 Matthew J. Grisewood
Matthew J. Grisewood James G. Ferry
James G. Ferry Costas D. Maranas
Costas D. Maranas- 1Department of Chemical Engineering, Pennsylvania State University, University Park, PA, United States
- 2Department of Biochemistry and Molecular Biology, Pennsylvania State University, University Park, PA, United States
The anaerobic oxidation of methane (AOM) by methanotrophic archaea offers a carbon- and electron- efficient route for the production of acetate, which can be further processed to yield liquid fuels. This acetate production pathway is initiated by methyl-coenzyme M reductase, but this enzyme can only oxidize trace amounts of methane ex situ. Efforts to improve the kinetics of methyl-coenzyme M reductase through enzyme engineering have been, in part, limited by low-throughput assays. Computational enzyme engineering can circumvent this limitation through the design of smaller, more focused libraries, which have a higher probability of success. By drawing from a new consensus reaction mechanism for Mcr and newly published data, the first complete kinetic characterization of the Mcr reaction mechanism is proposed. In the developed kinetic description, the rate of methyl-coenzyme M unbinding is proposed to limit Mcr overall kinetics. A revised computational method was devised to improve the rate of product release while not disrupting the reaction's activated complex. Large, hydrophobic amino acids that can assume multiple conformations were predicted to be most effective at reaching this design goal. Other rate-limiting scenarios were examined, such as (i) high-temperature (>45°C), (ii) methyltransferase-limiting, and (iii) ineffective cofactor F430 binding. A separate library of designs is put forth for each one of these cases. These efforts mark the first computational attempt at redesigning methyl-coenzyme M reductase for reversed or improved activity, which if experimentally validated, would have a cross-cutting impact across the biotechnology and biochemistry fields by debottlenecking anaerobic methane oxidation.
Introduction
Each year, marine sediments oxidize an estimated 70–300 teragrams of methane (Reeburgh, 1996; Hinrichs and Boetius, 2003) to form carbon dioxide (Knittel and Boetius, 2009), which is about 21 times less effective at trapping heat in the atmosphere (Ragsdale et al., 2017). Industrially mimicking this natural process enables the possibility of converting methane to liquid fuels in an efficient and environmentally friendly manner (Haynes and Gonzalez, 2014). Consortia of ANaerobic MEthanotrophic archaea (ANME) and sulfate-reducing bacteria are responsible for the anaerobic oxidation of methane (AOM) in these sediments (Knittel and Boetius, 2009; Shima et al., 2012). The key enzyme for methane activation is a homolog of methyl-coenzyme M reductase (Mcr)—an enzyme that catalyzes anaerobic methanogenesis—that runs in reverse (Hoehler et al., 1994; Krüger et al., 2003; Hallam et al., 2004; Moran et al., 2005; Scheller et al., 2010; Shima et al., 2012). ANME Mcr couples the endergonic oxidation of methane to methyl-coenzyme M (CH3-S-CoM) with the reduction of coenzyme M-coenzyme B heterodisulfide (CoM-S-S-CoB, HDS) to coenzyme B (HS-CoB, see Equation 1; Harmer et al., 2008; Thauer, 2011).
The ANME in marine sediments directly donate electrons to their syntrophic sulfate-reducing bacteria partner via nanowire-like structures (McGlynn et al., 2015; Wegener et al., 2015; Scheller et al., 2016). This electron transfer yields a thermodynamically favorable net reaction (Equation 2; Thauer, 2011), but this free energy change is unlikely to support growth of both organisms (Thauer and Shima, 2008).
Alternative electron acceptors [such as iron (III), manganese (IV), chromium (VI), and nitrate] are more energetically favorable than sulfate (Beal et al., 2009; Haroon et al., 2013; Mueller et al., 2015; Lu et al., 2016; Nazem-Bokaee et al., 2016; Soo et al., 2016) and can ensure thermodynamic feasibility of AOM.
Methanogenic Mcrs are (αβγ)2 hexamers that include two highly conserved active sites, where the nickel-containing cyclic tetrapyrrole prosthetic group known as cofactor F430 is non-covalently bound (Ermler et al., 1997; Grabarse et al., 2000). For methanogenesis, CH3-S-CoM must bind prior to HS-CoB to form a ternary complex (Wongnate and Ragsdale, 2015), and the nickel of cofactor F430 must be present in the Ni(I) state (Goubeaud et al., 1997). The ordered binding for Mcr is facilitated through numerous important enzyme conformational changes (Grabarse et al., 2001; Cedervall et al., 2010; Ebner et al., 2010). Mcr methanogenesis is initiated through homolytic cleavage of CH3-S-CoM to yield methyl radical and Ni(II)-thiolate intermediates (Chen et al., 2012; Scheller et al., 2013; Wongnate et al., 2016). This radical mechanism is also feasible for AOM, consistent with the “reverse methanogenesis” hypothesis (Krüger et al., 2003; Hallam et al., 2004; Moran et al., 2005, 2007; Nauhaus et al., 2005; Heller et al., 2008; Knittel and Boetius, 2009; Scheller et al., 2010; Chen et al., 2012; Wongnate et al., 2016). Though trace AOM in methanogens has been demonstrated (Moran et al., 2005, 2007; Scheller et al., 2010), the reported specific AOM rate of a methanogenic Mcr was 7-fold lower than that of ANME Mcr (Scheller et al., 2010). This is consistent with Mcr limiting overall AOM kinetics. By improving the activity of Mcr, the economics for the carbon- and energy-efficient bioconversion of methane to liquid fuels becomes more propitious (Haynes and Gonzalez, 2014).
Improving enzyme activity is typically attained through directed evolution approaches that mandate high-throughput screening of large variant libraries (Bloom et al., 2005; Packer and Liu, 2015). High-throughput screening is streamlined through the use of a simple assay, such as a chromogenic or fluorogenic substrate or sensor (see Xiao et al., 2015 for review). Such a simple assay for AOM by Mcr does not currently exist. AOM Mcr activity has only been monitored using limited throughput techniques that include isotopic labeling (Moran et al., 2005, 2007; Scheller et al., 2010; Soo et al., 2016) and cell growth on methane (Soo et al., 2016). The complexity of the Mcr system—exemplified by its many post-translational and cofactor modifications (Ermler et al., 1997; Grabarse et al., 2000; Shima et al., 2012; Allen et al., 2014), cofactor synthesis (Zheng et al., 2016), and oxidative inactivation (Goubeaud et al., 1997)—makes it extremely challenging to study in vitro and thus further limits the gamut of available assays. These limitations motivate the use of rational approaches to design small, focused libraries with a higher likelihood of success. Various rational approaches, such as site saturation mutagenesis or manual rational design, are also inapt for Mcr because of its complex chemistry and the inability to focus on one or two variable positions.
A robust approach to rationally engineer Mcr for improved catalytic activity is computational enzyme redesign. Several types of computational procedures have been successfully deployed for enzyme engineering, including de novo (Jiang et al., 2008; Röthlisberger et al., 2008; Faiella et al., 2009; Siegel et al., 2010; Richter et al., 2012; Garrabou et al., 2016), structure-based (Ashworth et al., 2006; Murphy et al., 2009; Grisewood et al., 2017), and sequence-based approaches (Moore and Maranas, 2004; Meyer et al., 2006; Pantazes et al., 2007) (see refs. Pantazes et al., 2011; Hilvert, 2013; Huang et al., 2016 for review). Structure-based redesign is best suited for the aim of improving AOM activity because Mcr, which has a known structure (Shima et al., 2012), naturally catalyzes this reaction (Moran et al., 2005, 2007; Scheller et al., 2010). The Iterative Protein Redesign and Optimization procedure (IPRO) is a structure-based protein redesign tool that incorporates (step 1) recursive random backbone perturbations, (step 2) deterministic rotational isomer (i.e., rotamer) optimizations, and structural refinements to improve enzyme performance toward a specific target (Saraf et al., 2006; Fazelinia et al., 2007; Grisewood et al., 2013; Pantazes et al., 2015). These structural refinements include (step 3) local ligand docking and (step 4) a force field energy minimization. Designs then have (step 5) their interaction energies with various ligands calculated, and based on these calculated energies, (step 6) the variant is accepted or rejected using the Metropolis criteria. IPRO offers key advantages over other available structure-based redesign procedures in that it can (i) handle multiple design criteria simultaneously, (ii) be easily manipulated for a problem-specific objective function, and (iii) maintain the geometry of catalytic residues using distance restraints.
In this work, we investigated the limiting steps for AOM kinetics and developed multiple case studies under which different steps may be limiting. For each of these case studies, Methanosarcina acetivorans serves as the host system because ANME methanotrophs have not yet been isolated (Scheller et al., 2010; Haynes and Gonzalez, 2014). M. acetivorans is phylogenetically closely related to ANME-2 (a specific clade of ANME; Mueller et al., 2015) archaea (Moran et al., 2007; Yan et al., 2018). The goal of improving AOM kinetics was subdivided into four separate case studies. The first case study (CS1) considered redesigning ANME-1 Mcr to accept the cofactor F430 found in methanogenic archaea in lieu of its native cofactor (Mayr et al., 2008; Shima et al., 2012). In a second case study (CS2), we considered that Mcr may be limited by formation of the methyl radical at high temperatures (>45°C; Wongnate et al., 2016). In the third study (CS3), published Mcr binding and reaction rates were used to postulate that Mcr kinetics is limited by CH3-S-CoM unbinding (Ellermann et al., 1988; Scheller et al., 2010; Chen et al., 2012; Wongnate and Ragsdale, 2015; Wongnate et al., 2016). A final investigation (CS4) examined the possibility that AOM is limited by the second step of the reverse aceticlastic pathway involving a methyl-tetrahydrosarcinapterin:coenzyme M (CH3-H4SPT:HS-CoM) methyltransferase (Mtr) (Benedict et al., 2012; Vepachedu and Ferry, 2012; Nazem-Bokaee et al., 2016). An overview of these case studies is provided in Figure 1.
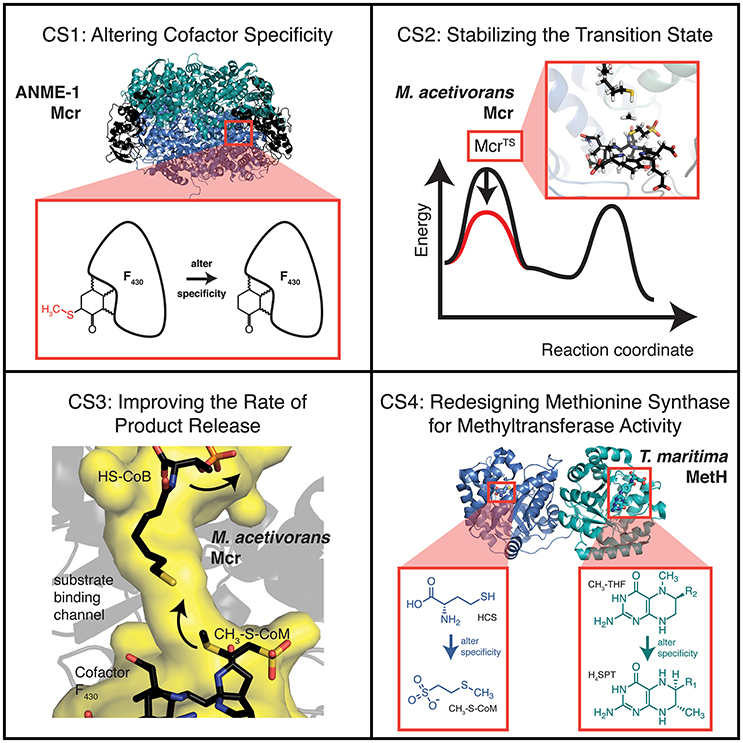
Figure 1. Overview of the various case studies examined for improving AOM activity. Substrate, intermediate, and transition state energies for the free energy diagram shown for CS2 are taken from density functional theory calculations (Chen et al., 2012). The substrate abbreviations for CS4 are homocysteine (HCS), methyl-coenzyme M (CH3-S-CoM), 5-methyltetrahydrofolate (CH3-THF), and tetrahydrosarcinapterin (H4SPT).
