 Jing Yan
Jing Yan Ronald Manelski
Ronald Manelski Bruce Vasilas
Bruce Vasilas Yan Jin
Yan Jin- Department of Plant and Soil Sciences, University of Delaware, Newark, DE, United States
Mobile colloids, 1–1,000 nm particles, are ubiquitous in every ecosystem. They have small size, large specific surface area, and high mobility in the subsurface, and thus can regulate the fate and transport of sorbing constituents such as nutrients, contaminants, and organic carbon (OC). The movement of colloids and colloidal OC (COC) through soils is an important process in mass and energy transport (including carbon) both within and between ecosystems, e.g., from terrestrial to aquatic ecosystems, and likely contribute to the ecosystem- or global-scale carbon balance given their ubiquitous distribution and unique environmental functions. However, despite their importance for terrestrial and aquatic carbon transport and balance, colloids, and COC have not been adequately accounted for because of the current operational definition uses 0.45 μm as the cutoff size for colloids. In this study, we quantified and characterized loadings of colloids and COC in aqueous samples collected from agricultural, forestry, freshwater wetland, and estuary ecosystems. Results reveal that, in all samples regardless of sampling sources, the total colloidal loads were underestimated by ≥50% and considered as “dissolved” solutes when the (cutoff size of 0.45 μm) was used. Together with a large number of data from the literature, our results further demonstrate that colloids are quantitatively substantial, carbon-dense and that as much as 8–19% of operationally defined DOC is in fact COC. Conservatively, this suggests that COC potentially accounts for 13.6 TgC year−1 as a riverine flux and 530 ± 25 TgC of global DOC pool in the ocean in global carbon cycles. In addition, freshwater wetland was found to be a hotspot, which released more colloids and COC compared to the other sampled ecosystems. These findings clearly demonstrate the limitations of using the operational definition for colloids and DOC and highlight the need for improving quantification and characterization of size-dependent colloidal and OC loads. Such effort will allow direct fundamental research into questions toward microbial access to “protected” carbon by minerals and more accurate assessment of global carbon cycles.
Introduction
Mobile colloids, i.e., particles with sizes between 1–1,000 nm (Everett, 1972; Wilkinson et al., 2007), are heterogeneous and complex mixtures of organic and inorganic entities in natural ecosystems. They include nano-size macromolecules and micro-size fragments of clay, metal oxides, and other solids. Mobile colloids have attracted much research attention due to their small size, large specific surface area, and the ability to facilitate transport of nutrients and contaminants in the subsurface environments (Vold and Vold, 1983; Mccarthy and Zachara, 1989; Kretzschmar et al., 1999; Baalousha et al., 2011). The movement of colloids and COC is an important process in mass and energy transport both within and between ecosystems, e.g., from terrestrial to aquatic ecosystems, and likely contribute to the ecosystem- or global-scale carbon balance given their ubiquitous distribution and unique environmental functions. An emerging view on soil organic matter (SOM) that favors a soil continuum model (SCM), as opposed to the traditional humification model, considers SOM as a continuum of progressively decomposing organic compounds that actively form intimate associations with soil minerals (Lehmann and Kleber, 2015). This further signifies the importance of COC in the formation of SOM continuum in the terrestrial ecosystems and its potential impacts on ecosystem- or global-scale carbon cycles. However, while the behaviors of colloids in some aquatic systems (e.g., ocean) have been extensively studied, quantitative information of environmental occurrence of colloids and colloid-associated constituents in other ecosystems, especially those with sizes < 0.45 μm, is scarce. The scarcity is mainly due to the practice that uses 0.45-μm filters to separate colloidal or particulate and dissolved phases and thus operationally defines the “dissolved” phase as filtrates < 0.45 μm. This practice overestimates the dissolved pool while underestimates colloidal loading (including any constituents associated with the colloids). It prevents accurate differentiation between colloidal and “dissolved” phases thus hinders our ability to provide accurate assessment of the environmental functions and fate and transport of colloids and colloid-associated-constituents, such as OC.
The mobility and stability of OC in soils and aquatic systems are closely related to the interactions with mobile colloids, such as sorption on or coagulation with colloids (Philippe and Schaumann, 2014). While coating of organic matter (OM) on mineral colloids produces negatively charged surfaces and affects colloid dispersion and OM/OC mobilization (Hu et al., 2010), OM transformation and decomposition can also be affected when associated with colloids (Amon and Benner, 1996; Hama et al., 2004; Guo and Macdonald, 2006; Kleber et al., 2007; Kang and Mitchell, 2013). Colloidal organic matter/carbon (COM/COC) in riverine and marine samples has been found to have higher bioavailability than dissolved organic matter/carbon (DOM/DOC), based on previous incubation experiments (Hama et al., 2004) and composition analyses, i.e., C/N ratios and nitrogen contents (McKnight et al., 1997; Rostad et al., 1997; Guo et al., 2003), functional groups and spectroscopic characteristics (Benner et al., 1992; Aluwihare et al., 1997, 2002; Repeta et al., 2002) and isotopic analyses (Aluwihare et al., 2002; Guo et al., 2003; Guo and Macdonald, 2006). These results indicate that colloid-OC interactions may have profound significance in carbon mobilization and stabilization. However, most previous studies monitoring DOM/DOC in natural systems, especially studies of inland aquatic or terrestrial systems, have in general followed the same practice as characterizing colloids, which operationally defines DOM/DOC as the fraction that passes a 0.45-μm filter. Therefore, the role of mobile colloids has not been considered and thus information on the quantitative contribution of colloid-associated carbon to the mobile carbon pool remains very limited. This may lead to inaccurate assessment of the actual mobility, reactivity, and environmental functions of OC in natural systems.
In addition, the ability of colloid-facilitated transport in regulating carbon retention and mobilization in ecosystems varies depending on changes in environmental conditions; such changes include, but are not limited to, hydrological, physicochemical, and biological perturbations. For example, the environmental characteristics of SOM continuum, e.g., mobility and bioavailability of organic fragments, continuously evolve with the variability and heterogeneity of surrounding environmental conditions (Lehmann and Kleber, 2015). The variabilities in colloid and COC concentrations ([COC]) and OC content on colloids (%OC) could have significant influence on carbon storage and stable carbon pool in different ecosystems, terrestrial, riverine, and marine, because colloids' role in possible allocation or input of COC from surface to soils (Ahrens et al., 2015), and from soils to inland water (Billett et al., 2004) and marine systems (Ward et al., 2017). Therefore, to better assess and predict colloids' impact on carbon cycling across different ecosystems, mobilization of colloids, and COC in multiple environmental systems need to be quantified. Additionally, understanding the dynamics of colloids and COC under different environmental conditions could have strong implications in ecosystem carbon models that consider vertical mixing and convection/dispersion of OC (Koven et al., 2013; Dwivedi et al., 2017).
This study focused on quantification of total colloidal load (< 1.0 μm) and COM/COC with samples collected from representative environmental systems, including agricultural, forest stream, freshwater wetland, to estuarine field sites. The specific objectives were to (1) quantify colloidal loads and total COC in different size fractions (10 kDa/0.1–0.45, 0.45–1.0 μm), with a special emphasis on < 0.45 μm fraction (the fraction currently being considered as part of the dissolved phase); (2) quantitatively characterize the relationships between colloid and COC concentrations and assess the variability in colloids' ability to bind OC; and (3) assess the contribution of COC to the operationally defined DOC pool in the SOM continuum based on results from this study along with a large number of data collected from the literature.
Materials and Methods
Sample Collection
Water samples were collected from an agricultural field, forest streams, a freshwater wetland, and an estuary, representing varied hydrological and physicochemical conditions. The locations and detailed descriptions of the study sites are presented in Figure S1 and Text S1 in Supplemental Material. Sampling at multiple wells at the different locations referred above was performed from 2013 to 2015; a detailed schedule is summarized in Table S1. Stream water samples (10–30 cm from the top) were collected directly from estuarine tributary, ditches, and wells at the agricultural field using either sampling buckets or a peristatic pump, while stream water was automatically sampled using ISCO auto-samplers when precipitation intensity was >2.54 mm per hour at the forest streams. For the freshwater wetland, special well installation and sampling techniques were designed for collecting water samples under anaerobic conditions. Briefly, each well annulus was filled with sand and sealed with polyurethane to eliminate bentonite contamination. After purging for 3 well volumes, sampling was performed with a peristatic pump (Geotech, Denver, CO) at ~100 mL/min, suggested by Ryan and Gschwend (1990), to minimize suspending colloids from surrounding soils. The water samples were collected in acid-washed 250-mL serum bottles, flushed with argon gas, and sealed with PTFE silicone septa to avoid oxidation. All samples were kept at 4°C during transportation to the lab and size fractionation was performed within 48 h of sample collection.
