 Patrick I. Bennett1,2,3
Patrick I. Bennett1,2,3 Javier F. Tabima3,4
Javier F. Tabima3,4 Anna L. Leon5John Browning5
Anna L. Leon5John Browning5 Michael J. Wingfield6
Michael J. Wingfield6 Jared M. LeBoldus3,7*
Jared M. LeBoldus3,7*- 1U.S. Department of Agriculture Forest Service, Rocky Mountain Research Station, Moscow, ID, United States
- 2U.S. Department of Agriculture Forest Service, Forest Health Protection, Northern Region, Missoula, MT, United States
- 3Department of Botany and Plant Pathology, Oregon State University, Corvallis, OR, United States
- 4Department of Biology, Clark University, The Lasry Center for Bioscience, Worcester, MA, United States
- 5Weyerhaeuser Company, Centralia, WA, United States
- 6Forestry and Agriculture Biotechnology Institute (FABI), University of Pretoria, Pretoria, South Africa
- 7Department of Forest Engineering, Resources, and Management, Oregon State University, Corvallis, OR, United States
Many fungi in the Ophiostomatales are vectored by bark beetles that introduce these fungi directly into their tree hosts. Most of these fungal associates have little effect on their hosts, but some can cause serious diseases. One such fungus, Leptographium wageneri, causes an economically and ecologically important tree disease known as black stain root disease (BSRD). For this study, 159 full genome sequences of L. wageneri were analyzed using a population genomics approach to investigate the epidemiology, dispersal capabilities, and reproductive biology of this fungus. Analyses were performed with SNP haplotypes from 155 isolates of L. wageneri var. pseudotsugae collected in 16 Douglas-fir stands in Oregon and 4 isolates of L. wageneri var. wageneri collected in pinyon pine stands in southern California. These two host-specific varieties appear to be evolutionarily divergent, likely due a combination of factors such as host differentiation and geographic isolation. We analyzed gene flow and population structure within and among Douglas-fir plantations in western Oregon to infer the relative importance of local vs. long distance dispersal in structuring populations of L. wageneri var. pseudotsugae. Long-distance gene flow has occurred between Douglas-fir plantations, contributing to diversity and population structure within stands, and likely reflecting the behavior of an important insect vector. Genetic clustering analyses revealed the presence of unique local clusters within stands and plantations in addition to those common among multiple stands or plantations. Although populations of L. wageneri var. pseudotsugae are primarily asexual, two mating types were present in many stands, suggesting that recombination is at least possible and may contribute to genetic diversity.
Introduction
Many fungi in the Ophiostomatales form symbiotic relationships with conifer-infesting bark beetles (Curculionidae, Scolytinae) (Paine et al., 1997; Six, 2012). The nature of these relationships is complex and varied, ranging from mutualistic to commensal to antagonistic (Paine et al., 1997; Six and Wingfield, 2011; Six, 2012; Davis et al., 2019). While many of these insects and their associated fungi are relatively innocuous, having little effect on forests, some have important roles in structuring the ecosystems they inhabit (Six, 2020). Relatively few fungi associated with bark beetles can be considered virulent pathogens or agents of mortality of their host trees (Six and Wingfield, 2011; Six, 2012). The few that are aggressive pathogens differ in that they may be carried by multiple beetle vectors (Gibbs, 1978; Witcosky and Hansen, 1985; Witcosky et al., 1986a), can spread through living tree tissues including root grafts (Hessburg and Hansen, 1986), and colonize the xylem tracheids exclusively (Hessburg and Hansen, 1987; Cobb, 1988). Other fungi in the Ophiostomatales such as Grosmannia clavigera (Rob.-Jeffr. and R.W. Davidson) Zipfel, Z.W. de Beer and M.J. Wingf., Ophiostoma minus (Hedgc.) Syd. and P. Syd, and O. ips (Rumbold) Nannf. primarily colonize ray parenchyma (Rumbold, 1931; Cobb, 1988; Six, 2020). This difference in growth habit may explain how fungi like Leptographium wageneri (W.B. Kendr.) M.J. Wingf., causal agent of black stain root disease (BSRD) of conifers (Wagener and Mielke, 1961), and O. ulmi (Buisman) Melin and Nannf. and O. novo-ulmi Brasier, causal agents of Dutch elm disease (Brasier, 1991), can cause serious vascular wilt diseases leading to mortality of the tree host.
BSRD caused by the fungus L. wageneri (hereafter referred to as Lw) affects a variety of conifer hosts in the Pinaceae within their western North American ranges. The disease was first described on ponderosa pine (Pinus ponderosa Dougl. ex P. and C. Laws.) in California in 1938 and was first reported as a mortality agent of single-leaf pinyon pine (P. monophylla Torr. and Frem.) in the San Bernardino National Forest in southern California in 1941 (Wagener and Mielke, 1961). It was determined at that time that the disease was generally distributed in pinyon pine stands in the area (Wagener and Mielke, 1961). The first formal description of the disease on Douglas-fir (Pseudotsuga menziesii (Mirb.) Franco) was published in 1967 (Cobb and Platt, 1967). Goheen and Hansen (1978) later performed field surveys and found that BSRD was generally widespread in Douglas-fir stands in western Oregon and Washington. Observational and anecdotal evidence suggest that it may be emerging as a serious threat to young Douglas-fir trees in economically important timber plantations. Most of the studies on the biology, ecology, and epidemiology of Lw were conducted in the 1980’s, and few have been published since.
The fungus that causes BSRD was originally described as a novel species (Verticicladiella wageneri) in 1962 (Kendrick, 1962), and subsequently moved to the genus Leptographium (Wingfield, 1985). Three host-specific varieties of the fungus are currently recognized: Lw var. wageneri (W.B. Kendr.) M.J. Wingf., which affects pinyon pines (Pinus edulis Engelm. and P. monophylla Torr. and Frem.), Lw var. ponderosum (T.C. Harr. and F.W. Cobb) T.C. Harr. and F.W. Cobb, which affects hard pines of the subgenus Diploxylon (mainly P. ponderosa, P. jeffreyi Grev. and Balf., and P. contorta Dougl. ex Loud.), and Lw var. pseudotsugae T.C. Harr. and F.W. Cobb, which affects Douglas-fir (Harrington and Cobb, 1984, 1987). These three varieties are morphologically very similar but can be distinguished based on key characteristics related to conidiophore morphology, optimal growth temperature, colony morphology, and host specificity (Harrington and Cobb, 1984, 1986, 1987). The varieties are also distinguishable based on isozyme phenotypes (Otrosina and Cobb, 1987; Zambino and Harrington, 1989; 1992) and nucleotide sequence polymorphisms (Witthuhn et al., 1997; Jacobs et al., 2001; Duong et al., 2012). These techniques have been used to investigate genetic variation among the varieties (Otrosina and Cobb, 1987; Zambino and Harrington, 1989, 1992; Witthuhn et al., 1997), as well as phylogenetic relationships among members of the genus Leptographium (Zambino and Harrington, 1992; Jacobs et al., 2001; Duong et al., 2012). However, the degree of evolutionary divergence between the three Lw varieties remains unclear and modern molecular approaches including analyses of whole-genome data are needed to address this.
The mycelium of Lw grows inside xylem tracheids, interfering with water transport and causing a vascular wilt disease (Hessburg and Hansen, 1987). The outward symptoms are comparable to those caused by other root diseases of conifers, including reduction in height and radial growth, discoloration and gradual loss of older foliage, and eventual death of the host tree (Wagener and Mielke, 1961; Cobb, 1988; Hansen et al., 1988). There is no cellular penetration, necrosis, or decay associated with BSRD (Hessburg and Hansen, 1987, 2000). The presence of darkly pigmented mycelium within the xylem tracheids causes the distinct brown to black staining in the sapwood from which the common name of the disease is derived (Wagener and Mielke, 1961; Hessburg and Hansen, 1987). The symptoms of BSRD generally progress over a period of approximately 1–8 years leading to the eventual mortality of the host tree (Wagener and Mielke, 1961; Cobb, 1988; Hansen et al., 1988). The symptoms associated with the three varieties of this fungus on their respective hosts appear similar by all accounts, though there may be minor differences in patterns of colonization, pathogenesis, and symptom expression among hosts (Cobb, 1988). The conidiophores of Lw are produced in the galleries of bark beetles and other root-feeding insects, with masses of conidia presented in sticky droplets. This morphology is particularly well-suited for insect dispersal (Witcosky and Hansen, 1985). Secondary spread of Lw generally occurs through root contacts or root grafts (Goheen and Hansen, 1978; Hessburg and Hansen, 1986). The fungus may also grow short distances through the soil between roots that are not in direct contact, entering through wounds or natural openings (Hessburg and Hansen, 1986, 2000).
The BSRD fungus is vectored primarily by bark beetles in the Scolytinae (Goheen and Cobb, 1978; Harrington et al., 1985; Witcosky and Hansen, 1985; Witcosky et al., 1986a; Hansen et al., 1988; Harrington, 1988). Insect vectors spread Lw locally and over longer distances, but their dispersal capabilities are not well characterized (Hessburg et al., 2001). The most important vector of Lw var. ponderosum is probably Hylastes macer LeConte, though condiophores of the fungus were also found in galleries formed by Dendroctonus valens LeConte and one buprestid (Goheen and Cobb, 1978; Harrington, 1988). The vectors of Lw var. wageneri on pinyon pines are mostly unknown, though Ips spp. are commonly associated with diseased trees and have been proposed as potential vectors (Cobb, 1988). The Douglas-fir variety of Lw has three known insect vectors, the weevils Pissodes fasciatus LeConte and Steremnius carinatus Boheman (Curculionidae, Molytinae), and the bark beetle H. nigrinus Mannerheim (Curculionidae, Scolytinae) (Harrington et al., 1985; Witcosky and Hansen, 1985; Witcosky et al., 1986a). The flying weevil P. fasciatus is the least common, and probably the least important, of these three vectors (Harrington et al., 1985; Hansen et al., 1988). The weevil S. carinatus is flightless, though it has been demonstrated through mark-recapture studies that it can crawl up to 100 m in a period of 7 weeks (Hansen et al., 1988). The root beetle H. nigrinus is likely the most important vector for long-distance dispersal and the establishment of new BSRD infection centers in Douglas-fir (Harrington et al., 1985). It is strongly attracted to cut stumps and stressed trees and has been known to feed on the roots of living Douglas-fir trees and would thus be capable of spreading the spores of Lw var. pseudotsugae from diseased trees to healthy trees (Harrington et al., 1985). Hylastes nigrinus is capable of flight (Zethner-Møller and Rudinsky, 1967; Harrington et al., 1985; Witcosky et al., 1986a; Jacobi, 1992), though its dispersal range is unknown.
Soil disturbance and compaction related to forest management activities such as precommercial thinning, road building, skid trails, and tractor logging are known to predispose trees to BSRD infection (Harrington et al., 1985; Witcosky et al., 1986b; Hessburg et al., 2001). The volatile compounds produced by injured and stressed trees (e.g., ethanol) attract the insects that vector Lw (Kelsey and Joseph, 1998). The insects that vector Lw are also attracted to the stumps left after thinning or regeneration harvests, resulting in local increases in vector populations where management activities have occurred (Harrington et al., 1983; Witcosky et al., 1986b; Hessburg et al., 2001). In western Oregon, much of the timber industry is focused on the production of Douglas-fir. BSRD is particularly damaging in this region where susceptible host trees are widely planted and precommercial and commercial thinning operations are regularly practiced (Hansen and Goheen, 1988; Hessburg et al., 2001). In the regions where BSRD is most active in Douglas-fir, summers are expected to become both hotter and drier due to climate change (Mote and Salathé, 2010), leading to further intensification of disease due to heat and drought stress (Agne et al., 2018).