Results and Discussion
For each case study, we constructed enzyme variant libraries to alleviate its particular kinetic limitation. Execution of various case studies is important for improving AOM kinetics because the precise relationship between physical conditions and the rate-limiting step is ill-defined. We describe general trends observed in the variant libraries for each case study and analyze differences between the libraries. The top results for each library are presented in each case.
Case Study 1: Altering Anme-1 Mcr Cofactor Specificity
The recently elucidated structure of ANME-1 Mcr (Shima et al., 2012) revealed key structural differences relative to methanogenic Mcrs. These differences included enriched regions of cysteine residues, a methylthiolation of cofactor F430 at C172, and a distinct set of post-translationally modified amino acids (Shima et al., 2012). While the significance of each of these differences has not been fully resolved, it is reasonable to assume that they arose from evolutionary divergence or fine-tuning of the enzyme for its specific function. It has been suggested that the 172-methylthiolation of cofactor F430 is catalytically non-essential because ANME-2 Mcrs contain the unmodified cofactor (Mayr et al., 2008). Moreover, the importance of Mcr's post-translational modifications has been questioned since many of these adaptations are not conserved (Kahnt et al., 2007). In CS1, the assumption is made that these post-translational and cofactor modifications help to maintain the proper active site geometry but are non-essential for catalytic activity.
Expression of ANME-1 Mcr into the M. acetivorans host and subsequent AOM was recently demonstrated (Soo et al., 2016). Although ANME-1 Mcr in M. acetivorans was not isolated and activity validated, wild-type (WT) M. acetivorans is unable to perform AOM in the absence of methanogenic substrates indicating methane consumption of the engineered strain is attributable to ANME-1 Mcr and not WT Mcr (Soo et al., 2016). However, the methane consumption by ANME-1 Mcr corresponds to an AOM specific activity of only ~20 nmol min−1 mg−1 (Soo et al., 2016), which is about 3-fold lower than the estimated in vivo activity of ANME Mcr (Scheller et al., 2010). The assumption that the 172-methylthiolation of cofactor F430 is crucial for locking the cofactor into its preferred orientation in ANME-1 Mcr implies a reduction in catalytic activity ensues if cofactor F430 is not correctly oriented. Therefore, the reduced activity of ANME-1 Mcr expressed in M. acetivorans may be partially explained by the unavailability of the methylthiolated cofactor in methanogens (Mayr et al., 2008; Allen et al., 2014). Improving ANME-1 Mcr binding to the unmodified cofactor could engender increased rates for AOM when the enzyme is expressed in M. acetivorans.
IPRO was used to predict ANME-1 Mcr variants with improved binding to the unmodified cofactor F430. Variable positions (i.e., design positions) were selected based on (i) proximity to C172, (ii) conservation amongst methanogens but not methanotrophs, and (iii) a review of existing literature, which suggested V419 is crucial for the C172 methylthiolation (Shima et al., 2012) (see Materials and Methods). The 10 selected design positions were Q72, L77, M78, N90, P149, I154, H157, H414, V419, and C423, which all reside within the α-subunit of ANME-1 Mcr. Five independent IPRO trajectories were simulated for 1000 iterations using ensemble structure refinements. Ensemble structure refinements are used within IPRO to sample multiple confirmations for a given protein sequence, thereby improving the accuracy of the energy calculations and quality of the results. During the five IPRO simulations, eight unique variants were identified. The top five variants are provided in Table 1.
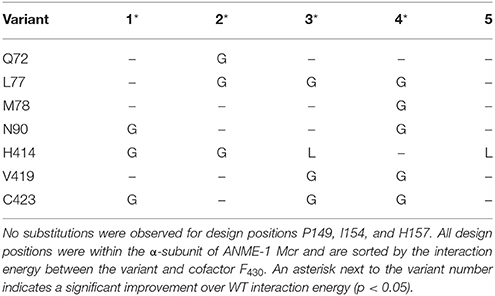
Table 1. Top five variants of CS1, sorted by interaction energy between the enzyme and unmodified cofactor F430.
In examining the top five variants presented in Table 1, a propensity for glycine substitutions was observed (see Figure 2). At first, it was thought that these substitutions were algorithmic artifacts due to unfavorable backbone conformations or to alleviate steric clashes within the active site. Despite introducing a Lennard-Jones softening term and reweighting the scoring function for the rotamer optimization step (i.e., step 2 of IPRO; Grisewood et al., 2017), this partiality for glycine persisted. Additionally, the same algorithmic architecture was employed for CS2–CS4 and for a separate enzyme system (Grisewood et al., 2017), but this glycine preference was not observed in those studies. Based on this and analyzing the top structures, it seems plausible that the glycine residues provide the required flexibility within the active site that allows other side chains to form beneficial contacts with the unmodified cofactor F430.
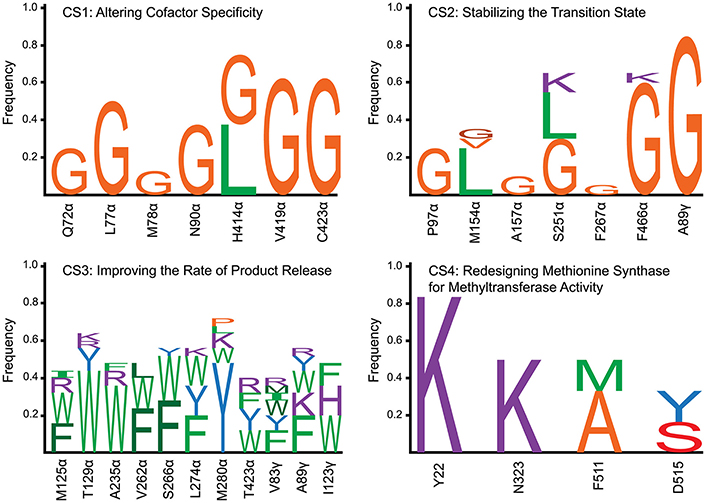
Figure 2. Frequency of amino acid substitutions for each of the four case studies. For CS1, CS2, and CS4, all of the identified variants were incorporated into the frequency calculation. Due to the high number of variants identified in CS3, only the top 25 results were included within the plot. Wild-type amino acids are not included within the barcharts, accounting for the sum of the individual frequencies not adding to 1.0. The design positions are labeled by the wild-type one-letter amino acid code, position, and subunit (except for CS4 which only has a single subunit). Amino acid types were classified into five different categories and colored according to category. These categories were (1) large [≥162 Å3 (Pommié et al., 2004)] with a non-polar side chain (green), (2) small (< 162 Å3) with a non-polar side chain (orange), (3) large with a polar side chain (blue), (4) small with a polar side chain (red), and charged (purple). Different shades of the same color were used to distinguish stacked one-letter codes of the same amino acid category.
The geometry of the top variants' active sites appears at an intermediate state between ANME-1 Mcr and Methanothermobacter thermautotrophicus (i.e., methanogenic) Mcr with their native cofactors (see Figure 3). It is unsurprising that the top ANME-1 variants do not bind cofactor F430 as tightly as M. thermautotrophicus because the methanogenic Mcr has naturally evolved to tightly bind its cofactor. Additionally, V419 is replaced with a methylated (presumably to increase the active site hydrophobicity) glutamine in the M. thermautotrophicus structure. IPRO is limited in that it can only replace the valine with canonical amino acids, and in this case, hydrophobic amino acids. This restricts the gamut of possible side chain conformations within the tightly packed and highly conserved Mcr active site. Despite not achieving the same level of tight binding to cofactor F430 relative to M. thermautotrophicus, the top variants demonstrate a noticeable improvement over ANME-1 Mcr in terms of the calculated interaction energies. The two closest design positions to C172 are H414 and V419, which constitute the methylthio- substituent's binding site in ANME-1 Mcr. Unlike M. thermautotrophicus Mcr that occupies the methylthio- binding site via a large side chain at position 419, IPRO suggests redesigns that shift the side chain of position 414 closer to C172 (see Figure 3). Variants that do not contain the H414G substitution (i.e., Variants 3 and 5, see Table 1), instead force V419 closer to C172, although not to the extent of the methyl-glutamine in M. thermautotrophicus. The effect of the other substitutions is more subtle since these design positions are more distant from the active site.
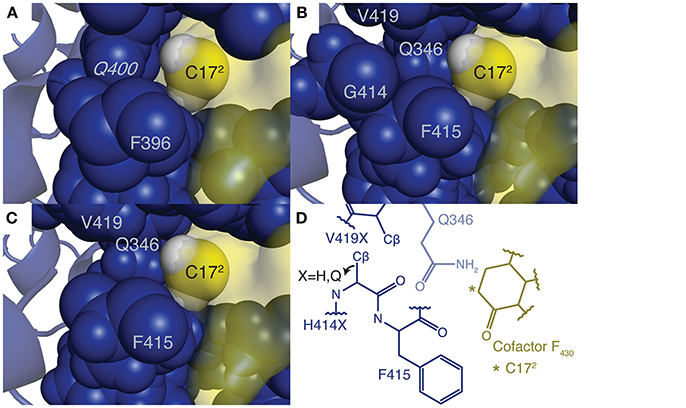
Figure 3. Active site structures of (A) M. thermautotrophicus Mcr, (B) the top CS1 variant, (C) ANME-1 Mcr, and (D) an overview of all three Mcrs in complex with cofactor F430. In subplots (A–C), the unmodified cofactor F430 is depicted as a yellow molecular surface with C172 and its two bonded hydrogen atoms (colored white) shown as spheres. Residues within six angstroms of C172 are displayed as blue spheres, while the remainder of the enzyme is represented by a blue cartoon diagram. G416 and G417 (G397 and G398 in M. thermautotrophicus Mcr) are not shown so that the spatial relationship between C172 and nearby amino acids is clearly visible. Residues constituting the methylthio- substituent binding site in the ANME-1 structure are labeled by their one letter amino acid abbreviation and sequence position, including the methylated glutamine (italicized). The C172 carbon is also labeled. Due to the highly conserved structures of the three Mcrs, subplot D was created, which depicts the general architecture of the active site. Residues forming the methylthio- moiety binding site are labeled using the numbering scheme of ANME-1 Mcr (Q346, H414, F415, and V419 correspond to Q332, Q395, F396, and methyl-Q400 in M. thermautotrophicus, respectively). Q346 is behind the other the amino acids, which is represented by its reduced opacity. The position of the Cβ is given for the second hydrogen atom in H414G (as in the top variant of CS1), but the Cβ position shifts away from C172 if a larger side chain is present.