Colloid Fractionation and Quantification
Water samples with suspended particles were hand shaken thoroughly and then sonicated for 5 min to homogenize the suspensions. Sonication was performed using a bath sonicator (Branson 5510R-MT, Branson Ultrasonics Corp. CT; 160 W output power, 40 kHz). The samples from the agricultural field, estuary and wetland were siphoned out from the supernatant after centrifugation (Thermo Scientific, MA) at 22,095 g for 8 min, 884 g for 10 min, 221 g for 8 min, respectively, to fractionate out colloids of < 0.1, < 0.45, and < 1.0 μm size fractions. Forest stream samples were prefiltered with 0.7 μm membrane filters, and then fractionated with centrifugation as mentioned above. The 0.7 and 1.0 μm cutoff sizes were selected to collect total or close-to-total colloidal pool below 1.0 μm, according to the IUPAC definition (Wilkinson et al., 2007). The 0.45 μm cutoff size was used to be consistent with the commonly used and operationally defined size to distinguish between colloidal and dissolved phases (Wilkinson et al., 2007). The 0.1 μm cutoff was selected to separate the nano-sized particles from the rest of the colloidal materials. Centrifugation speed and time duration were determined based on the Stokes' Law with an assumption of particle density of 2.65 g/cm3 (Gimbert et al., 2005). Although fractioned samples may include particles that are outside the target size range due to the heterogeneity in particle density, such as changes in density because of the changes in carbon loadings, we assumed that this impact was negligible. It should be also noted that the different methods used for colloid fractionation (i.e., filtration vs. centrifugation) could also lead to variations in the results, which was not accounted for in this study.
Colloid concentrations in most samples were determined by measuring turbidity in each size fractions and converting it to colloid mass concentration with predetermined colloid-turbidity relationships (Yan et al., 2017), while in the rest of the samples were determined gravimetrically. To determine the < 0.1 μm nano-size COC in samples from the estuary site, following centrifugation, 15-mL of < 0.1 μm aliquots were removed and subjected to ultrafiltration (10 kDa) at 53 psi with Amicon stir cell (EMD Millipore, NJ).
Chemical Analyses
Sample pH and electrical conductivity (EC) were measured with corresponding probes (Fisher Scientific, Hampton, NH) in the lab; measurements for wetland samples were performed in a glovebox and were done in the ambient air for samples from the other sites. Suspension OC concentration in each size fraction (< 10 kDa, 0.1, 0.45, 0.7, and 1.0 μm) after fractionation was measured using a catalyst-facilitated high temperature combustion following acidifying the samples with phosphoric acid (Teledyne Tekmar, OH). The [COC] in 0.45–1.0, 0.45–0.7, 0.1–0.45 μm size fraction were calculated from the difference between < 0.1, 0.45, 0.7, and 1.0 μm fractions. The OC content on colloid (%OC) of different size fractions was calculated by dividing the OC concentration by colloid concentration. It should be noted that part of the samples was not analyzed for OC measurement as shown in Table S1 because of low sample volume or low colloid and OC concentrations. Additionally, among samples measured for OC, some samples were contaminated during sample preparation and measurement for OC analyses, and thus these results were not presented.
Data and Statistical Analyses
Sample mean and standard deviation were calculated for each of the measured parameters, i.e., colloid concentration, OC concentration, %OC and the ratio of OC in < 10 kDa/0.1 μm and < 0.2–0.45 μm size fractions. Statistical analysis was performed for log-transformed results by Welch's one-way analysis of variance (Welch's ANOVA) combined with Games-Howell analysis to test their significance of differences (p < 0.05) among the different sampling sites as well as different size fractions by using R Version 3.42 with package of “userfriendlyscience” (Peters, 2017; R core team, 2017). The frequency distribution of log-transformed results of colloid and COC conc. and fitted normal distribution curves are presented in Figures S2, S3. Pearson's correlation coefficients between solution pH, EC, and OC and colloid concentration in different size fractions were provided. To examine the relationships between OC concentration and colloid mass concentration, linear regressions between OC and colloid mass concentration were obtained using the Origin (OriginLab, Northampton, MA).
Results and Discussion
Colloid and COC Concentration
Colloid concentrations in 0.1–0.45 and 0.45–0.7/1.0 μm fractions in the samples from different environmental systems were measured; < 0.1 μm fraction was assumed as the dissolved phase in this study. Colloid concentrations from the different ecosystems are summarized and shown in Figure 1A. While the absolute colloid concentrations differ significantly with environmental conditions or systems, the amounts of colloids in the < 0.45 μm fraction are relatively low but not negligible in samples from all the sampling sites and are comparable to those in 0.45–0.7/1.0 μm fraction. Specifically, Figure 1A and Table 1 show that colloid concentrations range from 0 to 20 mg L−1 in most samples but can be as high as 250 mg L−1 in some samples. The average concentrations of 0.1–0.45 μm and 0.45–0.7/1.0 μm colloids from all sampling sites, 6.26 ± 16.75 mg L−1 and 4.61 ± 19.55 mg L−1, respectively, are comparable, although statistically more colloids were observed in size of 0.1–0.45 μm. When compared within each sampling site, no statistically significant difference in colloid concentration was observed between 0.1–0.45 and 0.45–0.7/1.0 μm size fractions for most sampling sites, except for the samples from the estuary site where the colloid concentration was statistically higher in 0.1–0.45 μm size fraction (Figure 1A and Table 1). This means that ~50% (and in some cases more) of the total colloidal loads would have been considered as “dissolved” if the current operational practice were used.
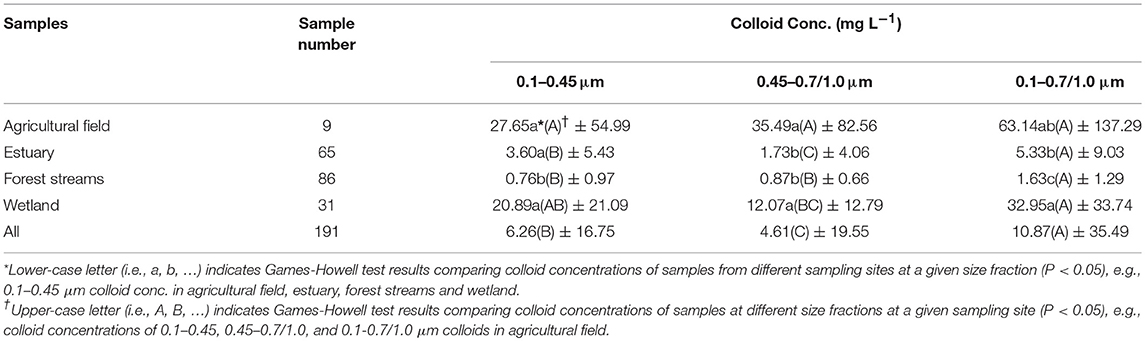
Table 1. Colloid concentrations in 0.1–0.45, 0.45–0.7/1.0, and 0.1–0.7/1.0 μm size fractions in samples from different sites.

Figure 1. Colloid and OC concentrations across ecosystems: (A) colloid concentrations in size fractions of 0.1–0.45, 0.45–0.7/1.0, and 0.1–0.7/1.0 μm; (B) OC concentrations in size fractions of < 10 kDa, 0.1, 0.45, and 0.7/1.0 μm in different natural systems; (C) COC concentration, and (D) %OC of colloids in size fractions of 0.1–0.45, 0.45–0.7/1.0, and 0.1–0.7/1.0 μm.
