 George Sharbeen
George Sharbeen Joshua McCarroll
Joshua McCarroll David Goldstein
David Goldstein Phoebe A. Phillips
Phoebe A. Phillips- 1Pancreatic Cancer Translational Research Group, Lowy Cancer Research Centre, Prince of Wales Clinical School, UNSW Australia, Sydney, NSW, Australia
- 2Children’s Cancer Institute, Lowy Cancer Research Centre, UNSW Australia, Sydney, NSW, Australia
Pancreatic ductal adenocarcinoma (PDA) is a highly chemoresistant and metastatic disease with a dismal 5-year survival rate of 6%. More effective therapeutic targets and approaches are urgently needed to tackle this devastating disease. The base excision repair (BER) pathway has been identified as a predictor of therapeutic response, prognostic factor, and therapeutic target in a variety of cancers. This review will discuss our current understanding of BER in PDA and its potential to improve PDA treatment.
Introduction
Pancreatic ductal adenocarcinoma (PDA) currently ranks as the fourth leading cause of cancer-related death in Western societies (1–3) and is projected to become the second leading cause by 2030 (4). Surgery is currently the best chance for a cure, but <15% of patients present with operable disease due to the lack of specific symptoms (2, 3). The remaining patients that present with unresectable tumors have a dismal prognosis, with an overall 5-year survival rate at 6% (5). This poor prognosis is largely due to the highly metastatic and chemoresistant nature of PDA (6, 7). Recent years have seen some improvement in treatments for PDA. Gemcitabine/abraxane combination therapy is a current standard of care for unresectable PDA, but this only extends median overall survival by 8 weeks over gemcitabine monotherapy (8). More aggressive polychemotherapeutic regimens, such as FOLFIRINOX, are also employed as treatments for PDA but only extend median survival by 17 weeks over gemcitabine monotherapy (9) and require careful selection of candidates for the treatment due to toxicity. There is clearly an urgent need for novel therapeutic approaches if we are to achieve a significant improvement in PDA patient survival.
A Mutagenic Environment: Impact on DNA Repair
Pancreatic ductal adenocarcinoma is an epithelial-derived tumor that is proposed to proceed in a step-wise manner from pancreatic intraepithelial neoplasia (PanIN) lesions to PDA (10). PDA tumors are highly fibrotic and consist of a complex microenvironment containing PDA cells, cancer-associated pancreatic stellate cells (CA-PSCs), immune cells, and extracellular matrix proteins (11–13). CA-PSCs play a major role in generating the extensive fibrotic response that is characteristic of PDA, by secreting excessive amounts of extracellular matrix proteins in response to signals from PDA cells (11, 12, 14–18). The fibrotic PDA microenvironment drives the highly chemoresistant and metastatic phenotype of this cancer. Fibrosis distorts the tumor vasculature, creating a hypoxic and nutrient-deprived microenvironment (11, 19–21). These two features are known to drive the Warburg effect, which is a switch from oxidative to glycolytic metabolism, as well as the transition of cancer cells from an epithelial phenotype to a more aggressive mesenchymal phenotype (22–25).
While this microenvironment selects for more aggressive pancreatic cancer cells, it also imposes mutagenic pressure on the cells, particularly in the form of oxidative stress (24, 26, 27). Hypoxia triggers the release of reactive oxygen species from mitochondria, initiating a signaling cascade that reprograms the cell to facilitate its survival (26, 27). In addition, the Warburg effect results in loss of potent anti-oxidant intermediates, reducing the ability of cells to deal with increased intracellular oxidative stress (24). These free radicals readily react with DNA bases, altering how DNA polymerases might recognize them. For example, the most common form of DNA damage induced by reactive oxygen species is 8-oxo-guanine (8-oxo-G) in guanine:cytosine (G:C) base pairs (28) (Figure 1). If left unrepaired, replication machinery can mis-insert adenine (A) opposite 8-oxo-G (29) (Figure 1). In subsequent rounds of replication, this can permanently convert a G:C base pair to a thymine:adenine (T:A) base pair (29) (Figure 1). Rapidly proliferating PDA cells must be able to tolerate this increased mutagenic load in order to replicate their genome, without accumulating genomic damage that can disrupt its most basic survival functions. Under these conditions, effective DNA repair becomes critical for PDA cell survival. The base excision repair (BER) pathway is responsible for repair of a variety of damaged DNA bases, and plays a prominent role in repair of oxidative DNA damage, which is particularly relevant to PDA as outlined above (30). Importantly, components of this pathway have increasingly been identified as predictors of cancer risk, prognosis, chemoresistance, and as direct therapeutic targets in a variety of cancers (31). This review will discuss components of the BER pathway that have been identified as therapeutic targets and predictors of therapeutic response in pancreatic cancer. It will also discuss the untapped potential for other PDA therapeutic targets in this pathway.
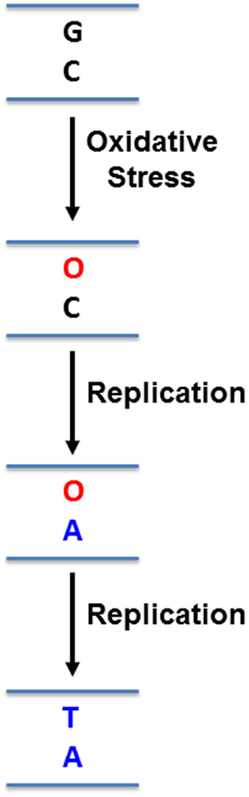
Figure 1. Mutation induced by 8-oxo-guanine. Reactive oxygen species can oxidize guanines (G) in DNA to 8-oxo-guanine (O). Replication machinery can then mis-insert adenine (A) opposite O. In a subsequent round of replication, a thymine (T) can be inserted opposite A, resulting in a G:C to T:A mutation.
The Base Excision Repair Pathway
The BER pathway consists of a number of specialized glycosylases, endonucleases, polymerases, and ligases that work together to repair a variety of damaged DNA bases (Figure 2). Generally speaking, BER of DNA damage proceeds through five steps. The first step is detection and removal of the damaged DNA base, which is carried out by glycosylases (Figure 2A). Glycosylases can be largely grouped by the types of damaged bases that they recognize and remove (Table 1): uracil/thymine excision enzymes (UNG, SMUG1, TDG, and MBD4) (32–37), 8-oxo-G repair enzymes (OGG1 and MYH) (38–45), oxidized pyrimidine repair enzymes (NTH1 and NEIL1–3) (46–56), and methyl-purine glycosylase (MPG) (57–59) (Table 1). Their substrates include many of the damaged bases generated by DNA-damaging chemotherapeutics (31).
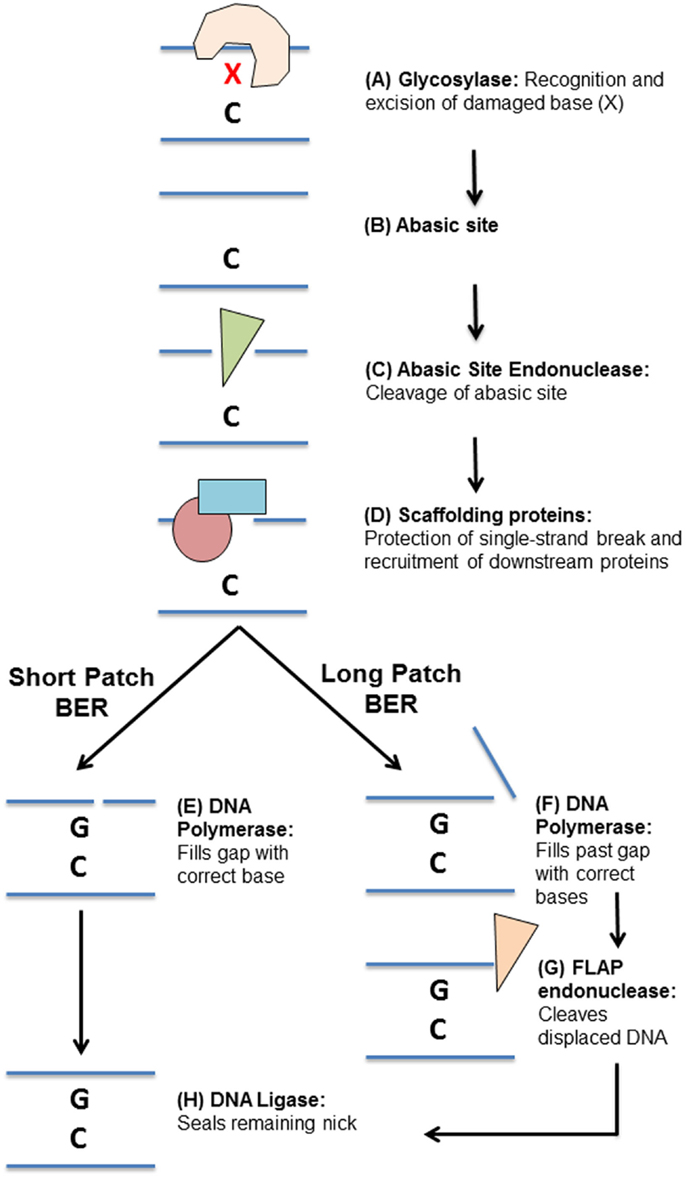
Figure 2. Overview of the base excision repair pathway. (A) DNA damage “X” is detected and excised by a specific glycosylase leaving an abasic site (B). (C) The abasic site processed by an apurinic/apyrimidinic endonuclease. (D) Scaffolding proteins bind the single-stranded DNA and recruit downstream base excision repair proteins. Repair can then proceed by (E) short-patch or (F) long-patch base excision repair. (E) A DNA polymerase fills in the missing DNA base. (F) DNA polymerases fill in the missing DNA base and continue synthesizing DNA past the initial damage site, displacing the original DNA strand. (G) A flap endonuclease then cleaves the flap of single-stranded DNA. (H) A DNA ligase seals the remaining DNA nick, completing the repair.
