracies and rank among the top algorithms submitted to the prediction challenge, indicating its broad applicability in toxicity prediction.
Introduction
The U.S. Toxicology in the twenty-first century (Tox21) initiative has been established in 2008 with the vision to support the transformation of toxicology into a predictive science (Krewski et al., 2010). In order to achieve this goal, a large library of compounds, including approximately 10K environmental chemicals and drugs, was screened for different mechanisms responsible for eliciting toxic effects. Among the screens were high-throughput assays for two important pathways, the nuclear receptor and the stress response pathway, which were the subject of the Tox21 Data Challenge 2014.
Interactions of chemicals with nuclear receptors represent a major health concern. In particular, binding of chemicals to steroid receptors can cause the disruption of the normal endocrine function and have an adverse effect on development, reproduction and metabolic homeostasis (Huang et al., 2014). A famous example of an endocrine disrupting chemical is bisphenol A, a compound which has been widely used, e.g. in plastic bottles and metal cans, but has only recently been associated with impairments of neurobehavioral development (Weiss, 2012). Bisphenol A and its derivatives have been shown to exhibit a promiscuous binding behavior involving, for instance, estrogen receptors (ER), androgen receptors (AR) and peroxisome proliferator-activated receptors (PPAR) of the γ subtype (Delfosse et al., 2014), all of which are subject of the Tox21 screening. Another current focus of the Tox21 screening is aromatase, an enzyme involved in the conversion of androgen to estrogen and therefore a target of endocrine disrupting chemicals (Chen et al., 2014), as well as the aryl hydrocarbon receptor (AhR), a nuclear receptor involved in the mediation of tumorgenesis induced by dioxin (Murray et al., 2014). Similarly, mechanisms related to cellular stress also play a role in toxicological pathways. For example, recent studies have shown that the impairment of mitochondrial function is associated with drug-induced adverse effects on the liver and cardiovascular system (Nadanaciva and Will, 2011; Attene-Ramos et al., 2015).
To assess the risks of new chemical entities, in vivo animal studies are required by regulatory agencies to evaluate various toxicological endpoints. However, in silico toxicology is gaining acceptance as an alternative method which can help to reduce the number of animal experiments performed. Computational predictions often rely on the observation or assumption that similar molecules manifest a similar biological effect. Similarity-based methods have been successfully applied to solve various research questions including predictions of targets (Campillos et al., 2008), therapeutic indications (Nickel et al., 2014) or side-effects (Lounkine et al., 2012). In particular, machine learning approaches such as k-nearest neighbors, naïve Bayes models, support vector machines, random forests or ensembles of different classification methods can use the similarity defined the molecular structure and properties to make predictions for novel compounds. This concept has also been frequently and successfully applied to predictions of various toxicological endpoints (Drwal et al., 2014; Gadaleta et al., 2014; Li et al., 2014; Liu et al., 2015).
Here, we describe the development of a fast and successful method for the prediction of different outcomes of the nuclear receptor and stress response pathway screening from the Tox21 Data Challenge 2014. The method is based on the combination of a simple molecular similarity calculation with a naïve Bayes machine learning algorithm. Three different two-dimensional (2D) molecular representation methods as well as their combination were compared and the prediction methods were optimized individually for every target. The evaluation of each model showed that all models can achieve good performance and prediction accuracies as well as rank among the top submissions among the Tox21 challenge participants.
Materials and Methods
Overview
An overview of the workflow used in this study is given in Figure 1. In the first step, all molecular structures were standardized and the duplicates as well as compounds with ambiguous activity values were removed. The training and test set provided by the Tox21 Data Challenge 2015 organizers were merged and used in a 13-fold cross-validation to optimize parameters for the classification algorithms. The optimized models were then used to predict the activities of the evaluation set compounds. All steps are described in detail in the following sections. For the majority of tasks, the open pipeline generation platform KNIME v.2.10.0 (Knime.com AG) was used.
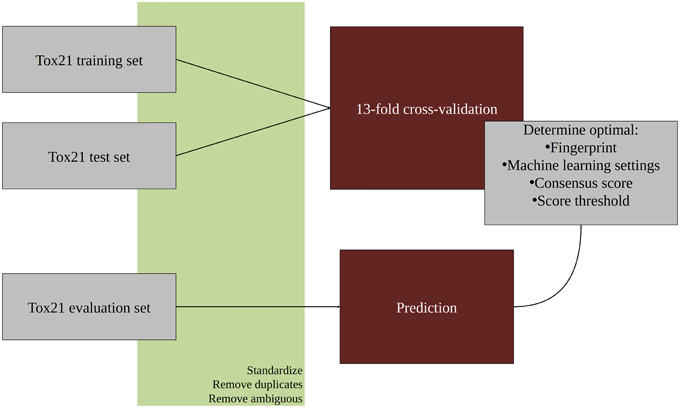
Figure 1. Workflow overview.