OC concentrations in size fractions of < 10 kDa, 0.1, 0.45, 0.7, and 1.0 μm were measured to examine the size distribution of OC and to evaluate the contribution of DOC and different size COC to the total mobile OC pool in samples from the different sites. Similar to colloid concentrations, we define dissolved, colloidal, and total mobile OC as DOC0.1 < 10 kDa or 0.1 μm, COC in 0.1–0.7/1.0 μm, and TOC < 0.7/1.0 μm, respectively. The quantitative contribution of COC to the TOC pool was found to be significant in all samples. Figure 1B shows that, across all sampling sites, COC generally accounted for ~10–20% of the TOC pool, whereas DOC0.1 consistently dominated, contributing 82–87% to the TOC pool (with 95% certainty). This indicates most of the organic molecules tends to partition into the dissolved phase, while a small portion of them forms intimate associations with mineral colloids. However, despite the relatively small percentage of COC compared to DOC0.1, the absolute concentrations of COC can be substantial, e.g., ~15 mgC/L in samples collected from the wetland and agricultural field. Similar to the distribution of colloid concentrations in different size fractions, Figure 1C and Table 2 show that the average [COC] in 0.1–0.45 μm fraction is similar to the concentration in 0.45–0.7/1.0 μm fraction (no statistically significant difference, Table 2). Although the COC pool may be smaller than DOC0.1 pool, these results demonstrate that the role of < 0.45 μm colloids in carbon retention and transport could be as significant as their larger size counterparts, and thus COC in < 0.45 μm fractions should be quantified.
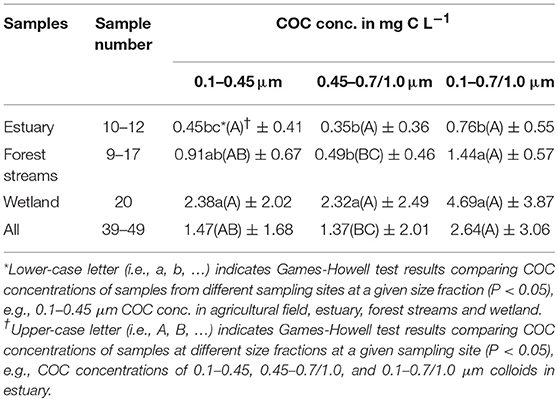
Table 2. COC concentration in 0.1–0.45, 0.45–0.7/1.0, and 0.1–0.7/1.0 μm size fractions from different sites.
Colloid concentrations in both 0.1–0.45 and 0.45–0.7/1.0 μm fractions vary significantly with sampling sites and related environmental conditions (Figure 1A). For the different systems/sites, total colloid concentration (0.1–0.7/1.0 μm) follows the order (high to low): agricultural field or wetland (63.14 ± 137.29 mg L−1 and 32.95 ± 33.74 mg L−1) > estuary (5.33 ± 9.03 mg L−1) > forest streams (1.63 ± 1.29 mg L−1) (p < 0.05, Table 1). This suggests that agricultural practices may enhance colloid release and mobilization, a result also observed in previous studies in other regions (de Jonge et al., 2004; Vendelboe et al., 2012). Higher colloid concentrations in the wetland samples were also observed, which could be induced by surface charge repulsion between charged colloids and soil grain surfaces after iron dissolution during prolonged periods of water saturation and inundation (Ryan and Gschwend, 1990; Chin W. C. et al., 1998; Thompson et al., 2006; De-Campos et al., 2009; Yan et al., 2016). This is reflected by the positive correlation between pH and colloid concentrations as proton consumption in reducing environment (p < 0.05, Table 3). Compared to those from the agricultural field and wetland site, samples of the forest stream and estuary site have much lower colloid concentrations, which could be due to dilution effect in those systems. Increase in EC could also decrease colloid concentration by suppressing the electrical double layer of the colloids thus increase their aggregation according to the DLVO (Derjaguin-Landau-Verwey-Overbeek) theory (Verwey et al., 1947; Derjaguin and Landau, 1993). This is indicated by the negative correlations between EC and colloid concentration in the agricultural field and estuary samples (p < 0.05 for most size fractions except 0.1–0.45/0.7 colloids for samples of estuary, Table 3). In the wetland samples, however, iron-reduction-promoted colloid release may have dominated colloid aggregation induced by EC increase, which is supported by the positive correlations found between EC and pH and EC and ferrous iron concentration (Yan et al., unpublished) with the correlation coefficient, r, of 0.72 and 0.44, respectively. Additionally, shifting in pH may have complicated the dynamics of colloid stability as well. Colloid dispersion is expected with increasing pH due to the increase in negative surface charge of colloids, which promotes repulsion between colloid-colloid or colloid-soil grains. This effect can be especially significant in the wetland samples in which mineral colloids with variable surface charges, e.g., iron-oxyhydroxides, are more abundant. This is evidenced by the positive correlations between pH and colloid concentration in the samples from agricultural field, estuary, and wetland, although statistically significant correlations were only observed for the wetland samples (p < 0.05, Table 3). Other factors, for example, variations in soil texture and mineralogy among the sampling sites, which were not considered in the current analysis, could have also contributed to the observed differences. Nevertheless, these results seem to imply that variability in total colloidal load largely depends on environmental conditions, and systems such as agricultural field and freshwater wetland could serve as the hot spots of colloid release.
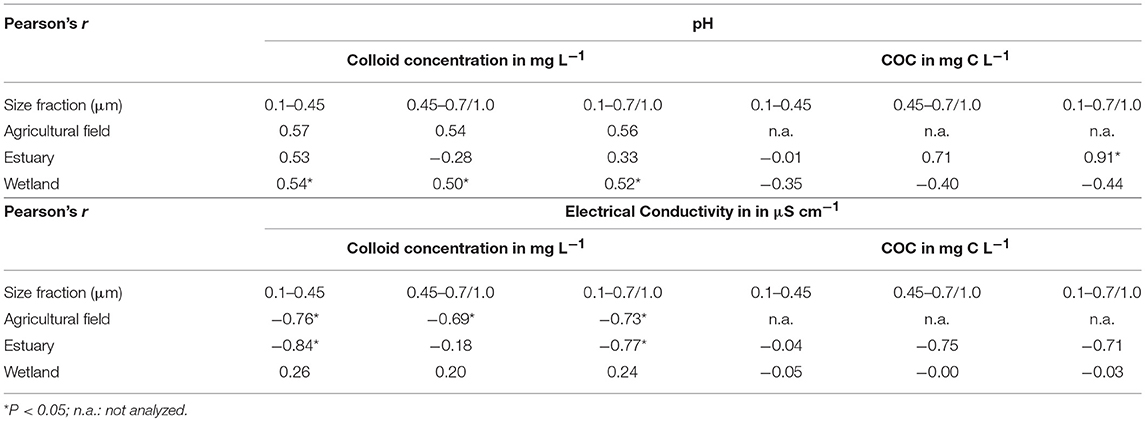
Table 3. Pearson's correlation coefficient between pH, EC and colloid and OC concentration among different size fractions.
Similar to colloid concentrations, [COC] changes significantly with changing environmental conditions as well (Figure 1C). Among the different sampling sites, the average [COC] in 0.1–0.7/1.0 μm fraction follows the order: wetland > forest streams > estuary. Consistently, comparable or higher [COC] were observed in the wetland samples than in the samples from the other sites (p < 0.05, Figure 1C, Table 2), indicating that COC release and transport are important processes in systems under dynamic redox conditions, which has been reported in previous studies (Ryan and Gschwend, 1990; Chin Y. P. et al., 1998; Thompson et al., 2006; Yan et al., 2016), and is likely caused by the release of colloidal organic matter from soil aggregates when solution chemistry changes during iron reduction. However, the mechanisms of COC release seemed to be more complex, as indicated by the negative correlations between pH and COC, for reasons not yet clear. One possible explanation could be variations in soil texture and mineralogy, which deserves to be investigated in the future research. In contrast to the wetland samples, [COC] in the estuarine samples decreased with increasing salinity as indicated by its slightly negative correlation with EC (Table 3). While selective mineralization and oxidation of COC may occur, it is also perceivable that COC formed either homo- or hetero- aggregates and settled out of the solution phase during their transport from inland upstream to offshore downstream where salinity is significantly higher (Sholkovitz, 1976; Sholkovitz et al., 1978). In addition, increase in pH may have promoted COC dispersion in the estuary due to surface charge repulsion as mentioned above, although erosion could also contribute to the results, given the observations of higher pH in the upper streams, e.g., ES-L (Supplemental data set), where higher erosion was expected due to more intense agricultural activities. These results suggest involvement of different factors and pathways that control COC release and transport in different systems. In general, consistent with results of colloid concentrations, field sites under reducing conditions, i.e., the wetland in this study, can be considered a hot spot of COC release and mobilization.