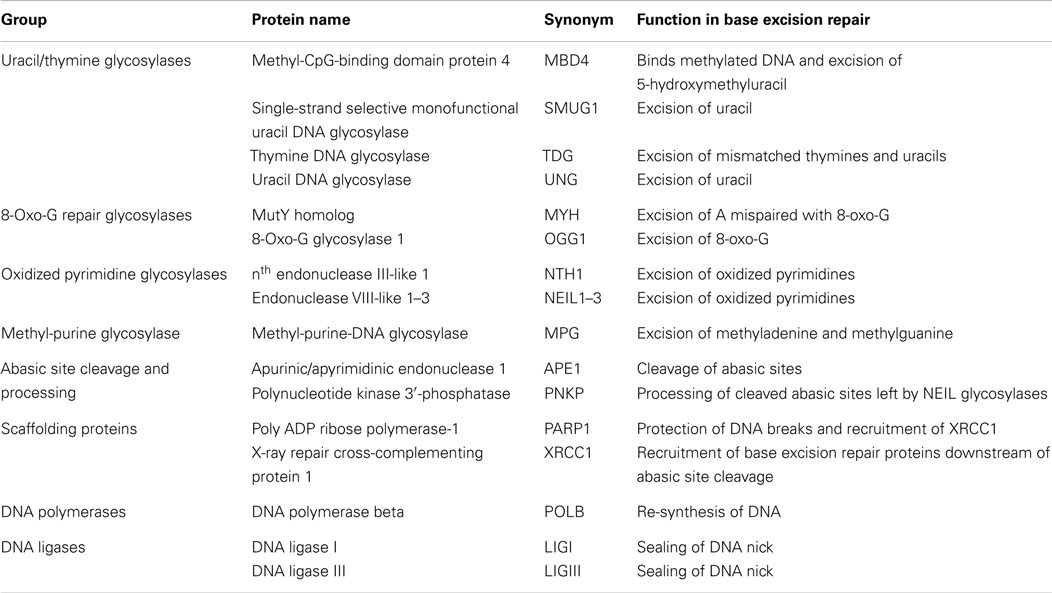
Table 1. Base excision repair proteins and function.
Once a glycosylase has removed a damaged base, it leaves behind a non-instructional abasic site (Figure 2B). This site can cause transcription stalling and must be cleaved and processed before repair can continue (60, 61). The second step in BER is thus cleavage of the abasic site (Figure 2C). Some glycosylases are bifunctional, meaning they can cleave the abasic site that they create (62–65). However, most glycosylases require cleavage of the abasic site by apurinic/apyrimidinic endonuclease 1 (APE1) (66, 67). Processing of an abasic site leaves a single-strand break, which needs to be protected to prevent further damage.
The third step in BER is binding of the single-strand break by scaffolding proteins (Figure 2D). There are two major scaffolding proteins in BER, poly ADP ribose polymerase-1 (PARP1) and X-ray repair cross-complementing protein 1 (XRCC1) (68–72). Once these proteins are bound to the single-strand DNA break, they recruit other BER proteins required to complete the repair (68–72).
The fourth step in BER is insertion of the missing base by a DNA polymerase, such as DNA polymerase β (POLB) (73). This can take the form of either single base repair, whereby only one base is inserted (short-patch repair, Figure 2E), or long-patch repair (Figure 2F), where a stretch of bases is inserted at and beyond the initial site of damage, displacing some of the nearby bases (74). In long-patch BER, an additional step is required involving the specialized flap-endonuclease, FEN1, to cleave the displaced stretch of DNA bases (Figure 2G) (75). The final step in BER is then sealing of DNA nicks left by these processes DNA ligase I (LIGI, long-patch BER) or DNA ligase III (LIGIII, short-patch BER) (Figure 2H) (76).
Cancer cells take advantage of BER’s ability to repair DNA damage in order to resist DNA-damaging chemotherapies and radiotherapy (31). BER proteins have thus been identified as potential therapeutic targets and chemoresistance factors in a variety of cancers (31). More specifically in PDA, the BER proteins APE1, XRCC1, and PARP1 provide well-studied examples of how BER proteins can be applied as therapeutic targets or predictors of therapeutic response (detailed below). They also highlight the complex roles that BER proteins can play outside of their primary DNA repair roles in the BER pathway, which can be exploited in therapeutic approaches to elicit a broader impact in PDA cells.
APE1 as a Therapeutic Target for Pancreatic Cancer
APE1 is an endonuclease that cleaves DNA abasic sites left by the activity of glycosylases (66, 67). This makes APE1 a central protein in the BER pathway, as its activity is required downstream of a variety of glycosylases. It is therefore not surprising that APE1 has been implicated in resistance to a diverse range of therapeutics. Moreover, APE1 can directly repair the replication-blocking abasic sites generated by alkylating agents (77). An analysis of APE1 activity in medulloblastomas and neuroectodermal tumors from patients treated with adjuvant radiation and multi-agent chemotherapy, found that increased APE1 activity correlated with poorer response to treatment (78). The same association has been observed in grade II–III gliomas, whereby increased APE1 activity correlated with increased resistance to radiation therapy and alkylating agents (79). Similar trends have been observed in head-and-neck cancer in relation to resistance to chemotherapy and radiotherapy (80). APE1 can also confer resistance to chemotherapeutics that may not directly generate substrates for the BER pathway, but may generate secondary types of DNA damage that BER is required to resolve. For example, APE1 upregulation has been associated with platinum resistance in ovarian cancer (81). This is likely due to the generation of reactive oxygen species by platinum-based therapies, which generates oxidized DNA bases that require BER to repair (82).
APE1’s involvement in chemoresistance and radioresistance in cancer cells has been functionally demonstrated using small molecule inhibitors and siRNA-based approaches. APE1 inhibitors have been shown to enhance the sensitivity of HeLa cells to the alkylating agent methyl methanesulfonate (MMS) (83). In an siRNA-based approach, APE1 silencing has been shown to increase the chemosensitivity of ovarian cancer cells to cisplatin via induction of apoptosis (81). APE1 inhibition has similarly been shown to sensitize PDA cells (Panc-1) to gemcitabine (84), while Xiong et al. (85) later reproduced these findings in SW1990 PDA cells. APE1 downregulation has since been shown to also radiosensitize PDA cells (86).
APE1 inhibition also highlights how BER proteins can represent broader intracellular targets that can both chemosensitize cancer cells and compromise cancer cell survival. APE1 is also known as Ref-1, as its N-terminal plays a role as a redox factor, responsible for regulating a variety of transcription factors that facilitate cell survival in response to oxidative stress (87–92). Several studies have identified APE1 as a survival factor in PDA cells. Jiang and colleagues (93) observed that APE1 silencing in PDA cells (Panc1 and MiaPaCa2), reduced proliferation/colony forming ability and increased apoptosis, by increasing DNA damage. Studies have similarly demonstrated this using a small molecule inhibitor of APE1 (E3330) (94, 95). Fishel et al. (94) showed that E3330 reduced PDA cell and cancer-associated endothelial cell proliferation and migration, decreased transcription factor activity for NFκB, AP-1, and HIF-1α, and reduced tumor growth in PDA xenograft mouse models. Recently, Fishel et al. (96) further demonstrated the complexity of signaling networks involving APE1. The group identified a novel interaction specifically between the redox component of APE1 and nuclear factor erythroid-related 2 (Nrf2) in PDA cells. Inhibition of APE1 using small molecule and genetics approaches increased activation of Nrf2, a protein known to play a role in protection from oxidative stress (96). This work identified a potential resistance mechanism that would need to be co-targeted with APE1 inhibitors. Cardoso and colleagues (95) demonstrated that dual targeting of APE1 and the transcription factor STAT3 synergistically reduced PDA cell survival and migration. The authors demonstrated that STAT3 DNA binding and transcriptional activity is under the control of APE1 (95), suggesting that the synergistic effects may have been due to enhancement of STAT3 inhibition by knocking-down APE1, or by inhibiting functions of each protein that were independent of the other, in addition of their co-dependent pathway. This also provides an important example of how the complex signaling roles of some BER proteins can have far reaching effects on how PDA cells interact with their microenvironment and migrate.
