 Jingying Xu1
Jingying Xu1 Van Liem-Nguyen
Van Liem-Nguyen Stefan Bertilsson
Stefan Bertilsson Erik Björn
Erik Björn Andrea G. Bravo
Andrea G. Bravo- 1Department of Ecology and Genetics, Limnology, Uppsala University, Uppsala, Sweden
- 2Department of Chemistry, Umeå University, Umeå, Sweden
- 3Swedish University of Agricultural Sciences, Uppsala, Sweden
- 4Institute of Marine Sciences, Spanish National Research Council, Spain
The potent neurotoxin methylmercury (MeHg) is a major concern due to its negative effects on wildlife and human health. Boreal wetlands play a crucial role in Hg cycling on a global scale, and therefore, it is crucial to understand the biogeochemical processes involved in MeHg formation in this landscape element. By combining high-throughput hgcA amplicon sequencing with molecular barcoding, we reveal diverse clades of potential HgII methylators in a wide range of wetland soils. Among Bacteria, Desulfuromonadota (14% of total reads), Desulfurobacterota_A, and Desulfurobacterota (up to 6% of total reads), previously classified as Deltaproteobacteria, were important members of the hgcA+ microbial community in the studied wetlands. We also identified Actinobacteriota (9.4% of total reads), Bacteroidota (2% of total reads), and Firmicutes (1.2% of total reads) as members of the hgcA+ microbial community. Within Archaea, Methanosarcinales represented up to 2.5% of the total reads. However, up to half of the hgcA+ community could not be resolved beyond domain Bacteria. Our survey also shows that local physicochemical conditions, such as pH, nutrient concentrations, water content, and prevailing redox states, are important for shaping the hgcA+ microbial community structure across the four studied wetlands. Furthermore, we observed a significant correlation between HgII methylation rate constants and the structure of the hgcA+ microbial community. Our findings expand the current knowledge on the hgcA+ microbial community composition in wetlands and the physicochemical factors underpinning spatial heterogeneity in such communities.
1 Introduction
Wetlands cover 6% of the world’s land surface and contain about 12% of the global carbon pool (Erwin, 2009). Hence, these systems play an important role in different global biogeochemical cycles, such as that of carbon and mercury (Hg) (Mitra et al., 2003; Tjerngren et al., 2012b). For example, wetlands take up and store carbon and release carbon dioxide and methane (Mitra et al., 2003). Also, wetlands have been identified as important sources or even sinks of the toxic methylmercury (MeHg) (St. Louis et al., 1994; Hall et al., 2008; Tjerngren et al., 2012b). St. Louis et al. (1994) found 26–79 times higher yields of MeHg from wetland portions of catchments than from purely upland areas. Mass-balance estimates have indicated that purely upland catchments and lakes are sites of MeHg retention or demethylation (St. Louis et al., 1994), and catchments with wetland areas are sites of net MeHg production (St. Louis et al., 1994; Mitchell et al., 2008b; Hall et al., 2008). It is also shown that the creation of wetlands by the flooding of boreal soils led to increased MeHg production (Gilmour et al., 1998; Hall et al., 2008; Mitchell et al., 2008a; Mitchell et al., 2008b). The wetland type, percentage wetland area, or annual water yield appear to have a consistent effect on the magnitude of MeHg production (Louis et al., 1996). Furthermore, because wetlands are parts of large watersheds and thereby are hydrologically connected to adjacent systems, there is a high risk for MeHg that is produced in wetlands to be transported to downstream waters. This is critical as MeHg bioaccumulates and biomagnifies along the aquatic food webs, and this poses severe risks to the environment and humans (Boening, 2000). Therefore, an understanding of the potential MeHg formation in boreal wetlands is crucial for robust risk assessment and rational landscape management.
The methylation of HgII to MeHg in the environment is governed by two factors: the activities of microbial communities (Bravo et al., 2017) and the availability of HgII for use by these methylators (Schaefer and Morel, 2009; Jonsson et al., 2014; Mazrui et al., 2016). Some members of sulfate-reducing bacteria (SRB) (Gilmour et al., 1992; King et al., 2001; Gilmour et al., 2013), iron-reducing bacteria (FeRB) (Fleming et al., 2006; Kerin et al., 2006; Bravo et al., 2018b), methanogens (Hamelin et al., 2011; Yu et al., 2013), Firmicutes (Gilmour et al., 2013), acetogens, and recently also obligate fermenting lineages (McDaniel et al., 2020b; Peterson et al., 2020) have so far been implicated in HgII methylation. The identification of two genes (hgcAB) essential for HgII methylation (Parks et al., 2013) provides the means to more directly characterize the diversity of potential HgII methylators in contrasting environmental niches, such as soils (Liu et al., 2014; Liu et al., 2018; Vishnivetskaya et al., 2018; Xu et al., 2019), lakes, reservoirs (Bravo et al., 2018a; Bravo et al., 2018b; Jones et al., 2019; Jones et al., 2020), coastal sediments, animal guts, extremes of pH, salinity environments (Gilmour et al., 2013), the global ocean (Podar et al., 2015; Villar et al., 2020), Baltic Sea (Capo et al., 2020), and even in the Southern Ocean (Gionfriddo et al., 2016). However, for most of the recently discovered microorganisms carrying the hgcA gene (hgcA+), their role in the environment and their capacity to actually methylate HgII has yet to be demonstrated. Progress in that direction has been made by testing the capacity to methylate HgII of hgcA+ isolates (Gilmour et al., 2018; Yu et al., 2018) or by amendment experiments with specific substrates or inhibitors (Schaefer et al., 2014; Christensen et al., 2017; Bravo et al., 2018a; Schaefer et al., 2020). However, although numerous studies show that hgcA+ microorganisms are present and potentially active in a wide range of environments, the ecological underpinnings of the hgcA+ microbial community remains to be elucidated across systems. In lakes, previous studies reveal the composition of organic matter and the structure on the whole microbial community (Bravo et al., 2018a) as important for the hgcA+ microbial community structure. However, still little is known on the factors influencing the structure and function of hgcA+ microbial communities in the environment or the effect of the hgcA+ microbial community composition on HgII methylation rate constants. Jones et al. (2020) amended freshwater lake sediments with different sulfate concentrations and observed that the experimental plots with the lowest sulfate concentrations presented the highest hgcA+ diversity and the highest HgII methylation rate, highlighting that diverse hgcA+ communities that do not reduce sulfate can methylate HgII as effectively as communities dominated by sulfate-reducing populations (Jones et al., 2020). Although this study emphasizes that the hgcA+ microbial community structure is relevant for the HgII methylation rate, further evidence is needed to confirm the effect of compositional changes of hgcA+ communities in HgII methylation rates.
In Swedish boreal wetlands, some studies have described the complex geochemical parameters determining HgII chemical speciation (Liem-Nguyen et al., 2017; Skyllberg, 2008), HgII methylation (Liem-Nguyen et al., 2021), and the organisms potentially involved in the process (Tjerngren et al., 2012b; Schaefer et al., 2014; Schaefer et al., 2020). A recent study performed a comprehensive thermodynamic speciation model, which provided new insights into the speciation of HgII in boreal wetland porewaters but did not demonstrate any clear relationship between HgII methylation rates and the calculated chemical speciation (Liem-Nguyen et al., 2021). In contrast, significant positive relationships were observed between HgII methylation rates and the sum of low molecular mass thiol compounds of biological origin. Although the authors could not conclude whether low molecular mass thiols might kinetically control the size and composition of the HgII pool available for microbial uptake and/or whether low molecular mass thiols were produced by HgII methylating microorganisms, their findings underline the role of microbial communities in determining HgII methylation rates. Knowledge about HgII methylating microbial composition, the factors controlling their activity, and their effects on HgII methylation rates will, thus, help us obtain a mechanistic understanding of these important ecosystem-level processes. The primary goal of the present paper is to expand on this and, more specifically, 1) map HgII methylating microbial communities in four boreal wetlands in Sweden, 2) identify geochemical characteristics important for shaping these communities, and 3) determine the interplay between hgcA+ microbial communities and HgII methylation rates. For this purpose, high-throughput Illumina sequencing of amplified hgcA genes combined with molecular barcoding and detailed geochemical characteristics were carried out to study hgcA+ microbial communities in 81 samples from boreal wetlands and, in this way, shed light on the organisms responsible for MeHg formation in the boreal landscape.
2 Materials and Methods
2.1 Site Description
Four boreal wetlands situated within predominantly forested watersheds in Sweden were selected for this study. These systems cover variable biogeochemical features with regards to climate, acidity, organic carbon, sulfide concentration, and nutrient status as well as ancillary chemistry. The selected wetlands were categorized as 1) a northern group of poor-fen type of wetlands Storkälsmyran (SKM) and Kroksjön (KSN), the latter with some open water, and 2) a southern group of bog-fen sites, including Långedalen (LDN) and the more nutrient-rich southern wetland Gästern (GTN). A brief map of sampling sites within the wetlands and more detailed wetland descriptions can be found in Tjerngren et al. (2012a), Liem-Nguyen et al. (2017), and Liem-Nguyen et al. (2021).
2.2 Soil and Porewater Sampling
In total, 81 soil samples were collected from 17 sites across four boreal wetlands at 5 cm intervals starting from the top of the water table to 25 cm below this interface. The top few centimeters of the dry topsoil above the water table was discarded. On average, five soil profiles (0–25 cm) were collected at each site to probe the spatial variability within the sites. Soil samples were preserved in an icebox in the field prior to being transferred to the lab within 1 h. Once in the laboratory, an aliquot was immediately frozen at −80°C for DNA extraction. Porewater samples were collected from the same depths as the soil samples using a custom device consisting of a vacuum pump and a perforated Teflon probe. During sampling, N2 gas was flushed through the tubing and bottle to keep the system anoxic.