The reproductive biology and the role of recombination in structuring populations of Lw is not well understood. Reproduction is predominately asexual, as conidiophores are regularly observed in insect galleries and in culture. The occurrence and frequency of sexual reproduction in natural Lw populations are not known, though other members of the Ophiostomatales regularly produce perithecia (Wingfield et al., 1993). The production of perithecia by Lw var. ponderosum has been reported once in a gallery formed by H. macer on a ponderosa pine tree in California (Goheen and Cobb, 1978). Analyses of mating type (MAT) loci in populations of Lw have indicated that it is heterothallic (Duong et al., 2016), which supports the possibility of sexual reproduction in nature. Elucidating the primary mode(s) of reproduction in plant pathogen populations is important for understanding population structure, as recombination influences genetic diversity and the rate of evolutionary change (Milgroom, 1996, 2017; McDonald and Linde, 2002; Grünwald et al., 2016). Understanding the reproductive biology of Lw is also important for describing its epidemiology. Mixed reproductive systems that include sexual recombination in addition to asexual reproduction may be particularly advantageous for plant pathogenic fungi (McDonald and Linde, 2002; Milgroom, 2017). Recombination allows for the generation of novel genotypes, while asexual reproduction could act to maintain beneficial combinations of alleles and allow them to spread (Milgroom, 1996, 2017; McDonald and Linde, 2002).
Population structure, gene flow, and epidemiology of bark beetle-associated fungi are strongly influenced by their relationships with insect vectors (Gibbs, 1978; Tsui et al., 2012, 2014; Bracewell et al., 2018; Six, 2020). Previous studies examining the epidemiology of BSRD have focused on trapping and identifying the insects that spread the fungus that causes the disease (Harrington et al., 1985; Witcosky and Hansen, 1985). However, population genetics and genomics have become valuable tools to investigate the epidemiology and dispersal dynamics of both native and introduced plant pathogens (Linde et al., 2002, 2009; Rivas et al., 2004; Banke and McDonald, 2005; Goss et al., 2009; Rieux et al., 2011; Garbelotto et al., 2013; Barnes et al., 2014; Brar et al., 2015, 2018; Grünwald et al., 2016; Bennett et al., 2019; Tabima et al., 2020, 2021). Information about population structure and genetic diversity is essential for understanding how factors such as landscape topology or forest management practices influence the epidemiology and evolutionary biology of pathogens and for developing effective strategies for managing plant diseases (Linde et al., 2002, 2009, McDonald and Linde, 2002; Banke and McDonald, 2005; Heinzelmann et al., 2012; McDonald and Mundt, 2016). The relative influences of local and long-distance dispersal on the landscape-level spatial genetic structure of Lw populations have not been investigated previously.
The overarching goal for this study was to apply the principles of population genomics to gain a better understanding of the epidemiology of a native insect-vectored conifer pathogen. The results of this investigation could inform the development of effective management strategies for BSRD and will provide a foundational knowledge enabling future studies of the factors influencing population structure and epidemiology of insect-vectored pathogens. To accomplish these goals, we analyzed full genome sequence data from two of the three Lw varieties and pursued the following objectives: (1) characterize the evolutionary divergence between Lw var. wageneri and Lw var. pseudotsugae; (2) examine the genetic structure of populations of Lw var. pseudotsugae; (3) infer the relative importance of local vs. long-distance dispersal in the epidemiology of BSRD; and (4) assess the potential for genetic recombination in Lw var. pseudotsugae by analyzing mating type (MAT) frequencies.
Materials and Methods
Field Sampling
In 2018 and 2019, young Douglas-fir stands (ages 4–10 years) were selected from areas in which BSRD had been previously observed. Five stands were selected in each of three Douglas-fir plantations including one tree farm near Forest Grove, OR (FG), a tree farm near Roseburg, OR (RB), and a tree farm near Springfield, OR (SP) (Table 1 and Figure 1). Samples were also collected from one symptomatic stand in a plantation in the foothills of Mary’s Peak near Philomath, OR (MP) (Table 1 and Figure 1). Additionally, two natural stands of single-leaf pinyon pine (P. monophylla) were sampled in the San Bernardino National Forest near Big Bear, California (BL, DC) (Table 1 and Figure 1). Trees within, or adjacent to, the focal stands with symptoms indicative of root disease (i.e., chlorosis or browning foliage, short leader length, wilting, stress cones) were examined for characteristic black staining in the vascular tissue near the root crown, and wood samples were collected if staining was present. Our sampling protocol called for collecting a minimum of 12 samples from each stand. However, this number varied due to disease incidence, availability of fresh material, time efficiency, and feasibility of sample collection. The wood samples were kept on ice and returned to the laboratory at Oregon State University within 48 h. The samples were then stored at 4°C for no more than 14 days.
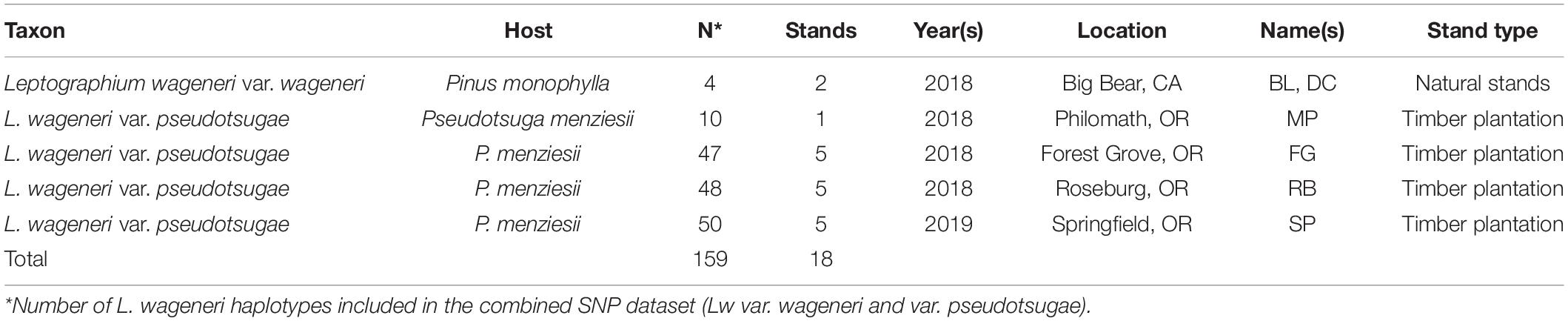
Table 1. Summary of Leptographium wageneri taxa, isolates, and sampling locations.
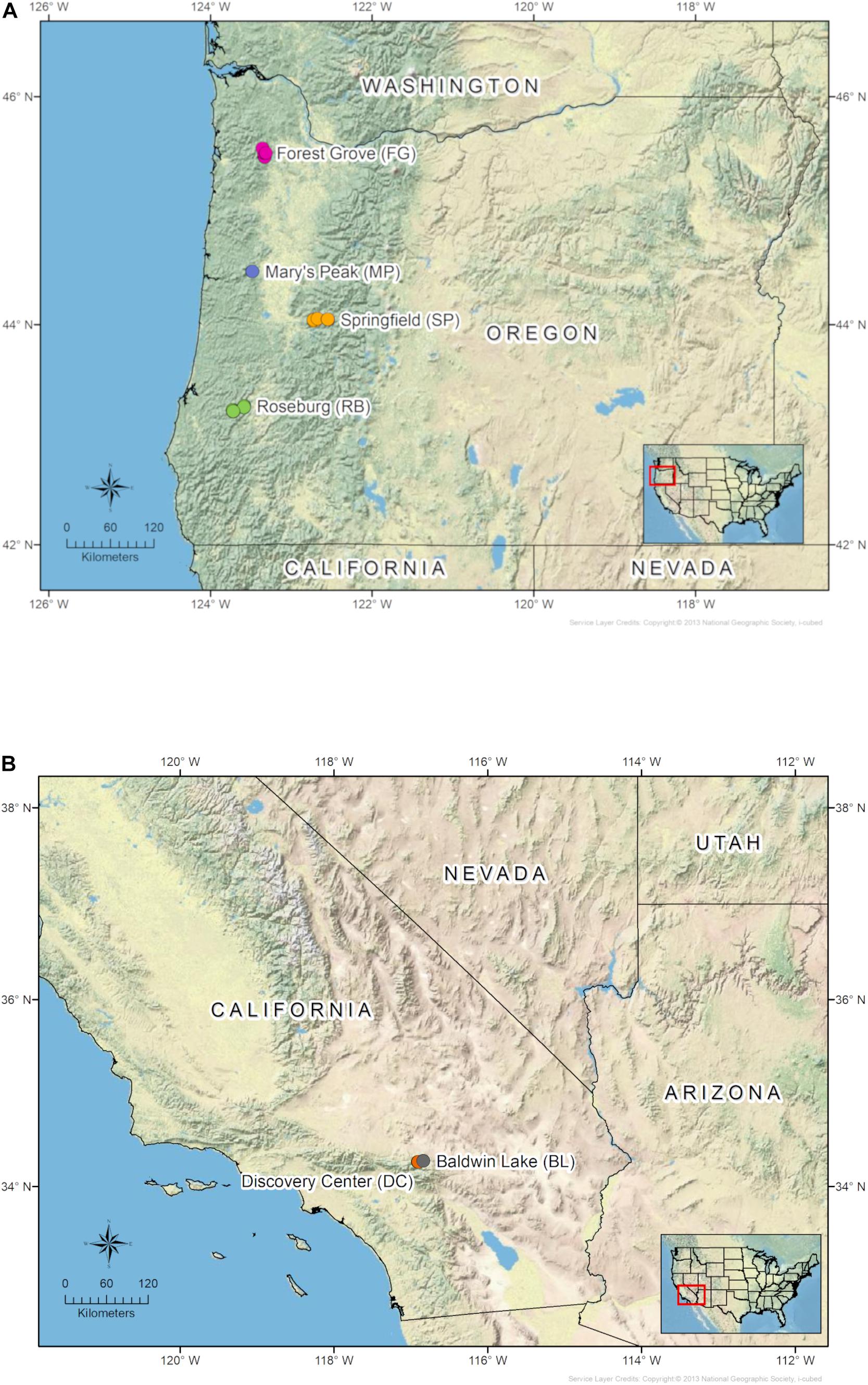
Figure 1. Maps of sampling locations in (A) Oregon Douglas-fir plantations, and (B) California pinyon pine stands.
Isolation of L. wageneri From Wood Samples
Stained wood pieces were surface sterilized by soaking for 30 s in 95% ethanol (EtOH) followed by 3 min in a 10% solution of household bleach (∼0.5% NaOCl) and rinsing with sterile deionized H2O. The surface-sterilized pieces were then embedded in 2% malt extract agar (MEA) amended with 200 ppm cycloheximide and 200 ppm streptomycin sulfate (CSMA) (Harrington, 1992). The characteristic serpentine hyphae of Lw often colonized the agar within 1–3 weeks. Conidiophores occasionally grew directly from the wood pieces within 10–14 days. Hyphal tips or conidia were aseptically transferred onto Petri dishes containing 2% MEA and incubated in the dark at 18°C for approximately 14 days. While over 200 samples were initially collected in the field and processed in the laboratory, some of the samples did not yield pure cultures of Lw or were lost due to culture contamination. To prepare mycelium for DNA extractions and downstream applications, each Lw isolate was grown in 2% malt extract broth (MEB) for 14–28 days at room temperature on a rotary shaker (150 rpm). Mycelium from each liquid culture was filtered through sterilized Miracloth (MilliporeSigma), rinsed with sterile deionized H2O, dried with sterile paper towel, and stored at –80°C.
DNA Extraction and Whole Genome Sequencing
A total of 164 samples were submitted to the Center for Genome Research and Biocomputing (CGRB) at Oregon State University for DNA extraction, Illumina library preparation, and whole genome sequencing. Approximately 30–50 mg of dried Lw mycelium from the liquid culture filtrate was placed in a 1.2 ml tube with a stainless-steel bead and frozen at –80°C. The tissue was homogenized with a Qiagen Tissue Lyser (Qiagen Inc., Hilden, Germany) at 30 hz for approximately 1 min. A magnetic particle extraction was performed with the Omega Bio-tek Mag-Bind Plant DNA DS kit (Omega Bio-tek Inc., Norcross, GA, United States) and the KingFisher Flex automated extraction platform (Thermo Fisher Scientific, Waltham, MA, United States). For the Illumina library prep, the purified DNA was mechanically sheared prior to the selection of 300 bp fragments. Illumina adapters were added to these fragments along with a unique barcode. The DNA samples were sequenced on two lanes of an Illumina HiSeq 3000 with 150 bp paired end reads.
