 Richard Eugene Frye
Richard Eugene Frye Daniel A. Rossignol
Daniel A. Rossignol- 1Department of Pediatrics, Arkansas Children’s Hospital Research Institute, University of Arkansas for Medical Sciences, Little Rock, AR, USA
- 2Rossignol Medical Center, Irvine, CA, USA
Recent studies point to the effectiveness of novel treatments that address physiological abnormalities associated with autism spectrum disorder (ASD). This is significant because safe and effective treatments for ASD remain limited. These physiological abnormalities as well as studies addressing treatments of these abnormalities are reviewed in this article. Treatments commonly used to treat mitochondrial disease have been found to improve both core and associated ASD symptoms. Double-blind, placebo-controlled (DBPC) studies have investigated l-carnitine and a multivitamin containing B vitamins, antioxidants, vitamin E, and co-enzyme Q10 while non-blinded studies have investigated ubiquinol. Controlled and uncontrolled studies using folinic acid, a reduced form of folate, have reported marked improvements in core and associated ASD symptoms in some children with ASD and folate related pathway abnormities. Treatments that could address redox metabolism abnormalities include methylcobalamin with and without folinic acid in open-label studies and vitamin C and N-acetyl-l-cysteine in DBPC studies. These studies have reported improved core and associated ASD symptoms with these treatments. Lastly, both open-label and DBPC studies have reported improvements in core and associated ASD symptoms with tetrahydrobiopterin. Overall, these treatments were generally well-tolerated without significant adverse effects for most children, although we review the reported adverse effects in detail. This review provides evidence for potentially safe and effective treatments for core and associated symptoms of ASD that target underlying known physiological abnormalities associated with ASD. Further research is needed to define subgroups of children with ASD in which these treatments may be most effective as well as confirm their efficacy in DBPC, large-scale multicenter studies.
Background
The autism spectrum disorders (ASD) are a group of behaviorally defined neurodevelopmental disorders with lifelong consequences. They are defined by impairments in communication and social interaction along with restrictive and repetitive behaviors (1). The definition of ASD has recently undergone revision. Previously, the Diagnostic Statistical Manual (DSM) Version IV Text Revision divided ASD into several diagnoses including autistic disorder, Asperger syndrome, and pervasive developmental disorder-not otherwise specified. The new revision of the DSM now does not differentiate between these ASD subtypes and considers communication and social impairments together in one symptom class (2). Complicating this change is the fact that over the past several decades, most research has used a framework from the former DSM versions.
Autism spectrum disorder has been recently estimated to affect 1 out of 68 individuals in the United States (3) with four times more males than females being affected (4). Over the past two decades, the prevalence of the ASDs has grown dramatically, although the reasons for this increase are continually debated. Despite decades of research on ASD, identification of the causes of and treatments for ASD remain limited. The standard-of-care treatment for ASD is behavioral therapy that requires full-time engagement of a one-on-one therapist typically requiring many years of treatment, and recent reviews have pointed out that controlled studies on commonly used behavior therapies are generally lacking (5). The only medical treatments approved by the United States of America Food and Drug Administration for ASD are antipsychotic medications. However, these medications only treat a symptom associated with ASD, irritability, but not any core ASD symptom. In children, these medications can be associated with significant adverse effects, including detrimental changes in body weight as well as triglyceride, cholesterol, and blood glucose concentrations within a short time (6) and they also increase the risk of type 2 diabetes (7). In some studies, the percentage of children experiencing these side effects is quite high. For example, one recent study reported that 87% of ASD children had side effects with risperidone, including drowsiness, weight gain, and rhinorrhea (8).
A great majority of ASD research has concentrated on genetic causes of ASD (9) despite the fact that inherited single gene and chromosomal defects are only found in the minority of cases (10). In fact, several recent studies that have conducted genome wide searches for common genetic defects across large samples of ASD children have only identified rare de novo mutations, thereby pointing to acquired mutations and/or mutations secondary to errors in DNA maintenance rather than inherited genetic syndromes (11, 12). As research in the field of ASD continues, it is becoming clear that the etiology of most ASD cases involves complicated interactions between genetic predisposition and environmental exposures or triggers. Indeed, a recent study of dizygotic twins estimated that the environment contributes a greater percentage of the risk of developing autistic disorder as compared to genetic factors (13). Another study of over two million children reported that environmental risk factors accounted for approximately 50% of ASD risk (14). Recent reviews have outlined the many environmental factors that are associated with ASD and have described how polymorphisms in specific genes can combine with the environment to cause neurodevelopmental problems (15).