Data Preparation
Standardization
All molecular structures were downloaded from the Tox21 Data Challenge 2014 website (https://tripod.nih.gov/tox21/challenge/index.jsp) and their molecular structures were standardized using the Instant JChem software (version 6.2, Chemaxon) with the following settings: Water molecules were removed, molecules were aromatized, adjacent positive and negative charges transformed into double/triple bonds, explicit hydrogens were added and the 3D conformation was generated and cleaned. After the standardization, InChIKeys were calculated using RDKit (http://www.rdkit.org) nodes in KNIME in order to identify and remove duplicates. In case duplicate molecules were found to have different activities (1 and 0) for a particular target, they were marked as ambiguous and removed from the training set of this target.
Additional Data
For each target, a search for additional known ligands was performed in the ChEMBL bioactivity database v.19 (Bento et al., 2014). A search was performed for the target name and EC50 or IC50 values in case of agonists or antagonists, respectively. Additional datasets were standardized and checked for duplicates as described above.
Calculation and Combination of Fingerprints
Different types of molecular representations were calculated for each compound: ToxPrint fingerprints were calculated using the ChemoTyper software (version 1.0, Molecular Networks GmbH). Extended-connectivity fingerprints (Rogers and Hahn, 2010) of the ECFP4 type were calculated using RDKit nodes in KNIME. 960-bit MACCS keys were calculated using the Discovery Studio 3.1 program (Accelrys Inc./BIOVIA). In addition, several topological properties indicating the three-dimensional (3D) structure were calculated using RDKit and CDK nodes in KNIME. The use of topological descriptors has been previously reported in a structure-toxicity relationship study (Pasha et al., 2009). Furthermore, topological descriptors have several advantages compared to 3D descriptors, including conformational independency, simplicity and low computational resources. A number of topological descriptors were calculated, but only those displaying values with considerable difference between active and inactive molecules were used further. These included the Chi0V, Chi1N, Kappa1 and HallKierAlpha descriptors (Hall and Kier, 1991) as well as the topological polar surface area. The descriptors were transformed into a binary vector by binning. For each descriptor, a number of “bins” (and bits in the fingerprint) was defined, representing different descriptor value ranges. Whenever the descriptor value was found in a specific range, the bit at the respective position was set to 1. Therefore, it was ensured that close values exhibited high fingerprint similarity. The combined fingerprint consisted of a concatenation of all four binary fingerprints with a length of 2929 bits—960 bits for MACCS keys, 1024 bits for ECFP4, 729 bits for ToxPrint and 216 bits for the property-based fingerprint, as indicated in Figure 2.
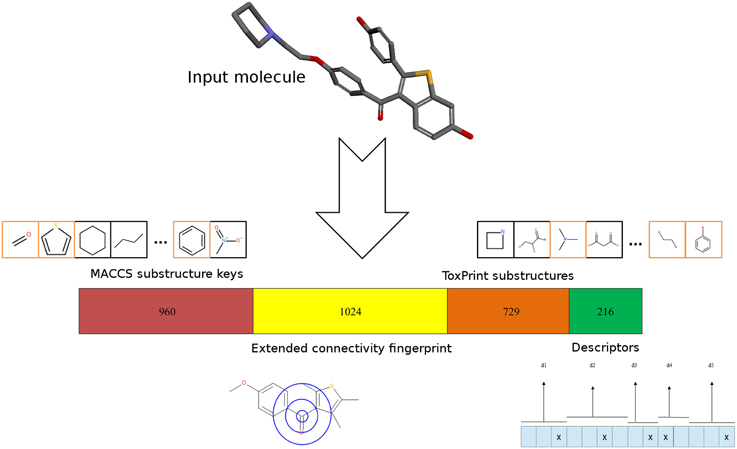
Figure 2. Molecular representation. For every input molecule from the Tox21 data set, different 2D-fingerprints are calculated and combined. The concatenation consists of MACCS keys (960 bits), the extended-connectivity fingerprint ECFP4 (1024 bits), ToxPrint (729 bits) and a fingerprint developed from topological descriptors (216 bits). Both MACCS as well as ToxPrint fingerprints encode the presence of specific substructures. Examples of MACCS and ToxPrint substructures are shown in boxes. Substructures present in a sample molecule taken from the Tox21 dataset are highlighted in orange boxes. ECFP4 encodes the connections of each atom within a 4-atom radius. The property-fingerprint encodes the presence of descriptor values in specific bins representing value ranges.