Bioinformatics and Data Processing
Illumina reads were mapped to an indexed reference genome from a Lw var. pseudotsugae ex-type isolate (CMW154) preserved in the culture collection (CMW) of the Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, South Africa. Alignments were performed with the Burrows-Wheeler aligner (BWA v. 0.7.17) (Li and Durbin, 2009) with default parameters. Post-processing was performed with SAMtools v. 1.12 (Conversion to BAM, Li et al., 2009) and PICARD tools (Marking duplicate reads, Broad Institute, 2019). Each sample was genotyped with the Haplotype Caller (HC) from the Genome Analysis Tool Kit (GATK v4.0) (McKenna et al., 2010) and combined into large variant call format (VCF) files, which were then filtered in R with the package vcfR v.1.0.0 (Knaus and Grünwald, 2017; R Core Team, 2020).
Two separate VCF files were created; one was a combined dataset with haplotypes from both Lw var. pseudotsugae and Lw var. wageneri isolates, and the other a single species dataset with only Lw var. pseudotsugae isolates. This allowed for a more accurate depiction of the evolutionary relationships among the two Lw varieties, and higher resolution in the analyses of spatial genetic variation in the Lw var. pseudotsugae populations. The filtering protocol for the combined dataset masked all SNP variants with sequencing depth (DP) less than 4x and minimum mapping quality (MQ) less than 50. Individual SNP variants were masked if they were present in only a single sample. A mask was also applied if more than 20% missing data occurred at either the variant or sample levels. Quality filtering on the combined dataset resulted in the removal of five samples. As a result, 155 samples of Lw var. pseudotsugae and four samples of Lw var. wageneri remained. The Lw var. pseudotsugae dataset was filtered similarly except that the minimum DP threshold was 10x. This filtering protocol resulted in the removal of one additional sample for a final sample size of 154 for the single-species dataset.
Evolutionary Divergence Among Two L. wageneri Varieties
The evolutionary relationship between the two host-specific Lw varieties and among the isolates of each variety was evaluated with a maximum-likelihood (ML) phylogeny. The MULTIGAMMAI algorithm from RAxML v. 8.1.2 was used with 1,000 bootstrap replicates to produce a phylogenetic reconstruction (Stamatakis, 2014). The resulting tree was visualized with the R package ggtree v. 2.4.1 (Yu et al., 2017). Average pairwise genetic differentiation (G’ST) (Hedrick, 2005) between the two taxa was calculated from the haplotypes of 155 Lw var. pseudotsugae and 4 Lw var. wageneri isolates with the R package vcfR v. 1.10.0 (Knaus and Grünwald, 2017). The proportion of SNP positions with G’ST (Hedrick, 2005) greater than 0.70 was calculated with the R package mmod v. 1.3.3 (Winter, 2012) to estimate the degree of differentiation between the two Lw taxa.
Diversity and Genetic Structure in L. wageneri var. pseudotsugae Populations
Discriminant analysis of principal components (DAPC) (Jombart et al., 2010) was performed in R with adegenet v. 2.1.1 (Jombart, 2008) with a priori designated populations representing the four Douglas-fir plantations to determine whether the observed genetic structure corresponded to the geographic sampling locations. If genetic structure corresponded precisely to the plantations that would be viewed as evidence supporting the existence of local clonal lineages with little or no gene flow at the regional scale. If no genetic structure was found in the population, or if it did not correspond to the four plantations, that would be viewed as evidence to support region-wide landscape-level gene flow that transcended individual plantations.
A K-means clustering algorithm from the R package adegenet v. 2.1.1 (Jombart, 2008; Jombart et al., 2010) was used to assign the Lw var. pseudotsugae haplotypes to genetic clusters without prior information about the plantations from which they originated. The optimal number of clusters (K) to describe the genetic variation within Lw var. pseudotsugae was chosen based on the identification of local minima in Bayesian information criterion (BIC) values (Jombart et al., 2010). Maps depicting the spatial distributions of the genetic clusters were produced with ArcMap 10.5.1 (ESRI, Redlands, California). Each stand was summarized with a pie chart representing the proportion of isolates belonging to each genetic cluster. The relationships among these clusters were also evaluated with DAPC.
Isolation by distance in the Lw var. pseudotsugae population was assessed with a Mantel test from the R package ade4 v. 1.7–16 (Dray and Dufour, 2007). This function implements a statistical test of the null hypothesis that no correlation exists between geographic and genetic distances. The genetic distance matrix consisted of pairwise dissimilarity values for the SNP haplotypes of Lw var. pseudotsugae isolates, calculated with the R package poppr v. 2.9 (Kamvar et al., 2014, 2015). The stats package v. 4.1.0 in R was used to calculate pairwise Euclidean distances among the geographic coordinates for each sample. Statistical significance was determined from a Monte-Carlo test with 10,000 random samples.
Expected heterozygosity (i.e., gene diversity) (Hs) was calculated in R with the package adegenet v. 2.1.1 (Jombart, 2008) to assess within-population genetic diversity at the stand and plantation levels. This value has also been called gene diversity and can be interpreted as the probability that two alleles selected at random from the population (or subpopulation) will be different (Nei, 1973). Genetic variation was partitioned among hierarchical levels of spatial scale including plantation, stand within plantations, and within-stand via analysis of molecular variance (AMOVA) (Excoffier et al., 1992), performed with the R package poppr v. 2.9 (Kamvar et al., 2014, 2015). This analysis included a calculation of phi statistics (ϕST) representing global estimates of genetic differentiation among sampling units within and between each of the hierarchical levels. Pairwise genetic differentiation among plantations, and among stands within plantations, was also estimated with G’ST (Hedrick, 2005) calculated with the R package mmod v. 1.3.3 (Winter, 2012).
Mating Type Assays
To identify the mating type of the reference isolate CMW154, a search for genes homologous to the reported MAT genes of other species of Leptographium was performed. The MAT1-1 idiomorph from L. procerum was used as reference locus (KC883456; Duong et al., 2013). A BLAST database (Altschul et al., 1990) was constructed using the MAT1-1-1, MAT1-1-2, and MAT1-1-3 amino acid sequences from L. procerum, as well as the upstream SLA gene of L. profanum (AGS32063.1; Duong et al., 2013). The blastp searches using the amino acid sequences predicted from the CMW154 reference genome were used as queries to detect homologous sequences, using an e-value of 10e–10 as the threshold.
Presence/absence of the MAT1-1 locus across the samples in this study was assayed by measuring the read depth at the region of the predicted MAT1-1 locus. The depth assays were performed on the BAM files from the read alignment to isolate CMW154 used for the population genetics analysis. Depth of coverage was calculated using SAMtools v. 1.12 depth (Li et al., 2009). Visualization of depth was done in the R package ggplot2 v. 3.3.1 (Wickham, 2016). Mating type idiomorphs were detected for each sample in the following manner- for MAT1-1: The presence and completeness of the SLA locus, the completeness of the MAT1-1-1 locus, and presence of MAT1-1-2 locus. For MAT1-2: The presence and completeness of the SLA locus, a truncated version of the MAT1-1-1 locus and the absence of a MAT1-1-2 locus (Duong et al., 2013; Tsui et al., 2013). Finally, to evaluate equal mating ratios, a chi-square test was performed for each site and location using the R statistical framework.
Results
The combined dataset containing 159 unique haplotypes (Lw var. wageneri = 4, Lw var. pseudotsugae = 155) consisted of 120,613 binary SNPs with 4.38% missing data overall. The single species dataset (Lw var. pseudotsugae only) consisted of 154 haplotypes with 96,088 binary SNPs and 4.04% missing data overall. The average number of reads per sample ranged from 7,665,531 to 23,658,824 and average sequencing depth per sample ranged from 17.63x to 77.66x (mean = 32.51x) (Supplementary Table 1). The percentage of reads mapped to the reference genome ranged from 82.99 to 97.71% (mean = 95.37%) (Supplementary Table 1).
Evolutionary Divergence Among Two L. wageneri Varieties
The topology of the rooted ML tree showed strong evolutionary divergence between the two Lw varieties. The varieties were grouped into two separate clades with relatively long associated branch length and robust statistical support (Figure 2). The average G’ST among isolates of Lw var. pseudotsugae and Lw var. wageneri was 0.411 and 44.9% of the SNPs in their genomes had G’ST greater than 0.7 suggesting strong genetic differentiation.

Figure 2. Maximum-likelihood (ML) phylogenetic reconstruction with 155 Leptographium wageneri var. pseudotsugae haplotypes and 4 Leptographium wageneri var. wageneri haplotypes. Data consisted of 120,613 binary SNPs. Bootstrap support values were calculated from 1,000 replicate trees.
Diversity and Genetic Structure in L. wageneri var. pseudotsugae Populations
In the ML tree, the Lw var. pseudotsugae group was represented by either local clonal lineages (i.e., isolates with similar haplotypes originating from a single plantation) or more diverse groupings consisting of closely related isolates from different plantations. Three distinct basal clades were observed among the Lw var. pseudotsugae isolates. One clade was represented by isolates collected in the SP plantation, one represented by isolates collected in the FG and SP plantations, and one represented by a mixture of isolates collected from the four Douglas-fir plantations (MP, FG, RB, and SP) (Figure 2).
The DAPC analyses produced from the single species data set revealed evidence for both local and regional genetic structure in Lw var. pseudotsugae (Figure 3). Four clusters were observed in the DAPC scatter plot (Figure 3A), representing the MP, FG, RB, and SP plantations. These four clusters appeared as distinct groupings, but with some overlap of isolates with similar haplotypes, a pattern that closely resembled the groupings observed in the phylogenetic tree (Figure 2). Isolates from the FG and MP plantations appeared most closely associated with the RB isolates, while SP appeared to be more divergent with little overlap with the cluster representing the RB isolates (Figure 3A). The haplotypes of isolates collected in the RB and MP plantations were unique in that they appeared to be admixed based on posterior probabilities of these individuals to the clusters representing each plantation (Figure 3B). These RB and MP haplotypes had at least 0.70 probability of assignment to the RB cluster and up to 0.30 probability of assignment to the MP cluster. Haplotypes similar to these were detected in all plantations (Figure 3B). The haplotypes of most isolates collected in the FG and SP plantations had a very high probability of membership in the genetic cluster represented by their respective plantation (i.e., most haplotypes from the FG plantation belonged to the FG population, and most haplotypes from the SP plantation belonged to SP population) (Figure 3B). Isolates collected at the MP and RB plantations, which are approximately 150 km apart (Figure 1), had similar genetic composition, with the posterior probabilities for each isolate reflecting partial membership in both the RB and MP clusters (Figure 3B).