Ziploc plastic bags were used to contain soil samples and were squeezed to minimize the remaining air in the bags and, hence, avoid further oxidation. Soil and porewater samples were handled inside a glovebox filled with N2 within 1 h of sampling at a field laboratory station located walking distance from the site. Within 1 h, porewater samples were vacuum filtrated through a 0.22 μm membrane filter (500 ml funnel filter, Sarstedt). More details on the sampling strategy can be found in Liem-Nguyen et al. (2017) and Liem-Nguyen et al. (2021).
2.3 Chemical Analyses
All the soil samples were analyzed for pH, water content, total Hg (THg), and MeHg. THg in soils was determined by solid combustion atomic absorption spectrometry using an AMA-254 Advanced Mercury Analyzer (LECO), and MeHg was acid extracted, ethylation derivatized, and determined by isotope dilution analysis (IDA) using gas chromatography inductively coupled plasma mass spectrometry (ICPMS) (Lambertsson et al., 2001). A detailed geochemical characterization of the studied wetland soils and porewater is presented in Supplementary Tables S1, S2 and in Liem-Nguyen et al. (2021).
The HgII methylation potential (km, d−1) was determined using two different enriched Hg isotope tracers, referred to as 198HgII(aq) and 200HgII–NOM(ads). Detailed information on the preparation of the enriched Hg stable isotopes can be found in Liem-Nguyen et al. (2021). THg in porewater was measured by IDA using Hg cold vapor generation coupled to ICPMS. MeHg in porewater was analyzed by IDA using purge-and-trap thermal desorption coupled to gas chromatography ICPMS (Lambertsson and Björn, 2004). Dissolved organic carbon was measured using a TOC-VCPH (Shimadzu), and FeII and FeIII were determined by UV absorption spectrophotometry (Viollier et al., 2000). The concentrations of SO42−, Cl−, PO43−, NH4+, and NO3− in porewater were measured by ion chromatography. The HgII concentrations in porewater were calculated by subtracting MeHg from THg. Total thiol in freeze-dried porewater were determined using Sulfur K-edge X-ray absorption near edge structure spectroscopy (S K-edge XANES) and Hg LIII-edge extended X-ray absorption fine structure (EXAFS) spectroscopy as described by Liem-Nguyen et al. (2017). Concentrations of 16 low molecular mass (LMM) thiols were determined in porewater using solid-phase extraction online preconcentration hyphenated to liquid chromatography tandem mass spectrometry (SPE/LC-MS/MS) (Liem-Nguyen et al., 2015).
The certified reference materials MESS-3 (0.091 ± 0.009 mg Hg/kg), ORMS-5 (26.2 ± 1.3 pg Hg/g), and ERM-CC580 (0.075 ± 0.004 mg/kg) were used for quality control of the total Hg and MeHg measurements in soil and porewater samples. The measured concentrations were in good agreement (95%–105%) with the reference materials. The chemical characteristics of the wetland sites can be also found elsewhere (Liem-Nguyen et al., 2017; Liem-Nguyen et al., 2021).
2.4 Microbiological Analyses
DNA was isolated from 0.2 g soil (wet weight) using the PowerSoil DNA isolation kit (Mo Bio Laboratories, Carlsbad, CA, United States). To target the functional gene hgcA, which plays an essential role in HgII methylation, previously published hgcA primers (hgcA_ 261F = CGGCATCAAYGTCTGGTGYGC, hgcA_912R = GGTGTAGGGGGTGCAGCCSGTRWARKT) (Schaefer et al., 2014) were adapted for high-throughput Illumina sequencing. Accordingly, HPLC-purified primers linked with generic adaptors were used for a first stage PCR. In a second stage PCR, primers binding to the generic adaptors but also featuring standard Illumina adaptors and sample-specific barcodes were used (Multiplex_fwd = AATGATACGGCGACCACCGAGA {TCTACAC}-[i5 barcode] ACACTCTTTCCCTACACGACG, Multiplex_rev = CAAGCAGAAGACGGCATACGAGAT-[i7 barcode] GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT), enabling us to include all the first stage PCR products on a single lane for parallelized Illumina sequencing.
The hgcA was first amplified in 50 μL volume with 1x Phusion GC Buffer, 0.2 mM dNTP mix, 5% DMSO, 0.1 μM of each adaptor-linked primer, 7 μg/μL BSA, 4 μL extracted DNA template, and 1.0 U Phusion high fidelity DNA polymerase (NEB, United Kingdom) for an initial denaturation of 2 min at 98°C followed by 35 amplification cycles (10 sat 96°C, 30 sat 56.5°C, and 45 sat 72°C) and a final extension at 72°C for 7 min. Following this initial step, a second PCR was conducted to add sample-specific molecular barcodes. Reactions were carried out in 20-μL volumes using 1x Q5 reaction buffer, 0.2 mM dNTP mix, 0.1 μM barcode primers, AMPure XP-purified first stage PCR products, and 1.0 U Q5 high-fidelity DNA polymerase (NEB, United Kingdom) for an initial denaturation of 30 s at 98°C followed by 18 cycles (10 s at 98°C, 30 s at 66°C, and 30 s at 72°C) and a final extension at 72°C for 2 min. Amplicons of hgcA genes were assessed by gel electrophoresis and GelRed visualization on a 1% agarose gel (Invitrogen, United States) prior to a final purification by Agencourt AMPure XP (Beckman Coulter, United States) after both steps of PCR. Quantification of purified amplicons from the second stage PCR was performed using the PicoGreen kit (Invitrogen).
Subsequent amplicon sequencing and sequence processing was carried out, largely following the protocol described in Sinclair et al. (2015) using the MiSeq instrument using illumina MiSeq sequencing performed by the SNP/SEQ SciLifeLab facility hosted by Uppsala University using 2x300bp chemistry. Chimera identification and operational taxonomic unit (OTU) clustering at 3% sequence dissimilarity was done using UPARSE. Only the forward-read sequences were used in data analysis as PCR products were too short to obtain robustly overlapping forward and reserve reads. Low-quality sequences were filtered and trimmed using SICKLE (Joshi and Fass, 2011), and adaptors were removed by using CUTADAPT (Martin, 2011). Subsequent processing of reads was performed by USEARCH version 8.0, and reads were clustered at 60% identity cutoff using cd-hit-est. We further performed a manual check of the protein sequences using the knowledge from the original paper of Parks et al. (2013) that described the unique motif from hgcA [NVWCA(A/G/S)GK] and hgcB protein sequences [C(M/I)EC(G/S)(A/G)C]. Last, pplacer (Matsen et al., 2010), and a likelihood cutoff of 0.8 search was used for taxonomical annotation with a manually curated a GTDB-derived database release 89 (McDaniel et al., 2020a).
2.5 Statistical Analyses
Microbial community compositions between samples were compared at the OTU level using nonmetric multidimensional scaling (nMDS) based on Bray-Curtis similarities using R 3.3.2 (https://www.r-project.org/). Relative dissimilarities (or distances) among the samples were computed according to the resemblance matrix using the vegan package (Oksanen et al., 2017). Information on the common set of samples from community composition based on Bray–Curtis similarities and that from geochemical variables based on Euclidean distance were presented in a single ordination. A combined nMDS plot with bubble and vector plots of geochemical factors projected on the same ordination of community composition was constructed to reveal the relationship between community compositions and correlated geochemical variables (Clarke and Gorley, 2015). Pearson’s correlation coefficients (R) between variables were tested using a significance level of alpha <0.05. The false discovery rate of significant correlations was corrected following Benjamini and Hochberg, (1995), and using the p.adjust function in R. ANOSIM, tests implemented in the vegan package were used to test for differences in microbial community structure across sites and depths. The function bioenv was used to find the best subset of environmental variables so that the Euclidean distances of scaled environmental variables have the maximum (rank) correlation with community dissimilarities (Bray–Curtis). Standard Mantel tests were used to investigate correlations between the hgcA+ microbial community and the most influential environmental variables. Euclidean distance metrics were used for the most influential environmental variables (squared-root transformed), and Bray–Curtis dissimilarities were used for hgcA+ microbial community composition data (square-root transformed). Both distance matrices were calculated with the vegdist function. Redundancy analysis (RDA) was performed using the rda function in the R package vegan to further explore the relationships between HgII-methylating microbial community composition and the studied environmental factors. RDA seeks a series of linear combinations of the explanatory variables that best explain the variation in the response matrix (i.e., HgII-methylating microorganisms) but requires the number of explanatory variables to be below or equal to the number of observations (i.e., number of wetland soil samples) and the explanatory variables to not be intercorrelated. Intercorrelations between physicochemical variables were inspected by computing the variance inflation factor (VIF). All the variables with a VIF of >10 were excluded from RDA analysis. Prior to this analysis, the variables had been scaled and centered.
3 Results
3.1 Distribution of hgcA+ Microbial Communities Across all Four Wetlands
A total of 4,535,861 high-quality hgcA sequences remained after quality control and chimera removal (2664–106,256 reads per sample). The data were rarefied to the remaining least covered sample (2664 sequences). The final combined sequence data set (215,784 reads) were clustered into 618 OTUs using a sequence identity threshold of 60%. In total, 24 taxa at the phyla level, 42 taxa at the class level, 45 taxa at the order level, and 53 taxa at the family level were detected from all the soil samples across the four wetlands. The combined richness detected for random subsets of analyzed samples approached saturation, suggesting that our subsampled data set covered the diverse hgcA+ microbial communities inhabiting the wetlands (Supplementary Figure S1).