The veracity of these results is dependent on two key assumptions. First, the ANME-1 Mcr post-translational modifications must be non-essential because the genes required to make these modifications may not exist within the host organism. Second, the 172-methylthiolation of cofactor F430 ought to be catalytically insignificant. Since these two assumptions cannot be tested a priori, we considered a second case study that focused on identifying the kinetic bottleneck and redesigning the native Mcr of M. acetivorans, where the concerns of heterologous expression are eliminated.
Developing a Complete Model for Mcr Kinetics
The complex chemistry undergone during Mcr catalysis has attracted numerous investigations into the enzyme's reaction mechanism, and recent findings have shown that Mcr follows a bi-bi radical-based reaction mechanism (Cedervall et al., 2010; Chen et al., 2012; Scheller et al., 2013; Wongnate and Ragsdale, 2015; Wongnate et al., 2016). This information, along with several assumptions, an existing IC50 value for HDS during methanogenesis (Ellermann et al., 1988), and the rate of 13CH3-S-CoM formation as a function of methane partial pressure (Scheller et al., 2010), yielded specific rate constants for each step in the mechanism. One key assumption in the development of this model is that Mcr kinetics is nearly invariant amongst various methanogenic archaea. This assumption, which is supported by very strong sequence conservation (Reeve et al., 1997) and nearly identical active site structures (Grabarse et al., 2000), enables integration of extensive data to fully characterize Mcr kinetics. These observed and estimated rate constants suggest the probable rate-limiting step of Mcr, which is the release of the produced CH3-S-CoM to regenerate the free enzyme.
The full reaction mechanism of Mcr, including substrate binding and product release, is depicted in Figure 4. The specific rate constant for step 1 was estimated using inhibition studies (Ellermann et al., 1988) and is discussed in greater detail below. Density functional theory calculations (Chen et al., 2012) and the Eyring equation were used to estimate the specific rate constants for step 2. While the transition state for step 3 was not found, the anionic intermediate (McrInt1) was only “transiently formed” and thus its kinetics must be rapid (Chen et al., 2012). The kinetics of step 4 is evaluated from fitting data to the Michaelis-Menten equation and is described in more detail below (Scheller et al., 2010). The specific rate constant for step 5 was also calculated from density functional theory calculations and the Eyring equation (Chen et al., 2012). Step 6 does not have an energy barrier and therefore also exhibits a fast reaction rate (Chen et al., 2012). The specific rate constants for steps 7 and 8 of the mechanism are taken from electron paramagnetic resonance (EPR) and fluorescence experiments (Wongnate and Ragsdale, 2015). Calculations for steps 2 and 5 assumed a temperature of 25°C to match the EPR and fluorescence conditions (Wongnate and Ragsdale, 2015).
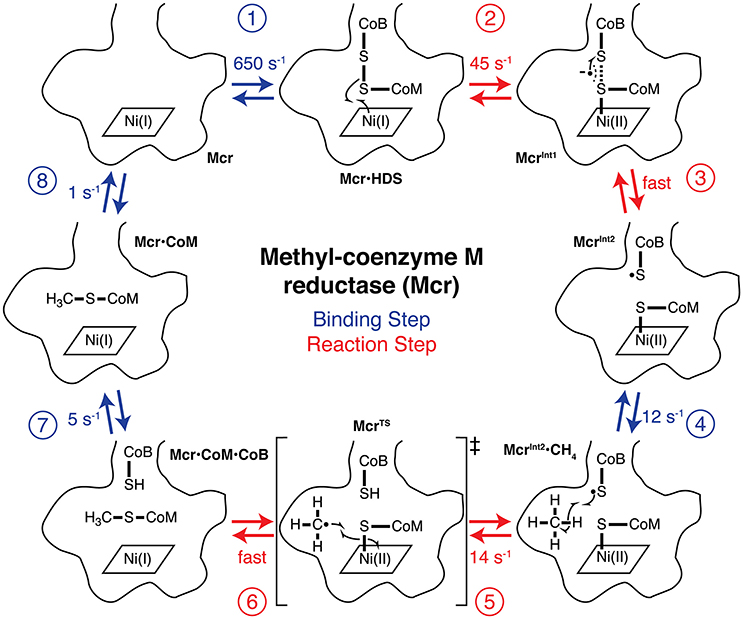
Figure 4. The complete catalytic cycle and associated specific rate constants for AOM by Mcr. The bottom of Mcr's narrow substrate channel and cofactor F430 binding are outlined for each step of the mechanism. Each of the reaction intermediates are numbered and labeled. The free state of Mcr first (step 1) binds HDS to form the Mcr·HDS intermediate. Ni(I) (step 2) donates an electron to HDS to form McrInt1, which rapidly (step 3) interconverts to form McrInt2. McrInt2 (step 4) binds methane to yield McrInt2·CH4, which has a C-H bond homolytically (step 5) cleaved to propagate the radical reaction. The methyl radical is formed as part of the reaction's transition state (McrTS). The Ni(II)-thiolate is homolytically (step 6) cleaved and donates an electron to the methyl radical, which terminates the radical mechanism. The product of step 6 is the product ternary complex (Mcr·CoM·CoB), where Mcr is bound to both CH3-S-CoM and HS-CoB simultaneously. HS-CoB is (step 7) released from the active site to generate Mcr·CoM, and CH3-S-CoM is subsequently (step 8) released to regenerate the free enzyme. Steps 1, 4, 7, and 8 are depicted in blue and are (un)binding steps of the reaction mechanism, while steps 2, 3, 5, and 6 are steps during the chemical reaction and are shown in red.
The kinetics of step 1 was estimated using competitive inhibition studies by HDS during methanogenesis, where an IC50 of 0.6 mM was reported (Ellermann et al., 1988). The mechanism for HDS inhibition is depicted in Figure 5. k12 in Figure 5 is equivalent to k1 (the rate constant for step 1 of methanotrophy) in Figure 4. Assuming Briggs-Haldane kinetics (i.e., reaction intermediate concentrations are time invariant) and that the concentration of Mcr·HDS formed via reaction 11 is negligible relative to that of reaction 12, k12 can be expressed as a function of the IC50 value (Equations 3, 4, see Supplementary Material).
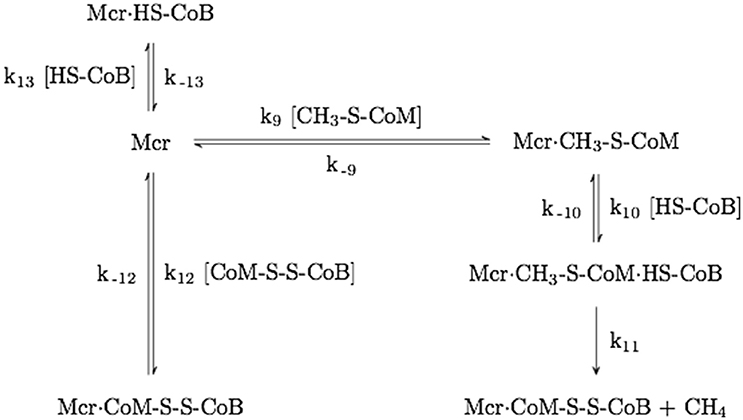
Figure 5. Mechanism of Mcr competitive inhibition by HDS. The rate constant numbering begins with k9 to avoid ambiguity with the mechanism shown in Figure 4.
The underlying assumption that Mcr·HDS is primarily formed through reaction 12 (see Figure 4) is justified because methane formation (i.e., the reaction co-product) is considerably reduced in the presence of HDS (Ellermann et al., 1988), and this assumption also implies that k−12 >18 s−1 (kcat for methanogenesis) (Wongnate and Ragsdale, 2015). The known HDS IC50 value (Ellermann et al., 1988), the corresponding substrate concentrations (Ellermann et al., 1988), and known specific rate constants (Wongnate and Ragsdale, 2015) establish a lower limit for k12 (1.08 × 106 M−1s−1). Using the IC50 value (0.6 mM) as an approximate HDS concentration, we can estimate the specific rate constant for step 1 as 650 s−1.
The specific rate constant for step 4 was determined using the hyperbolic dependence of reaction velocity on methane concentration (R2 = 0.998), which is indicative of Michaelis-Menten kinetics (Scheller et al., 2010). Methane partial pressures (Scheller et al., 2010) were converted to concentrations using the linear relationship between concentration and pressure under moderate conditions (1–20 bar, R2 = 1.000, see Supplementary Figure 1; Duan and Mao, 2006). The in vivo Mcr concentration (4.7 μM) was estimated from carbon monoxide-activated Methanothermobacter marburgensis cells (Zhou et al., 2013). Non-linear regression was used to estimate Michaelis-Menten parameters for the reaction (kcat = 0.12 s−1, KM = 2.5 mM, see Supplementary Figure 2). Established from the same experimental studies as steps 7 and 8, the lower limit for the koff rate is 20 s−1 (Wongnate and Ragsdale, 2015). Using the established koff, kcat, and KM values, the kon rate constant can be calculated (kon = 7.9 mM−1 s−1). At 25°C and atmospheric pressure, the methane concentration is ~1.5 mM (Duan and Mao, 2006). Therefore, the lower limit of the specific rate constant for step 4 is found to be 12 s−1.
Case Study 2: Stabilizing the Transition State of M. acetivorans Mcr
While the developed model suggests product release limits Mcr AOM kinetics, it is conceivable that under a different set of physical conditions, a separate step of the mechanism could constrain the net reaction rate. This theory gains credence due to the biphasic kinetics of methyl radical formation in Mcr, which is thought to be a result of a structural transition at 30°C (Wongnate et al., 2016). Above 30°C, the entropy of activation is ~-56 J mol−1K−1 (Wongnate et al., 2016). This indicates that the energy barrier for step 5 increases (slower reaction rate) with increasing temperature beyond 30°C. Alternatively, product dissociation from enzymes (i.e., steps 7 and 8 for Mcr) are expected to have a near-zero entropy of activation (Kamerlin et al., 2008), indicating that the energy barrier is insensitive to temperature changes. Since the specific rate constant for step 5 of the reaction mechanism is only marginally larger than that of the reported rate of product release (step 8), it seems plausible that the formation of the methyl radical would constrain the net rate for AOM at higher temperatures (>45°C). In Case Study 2, Mcr variants are identified that stabilize the transition state (McrTS), which corresponds to formation of the methyl radical.
Methanosarcina acetivorans Mcr variants were selected by IPRO with the objective function targeting an improvement in interaction energy with the transition state. Design positions were chosen on the basis of (i) proximity to the active site, and (ii) sequence diversity amongst methanogens (see Materials and Methods). The nine selected design positions were P97α, M154α, A157α, M163α, I245α, S251α, F267α, F466α, and A89γ. The transition state structure (Chen et al., 2012) was grafted into the M. acetivorans Mcr active site, with atoms from the resolved transition state structure fixed in place. Grafting the transition state structure resulted in an unfavorable Generalized Born implicit solvation energy term (Lee et al., 2003) for the enzyme that was not readily alleviated with a force field energy minimization. Interaction energies were used in lieu of complex energies to ensure that stabilization of the transition state does not also stabilize the ground state molecules, and render the energy barrier unaltered. Ten independent IPRO trajectories were simulated for 3000 iterations each, and 20 total variants were established. The top five variants are enumerated in Table 2.