Toxicity Prediction Methods
Cross-validation
In order to validate the prediction models, a 13-fold cross-validation was implemented in KNIME. The KNIME workflows are presented in Supplementary Figures S2, S3. A 13-fold validation was chosen in order to produce a test set similar in size to the final validation set of the Tox21 challenge. It was investigated whether the addition of external data (known ligands from the ChEMBL database, see Section Additional data) was able to improve the prediction rate. Different activity cut-offs for the ChEMBL compounds were considered for this purpose. Furthermore, it was also investigated whether reducing the actives in the training set to the most diverse compounds was able to increase the performance of the model. In this case, the RDKit Diversity Picker node was used using different thresholds. Finally, the effect of the removal of highly correlated fingerprint bits on the model performance was explored using the Correlation Filter node. To determine the best settings, the performance was evaluated using a receiver operating characteristic (ROC) analysis. The area under the curve (AUC) was calculated using the ROC curve node.
Naïve Bayes Learning
Naïve Bayes is a commonly applied stochastic classifier based on the Bayes theorem of conditional probability (Nidhi et al., 2006). The major characteristic of the classifier is the naïve assumption that all input features are independent. Main advantages of the method compared to other machine learning algorithms are fast computational time during training and prediction as well as a low parameter complexity and insusceptibility to irrelevant features. Furthermore, it has been suggested that the combination of molecular fingerprints with descriptors can be beneficial in the context of Bayesian modeling (Vogt and Bajorath, 2008).
Thus, we implemented a naïve Bayes predictor with the Tox21 training sets. The Fingerprint Bayesian Learner and Predictor nodes in KNIME were used for this purpose. The predictor received an input of active and inactive molecules and their fingerprints. The output consisted of two scores for each molecule, a score for being active (B1) and a score for being inactive (B0).
Molecular Similarity
The Tanimoto index is one of the most common metrics for fingerprint-based molecular similarity calculations and has recently been shown to be among the best choices for this purpose (Bajusz et al., 2015). For the comparison of molecular similarity, three Tanimoto coefficients were computed: the maximum Tanimoto coefficient to actives in the training set (T1), the average Tanimoto coefficient to actives in the training set (T2), and the maximum Tanimoto coefficient to all inactives in the training set (T3).
Combination of Methods
All scores and Tanimoto coefficients were normalized in KNIME using Z-score normalization to obtain scores following a Gaussian distribution and MinMax-normalization to obtain values between 0 and 1. Different combinations of the naïve Bayes scores B1 and (1-B0) as well as the Tanimoto scores T1, T2 and (1-T3) were examined, including the minimum, maximum and mean of the scores.
Determination of Score Threshold
For every target, a threshold of the final score was determined which was used to classify the compounds into active and inactive molecules. The score threshold was determined by choosing the threshold which resulted in the maximal balanced accuracy ((sensitivity+specificity)/2) over all rounds of cross-validation.
Results
The Tox21 Data Challenge 2014 consisted of the prediction of 12 different screening outcomes (targets): the activation or inhibition of nuclear receptors AhR, PPARγ, aromatase, ER and AR (full length and ligand binding domain, LBD) as well as the effect on stress response pathways consisting of the activation of the antioxidant response element (ARE), heat shock response (HSE) and p53 signaling, the disruption of mitochondrial membrane potential (MMP) and the induction of genotoxicity (ATAD5). Before building predictive models, all chemical structures were normalized as described in the Methods section and duplicates were removed. Only compounds explicitly marked as active or inactive were used for model development. Wherever available, additional active molecules were extracted from the ChEMBL database (Bento et al., 2014) and used for model development. As summarized in Supplementary Table S1, the proportion of unique active and inactive molecules as well as the presence of external actives differed considerably between targets.
Choice of Molecular Representation
How well a prediction model performs does not only depend on the underlying algorithm, but also the features used as input. In the case of predictions of small molecule toxicities and other biological activities, the performance thus depends on the molecular representation which ultimately influences the computed similarity between molecules (Floris et al., 2014). Here we compared the performance of three common molecular fingerprints as well as their combination. ECFP4 is a member of the extended-connectivity fingerprint type often used to analyze structure-activity relationships of small molecules (Rogers and Hahn, 2010). MACCS keys are another frequently used fingerprint type which encodes the presence of specific substructures and has been successfully used for predictions of acute oral toxicity (Li et al., 2014). The ToxPrint fingerprint (Yang et al., 2015a) is based on a library of more than 700 chemotypes which represent molecules in public chemical and toxicity databases and cover substructures associated with toxic effects and thus may be of particular importance for in silico toxicity predictions. We also evaluated the addition of a property-based fingerprint as has been suggested previously (Xue et al., 2003). Here, descriptors encoding the topology of the Tox21 compounds were calculated and translated into a binary fingerprint.