Among Bacteria, Desulfuromonadota (14% of total reads), Desulfurobacterota_A, and Desulfurobacterota (up to 6% of total reads) were abundant in all four studied sites (Table 1). These three phyla, previously classified as Deltaproteobacteria in the NCBI taxonomy, each contain confirmed HgII methylators (Ranchou-Peyruse et al., 2009; Yu et al., 2012; Table 1). Within these three phyla, Desulfuromonadales is the most abundant order, representing up to 13.8% of the total reads (Table 1). Noteworthy is that, within the Desulfuromonadales order, the Geopsychrobacteraceae family reached 10% of the total reads. Actinobacteriota (Actinobacteria in the NCBI taxonomy) were also quite abundant, representing up to 9.5% of the total hgcA+ reads. About 27% of the total sequences could not be taxonomically assigned (Unclassified), and 35% of the reads could not be assigned beyond the rank of Bacteria (Unclassified Bacteria) (Table 1). Within the Firmicutes, the Syntrophomonadales and the Desulfitobacteriales orders contributed to 2% of the total reads (Supplementary Table S3). Archaea and, in particular, the order Methanosarcinales represented about 3% of the total reads. At the OTU level, hgcA+ microbial communities were more similar within, as compared to between wetlands (Figure 1). Indeed, hgcA+ microbial community composition showed a significant clustering related to the studied site (SKM, KSN, LDN, and GTN; ANOSIMbySITER = 0.38, p < 0.001, Figure 1) with nMDS1 axis one reflecting a gradient from Northern (SKM and KSN) to Southern (LDN and GTN) wetlands. In contrast, no significant separation of communities was seen between different depth layers (ANOSIMbyDEPTHR = 0.01, p < 0.24, Figure 1A).
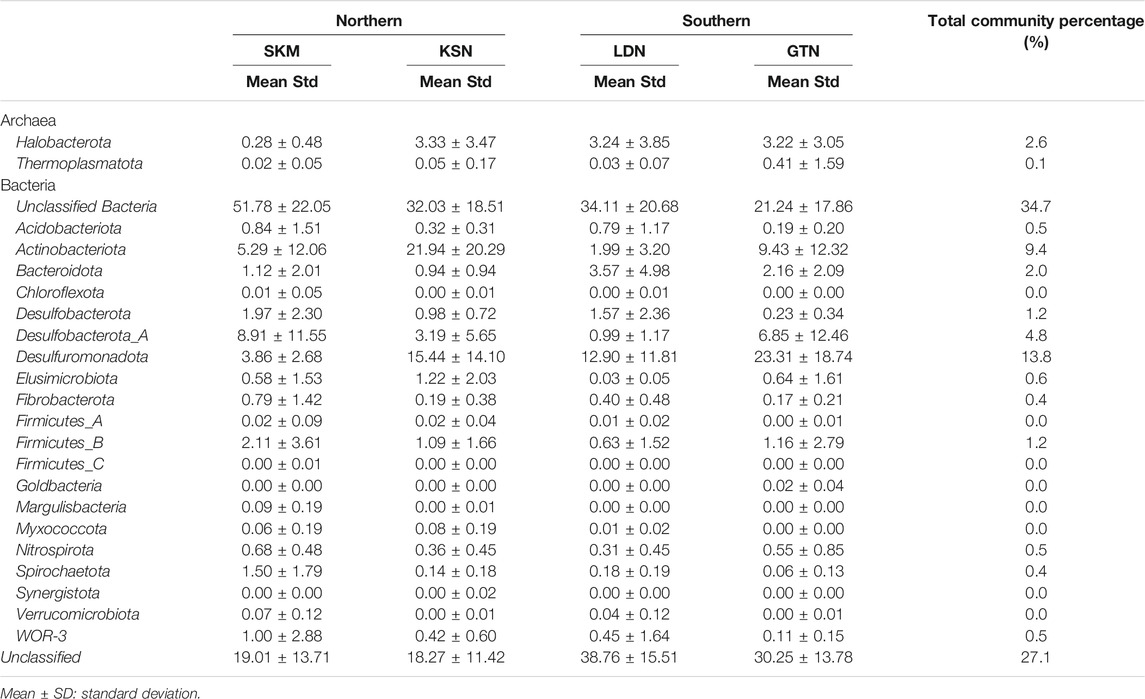
TABLE 1. Relative hgcA sequence abundances (%) at levels of phyla for the combined 81-sample data set that includes all four wetlands.
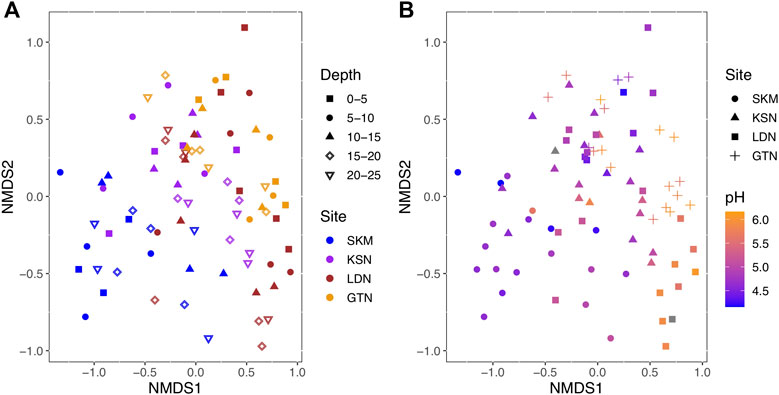
FIGURE 1. Nonmetric multidimensional scaling (nMDS) ordination of HgII methylating microbial communities based on the Bray–Curtis dissimilarity of community composition (stress = 0.1). (A) Sites and depth are color- and shape-coded, respectively. (B) Sites and pH are shape and color-coded, respectively.
3.2 Potential Mercury Methylators Within Individual Wetlands
Among the classified hgcA+ prokaryotes, Syntrophales, OPB41, Desulfovibrionales, and Desulfuromonadales, Syntrophomonadales were the predominant orders in all four individual wetlands. SKM had the highest number of unclassified Bacteria (up to 52%, Figure 2). Spirochaetales, Syntrophomonadales, Desulfovibrionales, Syntrophales, and Syntrophorhabdales featured higher abundances in SKM compared with the other wetlands (Supplementary Table S3 and Figure 2). In contrast, Desulfuromonadales reached their minimum relative abundance at SKM compared with the other wetlands. In SKM, hgcA+ microorganisms that grouped into SRB, including syntrophs, featured the highest relative abundances (Supplementary Table S3). In KSN, the hgcA+ community hosted more Actinobacteriota (∼22% of the total reads of KSN samples) compared with the other wetlands, and Bacteroidota were the least abundant among all the studied wetlands (Table 1). Compared to SKM, KSN presented a much higher relative abundance of Desulfuromonadales and Elusimicrobiales and also Methanosarcinales (Figure 2 and Supplementary Table S3). In the Southern wetlands, Desulfuromonadales, Desulfovibrionales, and Elusomicorbiales were more abundant in GTN compared to LDN (Figure 2 and Supplementary Table S3). In addition, the relative abundance of Methanosarcinales were similar in LDN and GTN and similar to that seen in KSN.
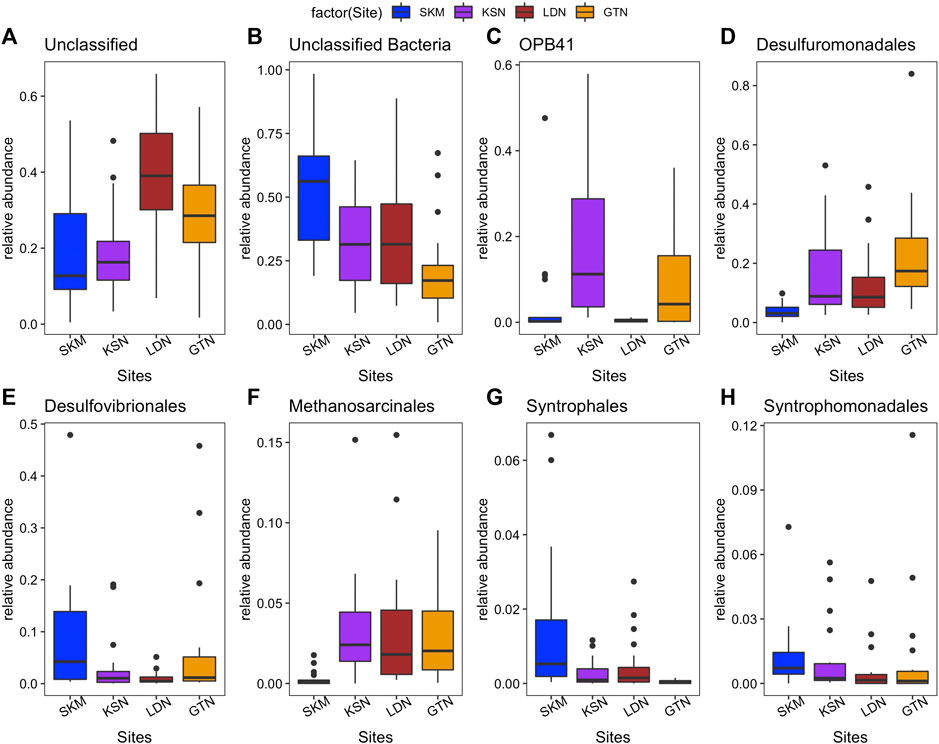
FIGURE 2. Relative abundance (%) of most abundant order-level groups of hgcA+ microorganisms in four studied wetlands based on high throughput hgcA amplicon sequencing [(A) Unclassified; (B) Unclassified Bacteria; (C) OPB41 (Actinobacteriota phylum); (D) Desulfuromonadales; (E) Desulfovibrionales; (F) Methanosarcinales, (G) Syntrophales, and (H) Syntrophomonadales.