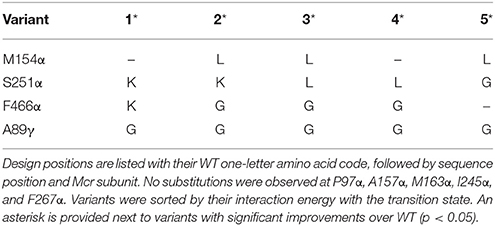
Table 2. Top five variants of CS2, sorted by interaction energy between the enzyme and grafted transition state.
Table 2 demonstrates that substitution with leucine at position M154α and a substitution with glycine at A89γ are ubiquitous. The WT Mcr structure reveals an unfavorable hydrophilic-hydrophobic contact between R152γ and A89γ (4.8 Å between the guanidino group of R152γ and Cβ of A89γ). The substitution to glycine alleviates this poor contact due to its lower hydrophobicity and longer distance to R152γ (5.2 Å). The A89γG substitution does not interact with the transition state. The other ubiquitous substitution, M154αL, indirectly removes a poor interaction with the transition state structure of cofactor F430 (unmodified in CS2–CS4). The leucine side chain forces Q244α into an alternate conformation with respect to the WT structure. In the WT structure, two amido groups are in close proximity (one from cofactor F430, one from Q244α), weakening nearby hydrogen bonds between Mcr and cofactor F430. When Q244α is in its alternate conformation, caused by M154αL, these hydrogen bonds remain intact, improving the interaction energy between Mcr and the grafted transition state.
The top designs listed in Table 2 suggest that the presence of S251αK, S251αL, and S251αG can all form beneficial interactions with the transition state. Though the nature of these side chains vary drastically, they all improve interaction energy by stabilizing a nearby loop of residues between M255α and G258α, which is immediately adjacent to cofactor F430. S251αK stabilizes this loop by forming a hydrogen bond between the S251αK ε-amino hydrogen and the unprotonated nitrogen of H145α. S251αL stabilizes the M255α-G258α loop by packing nicely within a binding site formed by M72, H73, T149, V159, and A252, all of which consitute the α-subunit. Finally, S251αG forms favorable dispersion forces with V159α, which is sandwiched between the loop and S251α. Design position F466α can accommodate substitutions to lysine or glycine in order to improve interaction energy with the grafted transition state. Similar to the S251α substitutions, both F466αK and F466αG stabilize the transition state by altering the conformation of residues lining the long, narrow substrate channel of Mcr. The residues with the most drastic changes are F463α, F464α, Q469α, N501α, and H504α, where F463α and F464α form part of the ·S-CoM binding site. Since phenylalainine is hydrophobic and ·S-CoM is largely hydrophilic, the altered conformation reduces unfavorable hydrophobic-hydrophilic interactions in the active site, thereby improving the interaction energy with the transition state. In this alternate conformation, more hydrophilic amino acids, such as Y346α, Y365β, and R121γ, can create favorable contacts with ·S-CoM in its binding pocket (see Figure 6).
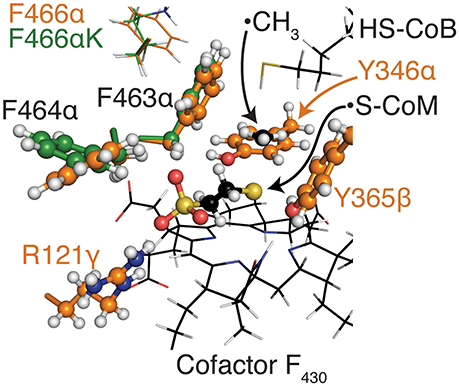
Figure 6. Alternate positions of F463α and F464α nearby the coenzyme M binding site for top variants in CS2. The CoM moiety and residues that form its binding pocket within Mcr are shown as balls and sticks, while other key residues are shown as thin sticks. All atoms are colored by atom type (H, white; N, blue; O, red; S, yellow), while carbon atoms are colored orange for WT M. acetivorans Mcr, green for the top variant of CS2, and black for HS-CoB, ·CH3, S-CoM, and cofactor F430. The conformations of F463α and F464α are largely invariant within the top CS2 variants so only the top variant is presented here.
In all, the top five CS2 designs converge to the same principles to improve interaction energy with the transition state. Substitutions at M154α (only leucine) help improve the hydrogen bonding network between Mcr and cofactor F430. The substitutions at S251α stabilize the conformation of the loop between M255α and G258α, which directly contacts cofactor F430. F466αK and F466αG reduce the hydrophobicity nearby the hydrophilic ·S-CoM. The substitution at position A89γ does not affect transition state binding but instead lessens an unfavorable contact formed with R152γ.
Case study 3: Engineering M. acetivorans Mcr for More Rapid Product Release
Case Study 3 was carried out to improve the rate of CH3-S-CoM and HS-CoB unbinding, which are proposed to be the first and second slowest steps, respectively, of the reaction mechanism at 25°C. Though a transition state structure is unattainable for (un)binding events, the energy barrier can be lowered by raising the energy of the ground state Mcr·CoM·CoB and Mcr·CoM complexes. Destabilization of the enzyme's ground state has been demonstrated to improve catalysis for other enzyme systems (Andrews et al., 2013; Ruben et al., 2013; Phillips et al., 2016) and is particularly applicable for improving the rate of product release, where finding a transition state structure is impractical. Mcr·CoM·CoB and Mcr·CoM exhibit similar enzyme topologies and CH3-S-CoM binding modes, with the key difference being increased flexibility in the Mcr substrate channel nearby the HS-CoB binding site (Cedervall et al., 2010). The low backbone root-mean-square deviation (RMSD) between Mcr·CoM·CoB and Mcr·CoM of 0.21 Å, with a ligand all-atom RMSD of 0.07 Å, supports this claim (Grabarse et al., 2001; Cedervall et al., 2010). Owing to this high structure similarity, variants that destabilize Mcr·CoM are expected to correspondingly destabilize Mcr·CoM·CoB. By destabilizing Mcr·CoM·CoB, the rates of both CH3-S-CoM and HS-CoB unbinding (i.e., steps 7 and 8) are expected to increase.
The structures of McrTS and Mcr·CoM·CoB are also similar, with a ligand all-atom RMSD of 1.7 Å. Due to this similarity, it was postulated that increasing the complex energy of Mcr·CoM·CoB might also destabilize McrTS. The variant structures from CS2 were used to test this hypothesis, and a strong correlation was observed between the two energies (r = 0.73). To account for this, IPRO's objective function was adjusted so as to minimize the difference in McrTS and Mcr·CoM·CoB complex energies. Additionally, an alternate conformation of the β-subunit between residues 364 and 370 persists in the free enzyme (Grabarse et al., 2001) near the HS-CoB binding site. A constraint was added to IPRO to prevent destabilization of the free enzyme (Mcr), while incorporating the structural differences in the β-subunit between Mcr and McrTS/Mcr·CoM·CoB.
A description of the revised MILP used within the second step of an IPRO iteration is provided below:
Sets
A binding assembly is a set of ligands that has its binding affinity for the design molecule (i.e., Mcr) altered by IPRO. Each structure within the Mcr mechanism (see Figure 4) signifies a separate binding assembly. For CS3, there are three binding assemblies: (1) McrTS, (2) Mcr·CoM·CoB, and (3) the unbound enzyme (i.e., Mcr). Only the first two binding assemblies are considered in the revised MILP. The third binding assembly is used within a subsequent MILP with its rotamers restricted to match the amino acid sequence from the first MILP's optimal solution (see Pantazes et al., 2015 for a more detailed description of the standard IPRO MILP). Thus, two MILPs are executed within a single IPRO iteration.
Binary Variables
Continuous Variables
Parameters
The rotamer-constant energy is the energy between a rotamer and any other non-rotamer atom within a single binding assembly (e.g., ligands and protein backbone). Using these defined sets, binary variables, continuous variables, and parameters, the MILP objective function is shown in Equation (5), subject to the constraints provided as Equations (6)–(9).
The objective function (Equation 5) minimizes the difference in complex energy between McrTS and Mcr·CoM·CoB. Equation (6) guarantees that the same amino acid type is used at each position in both binding assemblies. Equation (7) ensures that only one rotamer is selected at each position. The continuous variable, zirkjs, can be written as the product of the two binary variables (xirk × xjsk) and is linearized by Equations (8) and (9).
Since the designs from CS2 did not directly interact with the substrate, we reconsidered the design positions for CS3. The design positions for CS3 were selected based on (i) proximity to the active site or the β-subunit between residues 364 and 370, where the conformation varies between Mcr and McrTS/Mcr·CoM·CoB, (ii) amino acid diversity at the position, and (iii) proper orientation of the side chain toward either the multi-conformational β-subunit loop or the active site (see Materials and Methods). The 10 selected design positions for CS3 were M125α, T129α, A235α, V262α, S266α, L274α, M280α, T423α, V83γ, and A89γ. As was the case for CS2, transition state atoms for the first binding assembly were fixed in place. The third binding assembly that ensures E(Mcrvariant) ≤ E(McrWT) permitted the use of complex energies instead of interaction energies (that were used for CS2). Ten independent IPRO trajectories were executed for 100 iterations each, and 45 variants were identified. The decision to use a fewer number of iterations was made retroactively due to the large success rate and time-consuming nature of ensemble refinements (each refinement performed costs 250 additional IPRO iterations). Of the 45 identified variants, 15 showed simultaneous improvement in stabilizing McrTS and destabilizing Mcr·CoM·CoB. The top five of these 15 variants, which were sorting by ascending [E(McrTS) – E(Mcr·CoM·CoB)] values, are presented as Table 3.
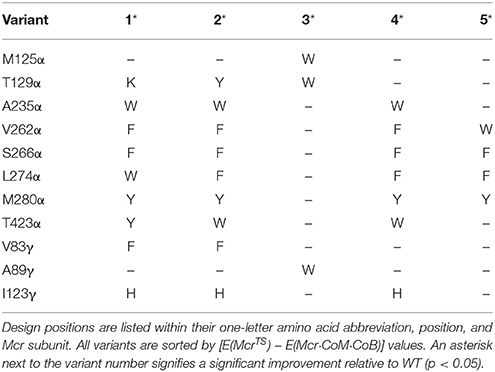
Table 3. Top five variants for simultaneously improving the rate of product release and stabilizing the transition state.