Correlation Between Organic Carbon and Colloids
Figure 2 shows results of linear regression analyses between [COC] and colloid concentration in different size fractions performed for samples combined from estuary, wetland, and forest stream sampling sites. Positive correlations are observed for all size fractions, although correlation coefficients, r, are relatively low and large variations in the correlations are observed (Figure 2; blue lines). This suggests that at least some released COC are in the form of mineral-OC complexes and are subject to the same influences as other types of colloids regardless of their sizes. The variations could be due to the heterogeneity in colloid composition having different affinity for OC, giving rise to different OC loading on colloids. Alternatively, colloids generated from different sources, e.g., mixtures of terrestrial and marine sources in estuary site, can also increase variability. For example, colloids collected from a clay layer in freshwater wetland (empty circles in red, Figure 2; supplemental data set) are primarily composed of mineral colloids and thus have much lower OC loadings compared to other samples. Indeed, by removing these “special” samples or outliers, the positive correlation between colloid concentration and [COC] improved significantly (Figure 2; red lines).
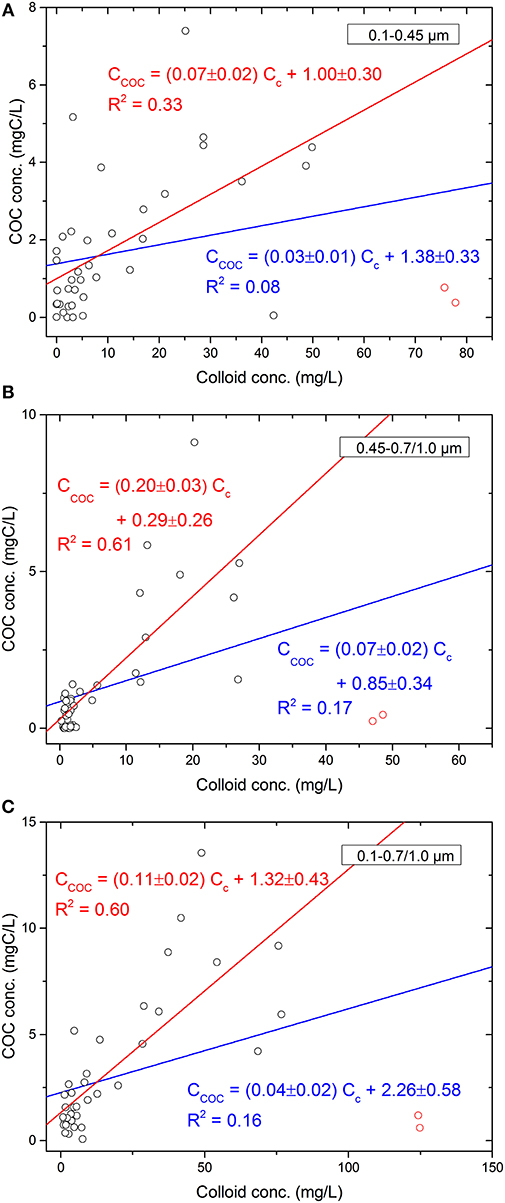
Figure 2. Linear regression analyses between COC and colloid conc. for (A) 0.1–0.45, (B) 0.45–0.7/1.0, and (C) 0.1–0.7/1.0 μm colloids without (blue lines) and with removing (red lines) the outliers (red circles).
The changes in size-dependent OC contents on colloids were examined by calculating OC contents on colloids (%OC), respectively, for size fractions of 0.1–0.45, 0.45–0.7/1.0, and 0.1–0.7/1.0 μm. Figure 1D shows that %OC values varied in all size fractions, ranging from 0 to 70% with a median value of ~20% for results between the lower and upper boundaries of the boxes, i.e., 25 and 75% percentiles. These results show that %OC on colloids are comparable to that of humic substances, i.e., ~35–76% carbon content (Nissenbaum and Kaplan, 1972; Rice and MacCarthy, 1991), while colloids are more carbon-dense than particulates, presumably because (1) they have larger specific surface area thus greater sorption capacity and (2) they form from detachment/transformation of particulate OM or from sorption of or coagulation with branched high molecular weight OM (Guo and Santschi, 1997). In fact, Schulten and Leinweber (2000) have confirmed that OC and total nitrogen content increase exponentially with decreasing particle size from soils with different clay and silt contents. Furthermore, the %OC on 0.1–1.0 μm colloids observed in this study are much higher than that on > 1.0 μm particulate matter reported in previous studies (Bergamaschi et al., 1997; Rostad et al., 1997; Dhillon and Inamdar, 2014). In particular, samples from the same forest streams were found to have much lower %OC on >0.45 μm particulate matters, i.e., 0.3–8.9% (Dhillon and Inamdar, 2014). In addition, Bergamaschi et al. (1997) found %OC of 2–500 μm sediment particles from the Peru Margin ranged from 1.5 to 9.8%, and Rostad et al. (1997) reported %OC on > 3.6 μm suspended particles in Mississippi River and its tributaries were 1.26–16.8%. These results suggest that %OC likely changes with sampling locations due to the differences in the sources of organic compounds in Bergamaschi et al. (1997) and Rostad et al. (1997), compared to this study. Therefore, a consistent difference in %OC across SOM continuum could occur, and more systematical sampling and analyses of %OC in the SOM continuum are needed.
Increase in carbon loading suggests changes in OM stability (Gu et al., 1994; Feng et al., 2014). According to the zonal model (Kleber et al., 2015), as carbon content or loading increase, weak organic-organic interactions become more dominant than strong organo-mineral interactions. Accordingly, the greater carbon loading could decrease the stability of the OC sorbed on 0.1–1.0 μm colloids, compare to >1.0 μm particulate matter. These results suggest that COC associated with different size colloids is likely to have different mineralization rates and stability (Guo and Santschi, 2007; Benner and Amon, 2015). Studies on bioavailability of COM from riverine and marine ecosystems through direct incubation experiments and indirect measurements of C/N ratios, functional groups and isotopic compositions have been conducted. Reported values of the C/N ratio of COM are in the range 10–50 with variations likely due to different sampling sources (i.e., fresh vs. marine water). In general, COM tends to be enriched in nitrogen, and thus serves as an important source for nitrogen mineralization and assimilation (McKnight et al., 1997; Rostad et al., 1997; Guo et al., 2003). Solid-state 1H or 13C NMR analysis of both fresh water and marine water samples have also shown that large percentages of the COM analyzed are composed of carbohydrates (Benner et al., 1992; Repeta et al., 2002), oligosaccharides and polysaccharides (Aluwihare et al., 1997, 2002), which are commonly considered as labile or metabolizable fractions. Additionally, the young 14C age of COM suggests a high reactivity or fast turnover rate of COM compared to DOM (Aluwihare et al., 2002; Guo et al., 2003; Guo and Macdonald, 2006). In addition to these indirect evidences, Hama et al. (2004) conducted an incubation experiment on algal-derived DOM and directly showed that the turnover time was shorter for COM than DOM. Although the information of biological reactivity or bioavailability of COC from terrestrial ecosystems is limited, our results, along with the observations reported in the literature, suggest that the reactive or bioavailable COC pool in the SOM continuum can be significantly underestimated as a result of treating COM in < 0.45 μm fractions as dissolved.