In order to determine the optimal fingerprint for the prediction, fingerprints were used individually as well as in combination and evaluated in cross-validation on one of the targets, namely ER-LBD. As summarized in Table 1, all three types of fingerprints showed a good performance using both the Bayesian classifier as well as the similarity search approach. In the majority of cases models built with individual fingerprints exhibited AUC values above 0.75 and a concatenation of all three fingerprints led to a slight increase in performance. Furthermore, a combination of the concatenated fingerprints with a property-based fingerprint encoding the topology of the molecules demonstrated the best prediction results and was thus used as a descriptor for all targets of the challenge.
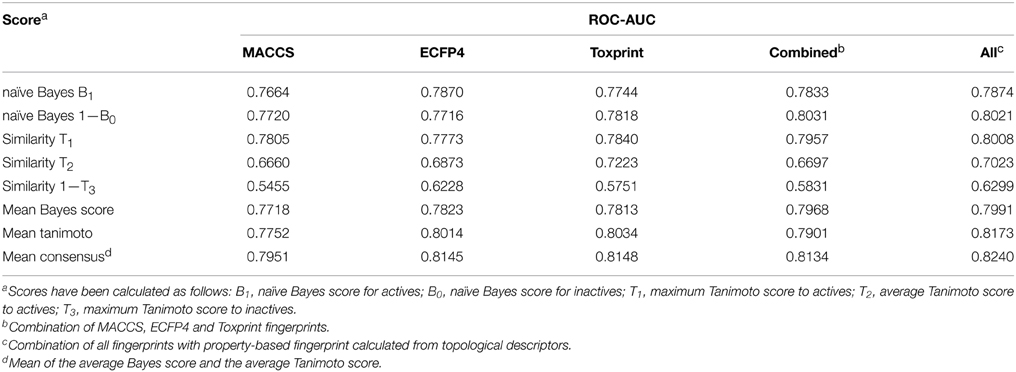
Table 1. Performance of different fingerprints in cross-validation of predictions for ER-LBD.
Model Optimization and Validation
In the preliminary evaluation of descriptors for ER-LBD, a common observation was that a consensus score consisting of a machine learning score and a similarity coefficient usually resulted in the best model performance (Table 1). Therefore, it was investigated which combination of scores led to the best prediction. In particular, the scores from the Bayesian classifier and the similarity search were combined into a consensus score using either a mean, maximum or minimum value. Since the optimal settings might differ depending on the target and its active and inactive molecules, the best parameters were determined individually for every target in a cross-validation study. The optimization involved the variation of the following parameters: the addition of active molecules from external sources (ChEMBL database) using different activity value thresholds, the addition of a correlation filter to remove highly correlated fingerprint features as well as the incorporation of a diversity picker to restrict the number of active to train a naïve Bayes model to the ones with highest diversity.
The best settings found for every Tox21 target are shown in Table 2. As indicated, similarity search gave the best performance for 4/12 targets when an average Tanimoto was calculated from the T1, T2, and (1-T3) scores indicating the similarity to active as well as the dissimilarity to inactive molecules (see Methods). For all other targets, a combination of the machine learning algorithm and a similarity scoring showed the best results. In most cases, a mean function was used to generate a consensus score combining the naïve Bayes and Tanimoto coefficients.
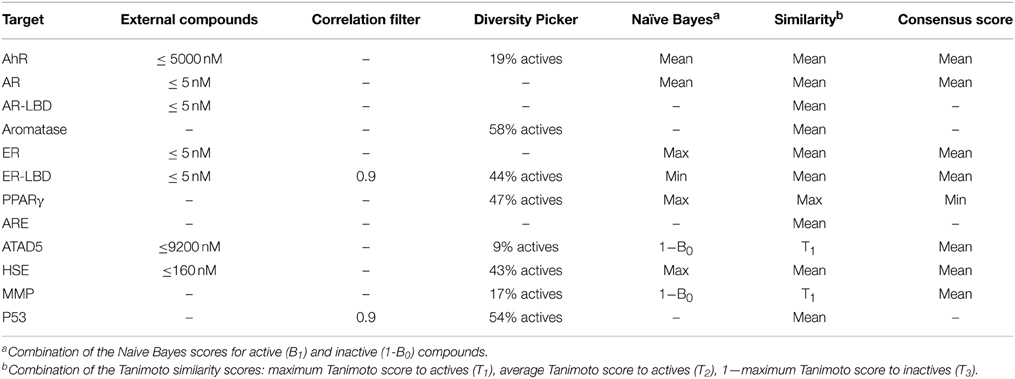
Table 2. Parameters of the most successful prediction models.