3.3 Geochemical Factors Affecting HgcA+ Microbial Communities Across Wetlands
We used RDA, a well-recognized multivariate analysis method in microbial ecology (Ramette, 2007), to link a matrix of multiple response variables (i.e., the abundance of the hgcA+ OTU) to a corresponding matrix of explanatory variables (i.e., the most influential geochemical parameters). However, as a prerequisite for RDA modeling, the geochemical variables selected should not be intercorrelated in order to optimize the precision in the prediction of the hgcA+ OTUs (Ramette, 2007). Hence, some variables were excluded, and in the final RDA model, the explanatory variables included phosphate (µg/L), FeII/FeIII, total_LMM thiols (nM), H2S (µM), pH, HgII (pM), Cl− (mg/L), NH4+(µg/L), SO42− (mg/L), water (%), DOC (mg/L), Si (mg/L), percentage of MeHg as THg in wetland soil, ionic strength, and K+ (mg/L). The RDA model (Figure 3, F = 3, p–value < 0.001) shows that the mentioned physicochemical variables explained 47% of the variation in the composition of hgcA+ microbial communities (RDA1 = 14%, p < 0.001; RDA2 = 9.5%, p < 0.001; RDA3 = 6.5% p < 0.001; RDA4 = 4.6%, p = 0.003) with an associated adjusted R2 of 31%. Hence, the RDA analysis emphasizes the importance of electron donors, such as SO42−; nutrients (phosphate); and organic matter (DOC and total LMM thiols) in shaping hgcA+ microbial communities and also points to a role of redox conditions (i.e., FeII/FeIII, water saturation, and H2S). These results suggest that the environmental variables had a strong effect on the potential HgII methylating microbial communities that is not merely due to geographical structuring. RDA and Mantel tests indicate that this large-scale spatial structuring of HgII methylating microbial community composition is highly influenced by pH (Mantel R = 0.23, p-value < 0.001, see Figure 1B), a physicochemical parameter that might act as an integrative factor of multiple chemical and landscape properties.
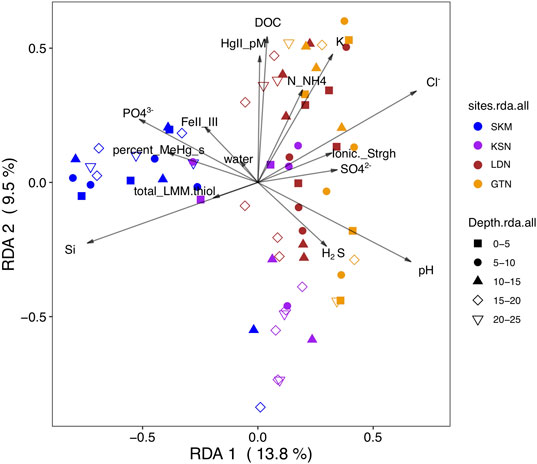
FIGURE 3. RDA biplot (2 first components) of the HgII methylating microbial community (hgcA gene, OTU level) (response variable) constrained by physicochemical variables (predicting variable) and the studied sites (SKM, KSN, LDN, and GTN). The color-coding in the legend indicates the different sites. The shapes correspond to wetland core depth. The arrows and text summarize explanatory variables.
Pearson correlation analyses between the different orders and the measured physicochemical variables also identify pH as one of the main factors (Supplementary Figure S4). Positive significant correlations were observed between Syntrophomonadales and phosphate, DOC, and MeHg concentration and LMM thiols from biological origin. Similar correlations were observed for Spirochaetales. Interestingly, unclassified Desulfobacterota (Deltaprotebacteria) correlated positively with FeIII and the ionic strength, indicating that FeIII might be an important geophysicochemical factor enhancing their metabolism.
3.4 Geochemical Factors Affecting hgcA+ Microbial Communities Within Each Wetland
At the SKM, GTN, and LDN sites, we observed a significant difference in hgcA+ microbial community composition as a function of the location within individual wetlands (ANOSIMbyLOCATIONatSKMR = 0.6, p < 0.001; ANOSIMbyLOCATIONatGTNR = 0.41, p < 0.001; ANOSIMbyLOCATIONatLDNR = 0.64, p < 0.001), reflecting high spatial heterogeneity of hgcA+ microbial community composition at these sites. The variability across the locations where the different cores were collected was higher than across depths as none of the ANOSIM analyses performed with depth as structuring factor was significant (i.e., ANOSIMbyDEPTHatSKM, ANOSIMbyDEPTHatGTN, ANOSIMbyDEPTHatLDN). In contrast, as illustrated in the NMDS (Supplementary Figure S2), hgcA+ microbial community composition was more similar across core replicates at KSN (ANOSIMbyLOCATIONatKSNR = 0.02, p = 0.35). Interestingly, at KSN, depth appeared to be an important factor shaping the hgcA+ microbial community composition (ANOSIMbyDEPTHatKSNR = 0.24, p = 0.003). NMDS of individual wetlands are presented in SI to visualize these differences (Supplementary Figure S2).
We used RDA analyses to investigate the geochemical factors influencing spatial and vertical variability in the hgcA+ microbial community composition across different locations within each individual wetland (Figure 4). Even if the explanatory variables shaping the hgcA+ microbial community composition differed across sites (Figure 4), RDA models identified nutrients (i.e., total-P, NO3−, or PO43−), electron acceptors for well-described HgII methylators (i.e., SO42− and FeIII), and local chemistry (e.g., pH, Al, K, etc.) as important factors predicting the hgcA+ community composition. Also, the ratio of FeII to FeIII, water saturation, and/or H2S, which can be indicators of contrasting redox conditions and/or iron-reduction and sulfate-reduction activity, emerged as important predictors. Moreover, the concentration of the sum of LMM thiols of biological origin also came up as an important factor structuring hgcA+ microbial community composition (Figure 4).
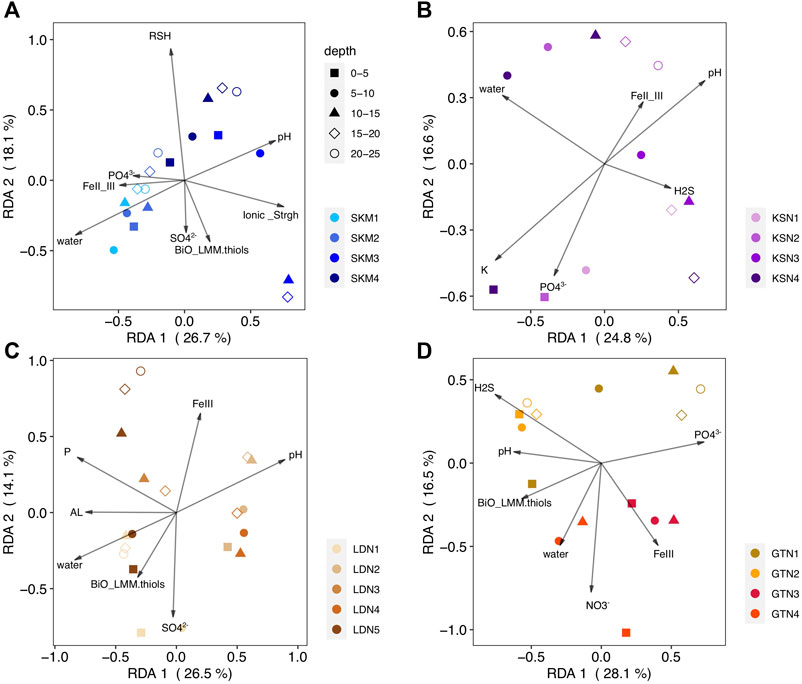
FIGURE 4. RDA biplots (2 first components) of the HgII methylating microbial community (hgcA gene, OTU level) (response variable) constrained by physicochemical variables (predicting variable) and the studied sites [(A) SKM, (B) KSN, (C) LDN, (D) GTN]. The color-coding in the legend indicates the sampling locations within a site. The shapes correspond to wetland core depth. The arrows and text summarize explanatory variables.
More precisely, the explanatory variables of RDASKM were total thiols (RSH), the FeII to FeIII ratio, total LMM thiols from a biological origin, SO42− (mg/L), pH, water content, ionic strength, and PO43−(µg/L). The RDASKM model (Figure 4A, F = 2.4, p–value < 0.001) shows that the mentioned physicochemical variables explained 68% of hgcA+ microbial community composition (RDA1 = 26.7% p < 0.001; RDA2 = 18.1% p < 0.001) and an associated adjusted R2 of 40%. The RDASKM model indicates that the hgcA+ microbial community composition of wetland soils at the SKM4 location (located closed to the inlet) was likely more influenced by pH (i.e., landscape/catchment properties) and also by low H2S and high FeIII concentrations (Supplementary Table S2). In contrast, at SKM1 (located close to a small dam), hgcA+ microbial community composition was more affected by nutrients and by local ferruginous geochemical conditions (FeII to FeIII ratio). The RDAKSN model (Figure 4B, F = 2, p–value = 0.003) shows that pH, water content (%), PO43− (µg/L), the FeII to FeIII ratio, H2S (µM), and K+ (mg/L) concentrations explained 71% of the variation in the composition of hgcA+ microbial communities (RDA1 = 26.7%, p < 0.001; RDA2 = 18.1%, p < 0.001) and an associated adjusted R2 of 36%. The RDALDN model (Figure 3C, F = 2.7, p–value < 0.001) shows total P, SO42- (mg/L), water content (%), FeIII (µM), pH, Al (mg/L), and LMM thiol of biological origin (BiO_LMM_thiols in nM) concentrations explained 63% (adjusted R2 of 39%) of the variation in the hgcA+ microbial community composition. Finally, the RDAGTN (Figure 4D, F = 2.3, p–value < 0.001) shows H2S (µM), water content (%), pH NO3− (µg/L), FeIII (µM), LMM thiols of biological origin, and PO43− (µg/L) concentrations explained 70% (adjusted R2 of 39%) of the variation in the hgcA+ microbial community composition.