Relationship Between COC and Operationally-Defined DOC
To extensively evaluate the contribution of COC to operationally-defined DOC, the ratios of DOC0.1 (< 10 kDa or 0.1 μm) to DOC0.45 (< 0.2–0.45 μm) were calculated for samples from this study and from data reported in the literature that include measurements of samples from numerous rivers, estuaries and marine sites across the globe. The OC concentration averaged for all the samples is 8.1 ± 14.6 mgC/L, and the DOC0.1 to DOC0.45 ratio is 86.7 ± 9.6% (Figure S4, Table 4). These results indicate that over 80% of OC in DOC0.45 is dissolved whereas 12–15% of DOC0.45 is COC, based on the classification definitions used in this study. The DOC0.1/DOC0.45 ratios are relatively constant, compared to the large variations in OC concentrations, as reflected by their variance coefficient, 0.11 and 1.81, respectively. This observation seems to suggest that the DOC0.1/DOC0.45 ratio is largely independent of variations in OC concentrations in the many samples analyzed (Figure 3, Table 4).
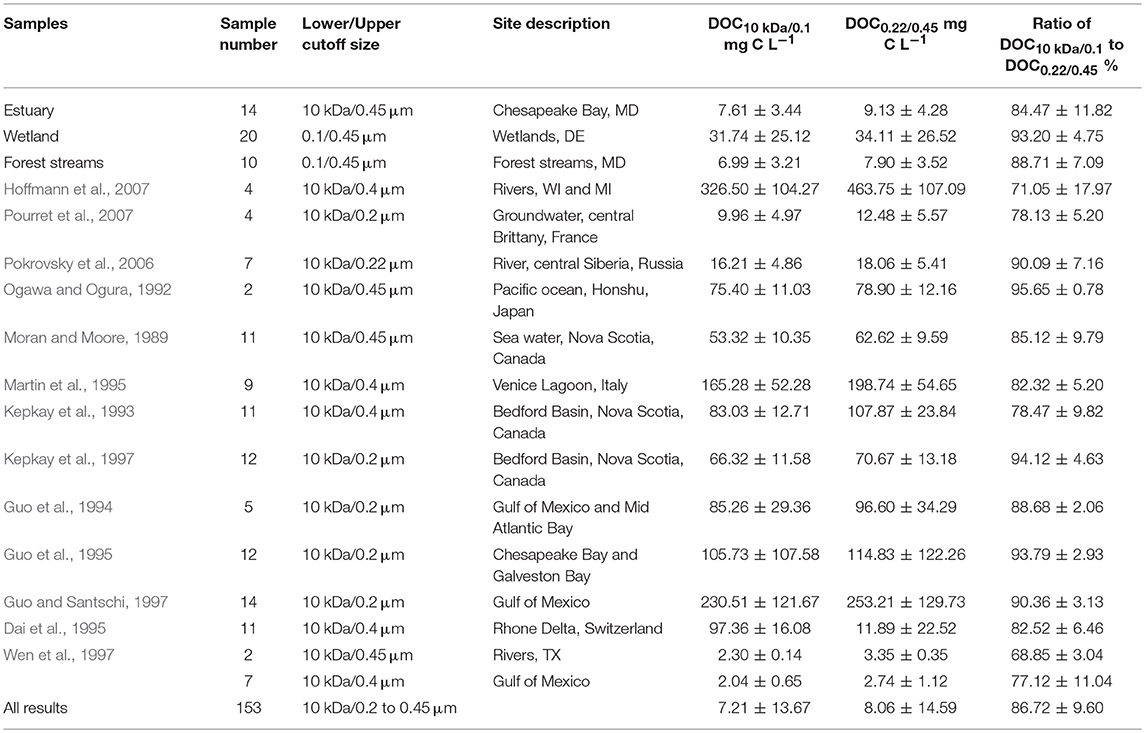
Table 4. The ratios of DOC10kDa/0.1 to DOC0.22/0.45 from this study and literature.
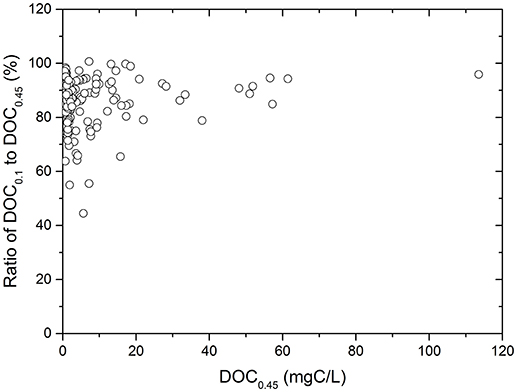
Figure 3. Relationship of ratio of DOC0.1 to DOC0.45 with DOC0.45 of samples from this study and literature.
Additionally, the linear relationship between DOC10kDa and DOC0.45 (< 10 kDa and < 0.4/0.45 μm OC) shows that majority of the data fall on the same regression line with a slope of 0.81 and a high R2 value of 0.96 (Figure 4). This seems to indicate the existence of a constant distribution between DOC10kDa (~80%) and COC (~20%) regardless of sampling source and location. The “universal” nature of this observation suggests the possibility of predicting [COC] from DOC0.45 concentrations. In other words, [COC] may be estimated using this relationship from data measured based on the current practice that uses the operational definition of 0.45 μm without additional fractionation and measurement effort.
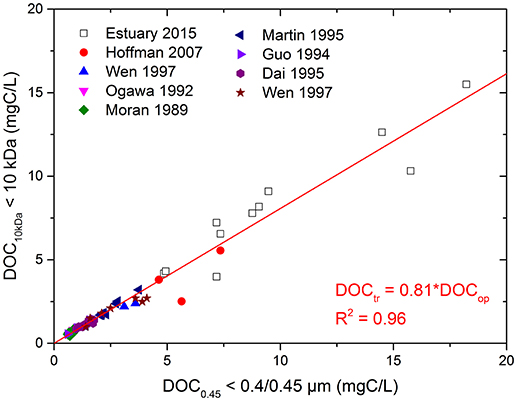
Figure 4. Relationships between DOC10kDa and DOC0.45 for < 10 kDa DOC and 0.4/0.45 μm DOC of samples from this study and literature.
Because the results presented in Table 4 are from different studies that used different cut-off sizes for colloid definition, additional regression analyses were performed using < 10 kDa OC as DOC10kDa and < 0.2/0.22 μm or the combination of < 0.2/0.22 and 0.4/0.45 μm OC, respectively, as DOC0.22 and DOC0.45. The DOC10kDa/DOC0.22 and DOC10kDa/DOC0.45 ratios are 0.88 for samples using < 0.2/0.22 μm OC, and 0.92 using the combination of < 0.2/0.22 and 0.4/0.45 μm OC, respectively, as the cut-off size for OC (R2 value are 0.97 and 0.96; Figure S5). Results in Figures 4 and Figure S5 collectively indicate that there is at least 8% but can be up to 12–19% of COC that has been regarded as DOC when using the operationally defined cut-off size of either 0.2/0.22 or 0.4/0.45 μm. This finding is comparable to Benner and Amon's estimation that ~77% of total OC in the oceans are low molecular weight OC (Benner and Amon, 2015). The slightly smaller contribution of COC to DOC0.45, compared to Benner and Amon (2015), could be either due to the difference in cut-off sizes (1 kDa-100 nm as COC in Benner and Amon's), or inclusion of samples added from terrestrial and riverine ecosystems in this study.
More interestingly, the seemingly “constant (~10%)” contribution of COC in DOC fraction of < 0.2 or 0.45 μm suggests that some of the organic colloids in our samples may be formed from a reversible assembly/dispersion equilibrium between COC and smaller-sized DOC, e.g., DOC10kDa and DOC0.1. This mechanism is supported by Chin W. C. et al. (1998) and Verdugo et al. (2004), who reported ~10% of DOC in surface sea water spontaneously assemble into colloidal gels. However, biological and chemical transformation of COC from >1.0 μm particulate OC (POC) and < 0.1 μm smaller-sized DOC, i.e., microbial degradation and synthesis, may also contribute to the formation of COC, as suggested by Benner and Amon (2015). Furthermore, compared to marine systems in previous studies (Chin Y. P. et al., 1998; Verdugo et al., 2004; Benner and Amon, 2015), more diverse terrestrial sources and interactions between substances and porous structures, such as soil and sediment, may also complicate the mechanisms. This suggests that COC partition from POC and DOC is under a complex equilibrium between colloid generation and source mixing in natural environments. In terms of SOM continuum, COC serves as important transition zone between POC and DOC pool and facilitates the exchange of essential resources between these pools. Therefore, to better understand the mechanisms of COC formation and its dynamic interactions with POC and DOC in ecosystem- or global-scale, more systematic studies focusing on COC in terrestrial systems should be performed.