Table 3 shows a preference for large hydrophobic amino acids (see Figure 2). The hydrophobic tendency is unsurprising because of the hydrophobic nature of the Mcr active site, but the large size of the substituted amino acids was unexpected due to the small accessible volume available within the active site. In analyzing the top structures presented in Table 3, these variants notably demonstrate alternate conformations when the rotamer in the McrTS binding assembly versus the Mcr·CoM·CoB binding assembly. In general, when the variant is in the McrTS state, the side chain does not extend toward the narrow substrate binding channel. However, when the variant is in the Mcr·CoM·CoB state, the side chain does extend toward the narrow substrate channel, partially occluding it. From a mechanistic point of view (see Figure 4), it is plausible that the conformational change of these residues helps to initiate product unbinding by “pushing” the products out of the active site via steric clashes. These side chains likely demonstrate a high degree of flexibility, evidenced by their different conformations in the two binding assemblies, and their overall non-specific interactions formed in both binding assemblies (see Figure 7). It is expected that the high flexibility of these residues drives product unbinding but similarly will likely slow the substrate binding of HDS (methane is likely small enough to still pass through the channel). Fortunately, since the predicted specific rate constant is two orders of magnitude higher for substrate binding (see Figure 4), these effects will likely go unnoticed since substrate binding still be unlikely to limit the overall AOM. Another key difference between McrTS and Mcr·CoM·CoB is the more compact aliphatic chain conformation of HS-CoB in the Mcr·CoM·CoB state of the enzyme. Finally, it is noted that the WT structures, even after refinement, do not demonstrate multiple conformations in a similar manner as the variant structures.
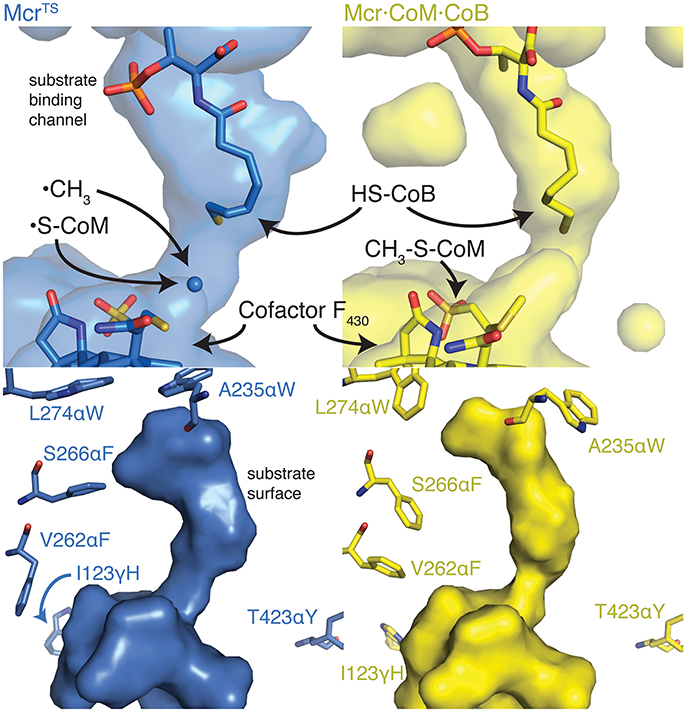
Figure 7. The alternate conformations observed for the top CS3 variant's design positions and their effect on the substrate binding channel. The top two panels show the cavities within Mcr nearby the active site. The long thin continuous surface is the substrate binding channel. The bottom two panels illustrate the substrate surface (i.e., not the cavity) and the design positions nearby the substrates. The enzyme structures depicted in blue (Left) and yellow (Right) represent the McrTS and Mcr·CoM·CoB structures, respectively. Key residues and molecules are shown as sticks, colored by atom type, and labeled. The conformation of the yellow side chains generally extend toward the substrate(s) and thus narrow the size of the substrate channel, forcing HS-CoB to assume a more compact conformation.
Case Study 4: Converting E. coli Methionine Synthase Into a Mtr
One final possibility that was considered is that Mcr does not limit the overall AOM kinetics, but instead, the second step of the reverse aceticlastic pathway catalyzed by Mtr may constrain the net reaction rate. Mtr, a transmembrane protein, catalyzes the transfer of a methyl group from CH3-H4SPT to HS-CoM and presumably catalyzes the reverse reaction for AOM. Although a soluble version of Mtr (CmtA) was discovered (Vepachedu and Ferry, 2012), this enzyme does not have a known structure and has not demonstrated reversibility, which is essential for inclusion in the AOM pathway. Moreover, homology modeling is unlikely to produce a reliable structure due to low sequence identity of available templates, where the best template covers < 60% of the CmtA sequence space and exhibits only 16% sequence overlap with CmtA (Arnold et al., 2006). Due to these problems associated with CmtA, an alternative approach is to redesign an enzyme homolog with an established structure.
One of the closest homologs to CmtA is methionine synthase (MetH), which catalyzes the transfer of a methyl group from 5-methyltetrahydrofolate to homocysteine to form tetrahydrofolate and methionine (Altschul et al., 1997). Both CmtA and MetH employ vitamin B12 as a cofactor for the reaction. CmtA catalyzes the transfer from an aliphatic methyl thioether to a pteridine ring, yielding a thiol, and methylated pteridine ring. Similarly, MetH transfers a methyl moiety from a methylated pteridine ring to a thiol, producing a methyl thioether and pteridine ring (see Figure 8). Additionally, MetH has a resolved crystal structure of its N-terminal domain, where 5-methyltetrahydrofolate and homocysteine bind (PDB 1Q8J) (Evans et al., 2004), and these binding sites are distant from the enzyme's catalytic machinery (Evans et al., 2004). The specific activity of MetH is about 2-fold higher than that of CmtA (Huang et al., 2007; Vepachedu and Ferry, 2012) and is approximately equal between the forward and reverse directions (Rüdiger and Jaenicke, 1969). The similarity of the chemical reaction to that of Mtr, its known crystal structure, cytosolic localization, and high specific activity make MetH a very attractive target for protein engineering. Case Study 4 aims at redesigning the MetH 5-methyltetrahydrofolate and homocysteine binding pockets to instead accommodate H4SPT and CH3-S-CoM, respectively.
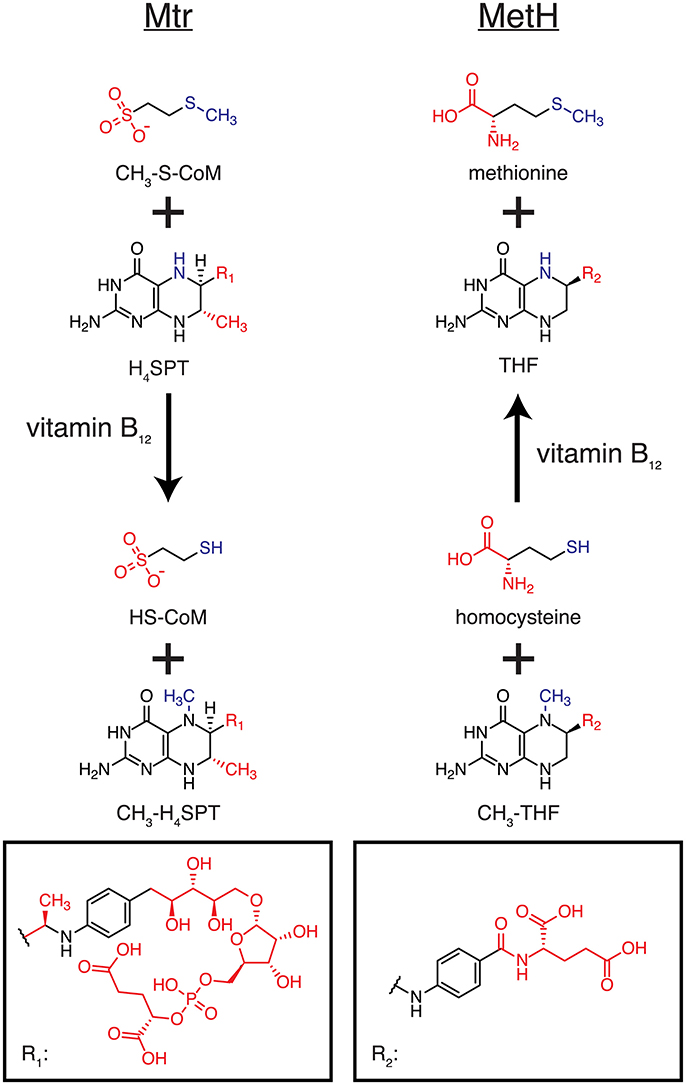
Figure 8. Homologous reactions catalyzed by Mtr and MetH. The left column illustrates the reaction catalyzed by Mtr (and CmtA), while the right columns shows the reaction catalyzed by MetH. The reactive portions of each molecule are colored blue, and the differences between Mtr and MetH substrates/products shown in red.
MetH variants that demonstrate improved binding to H4SPT and CH3-S-CoM were found by running IPRO for 1,500 iterations over 10 independent trajectories using ensemble structure refinements. Design positions were selected on the basis of (i) distance to atoms that differ between CH3-S-CoM and homocysteine or H4SPT and 5-methyltetrahydrofolate, (ii) sequence diversity as determined using family sequence alignments, and (iii) unfavorable contacts formed between the WT residue at this position and the novel substrates (see Materials and Methods). Twelve design positions were selected in total. A slightly larger number of design positions were permitted due to the large distance between the binding sites. Y22, K72 and D105 were selected from the CH3-S-CoM binding domain, while E320, N323, E345, N360, F511, D515, N538, D541, and E542 were selected from the H4SPT binding domain. Prior to performing a production run, the WT structure was refined with the new substrates to remove as many bad contacts as possible before redesigning the enzyme. In the subsequent production run, six variants were identified with improved binding to the new substrates. The top five designs are presented in Table 4.
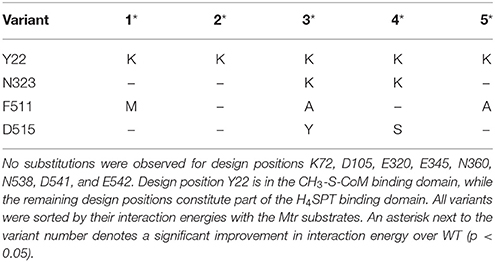
Table 4. Top five variants predicted to alter MetH specificity in efforts to mimic the reaction catalyzed by Mtr.
Table 4 reveals only one substitution made within the CH3-S-CoM binding domain of MetH – Y22K (see Figure 2). As shown in Figure 9, this substitution is introduced to help stabilize the negative charge of the CH3-S-CoM sulfonate group. In addition to the improved electrostatics, this conformation allows for hydrogen bonds to be formed between the protein backbone and the sulfonate. These hydrogen bonds are absent from the WT structure. The positive charge of Y22K is important because there are no other positively charged residues nearby to stabilize the sulfonate.