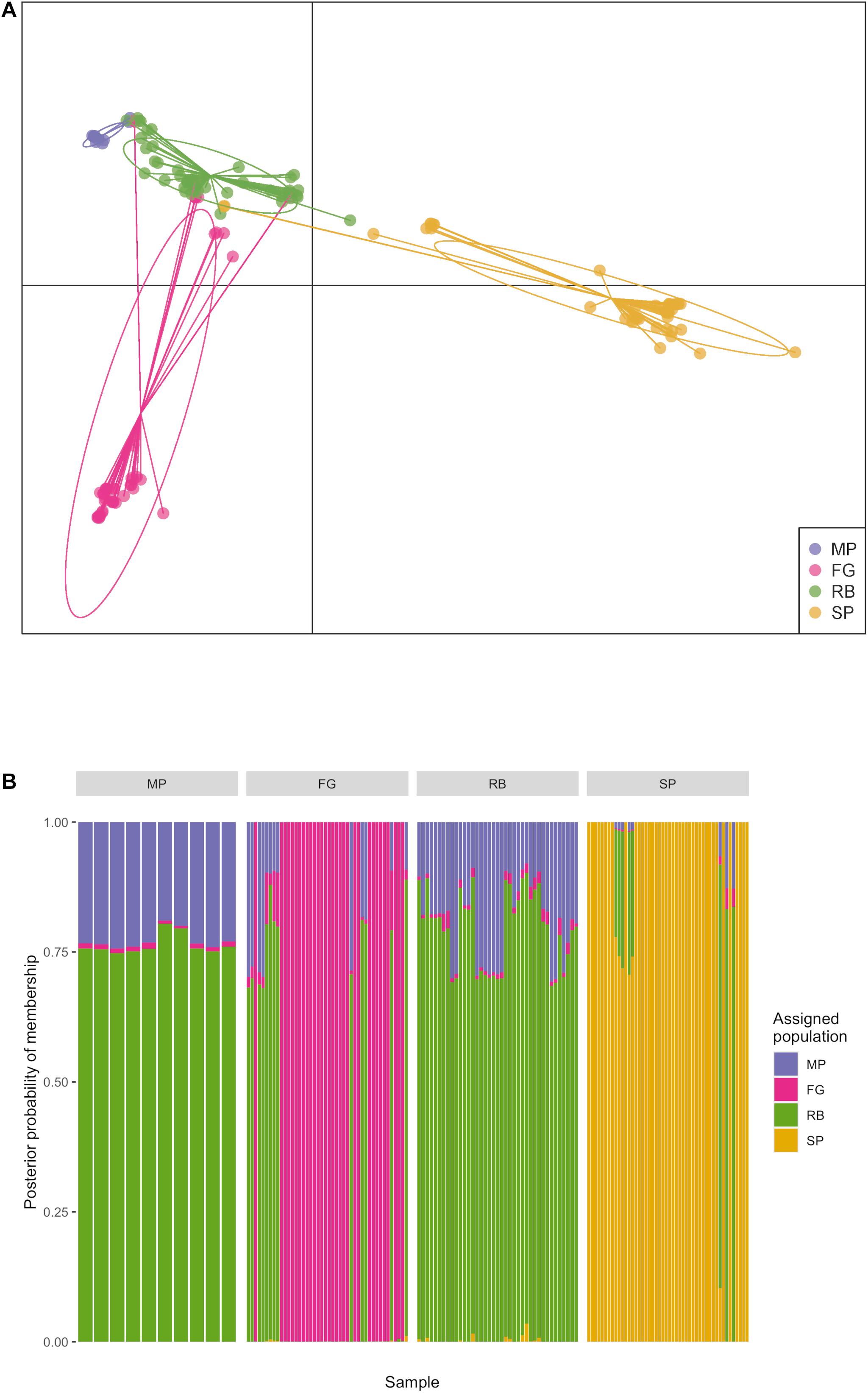
Figure 3. Discriminant analysis of principal components (DAPC) performed with 154 Leptographium wageneri var. pseudotsugae isolates from four Oregon Douglas-fir plantations. Plantation was used as a priori population designation. (A) Scatter plot showing clusters representing each plantation with ellipses, (B) composition plot showing probability of membership of each isolate haplotype in each of four genetic clusters representing the Douglas-fir plantations.
The average expected heterozygosity (Hs) served as an estimate of within-population genetic diversity (Nei, 1973). This metric was calculated for the entire sample population, and at the plantation and stand levels (Table 2). At the plantation level, the average expected heterozygosity ranged from 0.118 for the FG plantation to 0.243 for the SP plantation. The stand RB_5480 had the lowest diversity (Hs = 0.021) and SP_770 had the highest (Hs = 0.162) (Table 2). The gene diversity for the entire sample population of Lw var. pseudotsugae from Douglas-fir plantations in western Oregon was 0.209 (Table 2).
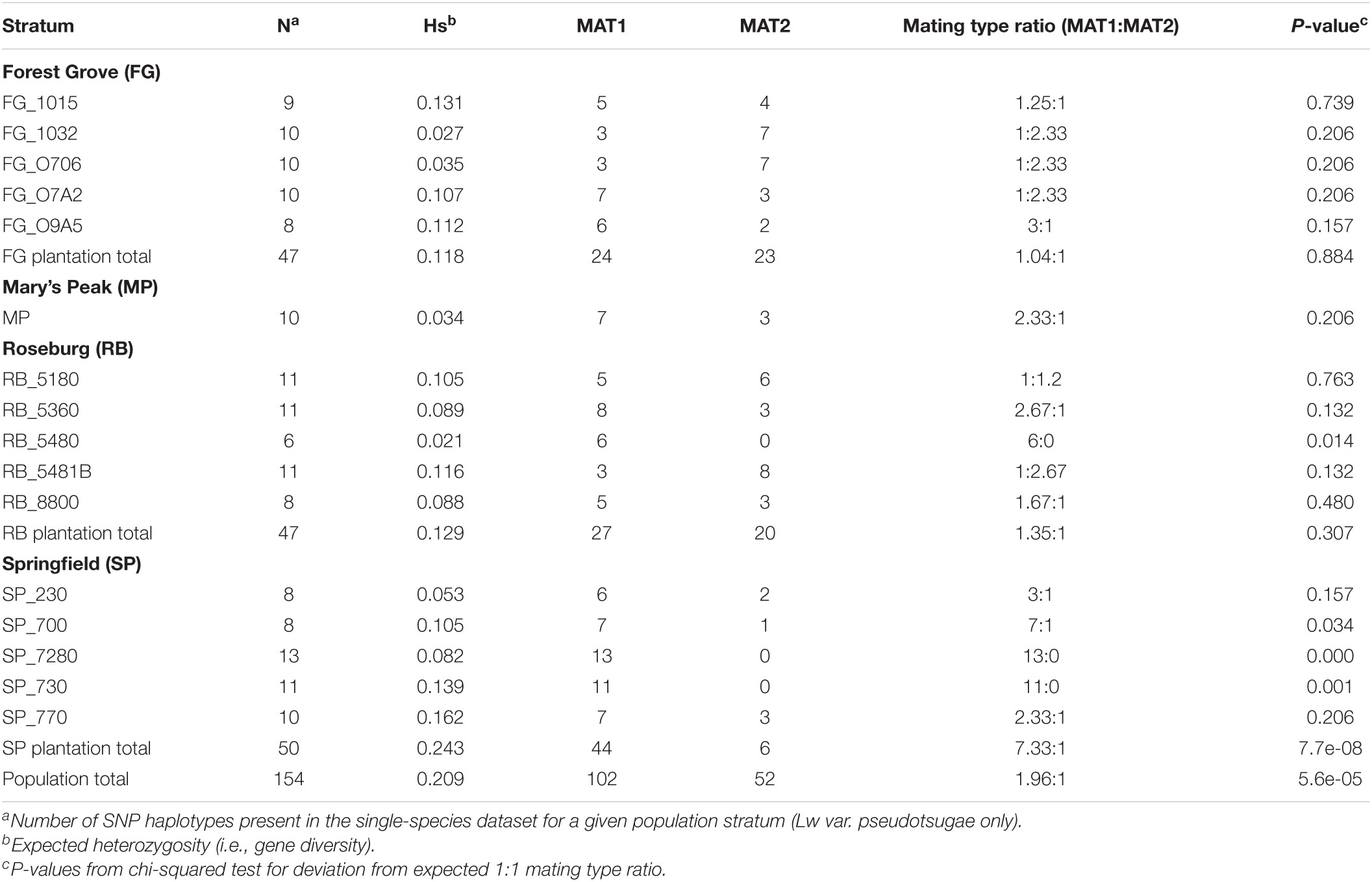
Table 2. Sample sizes, expected heterozygosity, and inferred mating type ratios for the stands and planations from which Leptographium wageneri var. pseudotsugae isolates were collected.
The AMOVA partitioned genetic variation among several levels of a spatial population hierarchy. A large proportion of the genetic variation in the Lw var. pseudotsugae population occurred within stands (43.812%, Table 3), with significant differentiation among plantations (ϕST = 0.213, P = 0.001), and among stands within plantations (ϕSS = 0.443, P = 0.001) (Table 3). Estimates of pairwise genetic differentiation among plantations, and among stands within plantations, provided additional support for these relationships (Figure 4). All plantations showed moderate-to-strong genetic differentiation from one another. The FG and MP plantations had the strongest plantation-level genetic differentiation (G’ST = 0.62), while the SP and RB plantations had the weakest (G’ST = 0.30) (Figure 4A). Stands within the FG and RB plantations were generally weakly differentiated from one another (Figure 4B), with a few exceptions. For instance, stand FG_1015 was more strongly differentiated from some other FG stands (e.g., FG_O706 and FG_1032) than from some stands in the more distant RB and SP plantations (Figure 4B). The isolates collected in stand FG_1015 shared the closest relationships with isolates collected in the RB stands 5180 (G’ST = 0.24), 8800 (G’ST = 0.25), and 5481B (G’ST = 0.26) (Figure 4B). Those stands are approximately 250 km apart (Figure 1). These G’ST values were similar to those calculated for pairwise comparisons among other stands within the FG plantation (Figure 4B). Stands within the SP plantation were very strongly differentiated from one another, with the exception of SP_230 and SP_770 (G’ST = 0.29) (Figure 4B). The Mantel test indicated a statistically significant relationship between geographic and genetic distances (r = 0.290, P < 0.001). Thus, isolates collected closer together were generally more closely related and those collected further apart were generally more distantly related.
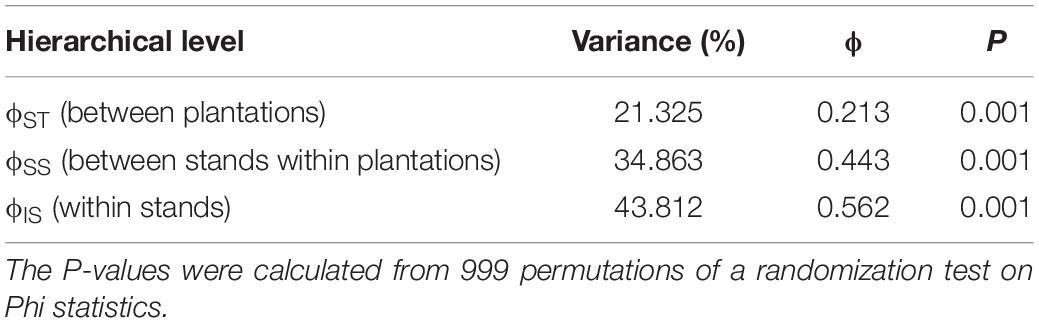
Table 3. Analysis of molecular variance (AMOVA) table showing the partitioning of total genetic variability in the Leptographium wageneri var. pseudotsugae population among levels of a population hierarchy.