3.5 Interplays Between hgcA+ Microbial Community Composition and HgII Methylation Rate Constants
HgII methylation rate constants at these sites have been reported elsewhere (Liem-Nguyen et al., 2021) and are presented in Supplementary Table S1. Liem-Nguyen et al. identified the concentration of HgII in porewater and the LMM thiol compounds of biological origin as the key factors controlling HgII methylation rate constants in SKM, KSN, LDN, and GTN wetlands. Here, we show a weak but still significant correlation between the hgcA+ community composition and HgII methylation rate constants when considering the combined data set that included the four wetland sites (Mantel R = 0.13, p-value = 0.003, Supplementary Figure S5). For visualization purposes, we present a correlation plot between hgcA+ microbial community (Bray–Curtis) dissimilarity and the Euclidean distance of HgII methylation rate constants (km of spiked inorganic-200Hg bound to organic matter, Supplementary Figure S5). Also, we performed principal coordinate analysis of the hgcA+ microbial community, and we show a positive significant correlation between the second component of principal coordinate analysis and HgII methylation rate constants, which supports the effect of the hgcA+ community structure on the resulting km (Supplementary Figure S5). In contrast, when considering each wetland separately, we did not observe any significant correlation between HgII methylating microbial community structure and HgII methylation rate constants.
4 Discussion
4.1 Community Composition of hgcA+ Microorganisms in Boreal Wetlands
The relationship between microbial community structure and function has emerged as an important yet elusive target in microbial ecology (Fuhrman, 2009). This is the first study to report a significant correlation between HgII methylation rate constants and the composition of hgcA+ microorganisms in wetland ecosystems. In particular, we show that more diverse hgcA+ communities are related to higher HgII methylation rates in wetlands. This has also been suggested by a recent study performed with freshwater lake sediments amended with different sulfate concentrations (Jones et al., 2020). Our study, therefore, emphasizes the relevance of describing the diversity of hgcA+ microbial communities in order to better understand the processes controlling MeHg formation in the environment.
Among the diverse hgcA+ microbial communities described at the studied wetlands, Deltaproteobacteria (Desulfuromonadota and Desulfobacterota_A) was the predominant class (Table 1). Within this large class of bacteria, Desulfuromonadales, Desulfovibrionales, and Syntrophales orders were the most abundantly classified hgcA+ bacteria (Supplementary Table S3). Similar dominance of Deltaproteobacteria is seen in wetland and paddy soils in previous studies (Bae et al., 2014; Liu et al., 2014; Schaefer et al., 2014). Both SRB and FeRB can be found within these taxa. SRB were the first bacteria identified as responsible for HgII methylation in aquatic systems and have been highlighted as the dominant HgII methylators in various environments (Compeau and Bartha, 1985; Gilmour et al., 1992; King et al., 2000; Ullrich et al., 2001; Yu et al., 2010; Achá et al., 2012; Bravo et al., 2014; Gascón Díez et al., 2016; Ma et al., 2017; Liu et al., 2018; Jones et al., 2019). SRB are also capable of syntrophic fermentation of simple organic acids in the absence of SO42− as the terminal electron acceptors (McInerney et al., 2008; Plugge et al., 2011). Syntrophales were abundant members of the hgcA+ microbial community, reaching the highest abundance in SKM among all the wetlands. This could have resulted from the low concentrations of SO42− observed at this site (Tables 1 and Supplementary Table S1). Moreover, a recent study performing laboratory studies pinpointed syntrophy as an important metabolic pathway involved in HgII methylation in nutrient-limited boreal peatlands (Hu et al., 2020), and we, therefore, suggest that this could be an important metabolic pathway involved in HgII methylation in the studied wetlands. All these findings combined hint at the importance of sulfate-reducing syntrophs as potential important HgII methylators in boreal wetlands.
FeRB were first implicated in HgII methylation in a study by Fleming et al. (2006) and have since then been confirmed as important Hg methylators in various ecosystems, e.g., lake sediments, paddy soils, and wetland soils (Bae et al., 2014; Liu et al., 2014b; Schaefer et al., 2014). Several Geobacter strains (as well as other studied FeRB) have been tested for HgII methylation in laboratory conditions and displayed high MeHg production rates (Fleming et al., 2006; Kerin et al., 2006; Schaefer and Morel, 2009). Moreover, this metabolic guild has been identified as important HgII methylators in both wetlands and sediments (Schaefer et al., 2014; Bravo et al., 2018b) and in forest soils (Xu et al., 2019). In this study, up to 10% of the total reads were associated to the Geopsychrobacteraceae family, which contains many Geobacter sp. already described as important methylators in isolates but also in the environment. The highest abundance of Geopsychrobacteraceae was observed at the more nutrient-rich site GTN and the lowest at the poor-fen SKM wetland (Supplementary Figure S6).
Firmicutes are relatively recently confirmed representatives of hgcA+ communities and have expanded the niches and metabolisms associated with MeHg production (Gilmour et al., 2013; Bae et al., 2014; Liu et al., 2014b). Methylation rates vary among the Firmicutes, and only a few species have been found to have methylation rates equal to some Deltaproteobacteria species living under similar conditions (Gilmour et al., 2013; Gilmour et al., 2018). Firmicutes represented only 1% of the total reads, although this exceeded 2% in one of the northern wetlands (Table 1). Methanogens were early on implicated in HgII methylation (Wood, 1975). More recent work demonstrates that several methanogens have HgII methylating properties (Gilmour et al., 2013; Yu et al., 2013) and that they may even be the primary methylators in some environments (Hamelin et al., 2011; Schaefer et al., 2014; Bravo et al., 2018b). HgcA genes from methanogens are important members of the hgcA+ microbial community in KSN, GTN, and LND (Table 1).
It is noteworthy that DNA extraction and PCR amplification are known to be prone to artifacts due to any combination of high primer mismatch, low abundance, and/or low DNA extraction efficiency that can significantly affect results. The primers inherently have preferences and also limitations in covering all species equally among different environmental samples. The primers used in this study (Schaefer et al. 2014) are predicted to cover less diversity than a more recently published primer set (Christensen et al., 2016; Gilmour et al., 2018). The high proportion of OTU attributed to unidentified species could be explained by the lack of identified organisms in the databases, primer mismatch, and/or the low conservation of the hgcA gene that is not ideal for biodiversity studies (Bravo and Cosio, 2020). Nevertheless, from our results, it is clear that hgcA+ microbial communities in wetlands are more diverse than previously thought. Furthermore, it is important to stress that the largest part of the microbial community could not be taxonomically identified, and we believe that efforts to develop techniques and increasing databases to enable a more extensive identification of the microorganisms involved in the formation of MeHg should be a research priority in the years to come.
4.2 Geochemical Factors Affecting Distribution of HgII Methylating Communities
The comparison between gene assemblages with rates of biogeochemical processes, such as HgII methylation rate constants, stems from the most fundamental level, where genes are prerequisites for any enzyme-catalyzed reaction and should, therefore, feature a correlation (Rocca et al., 2015). However, the relationship between gene and transcript abundances and the processes they facilitate can be obscured by various factors, such as habitat characteristics, complexity of biogeochemical pathways, and differences in the turnover time of nucleic acids and reaction products (Rocca et al., 2015). This study points to different physicochemical parameters as important factors determining the composition of the hgcA+ microbial community, which, in turn, also fundamentally controls HgII methylation rates. Across all investigated wetlands, nMDS plots and RDA analyses show that there was more spatial variation in the diversity of hgcA+ microorganisms between the different wetlands than between different depths (Figures 1, 3). The combined hgcA+ microbial community showed changes in composition mainly related to pH (Figure 1, Mantel R = 0.23, p–value < 0.001) and PO43− concentration (Mantel R = 0.17, p–value = 0.003). Previous work suggests that pH may act as an integrative factor of multiple landscape properties and could, thus, affect the hgcA+ microbial community in ways analogous to what has been shown for total bacterial communities (Niño-García et al., 2016). Indeed, in our study, we observe that changes in pH were strongly correlated to shifts in FeIII, Cl−, NH4+, ionic strength, reduced thiols, total LMM thiols, Al, Mg, Mg, Mn, Na, and S (Supplementary Figure S3).
Concerning PO43−, it is not surprising that there is a significant correlation between this typically limiting nutrient and the composition of the hgcA+ microbial community in boreal wetlands. PO43− was identified as being strongly correlated to the composition of the hgcA+ microbial communities (Mantel R = 0.17, p–value = 0.003) (Figure 3) and also when looking at variation within each individual wetland (Figure 4). Numerous studies have investigated the impact of nutrients, in particular P, for soil microbial community structure. The community composition of sulfate-reducing syntrophs and methanogens have, for example, been shown to depend on the position along nutrient gradients (Prechtel et al., 2001; Chauhan et al., 2004; Bae et al., 2015). Previous research has also repeatedly pointed out nutrient levels as strong regulators of bacterial growth and other metabolic activities, including MeHg production (Bigham et al., 2017; Bravo et al., 2017). Liem-Nguyen et al. (2016) also assessed the effects of variable nutrient N and P loadings to surface water and showed that increased nutrient loads caused increased phytoplankton biomass productivity and stimulated microbial activity and mercury methylation in sediments in which HgII was presumably highly bioavailable. From all these studies, it can be concluded that nutrient concentrations are an important factor shaping the hgcA+ microbial community.
Iron is also a key factor affecting HgII methylation (Bravo et al., 2014). FeIII can act as a direct electron acceptor for FeRB and has been found to significantly influence Hg cycling and methylation (Fleming et al., 2006; Si et al., 2015). In addition, FeIII, or the FeII to FeIII ratio, was significantly correlated to the composition of hgcA+ communities (Mantel R = 0.12, p–value = 0.02) with a particularly strong correlation in wetlands SKM and GTN (Figures 3, 4). The availability of inorganic HgII for uptake by methylating organisms has been found to limit the methylation process in laboratory conditions (Schaefer et al., 2011) and also in the studied sites (Liem-Nguyen et al., 2021). FeIII could directly affect hgcA+ communities by determining the availability of electron acceptors for FeRB and also indirectly via the relationship with FeII that may influence HgII availability, which can, in turn, be strongly affected by the interaction/complexation of Fe with organic material (Faganeli et al., 2012) and sulfur (Hsu-Kim et al., 2013). The relative abundance of the Geopsychrobacteraceae family correlated negatively with the FeII/FeIII in porewater, likely indicating that more strongly reducing conditions limit their growth. Furthermore, the fact that the abundant unclassified Desulfobacterota (Deltaprotebacteria) correlated positively with FeIII and its ionic strength suggests an important role of hgcA+ FeRB in the studied wetlands.