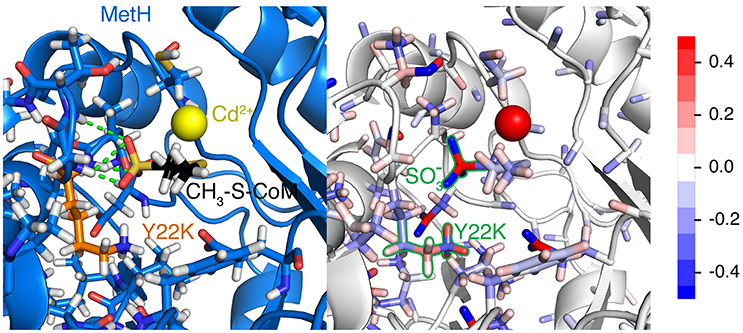
Figure 9. Y22K substitution observed in each of the top five variants for CS4. The (Left) panel depicts the hydrogen bonds (green) formed between the Mcr backbone and the sulfonate group. Mcr is shown as a blue cartoon, with active site residues colored by element and represented by sticks. Y22K and CH3-S-CoM are also shown as sticks with their carbon atoms colored orange and black, respectively. The cadmium ion in the active site is shown as a yellow sphere. The (Right) panel shows the same representation of the active site, but atoms are instead colored by partial charge. It is evident that there is a strong negative charge of the sulfonate group and a positive charge of the Y22K (keeping in mind that the positive charge is distributed amongst the H atoms). The scale for the color scheme is shown on the far right of the figure.
In the H4SPT-binding domain, N323K forms a salt bridge with D390. This interaction stabilizes the wall of the binding crevice and also provides additional volume in the binding site to accommodate one of the methyl substituents that exist in H4SPT but not THF. Both F511M and F511A open up a cavity in the binding site, similar to the N323K substitution. In addition, both F511M and F511A push N508 toward the carbonyl group of the H4SPT pteridine substituent. This movement allows a more favorable hydrogen bond to be formed between the carbonyl and N508. Both D515Y and D515S contribute to improving the stability of the binding site by creating a network of T-shaped π-π stacking interactions, including Y518 and Y519.
Materials and Methods
Modeling of Enzyme and Ligand Structures
Two of the initial enzyme structures were adopted from crystallography experiments. The initial structure of ANME-1 Mcr (CS1) was taken from PDB 3SQG (Shima et al., 2012). Post-translational modifications were removed from the structure, as there is no way to ensure the presence of these modifications when heterologously expressed. The initial structure of MetH (CS4) was adopted from the N-terminal domain (residues 1–566) of Thermotoga maritime expressed in Escherichia coli [PDB 1Q8J (Evans et al., 2004)]. The WT structure of M. acetivorans Mcr (CS2, CS3) was generated using homology modeling (Arnold et al., 2006), with M. barkeri Mcr as the template structure (each subunit ≥90% sequence identity; Grabarse et al., 2000). For the alternate conformation of the β-subunit between residues 364 and 370, the amino acid sequences of the red-1 silent form (free Mcr) of M. thermautotrophicus Mcr and M. acetivorans Mcr are identical. The two flanking positions on each side of the loop were superimposed to the existing M. acetivorans structure, creating the model for the unbound from of Mcr used within CS3.
For CS1, the position of cofactor F430 was determined by superimposing against the crystallized methylthiolated cofactor within ANME-1 Mcr. For CS2, the Mcr side chains that were included in transition state structure (Chen et al., 2012) were used to graft the transition state into the M. acetivorans Mcr structure by minimizing the RMSD between the transition state and homology modeled structure. The positions of the remaining atoms that were not included in the model of the transition state (i.e., atoms distant from the reactive portions of the molecules) were modeled using CHARMM's internal coordinate system. For CS3, the CH3-S-CoM, HS-CoB, and cofactor F430 coordinates were modeled by superimposing against the ox-1 silent version of M. thermautotrophicus Mcr (Grabarse et al., 2001). For CS4, the homologous portions between CH3-S-CoM and homocysteine, as well as between H4SPT and 5-methyltetrahydrofolate were superimposed. Docking tools were then used to further refine the initial placement of the structures.
Design Position Selection
One of the main criteria used for design position selection for all of the case studies was a family sequence alignment. All sequence alignments were performed using Clustal-Omega (Sievers et al., 2011). The sequences to be aligned were extracted from the conserved domain database for CS2–CS4 (Marchler-Bauer et al., 2015). For CS1, differences between methanogenic and methanotrophic archaea were used and therefore manually curated (since methanogenic Mcr and methanotrophic Mcrs are still homologous). The methanogenic Mcr sequences were taken from Uniprot codes P07962, P22948, A4PJ22, D3E050, P12971, P11558, O27232, Q49605, Q58256, P11559, P07961, and Q6LWZ5 (UniProt, 2015). The methanotrophic Mcr sequences were codes Q6VUA6, Q64E03, Q64EA1, Q648C5, Q64D16, D1JBK4, Q6MZD1, Q64CB7, Q64AN3, Q64EF1, Q649Z5, and Q64DN6. Only those positions that were observed in ≥75% of the methanotrophs and < 45% of the methanogens were considered for CS1. For CS2–CS4, designs positions were considered sufficiently diverse if their sequence conservation was ≤ 70%.
Distances calculated to the active site were measured from the nickel atom of cofactor F430, or the sulfur atoms of CH3-S-CoM or HS-CoB, whichever was closest. For CS3, the Cβ atoms of the altered loop in the β- subunit were also considered in the distance calculation, and for CS2, the atoms from the transition state model were used exclusively. For CS3, the dot product was taken at each position between the Cα–Cβ vector and the vector between the Cα carbon and closest atom from the aforementioned distance screen. Positions whose dot product was < 0.5 indicated that this residue was oriented away from the active site, and these positions were not permitted to serve as design positions for CS3. In CS4, unfavorable interactions associated with introducing a larger substrate into the MetH binding site were considered. The rotamer-constant energy was calculated at each position and only those with an unfavorable interaction (i.e., a positive value) remained a potential design position for CS4.
Case Study Details
The CHARMM force field parameters were determined using the parameter files of homologs and with the aid of CGenFF (Vanommeslaeghe and MacKerell, 2012; Vanommeslaeghe et al., 2012). The Lazaridis-Karplus solvation files were created using the parameters during the model's construction (Lazaridis and Karplus, 1999). Each of the case studies incorporated multiple IPRO trajectories, an ensemble of variants to reliably estimate CHARMM energies, and on the order of ~1,000 iterations. Fewer iterations were used for CS3 due to the high percentage of successful variants. The ensemble of structures provided distributions of energy values, which were statistically analyzed using Welch's t-test. Two copies of the α- subunit, one copy of the β- subunit, and one copy of the γ-subunit form an active site for Mcr. For CS1, only the α- subunits were considered because the β- and γ- subunits are not nearby the C172 atom. For CS2, all four polypeptides were incorporated as design molecules (i.e., molecules that can have their structures perturbed). For CS3, the multiple β- subunit conformations were used to take advantage of subtle active site differences between the free and bound enzymes. Therefore, the β- subunit was considered a target molecule (along with HS-CoB, CH3-S-CoM, and cofactor F430), while the two α- subunits and γ-subunit were still considered design molecules. MetH only consists of one chain and was modeled as such. CS1–CS3 incorporated IPRO's dimer constraint, which ensures that the design positions are equally perturbed and varied for each polypeptide chain. The remaining IPRO parameters were set to their default values. All IPRO parameters for each case study are provided in Table 5.
Table 5. IPRO input parameters for CS1–CS4.
Conclusions
The AOM by archaea is a complex reaction cascade that is still not fully elucidated (Thauer, 2011; Nazem-Bokaee et al., 2016). Though a consensus mechanism for AOM has been agreed upon, the role of conformational changes, post-translational modifications, the role of symbiotic partnerships, and the kinetics for the full reaction remain unknown. Adding to this complexity is the inherently challenging reaction catalyzed by Mcr, which breaks a stable C-H bond without the use of oxygen-derived radicals (Scheller et al., 2016). In this work, we revisited existing literature to compile a more complete understanding of Mcr kinetics and which steps are likely to limit the net rate of AOM. Though the temperature ranges considered in this study are industrially relevant (>20°C), the in vivo conditions do not mimic the in situ environment. The possibility remains that a separate step may limit Mcr kinetics at even lower temperatures that more closely resemble environmental conditions (~0°C) for AOM by ANME. The calculations and their underlying assumptions suggested that the rate of product, specifically CH3-S-CoM, release limits the overall AOM kinetics. A geometric model developed by Samson and Deutch (Samson and Deutch, 1978) was used to test whether Mcr kinetics was diffusion-limited, but the derived second-order rate constant for diffusion was two orders of magnitude higher than that of methane binding (step 4 of the mechanism). Four separate case studies for improving the net AOM rate were developed, partially on the basis of these calculations.
A methanotrophic Mcr was redesigned to accept a methanogenic cofactor, assuming that the modified cofactor was exclusively important for active site rigidity. Despite the deletion of the methylthio- substituent of cofactor F430 and its associated increase in binding cavity size, substitutions to small, hydrophobic amino acids (especially glycine) most effectively improved binding to the methanogenic cofactor F430. At high temperatures (>45°C), methane activation may limit the kinetics for AOM, and Mcr variants with diverse chemical properties, ranging from large and hydrophilic to small and hydrophobic, stabilize amino acids immediately adjacent to the transition state. At the low-to-mid temperature range (< 45°C), large hydrophobic residues that can assume multiple conformations are favored in order to improve the rate of product release. In a final scenario, where the second step along the reverse aceticlastic pathway (i.e., Mtr) limits AOM, redesigning the substrate specificity of the more active methionine synthase homolog is achieved by introducing a positively charged residue to stabilize the negatively charged sulfonate of CH3-S-CoM, and surprisingly few substitutions are required to accommodate the larger H4SPT substrate. Taking these findings, later efforts can be pursued to test these variants which have the ability to not only make the conversion of methane to liquid fuels more economically viable, but also provide a deeper understanding of Mcr kinetics.
Author Contributions
MG and CM conceived the case studies. MG performed the simulations and analyzed existing kinetic data from literature. JF provided guidance during the redesign procedures. CM oversaw the simulations. MG, JF, and CM wrote and edited the manuscript. MG, JF, and CM accepted the final submitted version of the manuscript.