As a therapeutic target for PDA, APE1 is ideal in its ability to confer sensitivity to a variety of drugs and to broadly interfere with the cellular response to oxidative stress (Figure 3). However, this involvement in a broad range of BER sub-pathways also makes systemic inhibition of APE1 risky, as off-target toxicity and the risk of generating cancers elsewhere in the body would not be unexpected. APE1 knockout is embryonic lethal (97, 98) and it has been demonstrated to be essential for mammalian cell survival (99, 100). An approach targeting APE1 should preferably be cancer cell-specific, for example, siRNA complexed to a nanoparticle, which has cancer cell-specific targeting moieties attached (101, 102).
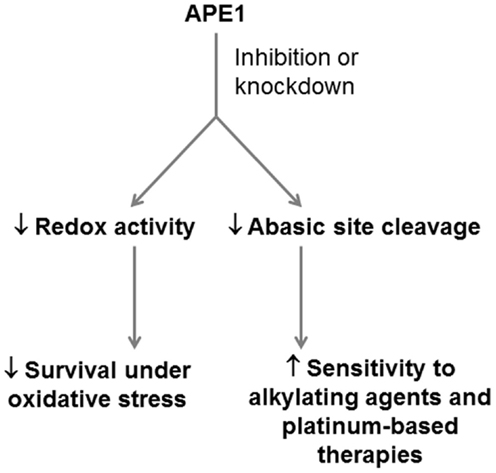
Figure 3. Potential application of APE1 as a therapeutic target in PDA. APE1 represents a potential dual target in PDA, whose inhibition can reduce the ability of PDA cells to respond to oxidative stress through its redox signaling role, and the ability of PDA cells to resist alkylating agents and platinum-based drugs through its role as an abasic site endonuclease.
XRCC1 and Chemotherapeutic Response in Pancreatic Cancer
XRCC1 is a scaffolding protein with no enzymatic activity, which plays an important role in recruiting and coordinating other BER proteins at sites of DNA damage (68–72). XRCC1 polymorphisms that interfere with PARP binding and cause broadly reduced BER efficiency have been implicated in resistance to platinum-based therapies in lung and colorectal cancer (103–105). Interestingly, these are an example of how reduced DNA repair efficiency can confer resistance to chemotherapeutics. This relationship might appear illogical as DNA adducts generated by platinum therapies are not direct targets of the BER pathway and are instead processed by the nucleotide excision repair pathway (106). However, studies by Kothandapi and colleagues (107, 108) provide a potential explanation for this relationship and highlight the complex interaction of DNA repair pathways. The study suggested that rather than acting directly on the primary DNA damage, that is, DNA inter-strand cross-links, or adducts, BER could act on DNA that was exposed and deaminated around platinum-induced DNA adducts (107, 108). However, BER at these sites would interfere with the nucleotide excision repair pathway that directly repairs the DNA adducts, thus maintaining platinum-induced toxicity (107, 108). XRCC1 polymorphisms that reduce BER recruitment to these sites would reduce interference with the nucleotide excision repair pathway, allowing more effective repair of the DNA adduct and increasing cancer cell resistance to the drug.
There is also evidence that lower XRCC1 expression is associated with increased sensitivity to platinum-based therapies in ovarian cancer cells, as demonstrated by increased accumulation of DNA double-strand breaks in response to platinum-based therapies (109). While these results may appear to be conflicting with the studies presented so far, they actually highlight two important points that we need to consider to effectively use BER proteins as a predictor of therapeutic response: (i) the biochemistry of the DNA damage induced by a therapeutic and (ii) cross-talk between DNA repair pathways that respond. In this case, we need to note that XRCC1 is also required for the final stages of the nucleotide excision repair pathway, which directly repairs the toxic DNA adducts generated by platinum drugs (110, 111). Thus, decreased XRCC1 interferes with the ability of nucleotide excision repair to remove the DNA adducts that facilitate cell death. On the other hand, polymorphisms that hinder recruitment of XRCC1 through the BER pathway reduce this interference with nucleotide excision repair and increase resistance to platinum-based therapies.
XRCC1 polymorphisms can similarly be applied in PDA to predict response to platinum-based drugs and to identify individuals with increased risk of pancreatic cancer incidence. In particular, a polymorphism in Arg399 of XRCC1 has been identified in pancreatic cancer studies as a risk factor and predictor of therapeutic response (112–114). This polymorphism occurs within a PARP binding domain of XRCC1 and has been associated with decreased BER function (112). Giovanetti and colleagues (113) identified a significant correlation between the Arg399 polymorphism of XRCC1 and a worse response to platinum-based therapeutic regimens (113), indicating that XRCC1 could be used in PDA to predict response to platinum-based therapies as in other cancers.
Moreover, XRCC1 polymorphisms can be used to predict PDA risk. Duell and colleagues (112) observed that carriers of this XRCC1 polymorphism who were smokers, had significantly higher risk of developing PDA than other members in the cohort of 309 PDA patients and 964 control individuals. Nakao et al. (114) similarly found a significant correlation between the XRCC1 Arg399 polymorphism and increased pancreatic cancer risk in a cohort that included 185 Japanese pancreatic cancer patients. As mentioned earlier, BER plays a major role in repairing oxidative DNA damage (30). Oxidative DNA damage occurs as a consequence of normal cell metabolism and can be increased by environmental factors, especially smoking (115–118). The increased pancreatic cancer risk in individuals carrying the Arg399 polymorphism of XRCC1 is potentially the result of a reduced cellular capacity to repair mutations that result from oxidative DNA damage. Thus, the XRCC1 Arg399 polymorphism, in combination with environmental factors, could potentially be used to identify high-risk individuals for early pancreatic cancer screening.
While the development of PARP inhibitors (discussed in next section) removes some incentive to therapeutically target XRCC1, this protein clearly represents an important predictive factor for cancer risk and platinum-based therapeutics in PDA (Figure 4).
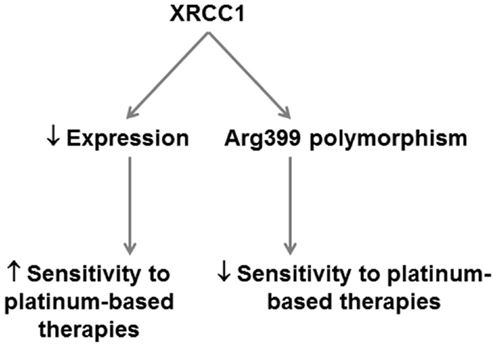
Figure 4. Potential application of XRCC1 as a predictor of therapeutic response in PDA. XRCC1 expression and polymorphisms can potentially be applied in PDA to predict therapeutic response. XRCC1 downregulation predicts increased sensitivity to platinum-based therapies. XRCC1 Arg399 polymorphisms that decrease base excision repair efficiency predict poorer response to platinum-based therapies.
PARP1 as a Therapeutic Target for Pancreatic Cancer
Poly(ADP)-ribose polymerase-1 binds single-strand and double-strand DNA breaks and interacts with XRCC1 to help recruit downstream BER proteins to facilitate repair (68, 72). However, unlike XRCC1 it can also directly regulate proteins, particularly histones and transcription factors, by adding poly(ADP)-ribose units to them (termed PARylation), which it synthesizes from NAD+ (119). PARP uses this activity to rapidly co-ordinate DNA repair in response to the DNA breaks (119). PARP inhibitors are the perfect example of how an understanding of BER and its interaction with other pathways can be effectively applied to tailor therapeutic approaches for cancer. This field was launched by two landmark studies published in 2005 (120, 121) demonstrating that PARP inhibition was an effective approach to treat BRCA1- and BRCA2-defective cancer cells (cells defective in homologous recombination). These studies not only identified a new therapeutic target but also highlighted the potency of “synthetic lethality” approaches – approaches that compound existing weaknesses in DNA repair in cancer cells by eliminating complimentary repair pathways, thus overwhelming cells with DNA damage. BRCA1/2-deficient cells are unable to carry out effective homologous recombination to repair DNA double-strand breaks (122). Inhibition of PARP-1 eliminates the remaining active DNA repair pathways that can respond to these breaks and potentially generates more breaks by inhibiting multiple DNA repair pathways. PARP inhibitors are now applied in the clinic for BRCA1/2 defective ovarian and breast cancers and are in clinical trial for a variety of other BRCA1/2-defective tumors (123).