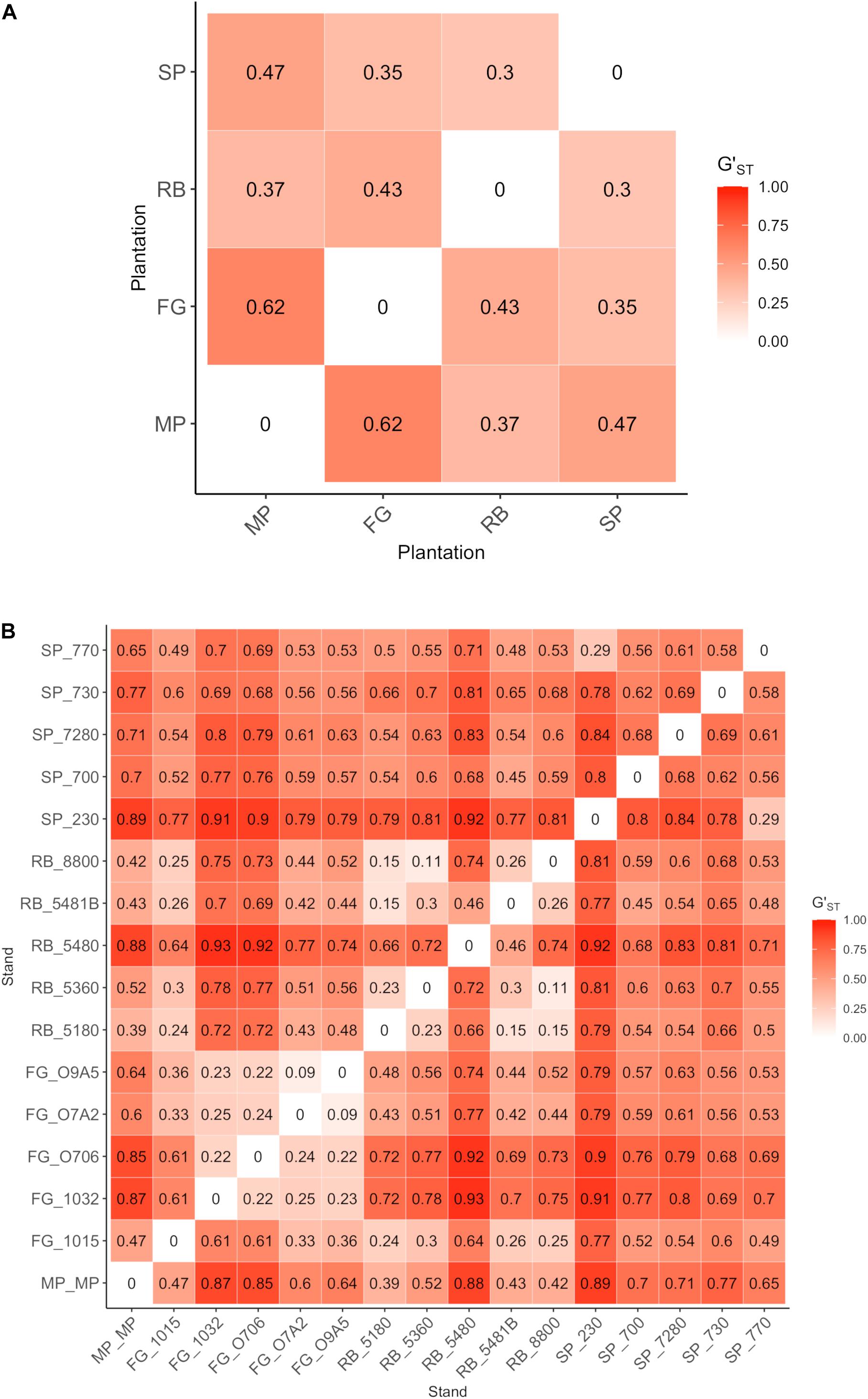
Figure 4. Pairwise genetic differentiation (G’ST) values for Leptographium wageneri var. pseudotsugae isolates in (A) plantations, and (B) stands within plantations.
The K-means clustering analysis with the 154 Lw var. pseudotsugae haplotypes yielded a local minimum BIC value at K = 9 clusters (Supplementary Table 2 and Supplementary Figure 1). Thus, nine was chosen as the optimal number of clusters necessary to describe the variation present in the Lw var. pseudotsugae haplotypes (Supplementary Figure 2). In some cases, the clusters were unique to a stand (e.g., Cluster 7, Figures 5, 6). We did not find evidence of Cluster 7 occurring outside of the stand where it was detected, despite having sampled 11 isolates from an adjacent stand (Figures 5D, 6). In other cases, isolates belonging to the same genetic cluster occurred in two or more stands within a single plantation (e.g., Cluster 3, Figures 5A, 6). One of the genetic clusters identified via K-means clustering (Cluster 8, Figures 5, 6) occurred in all four of the plantations sampled in western Oregon.
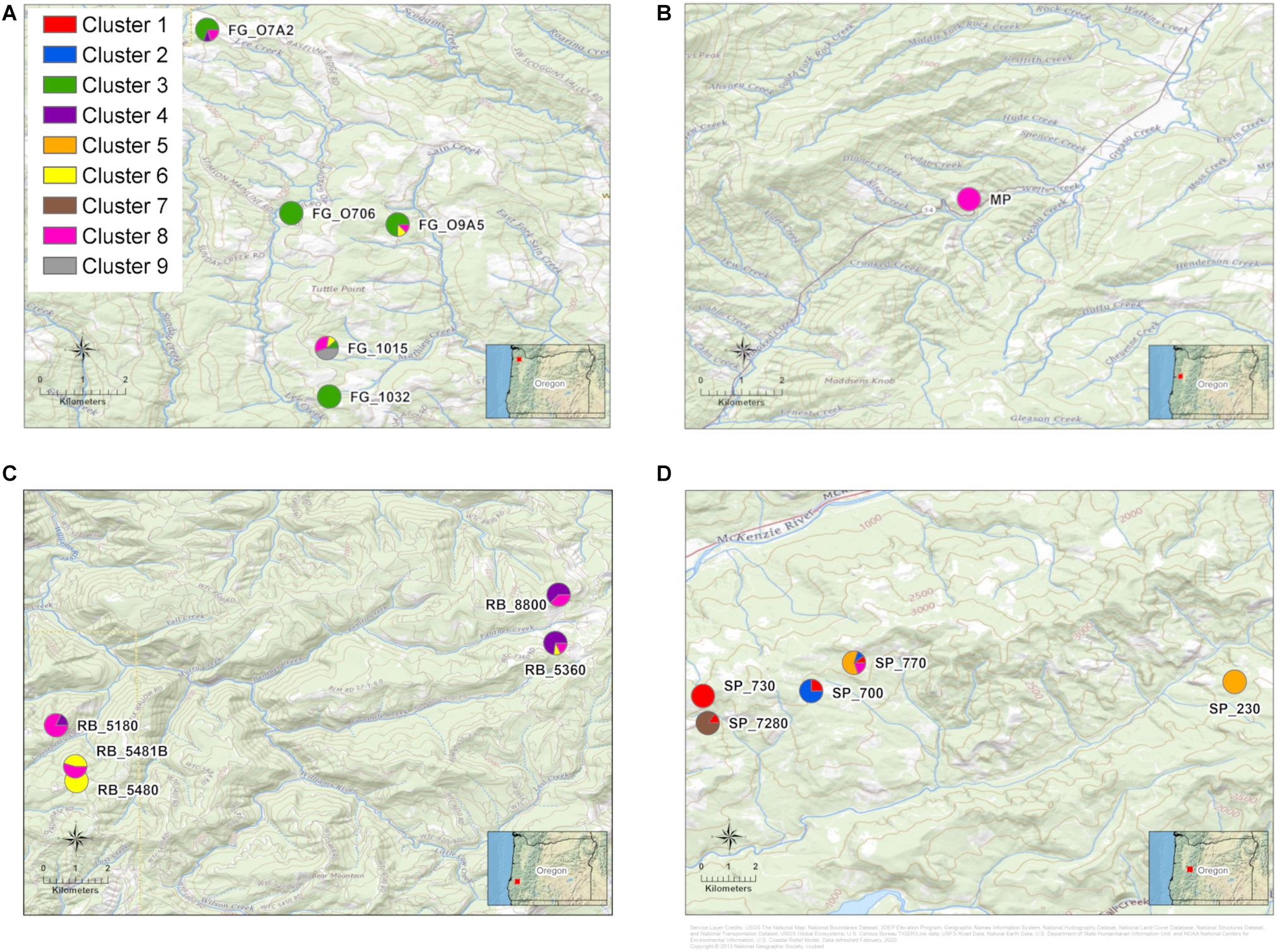
Figure 5. Maps of stand locations within each Douglas-fir plantation with pie charts representing proportion of isolates in each of nine genetic clusters. Genetic clusters were assigned for all Leptographium wageneri var. pseudotsugae isolates with a K-means clustering algorithm. (A) Forest Grove (FG) plantation, (B) Mary’s Peak (MP) plantation, (C) Roseburg (RB) plantation, (D) Springfield (SP) plantation. Labels next to the pie charts represent the abbreviation for each plantation followed by the stand number. See Table 2 for sample sizes within stands and plantations.
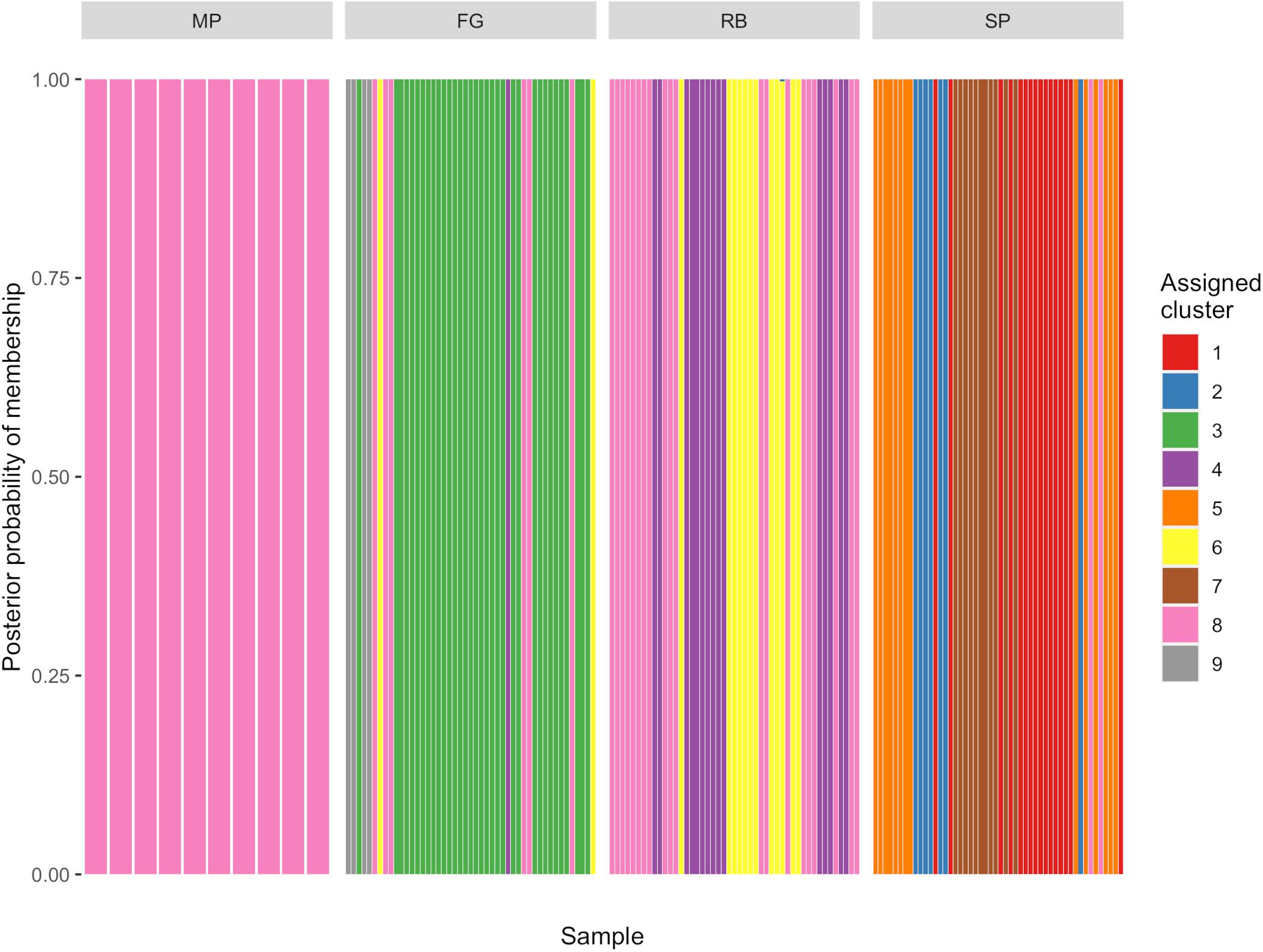
Figure 6. Discriminant analysis of principal components (DAPC) genotype composition plot showing probability of membership of each Leptographium wageneri var. pseudotsugae isolate haplotype in each of nine clusters assigned with K-means clustering algorithm.