Funding
Funding was provided by the Advanced Research Projects Agency-Energy (ARPA-E, DE-AR0000431). Additional funding was provided by The Center for Bioenergy Innovation, a U.S. Department of Energy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science (DE-AC05-00OR22725).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the Penn State Institute for Cyberscience for maintaining the supercomputers that were used to generate designs. We would also like to thank the members of the ARPA-E REMOTE (Reducing Emissions using Methanotrophic Organisms for Transportation Energy) program for their useful conversations.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2018.00084/full#supplementary-material
References
Allen, K. D., Wegener, G., and White, R. H. (2014). Discovery of multiple modified F(430) coenzymes in methanogens and anaerobic methanotrophic archaea suggests possible new roles for F(430) in nature. Appl. Environ. Microbiol. 80, 6403–6412. doi: 10.1128/AEM.02202-14
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Andrews, L. D., Fenn, T. D., and Herschlag, D. (2013). Ground state destabilization by anionic nucleophiles contributes to the activity of phosphoryl transfer enzymes. PLoS Biol. 11:e1001599. doi: 10.1371/journal.pbio.1001599
Arnold, K., Bordoli, L., Kopp, J., and Schwede, T. (2006). The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201. doi: 10.1093/bioinformatics/bti770
Ashworth, J., Havranek, J. J., Duarte, C. M., Sussman, D., Monnat, R. J. Jr., Stoddard, B. L., et al. (2006). Computational redesign of endonuclease DNA binding and cleavage specificity. Nature 441, 656–659. doi: 10.1038/nature04818
Beal, E. J., House, C. H., and Orphan, V. J. (2009). Manganese- and iron-dependent marine methane oxidation. Science 325, 184–187. doi: 10.1126/science.1169984
Benedict, M. N., Gonnerman, M. C., Metcalf, W. W., and Price, N. D. (2012). Genome-scale metabolic reconstruction and hypothesis testing in the methanogenic archaeon Methanosarcina acetivorans C2A. J. Bacteriol. 194, 855–865. doi: 10.1128/JB.06040-11
Bloom, J. D., Meyer, M. M., Meinhold, P., Otey, C. R., MacMillan, D., and Arnold, F. H. (2005). Evolving strategies for enzyme engineering. Curr. Opin. Struct. Biol. 15, 447–452. doi: 10.1016/j.sbi.2005.06.004
Cedervall, P. E., Dey, M., Pearson, A. R., Ragsdale, S. W., and Wilmot, C. M. (2010). Structural insight into methyl-coenzyme M reductase chemistry using coenzyme B analogues. Biochemistry 49, 7683–7693. doi: 10.1021/bi100458d
Chen, S.-L., Blomberg, M. R. A., and Siegbahn, P. E. M. (2012). How is methane formed and oxidized reversibly when catalyzed by ni-containing methyl-coenzyme M reductase? Chemistry 18, 6309–6315. doi: 10.1002/chem.201200274
Duan, Z. H., and Mao, S. D. (2006). A thermodynamic model for calculating methane solubility, density and gas phase composition of methane-bearing aqueous fluids from 273 to 523 K and from 1 to 2000 bar. Geochim. Cosmochim. Acta 70, 3369–3386. doi: 10.1016/j.gca.2006.03.018
Ebner, S., Jaun, B., Goenrich, M., Thauer, R. K., and Harmer, J. (2010). Binding of coenzyme B induces a major conformational change in the active site of methyl-coenzyme M reductase. J. Am. Chem. Soc. 132, 567–575. doi: 10.1021/ja906367h
Ellermann, J., Hedderich, R., Bocher, R., and Thauer, R. K. (1988) The final step in methane formation. Investigations with highly purified methyl-CoM reductase (component C) from Methanobacterium thermoautotrophicum (strain Marburg). Eur. J. Biochem. 172, 669–677. doi: 10.1111/j.1432-1033.1988.tb13941.x
Ermler, U., Grabarse, W., Shima, S., Goubeaud, M., and Thauer, R. K. (1997). Crystal structure of methyl coenzyme M reductase: the key enzyme of biological methane formation. Science 278, 1457–1462.
Evans, J. C., Huddler, D. P., Hilgers, M. T., Romanchuk, G., Matthews, R. G., and Ludwig, M. L. (2004). Structures of the N-terminal modules imply large domain motions during catalysis by methionine synthase. Proc. Natl. Acad. Sci. U.S.A. 101, 3729–3736. doi: 10.1073/pnas.0308082100
Faiella, M., Andreozzi, C., de Rosales, R. T., Pavone, V., Maglio, O., Nastri, F., et al. (2009). An artificial di-iron oxo-protein with phenol oxidase activity. Nat. Chem. Biol. 5, 882–884. doi: 10.1038/nchembio.257
Fazelinia, H., Cirino, P. C., and Maranas, C. D. (2007). Extending iterative protein redesign and optimization (IPRO) in protein library design for ligand specificity. Biophys. J. 92, 2120–2130. doi: 10.1529/biophysj.106.096016
Garrabou, X., Wicky, B. I., and Hilvert, D. (2016). Fast knoevenagel condensations catalyzed by an artificial schiff-base-forming enzyme. J. Am. Chem. Soc. 138, 6972–6974. doi: 10.1021/jacs.6b00816
Goubeaud, M., Schreiner, G., and Thauer, R. K. (1997). Purified methyl-coenzyme-M reductase is activated when the enzyme-bound coenzyme F430 is reduced to the Nickel(I) oxidation state by titanium(III) citrate. Eur. J. Biochem. 243, 110–114. doi: 10.1111/j.1432-1033.1997.00110.x
Grabarse, W., Mahlert, F., Duin, E. C., Goubeaud, M., Shima, S., Thauer, R. K., et al. (2001). On the mechanism of biological methane formation: structural evidence for conformational changes in methyl-coenzyme M reductase upon substrate binding1. J. Mol. Biol. 309, 315–330. doi: 10.1006/jmbi.2001.4647
Grabarse, W., Mahlert, F., Shima, S., Thauer, R. K., and Ermler, U. (2000). Comparison of three methyl-coenzyme M reductases from phylogenetically distant organisms: unusual amino acid modification, conservation and adaptation1. J. Mol. Biol. 303, 329–344. doi: 10.1006/jmbi.2000.4136
Grisewood, M. J., Gifford, N. P., Pantazes, R. J., Li, Y., Cirino, P. C., Janik, M. J., et al. (2013). OptZyme: computational enzyme redesign using transition state analogues. PLoS ONE 8:e75358. doi: 10.1371/journal.pone.0075358
Grisewood, M. J., Hernández-Lozada, N. J., Thoden, J. B., Gifford, N. P., Mendez-Perez, D., Schoenberger, H. A., et al. (2017). Computational redesign of acyl-ACP thioesterase with improved selectivity toward medium-chain-length fatty acids. ACS Catal. 7, 3837–3849. doi: 10.1021/acscatal.7b00408
Hallam, S. J., Putnam, N., Preston, C. M., Detter, J. C., Rokhsar, D., Richardson, P. M., et al. (2004). Reverse methanogenesis: testing the hypothesis with environmental genomics. Science 305, 1457-1462. doi: 10.1126/science.1100025
Harmer, J., Finazzo, C., Piskorski, R., Ebner, S., Duin, E. C., Goenrich, M., et al. (2008). A nickel hydride complex in the active site of methyl-coenzyme M reductase: implications for the catalytic cycle. J. Am. Chem. Soc. 130, 10907–10920. doi: 10.1021/ja710949e
Haroon, M. F., Hu, S., Shi, Y., Imelfort, M., Keller, J., Hugenholtz, P., et al. (2013). Anaerobic oxidation of methane coupled to nitrate reduction in a novel archaeal lineage. Nature 500, 567–570. doi: 10.1038/nature12375
Haynes, C. A., and Gonzalez, R. (2014). Rethinking biological activation of methane and conversion to liquid fuels. Nat. Chem. Biol. 10, 331–339. doi: 10.1038/nchembio.1509
Heller, C., Hoppert, M., and Reitner, J. (2008). Immunological localization of coenzyme M reductase in anaerobic methane-oxidizing archaea of ANME 1 and ANME 2 type. Geomicrobiol. J. 25, 149–156. doi: 10.1080/01490450802006884
Hilvert, D. (2013). Design of protein catalysts. Annu. Rev. Biochem. 82, 447–470. doi: 10.1146/annurev-biochem-072611-101825
Hinrichs, K.-U., and Boetius, A. (2003). “The anaerobic oxidation of methane: new insights in microbial ecology and biogeochemistry,” in Ocean Margin Systems, eds G. Wefer, D. Billett, D. Hebbeln, B. B. Jørgensen, M. Schlüter, and T. C. E. van Weering (Heidelberg: Springer Berlin Heidelberg), 457–477.
Hoehler, T. M., Alperin, M. J., Albert, D. B., and Martens, C. S. (1994). Field and laboratory studies of methane oxidation in an anoxic marine sediment - evidence for a methanogen-sulfate reducer consortium. Glob. Biogeochem. Cycles 8, 451–463. doi: 10.1029/94GB01800
Huang, P. S., Boyken, S. E., and Baker, D. (2016). The coming of age of de novo protein design. Nature 537, 320–327. doi: 10.1038/nature19946
Huang, S., Romanchuk, G., Pattridge, K., Lesley, S. A., Wilson, I. A., Matthews, R. G., et al. (2007). Reactivation of methionine synthase from thermotoga maritima (TM0268) requires the downstream gene product TM0269. Protein Sci. 16, 1588–1595. doi: 10.1110/ps.072936307
Jiang, L., Althoff, E. A., Clemente, F. R., Doyle, L., Rothlisberger, D., Zanghellini, A., et al. (2008). De novo computational design of retro-aldol enzymes. Science 319, 1387–1391. doi: 10.1126/science.1152692
Kahnt, J., Buchenau, B., Mahlert, F., Kruger, M., Shima, S., and Thauer, R. K. (2007). Post-translational modifications in the active site region of methyl-coenzyme M reductase from methanogenic and methanotrophic archaea. FEBS J. 274, 4913–4921. doi: 10.1111/j.1742-4658.2007.06016.x
Kamerlin, S. C. L., Florian, J., and Warshel, A. (2008). Associative versus dissociative mechanisms of phosphate monoester hydrolysis: on the interpretation of activation entropies. Chem. Phys. Chem. 9, 1767–1773. doi: 10.1002/cphc.200800356
Knittel, K., and Boetius, A. (2009). Anaerobic oxidation of methane: progress with an unknown process. Annu. Rev. Microbiol. 63, 311–334. doi: 10.1146/annurev.micro.61.080706.093130
Krüger, M., Meyerdierks, A., Glockner, F. O., Amann, R., Widdel, F., Kube, M., et al. (2003). A conspicuous nickel protein in microbial mats that oxidize methane anaerobically. Nature 426, 878–881. doi: 10.1038/nature02207
Lazaridis, T., and Karplus, M. (1999). Effective energy function for proteins in solution. Proteins 35, 133–152. doi: 10.1002/(SICI)1097-0134(19990501)35:2<133::AID-PROT1>3.0.CO;2-N
Lee, M. S., Feig, M., Salsbury, F. R., and Brooks, C. L. (2003). New analytic approximation to the standard molecular volume definition and its application to generalized born calculations. J. Comput. Chem. 24, 1821–1821. doi: 10.1002/jcc.10367
Lu, Y.-Z., Fu, L., Ding, J., Ding, Z.-W., Li, N., and Zeng, R. J. (2016). Cr(VI) reduction coupled with anaerobic oxidation of methane in a laboratory reactor. Water Res. 102, 445–452. doi: 10.1016/j.watres.2016.06.065