The performance of each model was evaluated using ROC-AUC values as well as balanced accuracies. The cross-validation results for the best settings as well as the external validation results provided by the challenge organizers are summarized in Figure 3. In cross-validation, all models exhibited excellent performance with AUC values between 0.78 and 0.9, with the best three models obtained for the targets AhR, AR-LBD, and MMP. For AhR, MMP, and p53, the results of the external validation set showed a very similar performance to the cross-validation, indicating good and universal models and scores. In the cross-validation, the balanced accuracies of the individual models ranged between 70 and 82% (see Figure 3). For several targets, including AhR, HSE, and p53, the balanced accuracy obtained in external validation remained constant or increased in comparison to the cross-validation results, illustrating broadly applicable models.
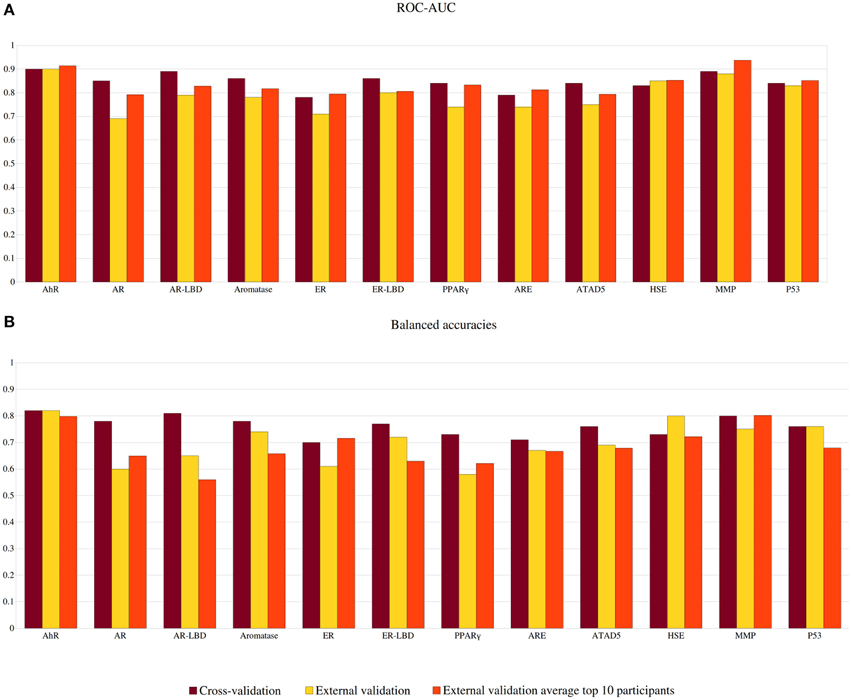
Figure 3. Performance of models predicting the outcome of the Tox21 screening outcomes. (A) Area under the curve (AUC) calculated in a ROC analysis. (B) Balanced accuracies (BAC). Results are shown for our models in cross-validation (dark red) and external validation (yellow) as well as the average external validation results among the top 10 challenge participants.
Comparison to Other Challenge Participants
All models submitted to the challenge were evaluated by the challenge organizers and ranked according to their AUC values for the external validation set. The prediction values for the top 10 participating teams are publicly available (https://tripod.nih.gov/tox21/challenge/leaderboard.jsp) and summarized in Figure 3, Supplementary Tables S1, S1. Taken together, 7 out of 12 models we submitted were found in the top 10 leaderboard. While our models were not nominated as the sub-challenge winners, in many cases their AUC value was found very close to the winning model. This was for instance observed for the target HSE, where the top 9 ranking models showed AUC values differing only by 0.02, suggesting that similarly good models can be obtained with various approaches. As indicated in Figure 3, our models for the targets AhR, ER-LBD and p53 were also very close to the average AUC of the leading models. Although most leaderboard models showed AUC values within a small range, large differences were observed for the prediction accuracies (between 49 and 90%). Interestingly, four of our models (targets: AR-LBD, ER-LBD, aromatase, and HSE) were the determined to be the most accurate amongst all submissions (see Figure 3 and Supplementary Table S1). Four additional models, developed for the targets AhR, ARE, ATAD5, and p53, displayed accuracies higher or equal to the average of the top 10 submitted models.