Mating Type Identification and Distribution
The homology assays to detect the mating type locus MAT1-1 in isolate CMW154 resulted in a region from 9,951 to 17,898 of Contig 94. This region contained sequences homologuous to the SLA gene (LWAP_08038, e-value = 0), MAT1-1-1 (LWAP_08037, e-value = 0) and MAT1-1-2 (LWAP_08037, e-value = 2.30e-87) genes. No homologuous sequences were found for MAT1-1-3 in the CMW154 reference genome. A total of 111 Lw var. pseudotsugae isolates were predicted to have the MAT1-1 idiomorph, characterized by the presence of full MAT1-1-1 and MAT1-1-2 loci (Duong et al., 2013). Fifty-three isolates were predicted to have the MAT1-2 idiomorph, characterized by a truncated MAT1-1-1 locus and the absence of the MAT1-1-2 locus (Duong et al., 2013; Supplementary Figure 3 and Supplementary Table 3).
For the majority of stands, the observed mating type ratios did not deviate significantly from the expected 1:1 ratio (Table 2). However, there were three stands in the SP plantation and one stand in the RB plantation that had mating ratios that differed significantly from the expectation (Table 2). At the plantation level, only the SP plantation differed significantly from the expectation (MAT1:MAT2 = 7:1, p < 0.01) (Table 2).
Discussion
We used a population genomics approach to investigate the epidemiology, dispersal capabilities, and reproductive biology of the BSRD fungus. The two varieties of Lw included in these analyses, Lw var. wageneri and Lw var. pseudotsugae, appear to be strongly divergent. Analyses of the population structure and diversity of Lw var. pseudotsugae in Douglas-fir plantations in western Oregon revealed signatures of clonality, but with mating type distributions suggesting heterothallism and the possibility of recombination via sexual reproduction. The spatial distribution of genetic variation in Lw var. pseudotsugae populations provided evidence for both local and long-distance dispersal, with implications for future management of BSRD.
Phylogenetic reconstruction and estimates of genetic differentiation indicated strong evolutionary divergence among isolates of Lw var. wageneri and Lw var. pseudotsugae. This level of divergence in populations of bark beetle symbionts can result from host differentiation, insect behavior, geographic isolation, or a combination of these factors (Alamouti et al., 2011; Bracewell et al., 2018). Cryptic speciation has occurred due to genetic isolation among two lineages of a closely related ophiostomatoid fungus, G. clavigera, co-occurring on mountain pine beetle (MPB) and/or jeffrey pine beetle (JPB) (Alamouti et al., 2011). The genetic isolation among these lineages was attributed to factors such as bark beetle population dynamics, tree host preferences, and host-tree physiology, all of which may have influenced genetic diversity and driven evolutionary divergence in the fungal symbiont populations (Alamouti et al., 2011). Similarly, Lw var. wageneri occurs on pinyon pine and is associated with an undescribed group of vectors that frequently invade that tree host, while Lw var. pseudotsugae is found only on Douglas-fir and is vectored by several species of insects that preferentially colonize that host. This suggests that similar factors to those described by Alamouti et al. (2011) may be influencing evolutionary dynamics and species divergence among lineages of Lw. Further phylogenetic analyses are needed to determine whether treatment of these fungi as separate species, rather than varieties of the same species, would be warranted. Future studies should include isolates of the third variety (Lw var. ponderosum) as well as additional taxa in the genus.
Population-level analyses of 154 SNP haplotypes of Lw var. pseudotsugae from 16 stands across four Douglas-fir plantations revealed spatial genetic structure suggesting a heterogeneous mosaic of genetic diversity at local and regional scales. Although Lw var. pseudotsugae populations are mainly clonal, consistent with asexual reproduction via mitospores, local populations are diverse, and the clonal lineages contained therein may occupy relatively large geographic areas. New clonal lineages likely arise due to mutation, recombination, and/or genetic drift, and may spread locally via root contact or disperse to new stands via insect vector. Although ascomata of Lw var. pseudotsugae have never been observed, there is evidence for heterothallism (i.e., the presence of two mating type idiomorphs) from this and previous studies (Duong et al., 2016). This, along with our observations that mating type ratios did not differ significantly from 1:1 in most stands (Table 2), suggests that this fungus is capable of sexual reproduction. However, the abundance of clonal lineages suggests that if recombination does occur, it is relatively infrequent. The occurrence of outcrossing via sexual reproduction, however infrequent, has likely played a role in evolutionary history of Lw and may influence current population genetic structure (Duong et al., 2016).
Isolates collected in the SP plantation were predominately MAT1 (Table 2) resulting in a highly clonal population structure in these stands (Figure 2). The results of this study precluded an understanding of the frequency with which sexual reproduction might occur in the presence of both mating types. Recombination may occur infrequently where both mating types are present and may account for the diversity of haplotypes observed within plantations. The primary asexual reproductive mode accompanied by less frequent sexual recombination appears to be a common strategy for ophiostomatoid fungi (Alamouti et al., 2011; Tsui et al., 2012, 2013, 2014; Duong et al., 2013, 2016) and other bark beetle ectosymbionts (Bracewell et al., 2018). This lifestyle may be particularly beneficial for these symbiotic fungi, as frequent asexual reproduction would maintain highly adaptive coevolutionary traits associated with beetle symbiosis and plant pathogenicity (Milgroom, 1996, 2017; McDonald and Linde, 2002; Möller and Stukenbrock, 2017; Bracewell et al., 2018). The relatively infrequent sexual reproduction would provide novel combinations of alleles, increasing the potential for adaptation and counteracting the effects of Muller’s ratchet (i.e., the accumulation of deleterious mutations) (Milgroom, 1996, 2017; McDonald and Linde, 2002; Möller and Stukenbrock, 2017; Bracewell et al., 2018). Future studies should focus on understanding the frequency of sexual reproduction in L. wageneri and its effects on population structure and the evolutionary dynamics of this species.
Gene flow within and among Douglas-fir plantations was assessed to infer the relative influence of local and long-distance dispersal on the genetic structure of the Lw var. pseudotsugae population. The observed spatial distribution of haplotypes suggests three possible modes of dispersal. Local spread of clonal haplotypes within a stand could occur via insect vector or by root contact/root grafting among adjacent trees (Goheen and Hansen, 1978; Harrington et al., 1985; Witcosky and Hansen, 1985; Hessburg and Hansen, 1986; Witcosky et al., 1986a; Hansen et al., 1988). This would result in small clusters of individuals with identical, or very similar, haplotypes. Mid-range dispersal could occur within and between stands in the same plantation, resulting in the spread of Lw var. pseudotsugae among trees where roots are not in direct contact, indicating vectoring by walking or flying insects (Harrington et al., 1985; Witcosky and Hansen, 1985; Hessburg and Hansen, 1986; Witcosky et al., 1986a; Hansen et al., 1988). This would result in identical or very similar haplotypes being detected between trees in the same stand that are not adjacent to one another, or those that are in different stands in the same plantation. The establishment of new infection centers would occur via long-distance dispersal by a flying insect such as H. nigrinus (Harrington et al., 1985) and would result in closely related Lw var. pseudotsugae haplotypes being detected in separate plantations. This long-distance dispersal may also occur by some other mechanism such as anthropogenic spread due to movement of infected seedlings, though there is currently no evidence to support this. These dispersal modes need not necessarily be mutually exclusive. Rather, a combination occurring simultaneously could explain the observed landscape-level distribution of genetic variation.
It is unclear whether the apparent admixture among isolates from the MP and RB plantations is due to gene flow and subsequent recombination among isolates or simply reflects shared ancestry. Either scenario could result in the observed patterns in the DAPC composition plot where MP and RB isolates had mixed population assignments (Figure 3B). Isolates with haplotypes similar to those from the RB and MP plantations were observed among the isolates from the FG and SP plantations, yet haplotypes characteristic of the SP or FG plantations were not observed in the RB or MP plantations (Figures 3, 5, 6). This suggests that unidirectional gene flow may have occurred from the RB and/or MP populations to the other plantations. When BSRD became established in these plantations is not known, but if it became established in the RB and/or MP plantations earlier than the others, that could be an indication that these plantations were the source for at least some of the Lw var. pseudotsugae subpopulations in plantations across western Oregon.
The spatial distribution of haplotypes suggests that vector-mediated dispersal plays a major role in the epidemiology of Lw var. pseudotsugae and is likely more common than previously recognized. The fact that isolates with similar haplotypes were detected at all four Douglas-fir plantations across the 250 km sampling range in Oregon (Figures 1, 3B, 5) suggests that gene flow occurs over long distances. The occurrence of a distinct genetic cluster in all of the sampled plantations (Cluster 8, Figures 5, 6) supports this hypothesis. This could be an indication of discrete long-range dispersal events by a flying vector such as H. nigrinus. However, it is more likely that this is an endemic haplotype that has come to occupy the range of Douglas-fir in western Oregon forming a contiguous distribution at the landscape scale through a series of dispersal events that occurred over shorter distances in a stepwise manner (Linde et al., 2002).
In addition to the haplotypes and genetic clusters that were similar or shared across plantations, we also observed unique haplotypes and genetic clusters that were found in only a single plantation or a single stand (e.g., Cluster 7 in the SP plantation) (Figures 5, 6). The observations that these clonal lineages are restricted to individual stands could be indicative of some dispersal limitations of the insect vectors. If the flying vectors were capable of regular long-distance dispersal, we would likely see less differentiation among stands and plantations. This differentiation among stands or plantations may also be responsible for the significant, albeit weak, relationship between geographic and genetic distance detected by the Mantel tests. These findings may indicate the presence of locally adapted clonal lineages that arose due to random mutation, genetic drift, or selection, or may reflect the introduction and establishment of migrants from unsampled populations. More intensive sampling could be performed in the surrounding areas to further investigate the spatial extent of these distinct clonal lineages and whether there are any relationships between genetic diversity and disease severity.
The observed genetic diversity in the SP plantation may provide additional support for the gene flow hypothesis. The isolates collected from the SP plantation had the greatest gene diversity (Table 2) and consisted of five genetic clusters, four of which were found only in that plantation (Figures 5D, 6). While this pattern could have been the result of several independent recombination events with subsequent clonal expansion, this seems unlikely given that MAT2 appears to be rare or absent in these stands (Table 2). It seems more likely that this high genetic diversity resulted from multiple independent introductions of Lw var. pseudotsugae into the plantation, possibly from unsampled source populations elsewhere in western Oregon. Insect dispersal or anthropogenic activity could have introduced a diverse collection of isolates from distant source populations into the SP plantation.
The spatial genetic structure observed in Lw populations is similar in many ways to that of G. clavigera and L. longiclavatum, vectored by the mountain pine beetle (D. ponderosae) (MPB). Tsui et al. (2012) found evidence of gene flow among some geographically separated populations of G. clavigera in the northern Rocky Mountains in the U.S. and Canada, and barriers to gene flow among others. The results of the Tsui et al. (2014) study showed differentiation among three genetic/geographic clusters in the L. longiclavatum population, but there was evidence of gene flow and admixture among isolates from these regional clusters. The congruent genetic structure and differentiation observed in the symbiont populations across their study sites was attributed to insect vector behavior as well as landscape geography, which imposed limitations on the dispersal of insects (Tsui et al., 2014). The similarities between those and our results suggest that there may be commonalities among the factors influencing the population structures of these bark beetle-vectored ophiostomatoid fungi.