Recent studies have suggested that ASD is associated with impairments in basic physiological processes such as redox (16) and mitochondrial (9) metabolism as well as abnormalities in regulating essential metabolites such as folate (17), tetrahydrobiopterin (18–20), glutathione (21–23), cholesterol (24), carnitine (25–28), and branch chain amino acids (29). Although many of these studies have based their findings on peripheral markers of abnormal metabolism, many studies have documented some of these same abnormalities in the brain of individuals with ASD, including mitochondrial dysfunction and oxidative stress (30) and one study has demonstrated a link between oxidative stress, inflammation, and mitochondrial dysfunction in the brain of individuals with ASD (23). Interestingly, several of these physiological abnormalities are also observed in genetic syndromes associated with ASD. For example, mitochondrial dysfunction is prevalent in both idiopathic ASD (31) and is associated with Rett syndrome (32–34), PTEN mutations (35), Phelan-McDermid syndrome (36), 15q11-q13 duplication syndrome (37, 38), Angelman syndrome (39), Septo-optic dysplasia (40), and Down syndrome (41, 42).
Identifying the metabolic or physiological abnormalities associated with ASD is important, as treatments for such abnormalities may be possible. Thus, a better understanding of these abnormalities may allow for the development of novel treatments for children with ASD. Below the evidence for metabolic abnormalities related to ASD that may be amenable to treatment are discussed along with the evidence of potential treatments for these disorders. Figure 1 provides a summary of the pathways and demonstrates which pathways are targeted by the better studied treatments. In addition, a section on the common adverse effects of these treatments follows the discussion of treatments.
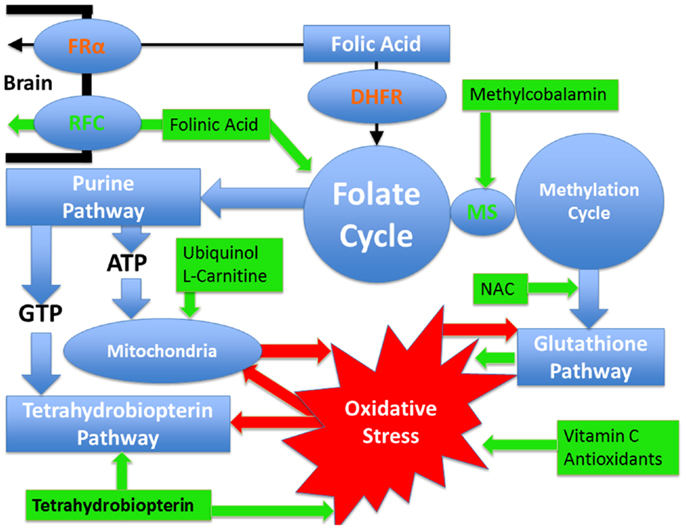
Figure 1. Pathways affected in autism spectrum disorder that are discussed in this article as well as the treatments discussed with their points of action. Pathways are outlined in blue while treatments are outlined in green. Oxidative stress is outlined in red and the red arrows demonstrate how it can negatively influence metabolic pathways. Certain pathways such as glutathione and tetrahydrobiopterin pathways have an antioxidant effect and a reciprocal relationship with oxidative stress such that they can improve oxidative stress but at the same time oxidative stress has a direct detrimental effect on them. Mitochondrial dysfunction and oxidative stress have mutually negative effects on each other such that oxidative stress causes mitochondrial dysfunction while mitochondrial dysfunction worsens oxidative stress. Dihydrofolate reductase (DHFR) is colored in red since polymorphisms in this gene, that are commonly seen in individuals with autism, have a detrimental effect on the reduction of folic acid such that the entry of folic acid into the folate cycle is decreased. Folinic acid enters the folate cycle without requiring this enzyme. Similarly the folate receptor alpha can be impaired in individuals with autism by autoantibodies and by mitochondrial dysfunction. In such cases, folinic acid can cross the blood–brain barrier by the reduced folate carrier. Methionine synthase (MS) connects the folate and methylation cycles and requires methylcobalamin as a cofactor.