Pre-clinical studies in PDA cells have similarly demonstrated the efficacy of PARP inhibitors in synthetic-lethal approaches and in sensitization of PDA cells to therapeutics. Drew and colleagues (124) demonstrated that BRCA2-defective PDA cells (CAPAN-1 line) were sensitive to the PARP1 inhibitor AG014699 in vitro and in sub-cutaneous mouse tumors. More recently, Chen et al. (125) combined inhibition of PARP family proteins by olaparib with inhibition of Bcl-2, an anti-apoptotic protein that is now recognized as a suppressor of non-homologous end-joining, which is an alternate pathway for double-strand DNA break repair (126, 127). This drug combination synergistically caused growth arrest and non-apoptotic cell death in PDA cells in vitro and in PDA xenografts (125). A possible explanation for these synergistic effects might be that PARP inhibition increases the persistence of double-strand breaks while Bcl-2 inhibition increases erroneous joining of DNA double-strand breaks by the non-homologous end-joining pathway, eventually leading to impairment of cell function by accumulation of translocations and mutations.
PARP inhibition need not only be applied on BRCA1/2-defective backgrounds but can also improve the efficacy of DNA-damaging agents by the same principle. For example, Jacob et al. (128) showed that PARP inhibition using 3-aminobenzamide (an inhibitor that competes with NAD+), could enhance the efficacy of gemcitabine in PDA cells. In an approach that indirectly targeted PARP1, Piao and colleagues (129) showed that silencing PARP1 binding protein (PARBP), a protein that enhances PARP1 activity, sensitized KLM-1 PDA cells to adriamycin, H2O2, and UV irradiation. Moreover, several studies have demonstrated the radiosensitizing effect of PARP inhibition in PDA cells (130–132). PARP inhibitors are currently in clinical trial for PDA (ClinicalTrials.gov identifiers: NCT00515866, NCT01286987, NCT00047307, NCT01989546, NCT02042378) (123, 133).
Poly ADP ribose polymerase 1 is a complex protein with roles in other DNA repair pathways, chromatin remodeling, transcriptional regulation, and cell death pathways (119). This extensive network of interactions means that PARP1 inhibition can affect more than just DNA repair. For example, Yuan et al. (134) found that PARP1 was elevated in pancreatic cancer cell lines resistant to TNF-related apoptosis-inducing ligand (TRAIL) antibody therapy (Panc1 and SUIT2) but low in TRAIL-sensitive lines (MiaPaCa2 and BxPC3). Silencing PARP1 in these resistant lines increased their sensitivity to TRAIL therapy in vitro and in a sub-cutaneous animal model (134). Klauschen et al. (135) investigated PARP expression and localization in 178 human PDA tissue specimens, using immunohistochemistry. The patients had undergone surgery for pancreatic masses, without use of chemotherapeutics prior to surgery. The group found that low-level nuclear expression of PARP significantly correlated with reduced median survival (135). As with XRCC1, these results are not necessarily conflicting with the proven effective application of PARP inhibitors in the clinic. A possible explanation is that PARP inhibitors, which inhibit specific functions or regions of PARP, produce very different effects to downregulation of total PARP protein and thus all PARP functions. It again highlights the importance of understanding all of the roles of a BER protein and its overlap with other repair pathways when designing a therapeutic approach. PARP inhibition has proven to be a successful therapeutic approach in many other cancers and is likely to become an effective treatment in synthetic-lethal approaches for PDA (Figure 5).
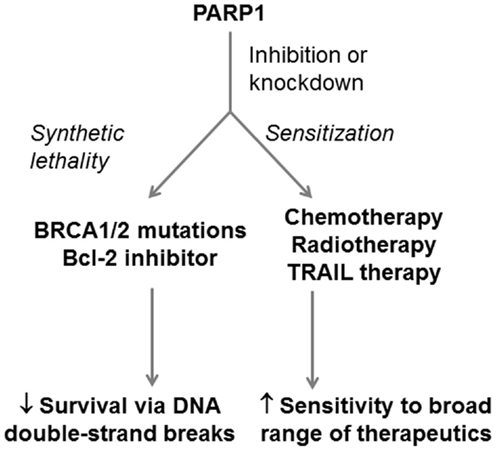
Figure 5. Potential application of PARP1 as a therapeutic target in PDA. PARP1 can be targeted in PDA in both synthetic-lethal approaches and in approaches that sensitizPARP1e PDA cells to therapeutics. PARP1 inhibition can be combined with BRCA1/2 defects or Bcl-2 inhibition to induce PDA cell death through double-strand DNA breaks. PARP1 inhibition can also be combined with DNA-damaging therapeutics, radiotherapies, and antibody-based TRAIL therapy to enhance their efficacy.
Future Directions: The Untapped Potential of Base Excision Repair
The information that we already have on the BER proteins APE1, XRCC1, and PARP1 can be applied to better tailor therapeutic approaches for PDA (Table 2). However, there is a lot of untapped potential for PDA therapeutic targets in this pathway. Glycosylases carry out excision of damaged DNA bases and are possible direct therapeutic targets for the chemosensitization of PDA cells. There are several examples of this in other cancers. For example, MPG inhibition is capable of sensitizing cancer cells to alkylating agents (136). The uracil excision glycosylase SMUG1 carries out the majority of the repair of 5-FU and could be targeted to increase sensitivity to this drug (137). There is also promise in BER proteins that are more downstream in the pathway, for example, inhibition of DNA polymerase beta POLB inhibition has been shown to sensitize cancer cells to the oxaliplatin, cisplatin, and the DNA methylating compound temozolomide (138–140).
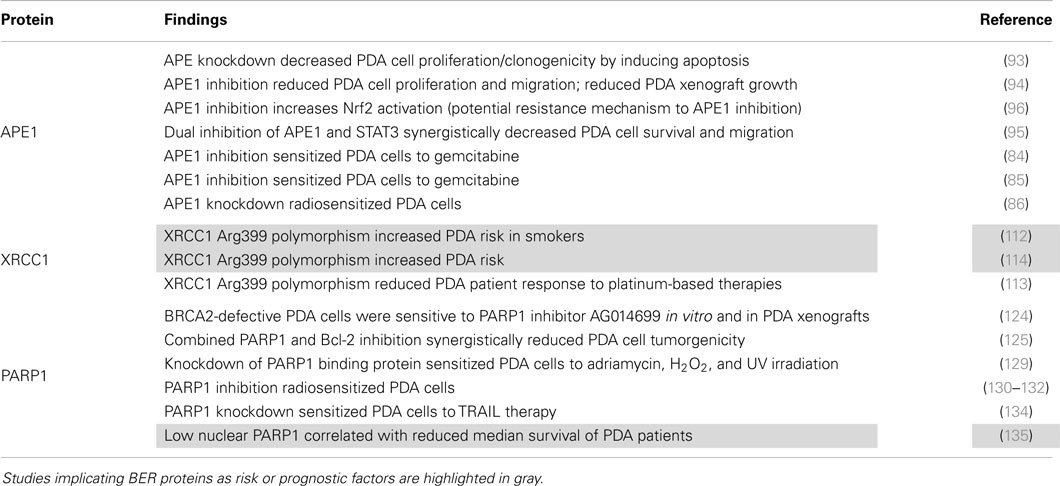
Table 2. Summary of APE1, XRCC1, and PARP1 studies in pancreatic cancer.
Moving forward, the examples in this review also provide valuable lessons for the effective targeting of BER proteins in therapeutic approaches. BER proteins can have complex roles outside of their primary role in the BER pathway. It is critical to understand these additional functions when deciding how to target a BER protein. Small molecule inhibitors may produce very different effects to knocking-down total protein, by leaving supplementary functions intact. BER proteins may also represent dual targets because of these additional roles, capable of both chemosensitizing cancer cells and interfering with basic survival functions in response to microenvironmental stress; it is essential to understand both the type of DNA damage induced by a chemotherapeutic (there may be multiple forms induced by one chemotherapeutic, either directly or indirectly), as well as the cross-talk between BER and other DNA repair pathways that can respond to the damage. The involvement of XRCC1 in response to platinum-based therapies demonstrates how DNA repair pathways can be linked and can even compete with each other in response to DNA damage (107, 108). By extension, an effective synthetic-lethal approach requires an understanding of how defects in one DNA repair pathway can make BER the Achille’s heal of a cancer cell, as demonstrated by PARP inhibitors. On this point, recent data from the Australian Pancreatic Genome Initiative showed that 14% of pancreatic cancers display high genomic instability and a further 36% display substantial “scattered” genomic instability (141). These have been linked to defects in genes involved in DNA double-strand break repair including BRCA1 and BRCA2 (known to be responsive to PARP1 inhibitors as discussed above) (141). The identification of pancreatic cancer sub-types based on their genomic instability, represents an exciting advance in our knowledge of PDA and could be exploited to personalize synthetic-lethal therapies using BER-targeted approaches.