The observed population structure and distribution of genetic diversity highlights the importance of insect dispersal, and may have direct implications for disease management. Those activities that injure trees, cause root compaction, and disturb soils attract the insect vectors leading to an increase in BSRD (Witcosky et al., 1986b; Hansen et al., 1988; Hessburg et al., 2001). Vectors may be moving between stands across intermediate distances (10–20 km, Figure 5) within a plantation or may move in from stands outside of the local plantation. Thus, management activities leading to tree stress can increase the risk of BSRD establishment even if there are no infections in nearby stands. The results of this study would also argue against the recommendations of Hessburg et al. (1995, 2001) that buffer zones be established around infection centers in which Douglas-fir thinning is avoided or non-host species are planted.
Many studies examining the epidemiology of forest diseases have focused on invasive pathogens (Goss et al., 2009; Garbelotto et al., 2013; Barnes et al., 2014; Brar et al., 2015; Bennett et al., 2019; Tabima et al., 2021) but there are relatively few studies that have focused on investigating the factors influencing the population structure of native insect-vectored conifer pathogens. The influences of landscape-level dispersal dynamics on population structure, described herein, provide insights into the epidemiology of Lw var. pseudotsugae. This analytical framework could be applied to other forest pathosystems where the pathogenic fungus, its insect vector, and their tree hosts are native. These studies can be useful for understanding and making predictions about how the ecology and epidemiology of biotic forest disturbance agents might be influenced by anthropogenic change.
Data Availability Statement
Raw sequence data are deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA): BioProjectID PRJNA722285. The code for processing the sequence data and reproducing the population genomic analyses is available in the following GitHub repository: https://github.com/Patrick-Bennett/leptographium_popgen. Additional data will be made available by the authors upon request.
Author Contributions
PB, JT, AL, JB, and JL designed the study. PB, JL, AL, and JB performed field collections. PB processed field samples, isolated L. wageneri cultures, and prepared samples for sequencing. JT and PB performed bioinformatics data processing. PB performed all population genomics analyses except for mating-type assays, which were performed by JT. PB, JT, and JL interpreted the results. PB created the figures and wrote the initial draft of the manuscript. All authors participated in preparing and approved the final manuscript.
Funding
This research was partially supported by the USDA Forest Service Pacific Northwest Region and Forest Health Monitoring Evaluation Monitoring grant WC-EM-17-05, Weyerhaeuser Co., and the Agricultural Research Foundation (ARF).
Conflict of Interest
AL and JB were employed by Weyerhaeuser Company and declare that this study received funding from Weyerhaeuser Company. The funder had the following involvement with the study: provided financial support for sequencing; provided access to field sites; helped collect samples; and participated in the study design.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank A. Bouche, R. Lenz, S. McMurtrey, and A. Rios for assisting with the field and/or laboratory work for this study. The analyses presented here would not have been possible without the reference genome for L. wageneri var. pseudotsugae, which was kindly provided by Dr. T. Duong and Prof. Brenda Wingfield at the Forestry and Agricultural Biotechnology Institute, University of Pretoria, South Africa. We are also grateful to K. Carter and M. Dasenko at the CGRB for providing technical support and J. McCafferty, C. Chase, C. Shoot, S. Barfield, and E. Sucre at Weyerhaeuser, and B. Reding at Stimson Lumber Co., for providing access to their forest lands and support in the field. Prof. Diana Six kindly reviewed an earlier version of the manuscript and provided constructive comments. We also thank the two reviewers whose feedback helped us to improve the quality of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/ffgc.2021.695981/full#supplementary-material
References
Agne, M. C., Beedlow, P. A., Shaw, D. C., Woodruff, D. R., Lee, E. H., Cline, S. P., et al. (2018). Interactions of predominant insects and diseases with climate change in Douglas-fir forests of western Oregon and Washington, U.S.A. For. Ecol. Manag. 409, 317–332. doi: 10.1016/j.foreco.2017.11.004
Alamouti, S. M., Wang, V., DiGuistini, S., Six, D. L., Bohlmann, J., Hamelin, R. C., et al. (2011). Gene genealogies reveal cryptic species and host preferences for the pine fungal pathogen Grosmannia clavigera. Mol. Ecol. 20, 2581–2602. doi: 10.1111/j.1365-294X.2011.05109.x
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Banke, S., and McDonald, B. A. (2005). Migration patterns among global populations of the pathogenic fungus Mycosphaerella graminicola. Mol. Ecol. 14, 1881–1896. doi: 10.1111/j.1365-294X.2005.02536.x
Barnes, I., Wingfield, M. J., Carbone, I., Kirisits, T., and Wingfield, B. D. (2014). Population structure and diversity of an invasive pine needle pathogen reflects anthropogenic activity. Ecol. Evol. 4, 3642–3661. doi: 10.1002/ece3.1200
Bennett, P. I., Hood, I. A., and Stone, J. K. (2019). The genetic structure of populations of the Douglas-fir Swiss needle cast fungus Nothophaeocryptopus gaeumannii in New Zealand. Phytopathology 109, 446–455. doi: 10.1094/PHYTO-06-18-0195-R
Bracewell, R. R., Vanderpool, D., Good, J. M., and Six, D. L. (2018). Cascading speciation among mutualists and antagonists in a tree–beetle–fungi interaction. Proc. Biol. Sci. 285:20180694. doi: 10.1098/rspb.2018.0694
Brar, S., Tabima, J. F., McDougal, R. L., Dupont, P.-Y., Feau, N., Hamelin, R. C., et al. (2018). Genetic diversity of Phytophthora pluvialis, a pathogen of conifers, in New Zealand and the west coast of the United States of America. Plant Pathol. 67, 1131–1139. doi: 10.1111/ppa.12812
Brar, S., Tsui, C. K., Dhillon, B., Bergeron, M.-J., Joly, D. L., Zambino, P. J., et al. (2015). Colonization history, host distribution, anthropogenic influence and landscape features shape populations of white pine blister rust, an invasive alien tree pathogen. PloS One 10:e0127916. doi: 10.1371/journal.pone.0127916
Brasier, C. M. (1991). Ophiostoma novo-ulmi sp. nov., causative agent of current Dutch elm disease pandemics. Mycopathologia 115, 151–161. doi: 10.1007/bf00462219
Broad Institute. (2019). Picard Tools. Available online at: http://broadinstitute.github.io/picard/
Cobb, F. W. Jr. (1988). “Leptographium wageneri, cause of black-stain root disease: a review of its discovery, occurrence and biology with emphasis on pinyon and ponderosa pine,” in Leptographium Root Diseases on Conifers, Symposium Series, American Phytopathological Society, eds T. C. Harrington and F. W. Cobb Jr. (St. Paul, MN: American Phytopathological Society Press), 41–62.
Cobb, F. W. Jr., and Platt, W. D. (1967). Pathogenicity of Verticicladiella wageneri to Douglas-fir. Phytopathology 57, 998–999.
Davis, T. S., Stewart, J. E., Mann, A., Bradley, C., and Hofstetter, R. W. (2019). Evidence for multiple ecological roles of Leptographium abietinum, a symbiotic fungus associated with the North American spruce beetle. Fungal Ecol. 38, 62–70. doi: 10.1016/j.funeco.2018.04.008
Dray, S., and Dufour, A.-B. (2007). The ade4 package: implementing the duality diagram for ecologists. J. Stat. Softw. 22, 1–20. doi: 10.18637/jss.v022.i04
Duong, T. A., de Beer, Z. W., Wingfield, B. D., and Wingfield, M. J. (2012). Phylogeny and taxonomy of species in the Grosmannia serpens complex. Mycologia 104, 715–732. doi: 10.3852/11-109
Duong, T. A., de Beer, Z. W., Wingfield, B. D., and Wingfield, M. J. (2013). Characterization of the mating-type genes in Leptographium procerum and Leptographium profanum. Fungal Biol. 117, 411–421. doi: 10.1016/j.funbio.2013.04.005
Duong, T. A., de Beer, Z. W., Wingfield, B. D., and Wingfield, M. J. (2016). Mating type markers reveal high levels of heterothallism in Leptographium sensu lato. Fungal Biol. 120, 538–546. doi: 10.1016/j.funbio.2016.01.001
Excoffier, L., Smouse, P. E., and Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491. doi: 10.1093/genetics/131.2.479
Garbelotto, M., Guglielmo, F., Mascheretti, S., Croucher, P. J. P., and Gonthier, P. (2013). Population genetic analyses provide insights on the introduction pathway and spread patterns of the North American forest pathogen Heterobasidion irregulare in Italy. Mol. Ecol. 22, 4855–4869. doi: 10.1111/mec.12452
Gibbs, J. N. (1978). Intercontinental epidemiology of Dutch elm disease. Annu. Rev. Phytopathol. 16, 287–307. doi: 10.1146/annurev.py.16.090178.001443
Goheen, D. J., and Cobb, F. W. (1978). Occurrence of Verticicladiella wagenerii and its perfect state, Ceratocystis wageneri sp. nov., in insect galleries. Phytopathology 68, 1192–1195. doi: 10.1094/Phyto-68-1192
Goheen, D. J., and Hansen, E. M. (1978). Black stain root disease in Oregon and Washington. Plant Dis. Rep. 62, 1098–1102.
Goss, E. M., Larsen, M., Chastagner, G. A., Givens, D. R., and Grünwald, N. J. (2009). Population genetic analysis infers migration pathways of Phytophthora ramorum in US nurseries. PLoS Pathog. 5:e1000583. doi: 10.1371/journal.ppat.1000583
Grünwald, N. J., McDonald, B. A., and Milgroom, M. G. (2016). Population genomics of fungal and oomycete pathogens. Annu. Rev. Phytopathol. 54, 323–346. doi: 10.1146/annurev-phyto-080614-115913
Hansen, E. M., Goheen, D. J., Hessburg, P. F., Witcosky, J. J., and Schowalter, T. D. (1988). “Biology and Management of Black-Stain Root Disease in Douglas-fir,” in Leptographium Root Diseases on Conifers, Symposium Series, eds T. C. Harrington and F. W. Cobb Jr. (St. Paul, MN: American Phytopathological Society Press), 63–80.
Hansen, E. M., and Goheen, D. J. (1988). Rate of increase of black-stain root disease in douglas-fir plantations in Oregon and Washington. Can. J. For. Res. 18, 942–946. doi: 10.1139/x88-143
Harrington, T. C. (1988). “Leptographium species, their distributions, hosts and insect vectors,” in Leptographium Root Diseases on Conifers, Symposium Series, eds T. C. Harrington and F. W. Cobb Jr. (St. Paul, MN: American Phytopathological Society Press), .1–39.
Harrington, T. C. (1992). “Leptographium,” in Methods for Research on Soilborne Phytopathogenic Fungi, eds L. L. Singleton, J. D. Mihail, and C. M. Rush (St. Paul, MN: American Phytopathological Society Press), 129–133.
Harrington, T. C., and Cobb, F. W. Jr. (1984). Host specialization of three morphological variants of Verticicladiella wageneri. Phytopathology 74, 286–290. doi: 10.1094/Phyto-74-286
Harrington, T. C., and Cobb, F. W. Jr. (1987). Leptographium wageneri var. pseudotsugae, var. nov., cause of black stain root disease on Douglas-fir. Mycotaxon 30, 501–507.
Harrington, T. C., Cobb, F. W. Jr., and Lownsbery, J. W. (1985). Activity of Hylastes nigrinus, a vector of Verticicladiella wageneri. in thinned stands of Douglas-fir. Can. J. For. Res. 15, 519–523. doi: 10.1139/x85-085
Harrington, T. C., and Cobb, F. W. (1986). Varieties of Verticicladiella wageneri. Mycologia 78, 562–567. doi: 10.2307/3807767
Harrington, T. C., Reinhart, C., Thornburgh, D. A., and Cobb, F. W. Jr. (1983). Notes: association of black-stain root disease with precommercial thinning of Douglas-fir. For. Sci. 29, 12–14.