Implications and Environmental Significance
These findings demonstrate that, for studies aiming at quantifying “truly” dissolved materials, interpretation of results based on measurements using the operational definition for colloids and DOM can be misleading. A possible and critical consequence of treating colloidal substances as dissolved is to interpret, erroneously, changes due to physical interactions of colloids, e.g., aggregation, disaggregation and settlement, as chemical or biological reactions. Furthermore, according to the “pulse-shunt” model proposed by Raymond et al. (2016), the input of substances from terrestrial to downstream aquatic systems is largely controlled by residence time, storage, and decomposition kinetics of the substances in the environments, which are different depending on whether the substances are colloidal or dissolved. In addition, the mobility of colloids can be higher than dissolved solutes due to size and charge exclusion (Jin and Flury, 2002), which is further promoted by preferential flow in soils where larger pores are usually observed. Therefore, accurate quantification of colloidal and dissolved phases will ultimately lead to more accurate assessment of the biological functions and environmental fate and transport of not only COC but also nutrients, trace elements, and contaminants. Additionally, our findings have implications for studies focusing on sampling dissolved constituents, such as soil pore water collected by rhizon samplers. The rhizon samplers with smaller pore size, i.e., 0.15 μm, are preferred to minimize the colloidal fractions in the “dissolved” phase.
Our findings fill the gap in the quantification of < 0.45 μm COC and provide the basis for considering COC as a separate phase in carbon cycling and assessment of carbon budget in different natural environments, as COC is likely to behave differently from DOC and POC in these systems (Guo and Santschi, 2007; Benner and Amon, 2015). For example, COM/COC have been shown to have higher bioavailability and biodegradability, as discussed in Correlation Between Organic Carbon and Colloids. More importantly, these results could benefit the development of SOM continuum models by providing quantitative characteristics of size distribution of OC in the continuum of SOM and DOM. The seemingly “constant” contribution of COC in DOC < 0.2 or 0.45 μm following the current conventional practice provides us a “tool” for estimating the underestimated COC pool in global carbon cycles. According to studies that quantify global DOC flux (Bauer and Bianchi, 2011; Bauer et al., 2013), riverine DOC flux to the ocean ranges from 0.17 to 0.4 PgC year−1 and the total DOC pool in the world's ocean is 662 ± 32 PgC (Hansell et al., 2009). Following our estimation that COC accounts for 8–19% of DOC pool, the amount of COC would be 13.6–32 TgC year−1 as a riverine flux and 530 ± 25–1258 ± 61 TgC of global DOC pool in the ocean. These numbers would be even larger if 1 kDa were used as the cut-off size for truly dissolved phase (Guo and Santschi, 2007). The significant, but unquantified amount of COC could represent a substantial and dynamic pool of biodegradable OC and thus contribute to carbon emission via mineralization and microbial respiration (Amon and Benner, 1996; Hama et al., 2004).
This study shows that COC could be a significant component in the global carbon pool with different fate and behavior from POC and DOC, and thus its contribution cannot be neglected and should be considered separately in models of carbon balance estimation. Such modification could potentially improve the accuracy of parameterization, e.g., advection velocity and diffusivity, in ecosystem carbon models that consider vertical mixing and advection/dispersion of OC (Koven et al., 2013; Dwivedi et al., 2017). In addition, it could improve mechanistic understanding of carbon storage in deep soil layers and aquatic continuum, as presented in several articles (e.g., Billett et al., 2004; Ahrens et al., 2015; Ward et al., 2017; Hicks Pries et al., 2018). Our findings further signify the importance in taking into account the impact of colloid-OC interactions on carbon cycles in systems that are subjected to variations in environmental conditions, such as redox status. The fate of colloids and COC can be sensitive to the physicochemical changes of the environments, as indicated by the higher colloid and COC concentrations in the freshwater wetland compared to the other sampling sites. Identification of such “hot spots” where could improve estimation of mobile and bioavailable carbon flux to the subsurface soil and downstream ecosystems. As climate change and anthropogenic perturbations intensify, the contribution of COC to the global carbon pool could be more pronounced in regions undergoing significant environmental changes, including intermittent anoxia in upland soils due to intensified precipitation, severe and frequent flooding due to deforestation, and coastal inundation undergoing sea level rise. These findings may aid our ability to more accurately assess and predict the influence of natural and anthropogenic factors on organic carbon dynamics, which have important implications in land use management and regulation of different environmental systems to sustain natural and anthropogenic future perturbations.
Author Contributions
JY, BV, and YJ conceived of the presented idea. JY and RM performed the sampling and experiments. BV and YJ supervised the findings of this work. JY initiated the manuscript, and all other authors discussed the interpretation and contributed to the final manuscript.
Funding
This research was part of the Christina River Basin Critical Zone Observatory (CRB-CZO) project, supported by the National Science Foundation (EAR 0724971) and National Integrated Water Quality Grant Program (NIWQP) no. 2013-67019-21361 from the USDA National Institute of Food and Agriculture.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge that the manuscript included content published in the dissertation of Yan (2016). The authors also acknowledge Kathryn Clark, Shreeram Inamdar, Deb Jaisi, Sunendra Joshi, Matt King, Jiying Li, and Zhixuan Qin of the University of Delaware for assistance with field sampling and Karen Gartley, Rovshan Mahmudov, Fang Tan of University of Delaware and Mingxin Guo at Delaware State University for assistance with organic carbon measurements. The data used are listed in the Supplemental Information.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2018.00148/full#supplementary-material
References
Ahrens, B., Braakhekke, M. C., Guggenberger, G., Schrumpf, M., and Reichstein, M. (2015). Contribution of sorption, DOC transport and microbial interactions to the14C age of a soil organic carbon profile: insights from a calibrated process model. Soil Biol. Biochem. 88, 390–402. doi: 10.1016/j.soilbio.2015.06.008
Aluwihare, L. I., Repeta, D. J., and Chen, R. F. (1997). A major biopolymeric component to dissolved organic carbon in surface sea water. Nature 387, 166–169. doi: 10.1038/387166a0
Aluwihare, L. I., Repeta, D. J., and Chen, R. F. (2002). Chemical composition and cycling of dissolved organic matter in the Mid-Atlantic Bight. Deep Sea Res. Part II Top. Stud. Oceanogr. 49, 4421–4437. https://doi.org/10.1016/S0967-0645(02)00124-8
Amon, R. M. W., and Benner, R. (1996). Bacterial utilization of different size classes of dissolved organic matter. Limnol. Oceanogr. 41, 41–51.
Baalousha, M., Lead, J. R., and Ju-Nam, Y. (2011). “3.05-Natural Colloids and Manufactured Nanoparticles in Aquatic and Terrestrial Systems,” in Treatise on Water Science, ed W. Editor-in-Chief: Peter (Oxford: Elsevier), 89–129.
Bauer, J. E., and Bianchi, T. S. (2011). “Dissolved organic carbon cycling and transformation,” in Treatise on Estuarine and Coastal Science Vol. 5. eds E. Wolanski, D. S. McLusky (Waltham, MA: Academic Press) 7–67.
Bauer, J. E., Cai, W.-J., Raymond, P. A., Bianchi, T. S., Hopkinson, C. S., and Regnier, P. A. G. (2013). The changing carbon cycle of the coastal ocean. Nature 504, 61–70. doi: 10.1038/nature12857
Benner, R., and Amon, R. M. W. (2015). The size-reactivity continuum of major bioelements in the ocean. Annu. Rev. Mar. Sci. 7, 185–205. doi: 10.1146/annurev-marine-010213-135126
Benner, R., Pakulski, J. D., McCarthy, M., Hedges, J. I., and Hatcher, P. G. (1992). Bulk chemical characteristics of dissolved organic matter in the ocean. Science 255, 1561–1564. doi: 10.1126/science.255.5051.1561
Bergamaschi, B. A., Tsamakis, E., Keil, R. G., Eglinton, T. I., Montlucon, D. B., and Hedges, J. I. (1997). The effect of grain size and surface area on organic matter, lignin and carbohydrate concentration, and molecular compositions in Peru Margin sediments. Geochim. Cosmochim. Acta 61, 1247–1260. doi: 10.1016/s0016-7037(96)00394-8
Billett, M. F., Palmer, S. M., Hope, D., Deacon, C., Storeton-West, R., Hargreaves, K. J., et al. (2004). Linking land-atmosphere-stream carbon fluxes in a lowland peatland system. Global Biogeochem. Cycles 18, 1–12. doi: 10.1029/2003GB002058
Chin, W. C., Orellana, M. V., and Verdugo, P. (1998). Spontaneous assembly of marine dissolved organic matter into polymer gels. Nature 391, 568–572.