Water content seems to be another important factor driving the composition of HgII methylating communities (Figures 3, 4) and HgII methylation rate constants (Supplementary Figure S3). Increasing moisture could result in elevated MeHg production from stimulation of microbial methylators, both by means of the organic material supplied via decaying vegetation and soils and from a concomitant oxygen depletion and lowering of the redox potential. It has previously been suggested that HgII methylating microbes are stimulated by enhanced water saturation in organic forest soils as this create conditions that provide readily available electron donors to drive the formation and buildup of MeHg (Kronberg et al., 2016). Eklöf et al. (2018) also found that the highest %MeHg was associated with water-filled cavities and water-logged soil environments, conditions that would favor methylating microorganisms and enhance MeHg formation. In wetland KSN, where water content was generally very high (Supplementary Table S1), depth appeared to play a more important role in shaping the local hgcA+ community (Supplementary Figure S2, ANOSIMbyDEPTHatKSNR = 0.24, p = 0.003). One explanation could be that the KSN soils provide vertically stratified heterogeneous habitats for microorganisms with varying fresh organic matter availability and nutrient levels (Fierer et al., 2003; Lorenz and Lal, 2005; Kramer and Gleixner, 2008). Furthermore, it is possible that other factors not considered in this study, such as the composition of the organic matter, may play a role in controlling the composition of the hgcA+ communities, and the strong influence of biologically derived LMM thiols support this.
5 Conclusion
Understanding the responses of bacterial communities to environmental factors is a major focus for research in microbial ecology, and this also applies to HgII methylating microbial communities. Combining high-throughput hgcA amplicon sequencing with molecular barcoding reveals diverse clades of hgcA+ microorganisms in a wide range of wetland soils. This study confirms the predominant role of Deltaproteobacteria as hgcA+ microorganisms in boreal wetland soils and also unveils the presence of Firmicutes and Archaea as abundant members of methylating communities. By uncovering relationships between the composition of such communities, HgII methylation rate constants, and the physicochemical factors, we propose an important role of pH, water saturation, nutrient status, Fe, and LMM thiols of biological origin for controlling the hgcA+ microbial community composition and their activities across the four wetlands. In the future, more attention should be paid to how specific groups of HgII methylating microbes respond to various levels of limiting nutrients. Furthermore, it would be worthwhile to carry out more in-depth studies on the interactions between various HgII methylating groups in contributing to the overall MeHg production. Our findings provide a better understanding of HgII methylating microbial communities in boreal wetland soils. As more than 50% of wetlands are located in the high northern latitudes, the results reported here are likely broadly relevant for a wide range of wetlands.
Data Availability Statement
The datasets generated for this study can be found in the EBI Archive under accession number PRJEB20882.
Author Contributions
EB, VL, JX, AB, SB, and JX. conceived the study. VL and EB, conducted the sampling campaigns. JX and AB performed the laboratory analyses with guidance from SB. MB performed the bioinformatics analyses. AB conducted all statistical analyses and built the figures with comments and suggestions from the rest of the co-authors. JX, AB, and SB. wrote the manuscript with significant assistance and comments from EB and VL.
Funding
This project was carried out within the Swedish-Sino SMaREF (2013-6978) funded by the Swedish Research Council. This study was also supported by the Swedish Research Council (Grants No. 2011–7192, 2012-3892 and 2017-04422), a Wennergren foundation project to LVN and the Kempe Foundations (SMK-2745, SMK-1243) and the Marie Curie Individual Fellowship (H2020-MSCA-IF-2016; project-749645).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Sequencing was carried out at the SciLifeLab SNP/SEQ facility hosted by Uppsala University, and we acknowledge the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) for access to storage and computational resources.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2020.518662/full#supplementary-material.
References
Achá, D., Hintelmann, H., and Pabón, C. A. (2012). Sulfate-reducing bacteria and mercury methylation in the water column of the lake 658 of the experimental lake area. Geomicrobiol. J. 29, 667–674. doi:10.1080/01490451.2011.606289
Bae, H. S., Dierberg, F. E., and Ogram, A. (2014). Syntrophs dominate sequences associated with the mercury methylation-related gene hgcA in the water conservation areas of the Florida Everglades. Appl. Environ. Microbiol. 80, 6517–6526. doi:10.1128/AEM.01666-14
Bae, H. S., Holmes, M. E., Chanton, J. P., Reddy, K. R., and Ogram, A. (2015). Distribution, activities, and interactions of methanogens and sulfate-reducing prokaryotes in the Florida everglades. Appl. Environ. Microbiol. 81, 7431–7442. doi:10.1128/AEM.01583-15
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289. doi:10.1111/j.2517-6161.1995.tb02031.x
Bigham, G. N., Murray, K. J., Masue-Slowey, Y., and Henry, E. A. (2017). Biogeochemical controls on methylmercury in soils and sediments: implications for site management. Integr. Environ. Assess. Manage. 13, 249–263. doi:10.1002/ieam.1822
Boening, D. W. (2000). Ecological effects, transport, and fate of mercury: a general review. Chemosphere 40, 1335–1351. doi:10.1016/S0045-6535(99)00283-0
Bravo, A. G., Bouchet, S., Tolu, J., Björn, E., Mateos-Rivera, A., and Bertilsson, S. (2017). Molecular composition of organic matter controls methylmercury formation in boreal lakes. Nat. Commun. 8, 14255. doi:10.1038/ncomms14255
Bravo, A. G., and Cosio, C. (2020). Biotic formation of methylmercury: a bio-physico-chemical conundrum. Limnol. Oceanogr. 65, 1010–1027. doi:10.1002/lno.11366
Bravo, A. G., Cosio, C., Amouroux, D., Zopfi, J., Chevalley, P. A., Spangenberg, J. E., et al. (2014). Extremely elevated methyl mercury levels in water, sediment and organisms in a Romanian reservoir affected by release of mercury from a chlor-alkali plant. Water Res. 49, 391–405. doi:10.1016/j.watres.2013.10.024
Bravo, A. G., Peura, S., Buck, M., Ahmed, O., Mateos-Rivera, A., Herrero Ortega, S., et al. (2018a). Methanogens and iron-reducing bacteria: the overlooked members of mercury-methylating microbial communities in boreal lakes. Appl. Environ. Microbiol. 84, e01774–18. doi:10.1128/AEM.01774-18
Bravo, A. G., Zopfi, J., Buck, M., Xu, J., Bertilsson, S., Schaefer, J. K., et al. (2018b). Geobacteraceae are important members of mercury-methylating microbial communities of sediments impacted by waste water releases. ISME J. 12, 802–812. doi:10.1038/s41396-017-0007-7
Capo, E., Bravo, A. G., Soerensen, A. L., Bertilsson, S., Pinhassi, J., Feng, C., et al. (2020). Deltaproteobacteria and spirochaetes-like bacteria are abundant putative mercury methylators in oxygen-deficient water and marine particles in the Baltic Sea. Front. Microbiol. 11, 574080. doi:10.3389/fmicb.2020.574080
Chauhan, A., Ogram, A., and Reddy, K. R. (2004). Syntrophic-methanogenic associations along a nutrient gradient in the Florida Everglades. Appl. Environ. Microbiol. 70, 3475–3484. doi:10.1128/AEM.70.6.3475-3484.2004
Christensen, G. A., Somenahally, A. C., Moberly, J. G., Miller, C. M., King, A. J., Gilmour, C. C., et al. (2017). Carbon amendments alter microbial community structure and net mercury methylation potential in sediments. Appl. Environ. Microbiol. 84, 1–14. doi:10.1128/AEM.01049-17
Christensen, G. A., Wymore, A. M., King, A. J., Podar, M., Hurt, R. A., Santillan, E. U., et al. (2016). Development and validation of broad-range qualitative and clade-specific quantitative molecular probes for assessing mercury methylation in the environment. Appl. Environ. Microbiol. 82, 6068–6078. doi:10.1128/AEM.01271-16
Compeau, G. C., and Bartha, R. (1985). Sulfate-reducing bacteria: principal methylators of mercury in anoxic estuarine sediment. Appl. Environ. Microbiol. 50, 498–502. doi:10.1128/AEM.50.2.498-502.1985
Eklöf, K., Bishop, K., Bertilsson, S., Björn, E., Buck, M., Skyllberg, U., et al. (2018). Formation of mercury methylation hotspots as a consequence of forestry operations. Sci. Total Env. 613, 1069–1078.