Discussion
Here, we describe a successful machine learning method for the prediction of different outcomes of the nuclear receptor and stress response pathway screening from the Tox21 Data Challenge 2014. The key to our method is the combination of different molecular fingerprints and descriptors as well as the integration of two different algorithms, a similarity-based approach and a naïve Bayes machine learning technique.
Combination of Features and Algorithms
The selection of features is a crucial and non-trivial part of development of predictive models. The features should be able to describe the differences between actives and inactives in the training set and allow extrapolating to other, yet untested compounds. Although several molecular fingerprints, such as extended-connectivity, substructure-based or path-based fingerprints are standards in the chemoinformatics field and have been successfully applied to prediction tasks, the results are dependent on the data and none of the methods is able to clearly outperform the others (Duan et al., 2010). To avoid the choice of the wrong descriptor, the combination of (independent) fingerprints has been suggested (Duan et al., 2010) and several studies have successfully applied combinations of path- and substructure-based fingerprints (Drwal et al., 2014; Banerjee et al., 2015). As we report here, the combination of different fingerprint types has also been of advantage for the prediction of estrogen receptor ligands. An associated problem, however, is that a combined fingerprint is likely to contain highly correlated features. We have thus investigated the use of a correlation filter to remove fingerprint bits with high correlation, but the filter was able to increase the prediction performance only for two targets. A more effective approach proved to be the use of a diverse subset of active molecules in the training set, though the size of the diverse subset giving the best results had to be optimized individually for every target. As the active molecules of the different Tox21 sub-challenges might contain different important molecular characteristics, the use of extensive cross-validation to optimize the feature selection for every sub-challenge could further improve the prediction performance. Automated feature selection using deep neural networks, as suggested by one of the other teams participating in the Tox21 challenge (Unterthiner et al., 2015), offers an alternative way to determine the most relevant features in the input molecules which can be advantageous for large sets of molecules, but is obviously associated with large computational costs.
Combinations of multiple machine learning algorithms, also referred to as hybrid or ensemble learning, are a well-described approach and have been applied to solve diverse research questions (Yang et al., 2015b). It is usually assumed that the use of multiple models can increase the prediction accuracy as compared to the use of a single model and help to manage high-dimensional and complex data sets. Similarly to our approach, several other studies have proven that merging a naïve Bayes classifier with a similarity-based approach such as k-nearest neighbors can result in highly predictive models for various applications including the prediction of molecular targets (Ferdousy et al., 2013; Liu et al., 2013). Future investigations could focus on the evaluation of other classification methods (logistic regression, random forests, etc.) and larger model ensembles for the purposes of toxicity prediction.
Conclusions
Our models use a combination of molecular fingerprints and algorithms and show consistently good performance for the 12 outcomes of the Tox21 screen, four of the models being the most accurate amongst the challenge participants. We are planning to make our models publicly available by incorporating them into our toxicity prediction platform ProTox (http://tox.charite.de) in the future.
The Tox21 Data Challenge 2014 has provided an excellent opportunity for academic and industrial groups to assess and directly compare the quality of their toxicity prediction methods. The results will be of great value to the scientific community and can help to pave the way toward the use of more in silico toxicity models as decision-making tools to evaluate potential health hazards of environmental chemicals and drugs.
Author Contributions
Data preparation and analysis: MND, VS, PB, MD, AG; Generation and validation of predictive models: MD, MND, VS, PB; Calculation and selection of descriptors: PB, VS, MD, MND; Writing of manuscript: MND; Project coordination: RP, MD, MND.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors kindly acknowledge the following funding sources: German Cancer Consortium (DKTK); Berlin-Brandenburg research platform BB3R (BMBF) [031A262C]; Immunotox project (BMBF) [031A268B]; research training group “Computational Systems Biology” [GRK1772].
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fenvs.2015.00054
Abbreviations
2D, two-dimensional; AhR, aryl hydrocarbon receptor; AR, androgen receptor; ARE, antioxidant response element; ATAD5, genotoxicity induction; AUC, area under the curve; BAC, balanced accuracy; ER, estrogen receptor 1; HSE, heat shock response; LBD, ligand binding domain; MMP, mitochondrial membrane potential; PPAR, peroxisome proliferator-activated receptor; ROC, receiver operating characteristic; Tox21, U.S. Toxicology in the twenty-first century initiative.