The paradox in targeting BER and, in fact, any DNA repair pathway is that defects in DNA repair is responsible for cancer initiation and progression. How can we be sure that targeting BER proteins to treat one cancer would not generate more aggressive and chemoresistant cancer cells? For a DNA-damaging therapeutic approach to be effective, it must induce enough damage to overcome any advantageous mutations that might arise as a consequence of the treatment – an advantageous mutation is useless if a cancer cell has accumulated so much genomic damage that it cannot maintain the basic functions it requires to stay alive. This is the principle behind DNA-damaging radiotherapies and chemotherapeutics. This is not to say that the recurrence of chemoresistant cancers is not a possibility and it has been documented in multiple cancers in response to DNA-damaging agents (142). Targeting BER proteins in combination with chemotherapeutics can improve the efficacy of existing treatments and potentially reduce the chances of leaving resistant cancer cells behind. Future therapeutic approaches for PDA may involve inhibiting two or more BER proteins responsible for repairing very different types of DNA damage, in combination with multiple structurally diverse chemotherapeutics to maximize genomic damage in PDA cells. If we are to reach that stage, a better understanding of BER proteins in PDA cells and their interactions with other DNA repair and signaling pathways is critical. An equally important consideration is the potential for off-target effects when systemically targeting DNA repair proteins. There is the potential that inhibition of DNA repair proteins could either cause toxicity in normal cells in the body or worse yet, generate an initiating mutation for a cancer elsewhere in the body. This is why a cancer cell-specific approach is preferable when targeting BER proteins.
In summary, the BER pathway holds a lot of potential as a therapeutic target in PDA that is still largely untapped. As we move toward more personalized treatments for patients, BER proteins could be inhibited in combination with existing mutations in cancer cells in synthetic-lethal approaches, in combination with microenvironment-induced stress to reduce cancer cell survival, or in combination with DNA-damaging agents to improve their efficacy. Further studies into the BER pathway in PDA are critical in our search for more effective therapeutic approaches for this devastating disease.
Author Contributions
GS and PP were involved in the conception, writing, and final approval of this review. JM and DG were involved in interpretation, revision, and final approval of the work. All authors agree to be accountable for all aspects of the review and ensure that questions related to accuracy or integrity of any part of this review are appropriately investigated and resolved.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Health and Medical Research Council (NHMRC; PP, JM, and DG), Cancer Institute New South Wales Career Development Fellowship (JM and GS), and NHMRC Career Development Fellowship (PP). PP, JM, and DG are also supported by a University of New South Wales Goldstar Award.
Abbreviations
APE1, apurinic/apyrimidinic endonuclease 1; BER, base excision repair; FEN1, flap-endonuclease 1; LIGI, DNA ligase I; LIGIII, DNA ligase III; MBD4, methyl-CpG-binding domain protein 4; MPG, methyl-purine glycosylase; MYH, MutY homologue; NEIL1–3, endonuclease VIII-like 1–3; NTH1, Nth endonuclease III-like 1; OGG1, 8-oxoguanine DNA glycosylase; PanIN, pancreatic intraepithelial neoplasia; PARP1, poly ADP ribose polymerase-1; PDA, pancreatic ductal adenocarcinoma; POLB, DNA polymerase β; SMUG1, single-strand selective monofunctional uracil DNA glycosylase; TDG, thymine DNA glycosylase; TRAIL, TNF-related apoptosis-inducing ligand; UNG, uracil DNA glycosylase; XRCC1, X-ray repair cross-complementing protein 1.
References
1. Raimondi S, Maisonneuve P, Lowenfels AB. Epidemiology of pancreatic cancer: an overview. Nat Rev Gastroenterol Hepatol (2009) 6:699–708. doi: 10.1038/nrgastro.2009.177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet (2011) 378:607–20. doi:10.1016/S0140-6736(10)62307-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res (2014) 74:2913–21. doi:10.1158/0008-5472.CAN-14-0155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin (2013) 63:11–30. doi:10.3322/caac.21166
6. Zalatnai A, Molnar J. Review. Molecular background of chemoresistance in pancreatic cancer. In vivo (2007) 21:339–47.
7. Wang Z, Li Y, Ahmad A, Banerjee S, Azmi AS, Kong D, et al. Pancreatic cancer: understanding and overcoming chemoresistance. Nat Rev Gastroenterol Hepatol (2011) 8:27–33. doi:10.1038/nrgastro.2010.188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Goldstein D, El-Maraghi RH, Hammel P, Heinemann V, Kunzmann V, Sastre J, et al. Nab-paclitaxel plus gemcitabine for metastatic pancreatic cancer: long-term survival from a phase III trial. J Natl Cancer Inst (2015) 107(2). doi:10.1093/jnci/dju413
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med (2011) 364:1817–25. doi:10.1056/NEJMoa1011923
10. Hruban RH, Maitra A, Goggins M. Update on pancreatic intraepithelial neoplasia. Int J Clin Exp Pathol (2008) 1:306–16.
11. Phillips P. Pancreatic stellate cells and fibrosis. In: Grippo PJ, Munshi HG, editors. Pancreatic Cancer and Tumor Microenvironment. Trivandrum: Transworld Research Network (2012).
12. McCarroll JA, Naim S, Sharbeen G, Russia N, Lee J, Kavallaris M, et al. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front Physiol (2014) 5:141. doi:10.3389/fphys.2014.00141
13. Rucki AA, Zheng L. Pancreatic cancer stroma: understanding biology leads to new therapeutic strategies. World J Gastroenterol (2014) 20:2237–46. doi:10.3748/wjg.v20.i9.2237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Apte MV, Park S, Phillips PA, Santucci N, Goldstein D, Kumar RK, et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas (2004) 29:179–87. doi:10.1097/00006676-200410000-00002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Bachem MG, Schunemann M, Ramadani M, Siech M, Beger H, Buck A, et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology (2005) 128:907–21. doi:10.1053/j.gastro.2004.12.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Vonlaufen A, Joshi S, Qu C, Phillips PA, Xu Z, Parker NR, et al. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res (2008) 68:2085–93. doi:10.1158/0008-5472.CAN-07-2477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Vonlaufen A, Phillips PA, Xu Z, Goldstein D, Pirola RC, Wilson JS, et al. Pancreatic stellate cells and pancreatic cancer cells: an unholy alliance. Cancer Res (2008) 68:7707–10. doi:10.1158/0008-5472.CAN-08-1132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Erkan M, Adler G, Apte MV, Bachem MG, Buchholz M, Detlefsen S, et al. StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut (2012) 61:172–8. doi:10.1136/gutjnl-2011-301220
19. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, Mcintyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (2009) 324:1457–61. doi:10.1126/science.1171362
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci U S A (2012) 109:15101–8. doi:10.1073/pnas.1213353109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut (2013) 62:112–20. doi:10.1136/gutjnl-2012-302529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Cannito S, Novo E, Compagnone A, Valfre Di Bonzo L, Busletta C, Zamara E, et al. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis (2008) 29:2267–78. doi:10.1093/carcin/bgn216
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi:10.1126/science.1160809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. El Sayed SM, Mahmoud AA, El Sawy SA, Abdelaal EA, Fouad AM, Yousif RS, et al. Warburg effect increases steady-state ROS condition in cancer cells through decreasing their antioxidant capacities (anticancer effects of 3-bromopyruvate through antagonizing Warburg effect). Med Hypotheses (2013) 81:866–70. doi:10.1016/j.mehy.2013.08.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Wu CA, Chao Y, Shiah SG, Lin WW. Nutrient deprivation induces the Warburg effect through ROS/AMPK-dependent activation of pyruvate dehydrogenase kinase. Biochim Biophys Acta (2013) 1833:1147–56. doi:10.1016/j.bbamcr.2013.01.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab (2005) 1:401–8. doi:10.1016/j.cmet.2005.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia. Exp Physiol (2006) 91:807–19. doi:10.1113/expphysiol.2006.033506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Kasai H, Nishimura S. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res (1984) 12:2137–45. doi:10.1093/nar/12.4.2137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Cheng KC, Cahill DS, Kasai H, Nishimura S, Loeb LA. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G–T and A–C substitutions. J Biol Chem (1992) 267:166–72.
30. Dianov GL, Hubscher U. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res (2013) 41:3483–90. doi:10.1093/nar/gkt076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Wallace SS, Murphy DL, Sweasy JB. Base excision repair and cancer. Cancer Lett (2012) 327:73–89. doi:10.1016/j.canlet.2011.12.038
32. Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature (1999) 401:301–4. doi:10.1038/45843
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Otterlei M, Warbrick E, Nagelhus TA, Haug T, Slupphaug G, Akbari M, et al. Post-replicative base excision repair in replication foci. EMBO J (1999) 18:3834–44. doi:10.1093/emboj/18.13.3834
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersen S, Slupphaug G, et al. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol Cell (2000) 5:1059–65. doi:10.1016/S1097-2765(00)80271-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, et al. hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J Biol Chem (2002) 277:39926–36. doi:10.1074/jbc.M207107200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Akbari M, Otterlei M, Pena-Diaz J, Aas PA, Kavli B, Liabakk NB, et al. Repair of U/G and U/A in DNA by UNG2-associated repair complexes takes place predominantly by short-patch repair both in proliferating and growth-arrested cells. Nucleic Acids Res (2004) 32:5486–98. doi:10.1093/nar/gkh872
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science (2011) 333:1303–7. doi:10.1126/science.1210944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Slupska MM, Baikalov C, Luther WM, Chiang JH, Wei YF, Miller JH. Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J Bacteriol (1996) 178:3885–92.