Erwin, K. L. (2009). Wetlands and global climate change: the role of wetland restoration in a changing world. Wetl. Ecol. Manag. 17, 71–84. doi:10.1007/s11273-008-9119-1
Faganeli, J., Hines, M. E., Covelli, S., Emili, A., and Giani, M. (2012). Mercury in lagoons: an overview of the importance of the link between geochemistry and biology. Estuar. Coast Shelf Sci. 113, 126–132. doi:10.1016/j.ecss.2012.08.021
Fierer, N., Schimel, J. P., and Holden, P. A. (2003). Variations in microbial community composition through two soil depth profiles. Soil Biol. Biochem. 35, 167–176. doi:10.1016/S0038-0717(02)00251-1
Fleming, E. J., Mack, E. E., Green, P. G., and Nelson, D. C. (2006). Mercury methylation from unexpected sources: molybdate-inhibited freshwater sediments and an iron-reducing bacterium. Appl. Environ. Microbiol. 72, 457–464. doi:10.1128/AEM.72.1.457-464.2006
Fuhrman, J. A. (2009). Microbial community structure and its functional implications. Nature, 459, 193. doi:10.1038/nature08058
Gascón Díez, E., Loizeau, J. L., Cosio, C., Bouchet, S., Adatte, T., Amouroux, D., et al. (2016). Role of settling particles on mercury methylation in the oxic water column of freshwater systems. Environ. Sci. Technol. 50, 11672–11679. doi:10.1021/acs.est.6b03260
Gilmour, C. C., Bullock, A. L., Mcburney, A., and Podar, M. (2018). Robust mercury methylation across diverse methanogenic archaea. mBio 9, e02403–e02417. doi:10.1128/mbio.02403-17
Gilmour, C. C., Henry, E. A., and Mitchell, R. (1992). Sulfate stimulation of mercury methylation in freshwater sediments. Environ. Sci. Technol. 26, 2281–2287. doi:10.1021/es00035a029
Gilmour, C. C., Podar, M., Bullock, A. L., Graham, A. M., Brown, S. D., Somenahally, A. C., et al. (2013). Mercury methylation by novel microorganisms from new environments. Environ. Sci. Technol. 47, 11810–11820. doi:10.1021/es403075t
Gilmour, C. C., Riedel, G. S., Ederington, M. C., Bell, J. T., Gill, G. A., Stordal, M. C., et al. (1998). Methylmercury concentrations and production rates across a trophic gradient in the northern Everglades. Biogeochemistry 40, 327–345.ST-Methylmercury concentrations and pro. doi:10.1023/a:1005972708616
Gionfriddo, C. M., Tate, M. T., Wick, R. R., Schultz, M. B., Zemla, A., Thelen, M. P., et al. (2016). Microbial mercury methylation in Antarctic sea ice. Nat. Microbiol. 1, 16127. doi:10.1038/nmicrobiol.2016.127
Hall, B. D., Aiken, G. R., Krabbenhoft, D. P., Marvin-DiPasquale, M., and Swarzenski, C. M. (2008). Wetlands as principal zones of methylmercury production in southern Louisiana and the Gulf of Mexico region. Environ. Pollut. 154, 124–134. doi:10.1016/j.envpol.2007.12.017
Hamelin, S., Amyot, M., Barkay, T., Wang, Y., and Planas, D. (2011). Methanogens: principal methylators of mercury in lake periphyton. Environ. Sci. Technol. 45, 7693–7700. doi:10.1021/es2010072
Hsu-Kim, H., Kucharzyk, K. H., Zhang, T., and Deshusses, M. A. (2013). Mechanisms regulating mercury bioavailability for methylating microorganisms in the aquatic environment: a critical review. Environ. Sci. Technol. 47, 2441–2456. doi:10.1021/es304370g
Hu, H., Wang, B., Bravo, A. G., Björn, E., Skyllberg, U., Amouroux, D., et al. (2020). Shifts in mercury methylation across a peatland chronosequence: from sulfate reduction to methanogenesis and syntrophy. J. Hazard. Mater. 387, 121967. doi:10.1016/j.jhazmat.2019.121967
Jones, D. S., Johnson, N. W., Mitchell, C. P. J., Walker, G. M., Bailey, J. V., Pastor, J., et al. (2020). Diverse communities of hgcAB+ microorganisms methylate mercury in freshwater sediments subjected to experimental sulfate loading. Environ. Sci. Technol., 54, 14265–14274. doi:10.1021/acs.est.0c02513
Jones, D. S., Walker, G. M., Johnson, N. W., Mitchell, C. P. J., Coleman Wasik, J. K., and Bailey, J. V. (2019). Molecular evidence for novel mercury methylating microorganisms in sulfate-impacted lakes. ISME J. 13, 1659. doi:10.1038/s41396-019-0376-1
Jonsson, S., Skyllberg, U., Nilsson, M. B., Lundberg, E., Andersson, A., and Björn, E. (2014). Differentiated availability of geochemical mercury pools controls methylmercury levels in estuarine sediment and biota, Nat. Commun. 5, 4624. doi:10.1038/ncomms5624
Joshi, N. A., and Fass, J. N. (2011). Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files. Available at: https://github.com/najoshi/sickle
Kerin, E. J., Gilmour, C. C., Roden, E., Suzuki, M. T., Coates, J. D., and Mason, R. P. (2006). Mercury methylation by dissimilatory Iron-reducing bacteria. Appl. Environ. Microbiol. 72, 7919–7921. doi:10.1128/AEM.01602-06
King, J. K., Kostka, J. E., Frischer, M. E., Saunders, F. M., and Jahnke, R. A. (2001). A Quantitative relationship that demonstrates mercury methylation rates in marine sediments are based on the community composition and activity of sulfate-reducing bacteria. Environ. Sci. Technol. 35, 2491–2496. doi:10.1021/es001813q
King, J. K., Kostka, J. E., Frischer, M. E., and Saunders, F. M. (2000). Sulfate-reducing bacteria methylate mercury at variable rates in pure culture and in marine sediments. Appl. Environ. Microbiol. 66, 2430–2437. doi:10.1128/AEM.66.6.2430-2437.2000
Kramer, C., and Gleixner, G. (2008). Soil organic matter in soil depth profiles: distinct carbon preferences of microbial groups during carbon transformation. Soil Biol. Biochem. 40, 425–433. doi:10.1016/j.soilbio.2007.09.016
Kronberg, R. M., Jiskra, M., Wiederhold, J. G., Björn, E., and Skyllberg, U. (2016). Methyl mercury formation in hillslope soils of boreal forests: the role of forest harvest and anaerobic microbes. Environ. Sci. Technol. 50, 9177–9186. doi:10.1021/acs.est.6b00762
Lambertsson, L., and Björn, E. (2004). Validation of a simplified field-adapted procedure for routine determinations of methyl mercury at trace levels in natural water samples using species-specific isotope dilution mass spectrometry. Anal. Bioanal. Chem. 380 871–875. doi:10.1007/s00216-004-2863-z
Lambertsson, L., Lundberg, E., Nilsson, M., and Frech, W. 2001). Applications of enriched stable isotope tracers in combination with isotope dilution GC-ICP-MS to study mercury species transformation in sea sediments during in situ. J. Anal. At. Spectrom. 16, 1296–1301. doi:10.1039/b106878b
Liem-Nguyen, V., Bouchet, S., and Björn, E. (2015). Determination of sub-nanomolar levels of low molecular mass thiols in natural waters by liquid chromatography tandem mass spectrometry after derivatization with p-(hydroxymercuri) benzoate and online preconcentration. Anal. Chem. 87, 1089–1096. doi:10.1021/ac503679y
Liem-Nguyen, V., Jonsson, S., Skyllberg, U., Nilsson, M. B., Andersson, A., Lundberg, E., et al. (2016). Effects of nutrient loading and mercury chemical speciation on the formation and degradation of methylmercury in estuarine sediment. Environ. Sci. Technol. 50, 6983–6990. doi:10.1021/acs.est.6b01567
Liem-Nguyen, V., Skyllberg, U., and Björn, E. (2021). Methylmercury formation in boreal wetlands in relation to chemical speciation of mercury(II) and concentration of low molecular mass thiols. Sci. Total Environ. 755, 142666. doi:10.1016/j.scitotenv.2020.142666
Liem-Nguyen, V., Skyllberg, U., and Björn, E. (2017). Thermodynamic modeling of the solubility and chemical speciation of mercury and methylmercury driven by organic thiols and micromolar sulfide concentrations in boreal wetland soils. Environ. Sci. Technol. 51, 3678–3686. doi:10.1021/acs.est.6b04622
Liu, X., Ma, A., Zhuang, G., and Zhuang, X. (2018). Diversity of microbial communities potentially involved in mercury methylation in rice paddies surrounding typical mercury mining areas in China. Microbiologyopen 7, e00577. doi:10.1002/mbo3.577
Liu, Y. R., Yu, R. Q., Zheng, Y. M., and He, J. Z. (2014). Analysis of the microbial community structure by monitoring an Hg methylation gene (hgcA) in paddy soils along an Hg gradient. Appl. Environ. Microbiol. 80, 2874–2879. doi:10.1128/AEM.04225-13
Lorenz, K., and Lal, R. (2005). The depth distribution of soil organic carbon in relation to land use and management and the potential of carbon sequestration in subsoil horizons. Adv. Agron. 88, 35–66. doi:10.1016/S0065-2113(05)88002-2
Louis, V. L. S. T., Rudd, J. W. M., Kelly, C. a., Beaty, K. G., Flett, R. J., and Roulet, N. T. (1996). Production and loss of methylmercury and loss of total mercury from boreal forest catchments containing different types of wetlands. Environ. Sci. Technol. 30, 2719–2729. doi:10.1021/es950856h
Ma, M., Du, H., Wang, D., Kang, S., and Sun, T. (2017). Biotically mediated mercury methylation in the soils and sediments of Nam Co Lake, Tibetan Plateau. Environ. Pollut. 227, 243–251. doi:10.1016/j.envpol.2017.04.037
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12. doi:10.14806/ej.17.1.200
Matsen, F. A., Kodner, R. B., and Armbrust, E. V. (2010). pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics. 11, 538. doi:10.1186/1471-2105-11-538
Mazrui, N. M., Jonsson, S., Thota, S., Zhao, J., and Mason, R. P. (2016). Enhanced availability of mercury bound to dissolved organic matter for methylation in marine sediments. Geochem. Cosmochim. Acta. 194, 153–162. doi:10.1016/j.gca.2016.08.019
McDaniel, E. A., Peterson, B., Stevens, S. L. R., Tran, P. Q., Anantharaman, K., and McMahon, K. D. (2020a). Expanded phylogenetic diversity and metabolic flexibility of microbial mercury methylation. bioRxiv. doi:10.1101/2020.01.16.909358
McDaniel, E. A., Peterson, B. D., Stevens, S. L. R., Tran, P. Q., Anantharaman, K., and McMahon, K. D. (2020b). Expanded phylogenetic diversity and metabolic flexibility of mercury-methylating microorganisms. mSystems, 5, e00299–20. doi:10.1128/msystems.00299-20
McInerney, M. J., Struchtemeyer, C. G., Sieber, J., Mouttaki, H., Stams, A. J., Schink, B., et al. (2008). Physiology, ecology, phylogeny, and genomics of microorganisms capable of syntrophic metabolism. Ann. N. Y. Acad. Sci. 1125, 58–72. doi:10.1196/annals.1419.005
Mitchell, C. P. J., Branfireun, B. A., and Kolka, R. K. (2008a). Assessing sulfate and carbon controls on net methylmercury production in peatlands: an in situ mesocosm approach. Appl. Geochem. 23, 503–518. doi:10.1016/j.apgeochem.2007.12.020
Mitchell, C. P., Branfireun, B. A., and Kolka, R. K. (2008b). Spatial characteristics of net methylmercury production hot spots in peatlands. Environ. Sci. Technol. 42, 1010–1016. doi:10.1021/es0704986
Mitra, S., Wassmann, R., and Vlek, P. L. G. (2003). Global inventory of wetlands and their role in the carbon cycle. Bonn: ZEF Discusson PapDev. Policy.