References
Attene-Ramos, M. S., Huang, R., Michael, S., Witt, K. L., Richard, A., Tice, R. R., et al. (2015). Profiling of the Tox21 chemical collection for mitochondrial function to identify compounds that acutely decrease mitochondrial membrane potential. Environ. Health Perspect. 123, 49–56. doi: 10.1289/ehp.1408642
Bajusz, D., Rácz, A., and Héberger, K. (2015). Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 7:20. doi: 10.1186/s13321-015-0069-3
Banerjee, P., Erehman, J., Gohlke, B. O., Wilhelm, T., Preissner, R., and Dunkel, M. (2015). Super Natural II–a database of natural products. Nucleic Acids Res. 43, D935–D939. doi: 10.1093/nar/gku886
Bento, A. P., Gaulton, A., Hersey, A., Bellis, L. J., Chambers, J., Davies, M., et al. (2014). The ChEMBL bioactivity database: an update. Nucleic Acids Res. 42, D1083–D1090. doi: 10.1093/nar/gkt1031
Campillos, M., Kuhn, M., Gavin, A. C., Jensen, L. J., and Bork, P. (2008). Drug target identification using side-effect similarity. Science 321, 263–266. doi: 10.1126/science.1158140
Chen, S., Zhou, D., Hsin, L. Y., Kanaya, N., Wong, C., Yip, R., et al. (2014). AroER tri-screen is a biologically relevant assay for endocrine disrupting chemicals modulating the activity of aromatase and/or the estrogen receptor. Toxicol. Sci. 139, 198–209. doi: 10.1093/toxsci/kfu023
Delfosse, V., Grimaldi, M., Le Maire, A., Bourguet, W., and Balaguer, P. (2014). Nuclear receptor profiling of bisphenol-A and its halogenated analogues. Vitam. Horm. 94, 229–251. doi: 10.1016/B978-0-12-800095-3.00009-2
Drwal, M. N., Banerjee, P., Dunkel, M., Wettig, M. R., and Preissner, R. (2014). ProTox: a web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 42, W53–W58. doi: 10.1093/nar/gku401
Duan, J., Dixon, S. L., Lowrie, J. F., and Sherman, W. (2010). Analysis and comparison of 2D fingerprints: insights into database screening performance using eight fingerprint methods. J. Mol. Graph. Model. 29, 157–170. doi: 10.1016/j.jmgm.2010.05.008
Ferdousy, E. Z., Islam, M., and Matin, M. (2013). Combination of naive bayes classifier and K-Nearest Neighbor (cNK) in the classification based predictive models. Comput. Inf. Sci. 6, 48. doi: 10.5539/cis.v6n3p48
Floris, M., Manganaro, A., Nicolotti, O., Medda, R., Mangiatordi, G. F., and Benfenati, E. (2014). A generalizable definition of chemical similarity for read-across. J. Cheminform. 6, 39. doi: 10.1186/s13321-014-0039-1
Gadaleta, D., Pizzo, F., Lombardo, A., Carotti, A., Escher, S. E., Nicolotti, O., et al. (2014). A k-NN algorithm for predicting the oral sub-chronic toxicity in the rat. ALTEX 31, 423–432. doi: 10.14573/altex.1405091s
Hall, L. H., and Kier, L. B. (1991). “The molecular connectivity chi indexes and kappa shape indexes in structure-property modeling,” in Reviews in Computational Chemistry, eds K. B. Lipkowitz and D. B. Boyd (Hoboken, NJ: John Wiley & Sons, Inc.), 367–422.