39. Aburatani H, Hippo Y, Ishida T, Takashima R, Matsuba C, Kodama T, et al. Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res (1997) 57:2151–6.
40. Arai K, Morishita K, Shinmura K, Kohno T, Kim SR, Nohmi T, et al. Cloning of a human homolog of the yeast OGG1 gene that is involved in the repair of oxidative DNA damage. Oncogene (1997) 14:2857–61. doi:10.1038/sj.onc.1201139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Radicella JP, Dherin C, Desmaze C, Fox MS, Boiteux S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A (1997) 94:8010–5. doi:10.1073/pnas.94.15.8010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Roldan-Arjona T, Wei YF, Carter KC, Klungland A, Anselmino C, Wang RP, et al. Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase. Proc Natl Acad Sci U S A (1997) 94:8016–20. doi:10.1073/pnas.94.15.8016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Slupska MM, Luther WM, Chiang JH, Yang H, Miller JH. Functional expression of hMYH, a human homolog of the Escherichia coli MutY protein. J Bacteriol (1999) 181:6210–3.
44. Wiederholt CJ, Delaney MO, Pope MA, David SS, Greenberg MM. Repair of DNA containing Fapy.dG and its beta-C-nucleoside analogue by formamidopyrimidine DNA glycosylase and MutY. Biochemistry (2003) 42:9755–60. doi:10.1021/bi034844h
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Pope MA, David SS. DNA damage recognition and repair by the murine MutY homologue. DNA Repair (Amst) (2005) 4:91–102. doi:10.1016/j.dnarep.2004.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Aspinwall R, Rothwell DG, Roldan-Arjona T, Anselmino C, Ward CJ, Cheadle JP, et al. Cloning and characterization of a functional human homolog of Escherichia coli endonuclease III. Proc Natl Acad Sci U S A (1997) 94:109–14. doi:10.1073/pnas.94.1.109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Hilbert TP, Chaung W, Boorstein RJ, Cunningham RP, Teebor GW. Cloning and expression of the cDNA encoding the human homologue of the DNA repair enzyme, Escherichia coli endonuclease III. J Biol Chem (1997) 272:6733–40. doi:10.1074/jbc.272.10.6733
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Ikeda S, Biswas T, Roy R, Izumi T, Boldogh I, Kurosky A, et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. J Biol Chem (1998) 273:21585–93. doi:10.1074/jbc.273.34.21585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Eide L, Luna L, Gustad EC, Henderson PT, Essigmann JM, Demple B, et al. Human endonuclease III acts preferentially on DNA damage opposite guanine residues in DNA. Biochemistry (2001) 40:6653–9. doi:10.1021/bi0028901
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Bandaru V, Sunkara S, Wallace SS, Bond JP. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst) (2002) 1:517–29. doi:10.1016/S1568-7864(02)00036-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, et al. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc Natl Acad Sci U S A (2002) 99:3523–8. doi:10.1073/pnas.062053799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Hazra TK, Kow YW, Hatahet Z, Imhoff B, Boldogh I, Mokkapati SK, et al. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J Biol Chem (2002) 277:30417–20. doi:10.1074/jbc.C200355200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Dou H, Mitra S, Hazra TK. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J Biol Chem (2003) 278:49679–84. doi:10.1074/jbc.M308658200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Hailer MK, Slade PG, Martin BD, Rosenquist TA, Sugden KD. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair (Amst) (2005) 4:41–50. doi:10.1016/j.dnarep.2004.07.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Zhang QM, Yonekura S, Takao M, Yasui A, Sugiyama H, Yonei S. DNA glycosylase activities for thymine residues oxidized in the methyl group are functions of the hNEIL1 and hNTH1 enzymes in human cells. DNA Repair (Amst) (2005) 4:71–9. doi:10.1016/j.dnarep.2004.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Liu M, Bandaru V, Bond JP, Jaruga P, Zhao X, Christov PP, et al. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc Natl Acad Sci U S A (2010) 107:4925–30. doi:10.1073/pnas.0908307107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Chakravarti D, Ibeanu GC, Tano K, Mitra S. Cloning and expression in Escherichia coli of a human cDNA encoding the DNA repair protein N-methylpurine-DNA glycosylase. J Biol Chem (1991) 266:15710–5.
58. O’Connor TR, Laval J. Human cDNA expressing a functional DNA glycosylase excising 3-methyladenine and 7-methylguanine. Biochem Biophys Res Commun (1991) 176:1170–7. doi:10.1016/0006-291X(91)90408-Y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Samson L, Derfler B, Boosalis M, Call K. Cloning and characterization of a 3-methyladenine DNA glycosylase cDNA from human cells whose gene maps to chromosome 16. Proc Natl Acad Sci U S A (1991) 88:9127–31. doi:10.1073/pnas.88.20.9127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Yu SL, Lee SK, Johnson RE, Prakash L, Prakash S. The stalling of transcription at abasic sites is highly mutagenic. Mol Cell Biol (2003) 23:382–8. doi:10.1128/MCB.23.1.382-388.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Tornaletti S, Maeda LS, Hanawalt PC. Transcription arrest at an abasic site in the transcribed strand of template DNA. Chem Res Toxicol (2006) 19:1215–20. doi:10.1021/tx060103g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Chen DS, Herman T, Demple B. Two distinct human DNA diesterases that hydrolyze 3’-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res (1991) 19:5907–14. doi:10.1093/nar/19.21.5907
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Winters TA, Henner WD, Russell PS, Mccullough A, Jorgensen TJ. Removal of 3’-phosphoglycolate from DNA strand-break damage in an oligonucleotide substrate by recombinant human apurinic/apyrimidinic endonuclease 1. Nucleic Acids Res (1994) 22:1866–73. doi:10.1093/nar/22.10.1866
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Suh D, Wilson DM III, Povirk LF. 3’-phosphodiesterase activity of human apurinic/apyrimidinic endonuclease at DNA double-strand break ends. Nucleic Acids Res (1997) 25:2495–500. doi:10.1093/nar/25.12.2495
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, et al. AP endonuclease-independent DNA base excision repair in human cells. Mol Cell (2004) 15:209–20. doi:10.1016/j.molcel.2004.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Demple B, Herman T, Chen DS. Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc Natl Acad Sci U S A (1991) 88:11450–4. doi:10.1073/pnas.88.24.11450
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Seki S, Hatsushika M, Watanabe S, Akiyama K, Nagao K, Tsutsui K. cDNA cloning, sequencing, expression and possible domain structure of human APEX nuclease homologous to Escherichia coli exonuclease III. Biochim Biophys Acta (1992) 1131:287–99. doi:10.1016/0167-4781(92)90027-W
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Masson M, Niedergang C, Schreiber V, Muller S, Menissier-De Murcia J, De Murcia G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol (1998) 18:3563–71.