Niño-García, J. P., Ruiz-González, C., and Del Giorgio, P. A. (2016). Interactions between hydrology and water chemistry shape bacterioplankton biogeography across boreal freshwater networks. ISME J. 10, 1755–1766. doi:10.1038/ismej.2015.226
Oksanen, A. J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., Mcglinn, D., et al. (2017). Package ‘vegan’ Software. Vienna, Austria: R Foundation for Statistical Computing.
Parks, J. M., Johs, A., Podar, M., Bridou, R., Hurt, R. A., Smith, S. D., et al. (2013). The genetic basis for bacterial mercury methylation. Science 339, 1332–1335. doi:10.1126/science.1230667
Peterson, B. D., McDaniel, E. A., Schmidt, A. G., Lepak, R. F., Tran, P. Q., Marick, R. A., et al. (2020). Microbial mercury methylation genes identified across diverse anaerobic microbial guilds in a eutrophic sulfate-enriched lake. Environ. Sci. Technol. 54, 15840–15851.
Plugge, C. M., Zhang, W., Scholten, J. C., and Stams, A. J. (2011). Metabolic flexibility of sulfate-reducing bacteria, Front. Microbiol. 2, 81–88. doi:10.3389/fmicb.2011.00081
Podar, M., Gilmour, C. C., Brandt, C. C., Soren, A., Brown, S. D., Crable, B. R., et al. (2015). Global prevalence and distribution of genes and microorganisms involved in mercury methylation. Sci. Adv. 1, e1500675. doi:10.1126/sciadv.1500675
Prechtel, A., Alewell, C., Armbruster, M., Bittersohl, J., Cullen, J. M., Evans, C. D., et al. (2001). Response of sulphur dynamics in European catchments to decreasing sulphate deposition. Hydrol. Earth Syst. Sci. 5, 311–326. doi:10.5194/hess-5-311-2001
Ramette, A. (2007). Multivariate analyses in microbial ecology, FEMS Microbiol. Ecol. 62, 142–160. doi:10.1111/j.1574-6941.2007.00375.x
Ranchou-Peyruse, M., Monperrus, M., Bridou, R., Duran, R., Amouroux, D., Salvado, J. C., et al. (2009). Overview of mercury methylation capacities among anaerobic bacteria including representatives of the sulphate-reducers: implications for environmental studies. Geomicrobiol. J. 26, 1–8. doi:10.1080/01490450802599227
Rocca, J. D., Hall, E. K., Lennon, J. T., Evans, S. E., Waldrop, M. P., Cotner, J. B., et al. (2015). Relationships between protein-encoding gene abundance and corresponding process are commonly assumed yet rarely observed. ISME J. 9, 1693–1699. doi:10.1038/ismej.2014.252
Schaefer, J. K., Kronberg, R. M., Björn, E., and Skyllberg, U. (2020). Anaerobic guilds responsible for mercury methylation in boreal wetlands of varied trophic status serving as either a methylmercury source or sink. Environ. Microbiol. 22, 3685. doi:10.1111/1462-2920.15134
Schaefer, J. K., Kronberg, R. M., Morel, F. M., and Skyllberg, U. (2014). Detection of a key Hg methylation gene, hgcA, in wetland soils. Environ. Microbiol. Rep. 6, 441–447. doi:10.1111/1758-2229.12136
Schaefer, J. K., and Morel, F. M. M. (2009). High methylation rates of mercury bound to cysteine by Geobacter sulfurreducens. Nat. Geosci. 2, 123–126. doi:10.1038/ngeo412
Schaefer, J. K., Rocks, S. S., Zheng, W., Liang, L., Gu, B., and Morel, F. M. (2011). Active transport, substrate specificity, and methylation of Hg(II) in anaerobic bacteria. Proc. Natl. Acad. Sci. U.S.A. 108, 8714–8719. doi:10.1073/pnas.1105781108
Si, Y., Zou, Y., Liu, X., Si, X., and Mao, J. (2015). Mercury methylation coupled to iron reduction by dissimilatory iron-reducing bacteria. Chemosphere 122, 206–212. doi:10.1016/j.chemosphere.2014.11.054
Sinclair, L., Osman, O. A., Bertilsson, S., and Eiler, A. (2015). Microbial community composition and diversity via 16S rRNA gene amplicons: evaluating the Illumina platform. PloS One. 10, e0116955. doi:10.1371/journal.pone.0116955
Skyllberg, U. (2008). Competition among thiols and inorganic sulfides and polysulfides for Hg and MeHg in wetland soils and sediments under suboxic conditions: illumination of controversies and implications for MeHg net production, J. Geophys. Res. 113. doi:10.1029/2008JG000745
St. Louis, V. L., Rudd, J. W. M., Kelly, C. A., Beaty, K. G., Bloom, N. S., and Flett, R. J. (1994). Importance of wetlands as sources of methyl mercury to boreal forest ecosystems. Can. J. Fish. Aquat. Sci. 51, 1065–1076. doi:10.1139/f94-106
Tjerngren, I., Karlsson, T., Björn, E., and Skyllberg, U. (2012a). Potential Hg methylation and MeHg demethylation rates related to the nutrient status of different boreal wetlands. Biogeochemistry 108, 335–350. doi:10.1007/s10533-011-9603-1
Tjerngren, I., Meili, M., Björn, E., and Skyllberg, U. (2012b). Eight boreal wetlands as sources and sinks for methyl mercury in relation to soil acidity, C/N ratio, and small-scale flooding. Environ. Sci. Technol. 46, 8052–8060. doi:10.1021/es300845x
Ullrich, S. M., Tanton, T. W., Abdrashitova, S. A., and Svetlana, A. (2001). Mercury in the aquatic environment: a review of factors affecting methylation. Crit. Rev. Environ. Sci. Technol. 31, 241–293. doi:10.1080/20016491089226
Villar, E., Cabrol, L., and Heimbürger‐Boavida, L. E. (2020). Widespread microbial mercury methylation genes in the global ocean. Environ. Microbiol. Rep. 12, 277. doi:10.1111/1758-2229.12829
Viollier, E., Inglett, P. W., Hunter, K., Roychoudhury, A. N., and Van Cappellen, P. (2000). The ferrozine method revisited: Fe(II)/Fe(III) determination in natural waters. Appl. Geochem. 15, 785–790. doi:10.1016/S0883-2927(99)00097-9
Vishnivetskaya, T. A., Hu, H., Van Nostrand, J. D., Wymore, A. M., Xu, X., Qiu, G., et al. (2018). Microbial community structure with trends in methylation gene diversity and abundance in mercury-contaminated rice paddy soils in Guizhou, China. Environ. Sci. Process. Impacts. 20, 673–685. doi:10.1039/C7EM00558J
Wood, J. M. (1975). Biological cycles for elements in the environment. Naturwissenschaften 62, 357–364. doi:10.1007/BF00625342
Xu, J., Buck, M., Eklöf, K., Ahmed, O. O., Schaefer, J. K., Bishop, K., et al. (2019). Mercury methylating microbial communities of boreal forest soils. Sci. Rep. 9, 518. doi:10.1038/s41598-018-37383-z
Yu, R. Q., Adatto, I., Montesdeoca, M. R., Driscoll, C. T., Hines, M. E., and Barkay, T. (2010). Mercury methylation in Sphagnum moss mats and its association with sulfate-reducing bacteria in an acidic Adirondack forest lake wetland. FEMS Microbiol. Ecol. 74, 655–668. doi:10.1111/j.1574-6941.2010.00978.x
Yu, R. Q., Flanders, J. R., MacK, E. E., Turner, R., Mirza, M. B., and Barkay, T. (2012). Contribution of coexisting sulfate and iron reducing bacteria to methylmercury production in freshwater river sediments. Environ. Sci. Technol. 46, 2684–2691. doi:10.1021/es2033718
Yu, R.-Q., Reinfelder, J. R., Hines, M. E., and Barkay, T. (2018). Syntrophic pathways for microbial mercury methylation. ISME J. 12, 1826. doi:10.1038/s41396-018-0106-0
Keywords: wetlands, methylmercury, mercury methylation, hgcA, community composition, bacteria, mercury, drivers
Citation: Xu J, Liem-Nguyen V, Buck M, Bertilsson S, Björn E and Bravo AG (2021) Mercury Methylating Microbial Community Structure in Boreal Wetlands Explained by Local Physicochemical Conditions. Front. Environ. Sci. 8:518662. doi: 10.3389/fenvs.2020.518662
Received: 09 December 2019; Accepted: 21 December 2020;
Published: 18 February 2021.
Edited by:
Vera I. Slaveykova, Université de Genève, SwitzerlandReviewed by:
Tamar Barkay, Rutgers, The State University of New Jersey, United StatesCaitlin Marissa Gionfriddo, Oak Ridge National Laboratory, United States
Cynthia C. Glimour, Smithsonian Environmental Research Center, United States
Copyright © 2021 Xu, Liem-Nguyen, Buck, Bertilsson, Björn and Bravo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea G. Bravo, amFuZHJpdWdhcmNpYUBnbWFpbC5jb20=