Review of Treatable Conditions and Their Potential Treatments
Mitochondrial Dysfunction
Recent studies suggested that 30–50% of children with ASD possess biomarkers consistent with mitochondrial dysfunction (31, 43) and that the prevalence of abnormal mitochondrial function in immune cells derived from children with ASD is exceedingly high (44, 45). Mitochondrial dysfunction has been demonstrated in the postmortem ASD brain (23, 30, 46–49) and in animal models of ASD (50). Novel types of mitochondrial dysfunction have been described in children with ASD (28, 51, 52) and in cell lines derived from children with ASD (53, 54). Several studies suggest that children with ASD and mitochondrial dysfunction have more severe behavioral and cognitive disabilities compared with children who have ASD but without mitochondrial dysfunction (55–57). Interestingly, a recent review of all of the known published cases of mitochondrial disease and ASD demonstrated that only about 25% had a known genetic mutation that could account for their mitochondrial disease (31).
Treatments that are typically used for patients with mitochondrial disease have been shown to improve functioning in some children with ASD (31). Several studies, including two double-blind, placebo-controlled (DBPC) studies (58, 59) and case reports (25, 37, 60–63) have reported improvements in core and associated ASD behaviors with l-carnitine treatment. Two DBPC studies using a multivitamin containing B vitamins, antioxidants, vitamin E, and co-enzyme Q10 reported various improvements in ASD symptoms compared to placebo (64, 65). Several other antioxidants (66), including vitamin C (67), methylcobalamin (68–70), N-acetyl-l-cysteine (71–73), ubiquinol (74), and carnosine (75), have also reported to demonstrate significant improvements in ASD behaviors and may function to improve mitochondrial function.
Thus, many treatments that are believed to improve mitochondrial function have been shown to be helpful for some children with ASD. However, none of these studies have specifically selected children with mitochondrial dysfunction or disease to study, so it is difficult to know if individuals with ASD and mitochondrial dysfunction would benefit the most from these treatments or whether these treatments are effective for a wider group of children with ASD. One study did demonstrate that the multivitamin used for treatment resulted in improvements in biomarkers of energy metabolism (as well as oxidative stress) suggesting that the effect of the multivitamin may have been at least partially related to improvements in mitochondrial function (65). Clearly, this is a fertile area for research but there remain several complications that could impede moving forward in a systematic way. For example, given the inconsistency in the prevalence estimates of mitochondrial disease and dysfunction across studies (ranging from about 5–80%), the notion that mitochondrial abnormalities are even associated with ASD is somewhat controversial. This may be, in part, due to the unclear distinction between mitochondrial disease and dysfunction. However, even the lower bound of the prevalence estimate of 5% is significant, as mitochondrial disease is only believed to affect <0.1% of individuals in the general population and given the current high prevalence of ASD, a disorder that affects even 5% of individuals with ASD would add up to millions of individuals who have the potential to have a treatable metabolic abnormality. Other complicating factors include the fact that there are many treatments for mitochondrial disease and these treatments have not been well-studied (76). Hopefully, the increased interest in treatments for mitochondrial disease will help improve our knowledge of how to best treat mitochondrial disease so that such information can be applied to children who have mitochondrial disease and dysfunction with ASD. Other recent approaches include the in vitro assessment of compounds that may improve mitochondrial function in individuals with ASD (53).
Folate Metabolism
Several lines of evidence point to abnormalities in folate metabolism in ASD. Several genetic polymorphisms in key enzymes in the folate pathway have been associated with ASD. These abnormalities can cause decreased production of 5-methyltetrahydrofolate, impair the production of folate cycle metabolites and decrease folate transport across the blood–brain barrier and into neurons. Indeed, genetic polymorphisms in methylenetetrahydrofolate reductase (22, 77–85), dihydrofolate reductase (86) and the reduced folate carrier (22) have been associated with ASD.