Marchler-Bauer, A., Derbyshire, M. K., Gonzales, N. R., Lu, S. N., Chitsaz, F., Geer, L. Y., et al. (2015). Cdd: NCBI's conserved domain database. Nucleic Acids Res. 43, D222–D226. doi: 10.1093/nar/gku1221
Mayr, S., Latkoczy, C., Kruger, M., Gunther, D., Shima, S., Thauer, R. K., et al. (2008). Structure of an F430 variant from archaea associated with anaerobic oxidation of methane. J. Am. Chem. Soc. 130, 10758–10767. doi: 10.1021/ja802929z
McGlynn, S. E., Chadwick, G. L., Kempes, C. P., and Orphan, V. J. (2015). Single cell activity reveals direct electron transfer in methanotrophic consortia. Nature 526, 531–535. doi: 10.1038/nature15512
Meyer, M. M., Hochrein, L., and Arnold, F. H. (2006). Structure-guided SCHEMA recombination of distantly related beta-lactamases. Protein Eng. Des.Selection 19, 563–570. doi: 10.1093/protein/gzl045
Moore, G. L., and Maranas, C. D. (2004). Computational challenges in combinatorial library design for protein engineering. AIChE J. 50, 262–272. doi: 10.1002/aic.10025
Moran, J. J., House, C. H., Freeman, K. H., and Ferry, J. G. (2005). Trace methane oxidation studied in several Euryarchaeota under diverse conditions. Archaea 1, 303–309. doi: 10.1155/2005/650670
Moran, J. J., House, C. H., Thomas, B., and Freeman, K. H. (2007). Products of trace methane oxidation during nonmethyltrophic growth by Methanosarcina. J. Geophys. Res. Biogeosci. 112, 1–7. doi: 10.1029/2006JG000268
Mueller, T. J., Grisewood, M. J., Nazem-Bokaee, H., Gopalakrishnan, S., Ferry, J. G., Wood, T. K., et al. (2015). Methane oxidation by anaerobic archaea for conversion to liquid fuels. J. Ind. Microbiol. Biotechnol. 42, 391–401. doi: 10.1007/s10295-014-1548-7
Murphy, P. M., Bolduc, J. M., Gallaher, J. L., Stoddard, B. L., and Baker, D. (2009). Alteration of enzyme specificity by computational loop remodeling and design. Proc. Natl. Acad. Sci. U.S.A. 106, 9215–9220. doi: 10.1073/pnas.0811070106
Nauhaus, K., Treude, T., Boetius, A., and Kruger, M. (2005). Environmental regulation of the anaerobic oxidation of methane: a comparison of ANME-I and ANME-II communities. Environ. Microbiol. 7, 98–106. doi: 10.1111/j.1462-2920.2004.00669.x
Nazem-Bokaee, H., Gopalakrishnan, S., Ferry, J. G., Wood, T. K., and Maranas, C. D. (2016). Assessing methanotrophy and carbon fixation for biofuel production by Methanosarcina acetivorans. Microb. Cell Fact. 15:10. doi: 10.1186/s12934-015-0404-4
Packer, M. S., and Liu, D. R. (2015). Methods for the directed evolution of proteins. Nat. Rev. Genet. 16, 379–394. doi: 10.1038/nrg3927
Pantazes, R. J., Grisewood, M. J., Li, T., Gifford, N. P., and Maranas, C. D. (2015). The iterative protein redesign and optimization (IPRO) suite of programs. J. Comput. Chem. 36, 251–263. doi: 10.1002/jcc.23796
Pantazes, R. J., Grisewood, M. J., and Maranas, C. D. (2011). Recent advances in computational protein design. Curr. Opin. Struct. Biol. 21, 467–472. doi: 10.1016/j.sbi.2011.04.005
Pantazes, R. J., Saraf, M. C., and Maranas, C. D. (2007). Optimal protein library design using recombination or point mutations based on sequence-based scoring functions. Protein Eng. Des. Sel. 20, 361–373. doi: 10.1093/protein/gzm030
Phillips, R. S., Vita, A., Spivey, J. B., Rudloff, A. P., Driscoll, M. D., and Hay, S. (2016). Ground-State destabilization by Phe-448 and Phe-449 contributes to tyrosine phenol-lyase catalysis. ACS Catal. 6, 6770–6779. doi: 10.1021/acscatal.6b01495
Pommié, C., Levadoux, S., Sabatier, R., Lefranc, G., and Lefranc, M. P. (2004). IMGT standardized criteria for statistical analysis of immunoglobulin V-REGION amino acid properties. J. Molecul. Recognit. 17, 17–32. doi: 10.1002/jmr.647
Ragsdale, S. W., Raugei, S., Ginovska, B., and Wongnate, T. (2017). “Biochemistry of methyl-coenzyme M reductase,” in The Biological Chemistry of Nickel, eds D. Zamble, M. Rowinska-Zyrek, and H. Kozlowski (Cambridge: The Royal Society of Chemistry), 149–169.
Reeburgh, W. S. (1996). “‘Soft Spots’ in the Global Methane Budget,” in Microbial Growth on C1 Compounds, eds M. E. Lidstrom and F. R. Tabita (Dordrecht: Kluwer Academic), 334–342.
Reeve, J. N., Nolling, J., Morgan, R. M., and Smith, D. R. (1997). Methanogenesis: genes, genomes, and who's on first? J. Bacteriol. 179, 5975–5986. doi: 10.1128/jb.179.19.5975-5986.1997
Richter, F., Blomberg, R., Khare, S. D., Kiss, G., Kuzin, A. P., Smith, A. J., et al. (2012). Computational design of catalytic dyads and oxyanion holes for ester hydrolysis. J. Am. Chem. Soc. 134, 16197–16206. doi: 10.1021/ja3037367
Röthlisberger, D., Khersonsky, O., Wollacott, A. M., Jiang, L., DeChancie, J., Betker, J., et al. (2008). Kemp elimination catalysts by computational enzyme design. Nature 453, U190–U194. doi: 10.1038/nature06879
Ruben, E. A., Schwans, J. P., Sonnett, M., Natarajan, A., Gonzalez, A., Tsai, Y., et al. (2013). Ground state destabilization from a positioned general base in the ketosteroid isomerase active site. Biochemistry 52, 1074–1081. doi: 10.1021/bi301348x
Rüdiger, H., and Jaenicke, L. (1969). Methionine synthesis: demonstration of the reversibility of the reaction. FEBS Lett. 4, 316–318. doi: 10.1016/0014-5793(69)80264-4
Samson, R., and Deutch, J. M. (1978). Diffusion-controlled reaction rate to a buried active site. J. Chem. Phys. 68, 285–290. doi: 10.1063/1.435494
Saraf, M. C., Moore, G. L., Goodey, N. M., Cao, V. Y., Benkovic, S. J., and Maranas, C. D. (2006). IPRO: an iterative computational protein library redesign and optimization procedure. Biophys. J. 90, 4167–4180. doi: 10.1529/biophysj.105.079277
Scheller, S., Goenrich, M., Boecher, R., Thauer, R. K., and Jaun, B. (2010). The key nickel enzyme of methanogenesis catalyses the anaerobic oxidation of methane. Nature 465, 606–608. doi: 10.1038/nature09015
Scheller, S., Goenrich, M., Thauer, R. K., and Jaun, B. (2013). Methyl-coenzyme M reductase from methanogenic archaea: isotope effects on the formation and anaerobic oxidation of methane. J. Am. Chem. Soc. 135, 14975–14984. doi: 10.1021/ja406485z
Scheller, S., Yu, H., Chadwick, G. L., McGlynn, S. E., and Orphan, V. J. (2016). Artificial electron acceptors decouple archaeal methane oxidation from sulfate reduction. Science 351, 703–707. doi: 10.1126/science.aad7154
Shima, S., Krueger, M., Weinert, T., Demmer, U., Kahnt, J., Thauer, R. K., et al. (2012). Structure of a methyl-coenzyme M reductase from black sea mats that oxidize methane anaerobically. Nature 481, 98–101. doi: 10.1038/nature10663
Siegel, J. B., Zanghellini, A., Lovick, H. M., Kiss, G., Lambert, A. R., St Clair, J. L., et al. (2010). Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science 329, 309–313. doi: 10.1126/science.1190239
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Soo, V. W., McAnulty, M. J., Tripathi, A., Zhu, F., Zhang, L., Hatzakis, E., et al. (2016). Reversing methanogenesis to capture methane for liquid biofuel precursors. Microb. Cell Fact. 15, 11. doi: 10.1186/s12934-015-0397-z
Thauer, R. K. (2011). Anaerobic oxidation of methane with sulfate: on the reversibility of the reactions that are catalyzed by enzymes also involved in methanogenesis from CO2. Curr. Opin. Microbiol. 14, 292–299. doi: 10.1016/j.mib.2011.03.003
Thauer, R. K., and Shima, S. (2008). Methane as fuel for anaerobic microorganisms. Ann. N. Y. Acad. Sci. 1125, 158–170. doi: 10.1196/annals.1419.000
UniProt, C. (2015). UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212. doi: 10.1093/nar/gku989
Vanommeslaeghe, K., and MacKerell, A. D. Jr. (2012). Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 52, 3144–3154. doi: 10.1021/ci300363c
Vanommeslaeghe, K., Raman, E. P., and MacKerell, A. D. Jr. (2012). Automation of the CHARMM general force field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 52, 3155–3168. doi: 10.1021/ci3003649
Vepachedu, V. R., and Ferry, J. G. (2012). Role of the fused corrinoid/methyl transfer protein CmtA during CO-dependent growth of Methanosarcina acetivorans. J. Bacteriol. 194, 4161–4168. doi: 10.1128/JB.00593-12
Wegener, G., Krukenberg, V., Riedel, D., Tegetmeyer, H. E., and Boetius, A. (2015). Intercellular wiring enables electron transfer between methanotrophic archaea and bacteria. Nature 526, 587–590. doi: 10.1038/nature15733
Wongnate, T., and Ragsdale, S. W. (2015). The reaction mechanism of methyl-coenzyme M reductase: how an enzyme enforces strict binding order. J. Biol. Chem. 290, 9322–9334. doi: 10.1074/jbc.M115.636761
Wongnate, T., Sliwa, D., Ginovska, B., Smith, D., Wolf, M. W., Lehnert, N., et al. (2016). The radical mechanism of biological methane synthesis by methyl-coenzyme M reductase. Science 352, 953–958. doi: 10.1126/science.aaf0616
Xiao, H., Bao, Z. H., and Zhao, H. M. (2015). High throughput screening and selection methods for directed enzyme evolution. Ind. Eng. Chem. Res. 54, 4011–4020. doi: 10.1021/ie503060a
Yan, Z., Joshi, P., Gorski, C. A., and Ferry, J. G. (2018). A biochemical framework for anaerobic oxidation of methane driven by Fe(III)-dependent respiration. Nat. Commun. 9:1642. doi: 10.1038/s41467-018-04097-9
Zheng, K., Ngo, P. D., Owens, V. L., Yang, X. P., and Mansoorabadi, S. O. (2016). The biosynthetic pathway of coenzyme F430 in methanogenic and methanotrophic archaea. Science 354, 339–342. doi: 10.1126/science.aag2947
Keywords: methyl-coenzyme M reductase, Mcr, product release, anaerobic oxidation of methane, IPRO, enzyme, redesign
Citation: Grisewood MJ, Ferry JG and Maranas CD (2018) Computationally Exploring and Alleviating the Kinetic Bottlenecks of Anaerobic Methane Oxidation. Front. Environ. Sci. 6:84. doi: 10.3389/fenvs.2018.00084
Received: 30 April 2018; Accepted: 05 July 2018;
Published: 27 July 2018.
Edited by:
Nidia S. Caetano, Instituto Superior de Engenharia do Porto (ISEP), PortugalReviewed by:
Naresh Singhal, University of Auckland, New ZealandSeung Gu Shin, Pohang University of Science and Technology, South Korea
Copyright © 2018 Grisewood, Ferry and Maranas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Costas D. Maranas, Y29zdGFzQHBzdS5lZHU=