Hedrick, P. (2005). A standardized genetic differentiation measure. Evolution 59, 1633–1638. doi: 10.1554/05-076.1
Heinzelmann, R., Rigling, D., and Prospero, S. (2012). Population genetics of the wood-rotting basidiomycete Armillaria cepistipes in a fragmented forest landscape. Fungal Biol. 116, 985–994. doi: 10.1016/j.funbio.2012.07.002
Hessburg, P. F., Goheen, D. J., and Bega, R. V. (1995). Black Stain Root Disease of Conifers. Forest Insect & Disease Leaflet 145 (revised). Washington, DC: USDA Forest Service, 9.
Hessburg, P. F., Goheen, D. J., and Koester, H. (2001). Association of black stain root disease with roads, skid trails, and precommercial thinning in Southwest Oregon. West. J. Appl. For. 16, 127–135. doi: 10.1093/wjaf/16.3.127
Hessburg, P. F., and Hansen, E. M. (1986). Mechanisms of intertree transmission of Ceratocystis wageneri in young Douglas-fir. Can. J. For. Res. 16, 1250–1254. doi: 10.1139/x86-221
Hessburg, P. F., and Hansen, E. M. (1987). Pathological anatomy of black stain root disease of Douglas-fir. Can. J. Bot. 65, 962–971. doi: 10.1139/b87-133
Hessburg, P. F., and Hansen, E. M. (2000). Infection of Douglas-fir by Leptographium wageneri. Can. J. Bot. 78, 1254–1261. doi: 10.1139/b00-097
Jacobi, W. R. (1992). Potential insect vectors of the black stain root disease pathogen on Southern Vancouver Island. J. Entomol. Soc. B. C. 89, 54–56.
Jacobs, K., Wingfield, M. J., and Wingfield, B. D. (2001). Phylogenetic relationships in Leptographium based on morphological and molecular characters. Can. J. Bot. 79, 719–732. doi: 10.1139/cjb-79-6-719
Jombart, T. (2008). Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405. doi: 10.1093/bioinformatics/btn129
Jombart, T., Devillard, S., and Balloux, F. (2010). Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11:94. doi: 10.1186/1471-2156-11-94
Kamvar, Z. N., Tabima, J. F., and Grünwald, N. J. (2014). Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. doi: 10.7717/peerj.281
Kamvar, Z. N., Brooks, J. C., and Grünwald, N. J. (2015). Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front. Genet. 6. doi: 10.3389/fgene.2015.00208
Kelsey, R. G., and Joseph, G. (1998). Ethanol in Douglas-fir with black-stain root disease (Leptographium wageneri). Can. J. For. Res. 28, 1207–1212. doi: 10.1139/x98-087
Kendrick, W. B. (1962). The Leptographium complex. Verticicladiella Hughes. Can. J. Bot. 40, 772–797. doi: 10.1139/b62-072
Knaus, B. J., and Grünwald, N. J. (2017). vcfr: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 17, 44–53. doi: 10.1111/1755-0998.12549
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Linde, C. C., Zala, M., and McDonald, B. A. (2009). Molecular evidence for recent founder populations and human-mediated migration in the barley scald pathogen Rhynchosporium secalis. Mol. Phylogenet. Evol. 51, 454–464. doi: 10.1016/j.ympev.2009.03.002
Linde, C. C., Zhan, J., and McDonald, B. A. (2002). Population structure of Mycosphaerella graminicola: from lesions to continents. Phytopathology 92, 946–955. doi: 10.1094/PHYTO.2002.92.9.946
McDonald, B. A., and Linde, C. (2002). Pathogen population genetics, evolutionary potential, and durable resistance. Annu. Rev. Phytopathol. 40, 349–379. doi: 10.1146/annurev.phyto.40.120501.101443
McDonald, B. A., and Mundt, C. C. (2016). How knowledge of pathogen population biology informs management of Septoria tritici blotch. Phytopathology 106, 948–955. doi: 10.1094/PHYTO-03-16-0131-RVW
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Milgroom, M. G. (1996). Recombination and the multilocus structure of fungal populations. Annu. Rev, Phytopathol. 34, 457–477. doi: 10.1146/annurev.phyto.34.1.457
Milgroom, M. G. (2017). Population Biology of Plant Pathogens: Genetics, Ecology, and Evolution. St. Paul, MN: The American Phytopathological Society, doi: 10.1094/9780890544525
Möller, M., and Stukenbrock, E. (2017). Evolution and genome architecture in fungal plant pathogens. Nat. Rev. Microbiol. 15, 756–771. doi: 10.1038/nrmicro.2017.76
Mote, P. W., and Salathé, E. P. (2010). Future climate in the Pacific Northwest. Clim. Change 102, 29–50. doi: 10.1007/s10584-010-9848-z
Nei, M. (1973). Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. U. S. A. 70, 3321–3323. doi: 10.1073/pnas.70.12.3321
Otrosina, W. J., and Cobb, F. W. (1987). Analysis of allozymes of three distinct variants of Verticicladiella wageneri isolated from conifers in Western North America. Phytopathology 77, 1360–1363. doi: 10.1094/Phyto-77-1360
Paine, T. D., Raffa, K. F., and Harrington, T. C. (1997). Interactions among scolytid bark beetles, their associated fungi, and live host conifers. Annu. Rev. Entomol. 42, 179–206. doi: 10.1146/annurev.ento.42.1.179
R Core Team. (2020). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rieux, A., Halkett, F., De Lapeyre de Bellaire, L., Zapater, M.-F., Rousset, F., Ravigne, V., et al. (2011). Inferences on pathogenic fungus population structures from microsatellite data: new insights from spatial genetics approaches. Mol. Ecol. 20, 1661–1674. doi: 10.1111/j.1365-294X.2011.05053.x
Rivas, G.-G., Zapater, M.-F., Abadie, C., and Carlier, J. (2004). Founder effects and stochastic dispersal at the continental scale of the fungal pathogen of bananas Mycosphaerella fijiensis. Mol. Ecol. 13, 471–482. doi: 10.1046/j.1365-294X.2003.02043.x
Rumbold, C. T. (1931). Two blue-staining fungi associated with bark beetle infestation of pines. J. Agric. Res. 43, 847–873.
Six, D. L. (2012). Ecological and evolutionary determinants of bark beetle– fungus symbioses. Insects 3, 339–366. doi: 10.3390/insects3010339
Six, D. L. (2020). A major symbiont shift supports a major niche shift in a clade of tree-killing bark beetles. Ecol. Entomol. 45, 190–201. doi: 10.1111/een.12786
Six, D. L., and Wingfield, M. J. (2011). The role of phytopathogenicity in bark beetle–fungus symbioses: a challenge to the classic paradigm. Annu. Rev. Entomol. 56, 255–272. doi: 10.1146/annurev-ento-120709-144839
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tabima, J. F., Gonen, L., Gómez-Gallego, M., Panda, P., Grünwald, N. J., Hansen, E. M., et al. (2021). Molecular phylogenomics and population structure of Phytophthora pluvialis. Phytopathology 111, 108–115. doi: 10.1094/PHYTO-06-20-0232-FI
Tabima, J. F., Søndreli, K. L., Keriö, S., Feau, N., Sakalidis, M. L., Hamelin, R. C., et al. (2020). Population genomic analyses reveal connectivity via human-mediated transport across populus plantations in North America and an undescribed subpopulation of Sphaerulina musiva. Mol. Plant Microbe Interact. 33, 189–199. doi: 10.1094/MPMI-05-19-0131-R
Tsui, C. K.-M., DiGuistini, S., Wang, Y., Feau, N., Dhillon, B., Bohlmann, J., et al. (2013). Unequal recombination and evolution of the mating-type (MAT) loci in the pathogenic fungus Grosmannia clavigera and relatives. G3 Genes Genomes Genetics 3, 465–480. doi: 10.1534/g3.112.004986
Tsui, C. K. M., Farfan, L., Roe, A. D., Rice, A. V., Cooke, J. E. K., El-Kassaby, Y. A., et al. (2014). Population structure of mountain pine beetle symbiont Leptographium longiclavatum and the implication on the multipartite beetle-fungi relationships. PLoS One 9:e105455. doi: 10.1371/journal.pone.0105455
Tsui, C. K. M., Roe, A. D., El-Kassaby, Y. A., Rice, A. V., Alamouti, S. M., Sperling, F. A. H., et al. (2012). Population structure and migration pattern of a conifer pathogen, Grosmannia clavigera, as influenced by its symbiont, the mountain pine beetle. Mol. Ecol. 21, 71–86. doi: 10.1111/j.1365-294X.2011.05366.x
Wagener, W. W., and Mielke, J. L. (1961). A staining-fungus root disease of ponderosa, Jeffrey, and pinyon pines. Plant Dis. Rep. 45, 831–835.
Wingfield, M. J. (1985). Reclassification of Verticicladiella based on conidial development. Trans. Br. Mycol. Soc. 85, 81–93. doi: 10.1016/S0007-1536(85)80157-1
Wingfield, M. J., Seifert, K. A., and Webber, J. F. (1993). Ceratocystis and Ophiostoma: Taxonomy, Ecology, and Pathogenicity. St. Paul, MN: American Phytopathological Society Press.
Winter, D. J. (2012). MMOD: an R library for the calculation of population differentiation statistics. Mol. Ecol. Resour. 12, 1158–1160. doi: 10.1111/j.1755-0998.2012.03174.x
Witcosky, J. J., and Hansen, E. M. (1985). Root-colonizing insects recovered from Douglas-Fir in various stages of decline due to black-stain root disease. Phytopathology 75, 399–402. doi: 10.1094/Phyto-75-399
Witcosky, J. J., Schowalter, T. D., and Hansen, E. M. (1986a). Hylastes nigrinus (Coleoptera: Scolytidae), Pissodes fasciatus, and Steremnius carinatus (Coleoptera: Curculionidae) as vectors of black-stain root disease of Douglas-fir. Environ. Entomol. 15, 1090–1095. doi: 10.1093/ee/15.5.1090
Witcosky, J. J., Schowalter, T. D., and Hansen, E. M. (1986b). The influence of time of precommercial thinning on the colonization of Douglas-fir by three species of root-colonizing insects. Can. J. For. Res. 16, 745–749. doi: 10.1139/x86-133
Witthuhn, R. C., Wingfield, B. D., Wingfield, M. J., and Harrington, T. C. (1997). Comparison of three varieties of Leptographium wageneri using random amplified polymorphic DNA. S. Afr. J. Bot. 63, 198–200. doi: 10.1016/S0254-6299(15)30744-4
Yu, G., Smith, D. K., Zhu, H., Guan, Y., and Lam, T. T.-Y. (2017). ggtree: an r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36. doi: 10.1111/2041-210X.12628
Zambino, P. J., and Harrington, T. C. (1989). isozyme variation within and among host-specialized varieties of Leptographium wageneri. Mycologia 81, 122–133. doi: 10.1080/00275514.1989.12025632
Zambino, P. J., and Harrington, T. C. (1992). Correspondence of isozyme characterization with morphology in the asexual genus Leptographium and taxonomic implications. Mycologia 84, 12–25. doi: 10.2307/3760398
Keywords: Douglas-fir, Leptographium wageneri, population genomics, epidemiology, emerging disease, plantation forest landscape
Citation: Bennett PI, Tabima JF, Leon AL, Browning J, Wingfield MJ and LeBoldus JM (2021) Spatial Genetic Structure of the Insect-Vectored Conifer Pathogen Leptographium wageneri Suggests Long Distance Gene Flow Among Douglas-fir Plantations in Western Oregon. Front. For. Glob. Change 4:695981. doi: 10.3389/ffgc.2021.695981
Received: 15 April 2021; Accepted: 07 July 2021;
Published: 28 July 2021.
Edited by:
Isabel Alvarez Munck, Forest Health Protection, State and Private Forestry, United States Forest Service (USDA), United StatesReviewed by:
Denita Hadziabdic, The University of Tennessee, Knoxville, United StatesBraham Dhillon, University of Florida, United States
Copyright © 2021 Bennett, Tabima, Leon, Browning, Wingfield and LeBoldus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jared M. LeBoldus, SmFyZWQuTGVCb2xkdXNAb3JlZ29uc3RhdGUuZWR1