Perhaps the most significant abnormalities in folate metabolism associated with ASD are autoantibodies to the folate receptor alpha (FRα). Folate is transported across the blood–brain barrier by an energy-dependent receptor-mediated system that utilizes the FRα (87). Autoantibodies can bind to the FRα and greatly impair its function. These autoantibodies have been linked to cerebral folate deficiency (CFD). Many cases of CFD carry a diagnosis of ASD (88–94) and other individuals with CFD are diagnosed with Rett syndrome, a disorder closely related to ASD within the pervasive developmental disorder spectrum (95–97). Given that the FRα folate transport system is energy-dependent and consumes ATP, it is not surprising that a wide variety of mitochondrial diseases (91, 94, 97–102) and novel forms of mitochondrial dysfunction related to ASD (52) have been associated with CFD. Recently, Frye et al. (17) reported that 60% and 44% of 93 children with ASD were positive for the blocking and binding FRα autoantibody, respectively. This high rate of FRα autoantibody positivity was confirmed by Ramaekers et al. (103) who compared 75 ASD children to 30 non-autistic controls with developmental delay. The blocking FRα autoantibody was positive in 47% of children with ASD but in only 3% of the control children.
Many children with ASD and CFD have marked improvements in clinical status when treated with folinic acid – a reduced form of folate that can cross the blood–brain barrier using the reduced folate carrier rather than the FRα transport system. Several case reports (89) and case series (90, 91) have described neurological, behavioral, and cognitive improvements in children with documented CFD and ASD. One case series of five children with CFD and low-functioning autism with neurological deficits found complete recovery from ASD symptoms with the use of folinic acid in one child and substantial improvements in communication in two other children (90). In another study of 23 children with low-functioning regressive ASD and CFD, 2 younger children demonstrated full recovery from ASD and neurological symptoms, 3 older children demonstrated improvements in neurological deficits but not in ASD symptoms, and the remainder demonstrated improvements in neurological symptoms and partial improvements in some ASD symptoms with folinic acid; the most prominent improvement was in communication (91). Recently, in a controlled open-label study, Frye et al. (17) demonstrated that ASD children who were positive for at least one of the FRα autoantibodies experienced significant improvements in verbal communication, receptive and expressive language, attention, and stereotypical behavior with high-dose (2 mg/kg/day in two divided doses; maximum 50 mg/day) folinic acid treatment with very few adverse effects reported.
Thus, there are several lines of converging evidence suggesting that abnormalities in folate metabolism are associated with ASD. Evidence for treatment of these disorders is somewhat limited but it is growing. For example, treatment studies have mostly concentrated on the subset of children with ASD who also possess the FRα autoantibodies. These studies have only examined one form of reduced folate, folinic acid, and have only examined treatment response in limited studies. Thus, large DBPC studies would be very helpful for documenting efficacy of this potentially safe and effective treatment. In addition, the role of other abnormalities in the folate pathway beside FRα autoantibodies, such as genetic polymorphisms, in treatment response needs to be investigated. It might also be important to investigate the role of treatment with other forms of folate besides folinic acid, but it might also be wise to concentrate research on one particular form of folate for the time being so as to optimize the generalizability of research studies in order to have a more solid understanding of the role of folate metabolism in ASD. Given the ubiquitous role of folate in many metabolic pathways and the fact that it has a role in preventing ASD during the preconception and prenatal periods (104), this line of research has significant potential for being a novel treatment for many children with ASD.
Redox Metabolism
Several lines of evidence support the notion that some children with ASD have abnormal redox metabolism. Two case-control studies have reported that redox metabolism in children with ASD is abnormal compared to unaffected control children (22, 105). This includes a significant decrease in reduced glutathione (GSH), the major intracellular antioxidant, and mechanism for detoxification, as well as a significant increase in the oxidized disulfide form of glutathione (GSSG). The notion that abnormal glutathione metabolism could lead to oxidative damage is consistent with studies which demonstrate oxidative damage to proteins and DNA in peripheral blood mononuclear cells and postmortem brain from ASD individuals (23, 30, 106), particularly in cortical regions associated with speech, emotion, and social behavior (30, 107).