Huang, R., Sakamuru, S., Martin, M. T., Reif, D. M., Judson, R. S., Houck, K. A., et al. (2014). Profiling of the Tox21 10K compound library for agonists and antagonists of the estrogen receptor alpha signaling pathway. Sci. Rep. 4:5664. doi: 10.1038/srep05664
Krewski, D., Acosta, D. Jr. Andersen, M., Anderson, H., Bailar, J. C. 3rd Boekelheide, K., et al. (2010). Toxicity testing in the 21st century: a vision and a strategy. J. Toxicol. Environ. Health B Crit. Rev. 13, 51–138. doi: 10.1080/10937404.2010.483176
Li, X., Chen, L., Cheng, F., Wu, Z., Bian, H., Xu, C., et al. (2014). In silico prediction of chemical acute oral toxicity using multi-classification methods. J. Chem. Inf. Model. 54, 1061–1069. doi: 10.1021/ci5000467
Liu, J., Mansouri, K., Judson, R. S., Martin, M. T., Hong, H., Chen, M., et al. (2015). predicting hepatotoxicity using toxcast in vitro bioactivity and chemical structure. Chem. Res. Toxicol. 28, 738–751. doi: 10.1021/tx500501h
Liu, X., Vogt, I., Haque, T., and Campillos, M. (2013). HitPick: a web server for hit identification and target prediction of chemical screenings. Bioinformatics 29, 1910–1912. doi: 10.1093/bioinformatics/btt303
Lounkine, E., Keiser, M. J., Whitebread, S., Mikhailov, D., Hamon, J., Jenkins, J. L., et al. (2012). Large-scale prediction and testing of drug activity on side-effect targets. Nature 486, 361–367. doi: 10.1038/nature11159
Murray, I. A., Patterson, A. D., and Perdew, G. H. (2014). Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat. Rev. Cancer 14, 801–814. doi: 10.1038/nrc3846
Nadanaciva, S., and Will, Y. (2011). Investigating mitochondrial dysfunction to increase drug safety in the pharmaceutical industry. Curr. Drug Targets 12, 774–782. doi: 10.2174/138945011795528985
Nickel, J., Gohlke, B. O., Erehman, J., Banerjee, P., Rong, W. W., Goede, A., et al. (2014). SuperPred: update on drug classification and target prediction. Nucleic Acids Res. 42, W26–W31. doi: 10.1093/nar/gku477
Nidhi, Glick, M., Davies, J. W., and Jenkins, J. L. (2006). Prediction of biological targets for compounds using multiple-category Bayesian models trained on chemogenomics databases. J. Chem. Inf. Model. 46, 1124–1133. doi: 10.1021/ci060003g
Pasha, F. A., Neaz, M. M., Cho, S. J., Ansari, M., Mishra, S. K., and Tiwari, S. (2009). In silico quantitative structure-toxicity relationship study of aromatic nitro compounds. Chem. Biol. Drug Des. 73, 537–544. doi: 10.1111/j.1747-0285.2009.00799.x
Rogers, D., and Hahn, M. (2010). Extended-connectivity fingerprints. J. Chem. Inf. Model. 50, 742–754. doi: 10.1021/ci100050t
Unterthiner, T., Mayr, A., Klambauer, G., and Hochreiter, S. (2015). Toxicity Prediction Using Deep Learning. Machine Learning. Available online at:http://arxiv.org/abs/1503.01445
Vogt, M., and Bajorath, J. (2008). Bayesian screening for active compounds in high-dimensional chemical spaces combining property descriptors and molecular fingerprints. Chem. Biol. Drug Des. 71, 8–14. doi: 10.1111/j.1747-0285.2007.00602.x
Weiss, B. (2012). The intersection of neurotoxicology and endocrine disruption. Neurotoxicology 33, 1410–1419. doi: 10.1016/j.neuro.2012.05.014
Xue, L., Godden, J. W., Stahura, F. L., and Bajorath, J. (2003). Design and evaluation of a molecular fingerprint involving the transformation of property descriptor values into a binary classification scheme. J. Chem. Inf. Comput. Sci. 43, 1151–1157. doi: 10.1021/ci030285+
Yang, C. H., Tarkhov, A., Marusczyk, J., Bienfait, B., Gasteiger, J., Kleinoeder, T., et al. (2015a). New publicly available chemical query language, csrml, to support chemotype representations for application to data mining and modeling. J. Chem. Inf. Model. 55, 510–528. doi: 10.1021/ci500667v
Keywords: molecular fingerprints, molecular similarity, machine learning, toxicity prediction, Tox21 Data Challenge 2014
Citation: Drwal MN, Siramshetty VB, Banerjee P, Goede A, Preissner R and Dunkel M (2015) Molecular similarity-based predictions of the Tox21 screening outcome. Front. Environ. Sci. 3:54. doi: 10.3389/fenvs.2015.00054
Received: 04 May 2015; Accepted: 14 July 2015;
Published: 30 July 2015.
Edited by:
Ruili Huang, NIH National Center for Advancing Translational Sciences, USAReviewed by:
Luis Gomez, University of Las Palmas de Gran Canaria, SpainIjaz Hussain, Quaid-i-Azam University, Islamabad, Pakistan
Copyright © 2015 Drwal, Siramshetty, Banerjee, Goede, Preissner and Dunkel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mathias Dunkel, Structural Bioinformatics Group, Institute for Physiology, Charité – University Medicine Berlin, Lindenberger Weg 80, 13125 Berlin, Germany,bWF0aGlhcy5kdW5rZWxAY2hhcml0ZS5kZQ==