69. Dianova II, Sleeth KM, Allinson SL, Parsons JL, Breslin C, Caldecott KW, et al. XRCC1-DNA polymerase beta interaction is required for efficient base excision repair. Nucleic Acids Res (2004) 32:2550–5. doi:10.1093/nar/gkh567
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Nazarkina ZK, Khodyreva SN, Marsin S, Lavrik OI, Radicella JP. XRCC1 interactions with base excision repair DNA intermediates. DNA Repair (Amst) (2007) 6:254–64. doi:10.1016/j.dnarep.2006.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Odell ID, Barbour JE, Murphy DL, Della-Maria JA, Sweasy JB, Tomkinson AE, et al. Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol Cell Biol (2011) 31:4623–32. doi:10.1128/MCB.05715-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Campalans A, Kortulewski T, Amouroux R, Menoni H, Vermeulen W, Radicella JP. Distinct spatiotemporal patterns and PARP dependence of XRCC1 recruitment to single-strand break and base excision repair. Nucleic Acids Res (2013) 41:3115–29. doi:10.1093/nar/gkt025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Beard WA, Prasad R, Wilson SH. Activities and mechanism of DNA polymerase beta. Methods Enzymol (2006) 408:91–107. doi:10.1016/S0076-6879(06)08007-4
74. Robertson AB, Klungland A, Rognes T, Leiros I. DNA repair in mammalian cells: base excision repair: the long and short of it. Cell Mol Life Sci (2009) 66:981–93. doi:10.1007/s00018-009-8736-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Asagoshi K, Tano K, Chastain PD II, Adachi N, Sonoda E, Kikuchi K, et al. FEN1 functions in long patch base excision repair under conditions of oxidative stress in vertebrate cells. Mol Cancer Res (2010) 8:204–15. doi:10.1158/1541-7786.MCR-09-0253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Sleeth KM, Robson RL, Dianov GL. Exchangeability of mammalian DNA ligases between base excision repair pathways. Biochemistry (2004) 43:12924–30. doi:10.1021/bi0492612
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Drablos F, Feyzi E, Aas PA, Vaagbo CB, Kavli B, Bratlie MS, et al. Alkylation damage in DNA and RNA – repair mechanisms and medical significance. DNA Repair (Amst) (2004) 3:1389–407. doi:10.1016/j.dnarep.2004.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Bobola MS, Finn LS, Ellenbogen RG, Geyer JR, Berger MS, Braga JM, et al. Apurinic/apyrimidinic endonuclease activity is associated with response to radiation and chemotherapy in medulloblastoma and primitive neuroectodermal tumors. Clin Cancer Res (2005) 11:7405–14. doi:10.1158/1078-0432.CCR-05-1068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Bobola MS, Emond MJ, Blank A, Meade EH, Kolstoe DD, Berger MS, et al. Apurinic endonuclease activity in adult gliomas and time to tumor progression after alkylating agent-based chemotherapy and after radiotherapy. Clin Cancer Res (2004) 10:7875–83. doi:10.1158/1078-0432.CCR-04-1161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Koukourakis MI, Giatromanolaki A, Kakolyris S, Sivridis E, Georgoulias V, Funtzilas G, et al. Nuclear expression of human apurinic/apyrimidinic endonuclease (HAP1/Ref-1) in head-and-neck cancer is associated with resistance to chemoradiotherapy and poor outcome. Int J Radiat Oncol Biol Phys (2001) 50:27–36. doi:10.1016/S0360-3016(00)01561-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Zhang Y, Wang J, Xiang D, Wang D, Xin X. Alterations in the expression of the apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE1/Ref-1) in human ovarian cancer and indentification of the therapeutic potential of APE1/Ref-1 inhibitor. Int J Oncol (2009) 35:1069–79. doi:10.3892/ijo_00000422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Kelley MR, Jiang Y, Guo C, Reed A, Meng H, Vasko MR. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PLoS One (2014) 9:e106485. doi:10.1371/journal.pone.0106485
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Simeonov A, Kulkarni A, Dorjsuren D, Jadhav A, Shen M, Mcneill DR, et al. Identification and characterization of inhibitors of human apurinic/apyrimidinic endonuclease APE1. PLoS One (2009) 4:e5740. doi:10.1371/journal.pone.0005740
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Lau JP, Weatherdon KL, Skalski V, Hedley DW. Effects of gemcitabine on APE/ref-1 endonuclease activity in pancreatic cancer cells, and the therapeutic potential of antisense oligonucleotides. Br J Cancer (2004) 91:1166–73. doi:10.1038/sj.bjc.6602080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Xiong GS, Sun HL, Wu SM, Mo JZ. Small interfering RNA against the apurinic or apyrimidinic endonuclease enhances the sensitivity of human pancreatic cancer cells to gemcitabine in vitro. J Dig Dis (2010) 11:224–30. doi:10.1111/j.1751-2980.2010.00442.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Chen S, Xiong G, Wu S, Mo J. Downregulation of apurinic/apyrimidinic endonuclease 1/redox factor-1 enhances the sensitivity of human pancreatic cancer cells to radiotherapy in vitro. Cancer Biother Radiopharm (2013) 28:169–76. doi:10.1089/cbr.2012.1266
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Xanthoudakis S, Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J (1992) 11:653–65.
88. Huang RP, Adamson ED. Characterization of the DNA-binding properties of the early growth response-1 (Egr-1) transcription factor: evidence for modulation by a redox mechanism. DNA Cell Biol (1993) 12:265–73. doi:10.1089/dna.1993.12.265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Walker LJ, Robson CN, Black E, Gillespie D, Hickson ID. Identification of residues in the human DNA repair enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun DNA binding. Mol Cell Biol (1993) 13:5370–6.
90. Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunit. J Biol Chem (1996) 271:32253–9. doi:10.1074/jbc.271.50.32253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Jayaraman L, Murthy KG, Zhu C, Curran T, Xanthoudakis S, Prives C. Identification of redox/repair protein Ref-1 as a potent activator of p53. Genes Dev (1997) 11:558–70. doi:10.1101/gad.11.5.558
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Nishi T, Shimizu N, Hiramoto M, Sato I, Yamaguchi Y, Hasegawa M, et al. Spatial redox regulation of a critical cysteine residue of NF-kappa B in vivo. J Biol Chem (2002) 277:44548–56. doi:10.1074/jbc.M202970200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Jiang Y, Zhou S, Sandusky GE, Kelley MR, Fishel ML. Reduced expression of DNA repair and redox signaling protein APE1/Ref-1 impairs human pancreatic cancer cell survival, proliferation, and cell cycle progression. Cancer Invest (2010) 28:885–95. doi:10.3109/07357907.2010.512816
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Fishel ML, Jiang Y, Rajeshkumar NV, Scandura G, Sinn AL, He Y, et al. Impact of APE1/Ref-1 redox inhibition on pancreatic tumor growth. Mol Cancer Ther (2011) 10:1698–708. doi:10.1158/1535-7163.MCT-11-0107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Cardoso AA, Jiang Y, Luo M, Reed AM, Shahda S, He Y, et al. APE1/Ref-1 regulates STAT3 transcriptional activity and APE1/Ref-1-STAT3 dual-targeting effectively inhibits pancreatic cancer cell survival. PLoS One (2012) 7:e47462. doi:10.1371/journal.pone.0047462
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Fishel ML, Wu X, Devlin CM, Logsdon DP, Jiang Y, Luo M, et al. Apurinic/apyrimidinic endonuclease/redox factor-1 (APE1/Ref-1) redox function negatively regulates NRF2. J Biol Chem (2015) 290:3057–68. doi:10.1074/jbc.M114.621995
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci U S A (1996) 93:8919–23. doi:10.1073/pnas.93.17.8919
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Ludwig DL, Macinnes MA, Takiguchi Y, Purtymun PE, Henrie M, Flannery M, et al. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat Res (1998) 409:17–29. doi:10.1016/S0921-8777(98)00039-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Fung H, Demple B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell (2005) 17:463–70. doi:10.1016/j.molcel.2004.12.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Izumi T, Brown DB, Naidu CV, Bhakat KK, Macinnes MA, Saito H, et al. Two essential but distinct functions of the mammalian abasic endonuclease. Proc Natl Acad Sci U S A (2005) 102:5739–43. doi:10.1073/pnas.0500986102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Kullberg M, Mccarthy R, Anchordoquy TJ. Systemic tumor-specific gene delivery. J Control Release (2013) 172:730–6. doi:10.1016/j.jconrel.2013.08.300
102. McCarroll J, Teo J, Boyer C, Goldstein D, Kavallaris M, Phillips PA. Potential applications of nanotechnology for the diagnosis and treatment of pancreatic cancer. Front Physiol (2014) 5:2. doi:10.3389/fphys.2014.00002
103. Stoehlmacher J, Ghaderi V, Iobal S, Groshen S, Tsao-Wei D, Park D, et al. A polymorphism of the XRCC1 gene predicts for response to platinum based treatment in advanced colorectal cancer. Anticancer Res (2001) 21: 3075–9.
104. Chung HH, Kim MK, Kim JW, Park NH, Song YS, Kang SB, et al. XRCC1 R399Q polymorphism is associated with response to platinum-based neoadjuvant chemotherapy in bulky cervical cancer. Gynecol Oncol (2006) 103:1031–7. doi:10.1016/j.ygyno.2006.06.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Cui Z, Yin Z, Li X, Wu W, Guan P, Zhou B. Association between polymorphisms in XRCC1 gene and clinical outcomes of patients with lung cancer: a meta-analysis. BMC Cancer (2012) 12:71. doi:10.1186/1471-2407-12-71
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Reardon JT, Vaisman A, Chaney SG, Sancar A. Efficient nucleotide excision repair of cisplatin, oxaliplatin, and Bis-aceto-ammine-dichloro-cyclohexylamine-platinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res (1999) 59:3968–71.
107. Kothandapani A, Dangeti VS, Brown AR, Banze LA, Wang XH, Sobol RW, et al. Novel role of base excision repair in mediating cisplatin cytotoxicity. J Biol Chem (2011) 286:14564–74. doi:10.1074/jbc.M111.225375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Kothandapani A, Sawant A, Dangeti VS, Sobol RW, Patrick SM. Epistatic role of base excision repair and mismatch repair pathways in mediating cisplatin cytotoxicity. Nucleic Acids Res (2013) 41:7332–43. doi:10.1093/nar/gkt479
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Abdel-Fatah T, Sultana R, Abbotts R, Hawkes C, Seedhouse C, Chan S, et al. Clinicopathological and functional significance of XRCC1 expression in ovarian cancer. Int J Cancer (2013) 132:2778–86. doi:10.1002/ijc.27980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Moser J, Kool H, Giakzidis I, Caldecott K, Mullenders LH, Fousteri MI. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol Cell (2007) 27:311–23. doi:10.1016/j.molcel.2007.06.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Bowden NA. Nucleotide excision repair: why is it not used to predict response to platinum-based chemotherapy? Cancer Lett (2014) 346:163–71. doi:10.1016/j.canlet.2014.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Duell EJ, Holly EA, Bracci PM, Wiencke JK, Kelsey KT. A population-based study of the Arg399Gln polymorphism in X-ray repair cross-complementing group 1 (XRCC1) and risk of pancreatic adenocarcinoma. Cancer Res (2002) 62:4630–6.