Treatments for oxidative stress have been shown to be of benefit for children with ASD. In children with ASD, studies have demonstrated that glutathione metabolism can be improved with subcutaneously injected methylcobalamin and oral folinic acid (69, 105), a vitamin and mineral supplement that includes antioxidants, co-enzyme Q10, and B vitamins (65) and tetrahydrobiopterin (20). Interestingly, recent DBPC studies have demonstrated that N-acetyl-l-cysteine, a supplement that provides a precursor to glutathione, was effective in improving symptoms and behaviors associated with ASD (72, 73). However, glutathione was not measured in these two studies.
Small (64, 67), medium (72, 73), and large (108) sized DPBC trials and small and medium-sized open-label clinical trials (68, 70) demonstrate that novel treatments for children with ASD, which can address oxidative stress are associated with improvements in core ASD symptoms (68, 70, 72), sleep and gastrointestinal symptoms (64), hyperactivity, tantruming, and parental impression of general functioning (108), sensory-motor symptoms (67), and irritability (72, 73). These novel treatments include N-acetyl-l-cysteine (72, 73), methylcobalamin with (69, 70) and without (68) oral folinic acid, vitamin C (67), and a vitamin and mineral supplement that includes antioxidants, co-enzyme Q10, and B vitamins (64, 65).
Several other treatments that have antioxidant properties (66), including carnosine (75), have also been reported to significantly improve ASD behaviors, suggesting that treatment of oxidative stress could be beneficial for children with ASD. Many antioxidants can also help improve mitochondrial function (31), suggesting that clinical improvements with antioxidants may occur through a reduction of oxidative stress and/or an improvement in mitochondrial function.
These studies suggest that treatments that address oxidative stress may improve core and associated symptoms of ASD. Furthermore, these treatments are generally regarded as safe with a low prevalence of adverse effects. Unfortunately many studies that have looked at antioxidants and treatments that potentially support the redox pathway did not use biomarkers to measure redox metabolism status in the participants or the effect of treatment on redox pathways. Including biomarkers in future studies could provide important information regarding which patients may respond to treatments that address redox metabolism and can help identify the most effective treatments. Since there are many treatments used to address oxidative stress and redox metabolism abnormalities in clinical practice and in research studies, the most effective treatments need to be carefully studied in DBPC studies to document their efficacy and effectiveness. Overall, the treatments discussed above have shown some promising results and deserve further study.
Tetrahydrobiopterin Metabolism
Tetrahydrobiopterin (BH4) is a naturally occurring molecule that is an essential cofactor for several critical metabolic pathways, including those responsible for the production of monoamine neurotransmitters, the breakdown of phenylalanine, and the production of nitric oxide (19). BH4 is readily oxidized by reactive species, leading it to be destroyed in the disorders where oxidative stress is prominent such as ASD (18). Abnormalities in several BH4 related metabolic pathways or in the products of these pathways have been noted in some individuals with ASD, and the cerebrospinal fluid concentration of BH4 has been reported to be depressed in some individuals with ASD (19). Clinical trials conducted over the past 25 years have reported encouraging results using sapropterin, a synthetic form of BH4, to treat children with ASD (19). Three controlled (109–111) and several open-label trials have documented improvements in communication, cognitive ability, adaptability, social abilities, and verbal expression with sapropterin treatment in ASD, especially in children younger than 5 years of age and in those who are relatively higher functioning at the beginning of the trial (19).
Frye has shown that the ratio of serum citrulline-to-methionine is related to the BH4 concentration in the cerebrospinal fluid, suggesting that abnormalities in both oxidative stress and nitric oxide metabolism may be related to central BH4 deficiency (18). More recently, Frye et al. demonstrated, in an open-label study, that sapropterin treatment improves redox metabolism and fundamentally alters BH4 metabolism in children with ASD. Interestingly, serum biomarkers of nitric oxide metabolism were found to predict response to sapropterin treatment in children with ASD (20), thereby suggesting that the therapeutic effect of BH4 supplementation may be specific to its effect on nitric oxide metabolism.