Chin, Y. P., Traina, S. J., Swank, C. R., and Backhus, D. (1998). Abundance and properties of dissolved organic matter in pore waters of a freshwater wetland. Limnol. Oceanogr. 43, 1287–1296.
Dai, M., Martin, J. M., and Cauwet, G. (1995). The significant role of colloids in the transport and transformation of organic carbon and associated trace metals (Cd, Cu and Ni) in the Rhône delta (France). Mar. Chem. 51, 159–175. doi: 10.1016/0304-4203(95)00051-r
de Jonge, L. W., Moldrup, P., Rubaek, G. H., Schelde, K., and Djurhuus, J. (2004). Particle leaching and particle-facilitated transport of phosphorus at field scale. Vadose Zo. J. 3, 462–470. doi: 10.2113/3.2.462
De-Campos, A. B., Mamedov, A. I., and Huang, C.-H. (2009). Short-term reducing conditions decrease soil aggregation. Soil Sci. Soc. Am. J. 73, 550–559. doi: 10.2136/sssaj2007.0425
Derjaguin, B., and Landau, L. (1993). Theory of the stability of strongly charged lyophobic sols and of the adhesion of strongly charged particles in solutions of electrolytes. Prog. Surf. Sci. 43, 30–59. doi: 10.1016/0079-6816(93)90013-L
Dhillon, G. S., and Inamdar, S. (2014). Storm event patterns of particulate organic carbon (POC) for large storms and differences with dissolved organic carbon (DOC). Biogeochemistry 118, 61–81. doi: 10.1007/s10533-013-9905-6
Dwivedi, D., Riley, W. J., Torn, M. S., Spycher, N., Maggi, F., and Tang, J. Y. (2017). Mineral properties, microbes, transport, and plant-input profiles control vertical distribution and age of soil carbon stocks. Soil Biol. Biochem. 107, 244–259. doi: 10.1016/j.soilbio.2016.12.019
Everett, D. H. (1972). Manual of symbols and terminology for physicochemical quantities and units, Appendix II: definitions, terminology and symbols in colloid and surface chemistry. Pure Appl. Chem. 31, 577–638. doi: 10.1351/pac197231040577
Feng, W., Plante, A. F., Aufdenkampe, A. K., and Six, J. (2014). Soil organic matter stability in organo-mineral complexes as a function of increasing C loading. Soil Biol. Biochem. 69, 398–405. doi: 10.1016/j.soilbio.2013.11.024
Gimbert, L. J., Haygarth, P. M., Beckett, R., and Worsfold, P. J. (2005). Comparison of centrifugation and filtration techniques for the size fractionation of colloidal material in soil suspensions using sedimentation field-flow fractionation. Environ. Sci. Technol. 39, 1731–1735. doi: 10.1021/es049230u
Gu, B. H., Schmitt, J., Chen, Z. H., Liang, L. Y., and McCarthy, J. F. (1994). Adsorption and desorption of natural organic matter on iron oxide: mechanisms and models. Environ. Sci. Technol. 28, 38–46. doi: 10.1021/es00050a007
Guo, L., and Macdonald, R. W. (2006). Source and transport of terrigenous organic matter in the upper Yukon River: evidence from isotope (delta C-13, Delta C-14, and delta N-15) composition of dissolved, colloidal, and particulate phases. Global Biogeochem. Cycles 20, 1–12. doi: 10.1029/2005gb002593
Guo, L., and Santschi, P. H. (2007). “4 Ultrafiltration and its Applications to Sampling and Characterisation of Aquatic Colloids,” in Environmental Colloids and Particles: Behaviour, Separation and Characterization, eds K. J. Wilkinson and J. R. Lead (West Sussex: Wiley), 159–221.
Guo, L. D., Coleman, C. H., and Santschi, P. H. (1994). The distribution of colloidal and dissolved organic-carbon in the Gulf-Of-Mexico. Mar. Chem. 45, 105–119. doi: 10.1016/0304-4203(94)90095-7
Guo, L. D., Lehner, J. K., White, D. M., and Garland, D. S. (2003). Heterogeneity of natural organic matter from the Chena River, Alaska. Water Res. 37, 1015–1022. doi: 10.1016/s0043-1354(02)00443-8
Guo, L. D., and Santschi, P. H. (1997). Isotopic and elemental characterization of colloidal organic matter from the Chesapeake Bay and Galveston Bay. Mar. Chem. 59, 1–15. doi: 10.1016/s0304-4203(97)00072-8
Guo, L. D., Santschi, P. H., and Warnken, K. W. (1995). Dynamics of dissolved organic carbon (DOC) in oceanic environments. Limnol. Oceanogr. 40, 1392–1403.
Hama, T., Yanagi, K., and Hama, J. (2004). Decrease in molecular weight of photosynthetic products of marine phytoplankton during early diagenesis. Limnol. Oceanogr. 49, 471–481. doi: 10.4319/lo.2004.49.2.0471
Hansell, D. A., Carlson, C. A., Repeta, D. J., and Schlitzer, R. (2009). Dissolved organic matter in the ocean: a controversy stimulates new insights. Oceanography. 22, 202–211. doi: 10.5670/oceanog.2009.109
Hicks Pries, C. E., Sulman, B. N., West, C., O'Neill, C., Poppleton, E., Porras, R. C., et al. (2018). Root litter decomposition slows with soil depth. Soil Biol. Biochem. 125, 103–114. doi: 10.1016/J.SOILBIO.2018.07.002
Hoffmann, S. R., Shafer, M. M., and Armstrong, D. E. (2007). Strong colloidal and dissolved organic ligands binding copper and zinc in rivers. Environ. Sci. Technol. 41, 6996–7002. doi: 10.1021/es070958v
Hu, J.-D., Zevi, Y., Kou, X.-M., Xiao, J., Wang, X.-J., and Jin, Y. (2010). Effect of dissolved organic matter on the stability of magnetite nanoparticles under different pH and ionic strength conditions. Sci. Total Environ. 408, 3477–3489. doi: 10.1016/j.scitotenv.2010.03.033
Jin, Y., and Flury, M. (2002). Fate and transport of viruses in porous media. Adv. Agron. 77, 39–102. doi: 10.1016/s0065-2113(02)77013-2
Kang, P.-G., and Mitchell, M. J. (2013). Bioavailability and size-fraction of dissolved organic carbon, nitrogen, and sulfur at the Arbutus Lake watershed, Adirondack Mountains, NY. Biogeochemistry 115, 213–234. doi: 10.1007/s10533-013-9829-1
Kepkay, P. E., Niven, S. E. H., and Jellet, J. F. (1997). Colloidal organic carbon and phytoplankton speciation during a coastal bloom. J. Plankton Res. 19, 369–389. doi: 10.1093/plankt/19.3.369
Kepkay, P. E., Niven, S. E. H., and Milligan, T. G. (1993). Low molecular weight and colloidal DOC production during a phytoplankton bloom. Mar. Ecol. Prog. Ser. 100, 233–244. doi: 10.3354/meps100233
Kleber, M., Eusterhues, K., Keiluweit, M., Mikutta, C., Mikutta, R., and Nico, P. S. (2015). “Chapter one - mineral–organic associations: formation, properties, and relevance in soil environments,” in Advances in Agronomy, ed L. S. Donald (Academic Press), 1–140.
Kleber, M., Sollins, P., and Sutton, R. (2007). A conceptual model of organo-mineral interactions in soils: self-assembly of organic molecular fragments into zonal structures on mineral surfaces. Biogeochemistry 85, 9–24. doi: 10.1007/s10533-007-9103-5
Koven, C. D., Riley, W. J., Subin, Z. M., Tang, J. Y., Torn, M. S., Collins, W. D., et al. (2013). The effect of vertically resolved soil biogeochemistry and alternate soil C and N models on C dynamics of CLM4. Biogeosciences 10, 7109–7131. doi: 10.5194/bg-10-7109-2013.