113. Giovannetti E, Pacetti P, Reni M, Leon LG, Mambrini A, Vasile E, et al. Association between DNA-repair polymorphisms and survival in pancreatic cancer patients treated with combination chemotherapy. Pharmacogenomics (2011) 12:1641–52. doi:10.2217/pgs.11.109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Nakao M, Hosono S, Ito H, Watanabe M, Mizuno N, Sato S, et al. Selected polymorphisms of base excision repair genes and pancreatic cancer risk in Japanese. J Epidemiol (2012) 22:477–83. doi:10.2188/jea.JE20120010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Ozguner F, Koyu A, Cesur G. Active smoking causes oxidative stress and decreases blood melatonin levels. Toxicol Ind Health (2005) 21:21–6. doi:10.1191/0748233705th211oa
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Isik B, Ceylan A, Isik R. Oxidative stress in smokers and non-smokers. Inhal Toxicol (2007) 19:767–9. doi:10.1080/08958370701401418
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Puri BK, Treasaden IH, Cocchi M, Tsaluchidu S, Tonello L, Ross BM. A comparison of oxidative stress in smokers and non-smokers: an in vivo human quantitative study of n-3 lipid peroxidation. BMC Psychiatry (2008) 8(Suppl 1):S4. doi:10.1186/1471-244X-8-S1-S4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Tavilani H, Nadi E, Karimi J, Goodarzi MT. Oxidative stress in COPD patients, smokers, and non-smokers. Respir Care (2012) 57:2090–4. doi:10.4187/respcare.01809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Kim MY, Zhang T, Kraus WL. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev (2005) 19:1951–67. doi:10.1101/gad.1331805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature (2005) 434:913–7. doi:10.1038/nature03443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Farmer H, Mccabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature (2005) 434:917–21. doi:10.1038/nature03445
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Powell SN, Kachnic LA. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene (2003) 22:5784–91. doi:10.1038/sj.onc.1206678
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Sonnenblick A, De Azambuja E, Azim HA Jr, Piccart M. An update on PARP inhibitors-moving to the adjuvant setting. Nat Rev Clin Oncol (2014) 12(1):27–41. doi:10.1038/nrclinonc.2014.163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Drew Y, Mulligan EA, Vong WT, Thomas HD, Kahn S, Kyle S, et al. Therapeutic potential of poly(ADP-ribose) polymerase inhibitor AG014699 in human cancers with mutated or methylated BRCA1 or BRCA2. J Natl Cancer Inst (2011) 103:334–46. doi:10.1093/jnci/djq509
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Chen S, Wang G, Niu X, Zhao J, Tan W, Wang H, et al. Combination of AZD2281 (olaparib) and GX15-070 (obatoclax) results in synergistic antitumor activities in preclinical models of pancreatic cancer. Cancer Lett (2014) 348:20–8. doi:10.1016/j.canlet.2014.02.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Wang Q, Gao F, May WS, Zhang Y, Flagg T, Deng X. Bcl2 negatively regulates DNA double-strand-break repair through a nonhomologous end-joining pathway. Mol Cell (2008) 29:488–98. doi:10.1016/j.molcel.2007.12.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Kumar TS, Kari V, Choudhary B, Nambiar M, Akila TS, Raghavan SC. Anti-apoptotic protein BCL2 down-regulates DNA end joining in cancer cells. J Biol Chem (2010) 285:32657–70. doi:10.1074/jbc.M110.140350
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Jacob DA, Bahra M, Langrehr JM, Boas-Knoop S, Stefaniak R, Davis J, et al. Combination therapy of poly (ADP-ribose) polymerase inhibitor 3-aminobenzamide and gemcitabine shows strong antitumor activity in pancreatic cancer cells. J Gastroenterol Hepatol (2007) 22:738–48. doi:10.1111/j.1440-1746.2006.04496.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Piao L, Nakagawa H, Ueda K, Chung S, Kashiwaya K, Eguchi H, et al. C12orf48, termed PARP-1 binding protein, enhances poly(ADP-ribose) polymerase-1 (PARP-1) activity and protects pancreatic cancer cells from DNA damage. Genes Chromosomes Cancer (2011) 50:13–24. doi:10.1002/gcc.20828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Porcelli L, Quatrale AE, Mantuano P, Leo MG, Silvestris N, Rolland JF, et al. Optimize radiochemotherapy in pancreatic cancer: PARP inhibitors a new therapeutic opportunity. Mol Oncol (2013) 7:308–22. doi:10.1016/j.molonc.2012.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Karnak D, Engelke CG, Parsels LA, Kausar T, Wei D, Robertson JR, et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin Cancer Res (2014) 20:5085–96. doi:10.1158/1078-0432.CCR-14-1038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Tuli R, Surmak AJ, Reyes J, Armour M, Hacker-Prietz A, Wong J, et al. Radiosensitization of pancreatic cancer cells in vitro and in vivo through poly (ADP-ribose) polymerase inhibition with ABT-888. Transl Oncol (2014). doi:10.1016/j.tranon.2014.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol (2014) 33(3):244–50. doi:10.1200/JCO.2014.56.2728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Yuan K, Sun Y, Zhou T, Mcdonald J, Chen Y. PARP-1 regulates resistance of pancreatic cancer to TRAIL therapy. Clin Cancer Res (2013) 19:4750–9. doi:10.1158/1078-0432.CCR-13-0516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Klauschen F, Von Winterfeld M, Stenzinger A, Sinn BV, Budczies J, Kamphues C, et al. High nuclear poly-(ADP-ribose)-polymerase expression is prognostic of improved survival in pancreatic cancer. Histopathology (2012) 61:409–16. doi:10.1111/j.1365-2559.2012.04225.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Agnihotri S, Burrell K, Buczkowicz P, Remke M, Golbourn B, Chornenkyy Y, et al. ATM regulates 3-methylpurine-DNA glycosylase and promotes therapeutic resistance to alkylating agents. Cancer Discov (2014) 4, 1198–213. doi:10.1158/2159-8290.CD-14-0157
137. An Q, Robins P, Lindahl T, Barnes DE. 5-Fluorouracil incorporated into DNA is excised by the Smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res (2007) 67:940–5. doi:10.1158/0008-5472.CAN-06-2960
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Boudsocq F, Benaim P, Canitrot Y, Knibiehler M, Ausseil F, Capp JP, et al. Modulation of cellular response to cisplatin by a novel inhibitor of DNA polymerase beta. Mol Pharmacol (2005) 67:1485–92. doi:10.1124/mol.104.001776
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Stachelek GC, Dalal S, Donigan KA, Campisi Hegan D, Sweasy JB, Glazer PM. Potentiation of temozolomide cytotoxicity by inhibition of DNA polymerase beta is accentuated by BRCA2 mutation. Cancer Res (2010) 70:409–17. doi:10.1158/0008-5472.CAN-09-1353
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Yang J, Parsons J, Nicolay NH, Caporali S, Harrington CF, Singh R, et al. Cells deficient in the base excision repair protein, DNA polymerase beta, are hypersensitive to oxaliplatin chemotherapy. Oncogene (2010) 29:463–8. doi:10.1038/onc.2009.327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature (2015) 518:495–501. doi:10.1038/nature14169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Cheung-Ong K, Giaever G, Nislow C. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol (2013) 20:648–59. doi:10.1016/j.chembiol.2013.04.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: base excision repair, pancreatic cancer, chemoresistance, therapeutic targets, prognostic factors
Citation: Sharbeen G, McCarroll J, Goldstein D and Phillips PA (2015) Exploiting base excision repair to improve therapeutic approaches for pancreatic cancer. Front. Nutr. 2:10. doi: 10.3389/fnut.2015.00010
Received: 30 January 2015; Paper pending published: 28 February 2015;
Accepted: 10 March 2015; Published online: 27 March 2015.
Edited by:
Marco Falasca, Curtin University, AustraliaReviewed by:
Michele Milella, Regina Elena National Cancer Institute, ItalyEdwin Charles Thrower, Yale University, USA
Jens Thomas Siveke, Technical University of Munich, Germany
Copyright: © 2015 Sharbeen, McCarroll, Goldstein and Phillips. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Phoebe A. Phillips, Pancreatic Cancer Translational Research Group, Room 210, Level 2, Lowy Cancer Research Centre, Prince of Wales Clinical School, UNSW Australia, Sydney, NSW 2052, Australia e-mail:cC5waGlsbGlwc0B1bnN3LmVkdS5hdQ==