Kretzschmar, R., Borkovec, M., Grolimund, D., and Elimelech, M. (1999). Mobile subsurface colloids and their role in contaminant transport. Adv. Agron. 66, 121–193.
Lehmann, J., and Kleber, M. (2015). The contentious nature of soil organic matter. Nature 528, 60–68. doi: 10.1038/nature16069
Martin, J. M., Dai, M. H., and Cauwet, G. (1995). Significance of colloids in the biogeochemical cycling of organic-carbon and trace-metals in the venice lagoon (italy). Limnol. Oceanogr. 40, 119–131.
Mccarthy, J. F., and Zachara, J. M. (1989). Subsurface transport of contaminants - mobile colloids in the subsurface environment may alter the transport of contaminants. Environ. Sci. Technol. 23, 496–502.
McKnight, D. M., Harnish, R., Wershaw, R. L., Baron, J. S., and Schiff, S. (1997). Chemical characteristics of particulate, colloidal, and dissolved organic material in loch vale watershed, rocky mountain national park. Biogeochemistry 36, 99–124. doi: 10.1023/a:1005783812730
Moran, S. B., and Moore, R. M. (1989). The distribution of colloidal aluminum and organic carbon in coastal and open ocean waters off Nova Scotia. Geochim. Cosmochim. Acta 53, 2519–2527. doi: 10.1016/0016-7037(89)90125-7
Nissenbaum, A., and Kaplan, I. R. (1972). Chemical and isotopic evidence for the in situ origin of marine humic substances. Limnol. Oceanogr. 17, 570–582. doi: 10.4319/lo.1972.17.4.0570
Ogawa, H., and Ogura, N. (1992). Comparison of two methods for measuring dissolved organic carbon in sea water. Nature 356, 696–698. doi: 10.1038/356696a0
Peters, G.-J. Y. (2017). Userfriendlyscience: Quantitative Analysis Made Accessible. R Package. Version 0.6–1. doi: 10.17605/OSF.IO/TXEQU
Philippe, A., and Schaumann, G. E. (2014). Interactions of dissolved organic matter with natural and engineered inorganic colloids: a review. Environ. Sci. Technol. 48, 8946–8962. doi: 10.1021/es502342r
Pokrovsky, O. S., Schott, J., and Dupre, B. (2006). Trace element fractionation and transport in boreal rivers and soil porewaters of permafrost-dominated basaltic terrain in Central Siberia. Geochim. Cosmochim. Acta 70, 3239–3260. doi: 10.1016/j.gca.2006.04.008
Pourret, O., Dia, A., Davranche, M., Gruau, G., Henin, O., and Angee, M. (2007). Organo-colloidal control on major- and trace-element partitioning in shallow groundwaters: confronting ultrafiltration and modelling. Appl. Geochem. 22, 1568–1582. doi: 10.1016/j.apgeochem.2007.03.022
R core team (2017). R: A Language and Environment for Statistical Computing. Vienna: The R Project for Statistical Computing. Available online at: http://www.R-project.org/
Raymond, P. A., Saiers, J. E., and Sobczak, W. V. (2016). Hydrological and biogeochemical controls on watershed dissolved organic matter transport: pulse-shunt concept. Ecology 97, 5–16. doi: 10.1890/14-1684.1
Repeta, D. J., Quan, T. M., Aluwihare, L. I., and Accardi, A. (2002). Chemical characterization of high molecular weight dissolved organic matter in fresh and marine waters. Geochim. Cosmochim. Acta 66, 955–962. https://doi.org/10.1016/S0016-7037(01)00830-4
Rice, J. A., and MacCarthy, P. (1991). Statistical evaluation of the elemental composition of humic substances. Org. Geochem. 17, 635–648. doi: 10.1016/0146-6380(91)90006-6
Rostad, C. E., Leenheer, J. A., and Daniel, S. R. (1997). Organic carbon and nitrogen content associated with colloids and suspended particulates from the Mississippi River and some of its tributaries. Environ. Sci. Technol. 31, 3218–3225. doi: 10.1021/es970196b
Ryan, J. N., and Gschwend, P. M. (1990). Colloid mobilization in 2 atlantic coastal-plain aquifers - field studies. Water Resour. Res. 26, 307–322.
Schulten, H. R., and Leinweber, P. (2000). New insights into organic-mineral particles: composition, properties and models of molecular structure. Biol. Fertil. Soils 30, 399–432. doi: 10.1007/s003740050020
Sholkovitz, E. R. (1976). Flocculation of dissolved organic and inorganic matter during the mixing of river water and seawater. Geochim. Cosmochim. Acta 40, 831–845. doi: 10.1016/0016-7037(76)90035-1
Sholkovitz, E. R., Boyle, E. A., and Price, N. B. (1978). The removal of dissolved humic acids and iron during estuarine mixing. Earth Planet. Sci. Lett. 40, 130–136. doi: 10.1016/0012-821X(78)90082-1
Thompson, A., Chadwick, O. A., Boman, S., and Chorover, J. (2006). Colloid mobilization during soil iron redox oscillations. Environ. Sci. Technol. 40, 5743–5749. doi: 10.1021/es061203b
Vendelboe, A. L., Schjonning, P., Moldrup, P., Jin, Y., Merbach, I., and de Jonge, L. W. (2012). Colloid release from differently managed loess soil. Soil Sci. 177, 301–309. doi: 10.1097/SS.0b013e3182506dd3
Verdugo, P., Alldredge, A. L., Azam, F., Kirchman, D. L., Passow, U., and Santschi, P. H. (2004). The oceanic gel phase: a bridge in the DOM-POM continuum. Mar. Chem. 92, 67–85. doi: 10.1016/j.marchem.2004.06.017
Verwey, E. J. W., Overbeek, J. T. G., and Nes, K. (1947). Theory of The Stability of Lyophobic Colloids: The Interaction of Sol Particles Having an Electric Double Layer. New York, NY: Elsevier Pub. Co.
Ward, N. D., Bianchi, T. S., Medeiros, P. M., Seidel, M., Richey, J. E., Keil, R. G., et al. (2017). Where carbon goes when water flows: carbon cycling across the aquatic continuum. Front. Mar. Sci. doi: 10.3389/fmars.2017.00007
Wen, L. S., Santschi, P. H., Gill, G. A., Paternostro, C. L., and Lehman, R. D. (1997). Colloidal and particulate silver in river and estuarine waters of Texas. Environ. Sci. Technol. 31, 723–731. doi: 10.1021/es9603057
Wilkinson, K. J., and Lead, J. R., and Wiley InterScience (2007). Environmental colloids and particles: Behaviour, separation and characterisation. Chichester: John Wiley & Sons.
Yan, J. (2016). Quantification and Characterization of Mobile Colloids: Their Potential Role in Carbon Cycling Under Varying Redox Conditions. Doctoral dissertation, University of Delaware: Newark, DE.
Yan, J., Lazouskaya, V., and Jin, Y. (2016). Soil colloid release affected by dissolved organic matter and redox conditions. Vadose Zo. J. 15, 1–10. doi: 10.2136/vzj2015.02.0026
Keywords: colloids, colloid size distribution, colloidal organic carbon, dissolved organic carbon, soil organic matter continuum, carbon cycles, multiple ecosystems
Citation: Yan J, Manelski R, Vasilas B and Jin Y (2018) Mobile Colloidal Organic Carbon: An Underestimated Carbon Pool in Global Carbon Cycles? Front. Environ. Sci. 6:148. doi: 10.3389/fenvs.2018.00148
Received: 06 June 2018; Accepted: 22 November 2018;
Published: 14 December 2018.
Edited by:
Balwant Singh, University of Sydney, AustraliaReviewed by:
Steven Sleutel, Ghent University, BelgiumSanjai J. Parikh, University of California, Davis, United States
Copyright © 2018 Yan, Manelski, Vasilas and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Jin, eWppbkB1ZGVsLmVkdQ==
†Present Address: Jing Yan, Life and Environmental Sciences, University of California, Merced, Merced, CA, United States