The potential positive effects on nitric oxide metabolism by BH4 supplementation could be significant for several reasons. The literature supports an association between ASD and abnormalities in nitric oxide metabolism. Indeed studies have documented alterations in nitric oxide synthase genes in children with ASD (112, 113). In the context of low BH4 concentrations, nitric oxide synthase produces peroxynitrite, an unstable reactive nitrogen species that can result in oxidative cellular damage. Indeed, nitrotyrosine, a biomarker of reactive nitrogen species, has been shown to be increased in multiple tissues in children with ASD, including the brain (22, 23, 107, 114, 115). Thus, BH4 supplementation could help stabilize nitric oxide synthase as well as act as an antioxidant and improve monoamine neurotransmitter production. Further DBPC studies using biomarkers of metabolic pathways related to BH4 metabolism will be needed to determine which children with ASD will most benefit from formulations of BH4 supplementation like sapropterin.
Potential Adverse Effects
Although many of the treatments discussed within this manuscript are considered safe and are generally well-tolerated, it is important to understand that these treatments are not without potential adverse effects. In general, these treatments are without serious adverse effects but some children may not tolerate all treatments well. Systematic and controlled studies are best at providing data on adverse effects, so the true adverse effects of the supplements discussed will only be based on the limited treatments that have been studied in such a fashion. It is also important to understand that because of the complicated nature of the effects of these treatments, they should only be used under the care of a medical professional with appropriate expertise and experience.
Controlled studies for treatments that address mitochondrial disorders include l-carnitine and a multivitamin with various mitochondrial supplements. In one small DBPC study, there were no significant adverse events reported in the 16 children treated with l-carnitine (59) while a second small DBPC trial reported no differences between the adverse effects reported by the treatment and placebo groups; notably, more patients in the placebo group withdrew from the study because of adverse effects (58). Thus, there is no data to suggest that l-carnitine has any significant adverse effects. In the large DBPC multivitamin study, about equal numbers of children in the treatment and placebo groups withdrew from the study because of behavior or gastrointestinal issues (65). In another small DBPC study, the investigators noted that two children began to have nausea and emesis when they started receiving the treatment at nighttime on an empty stomach (64). This adverse effect resolved when the timing of the treatment was adjusted. Thus, with proper dosing of this multivitamin, it appears rather safe and well-tolerated.
Controlled studies for folate pathway abnormalities only include folinic acid. In a medium-sized, open-label controlled study, 44 children with ASD and the FRα autoantibody were treated with high-dose folinic acid (2 mg/kg/day in two divided doses; maximum 50 mg/day) and four children discontinued the treatment because of an adverse effect (17). Of the four children who discontinued the treatment, three children, all being concurrently treated with risperidone, demonstrated increased irritability soon after starting the high-dose folinic acid while the other child experienced increased insomnia and gastroesophageal reflux after 6 weeks of treatment. Since there was no placebo in this study, the significance of these adverse effects is difficult to determine. For example, it is not clear whether this was related to concurrent risperidone treatment or was related to a baseline high irritability resulting in the needed for risperidone. All other participants completed the trial without significant adverse effects. Due to the timing of the adverse events in the children on risperidone in this trial, to be safe, the authors suggested caution when using folinic acid in children already on antipsychotic medications.
Clinical studies for treatments that could address redox metabolism include N-acetyl-l-cysteine, methylcobalamin, methylcobalamin combined with oral folinic acid and a multivitamin (as previous mentioned). One small open-label study that provided 25–30 μg/kg/day (1500 μg/day maximum) of methylcobalamin to 13 patients found no adverse effects (68) while a medium-sized, open-label trial that provided 75 μg/kg subcutaneously injected methylcobalamin given every 3 days along with twice daily oral low-dose (800 μg/day) folinic acid to 44 children noted some mild adverse effects (69, 70). Four children discontinued the treatment, two because their parents were uncomfortable given injections and two because of hyperactivity and reduced sleep. The most common adverse effect in the participants that remained in the study was hyperactivity, which resolved with a decrease in the folinic acid to 400 μg/day. Lastly, two medium sized, DBPC studies examined N-acetyl-l-cysteine, one as a primary treatment and another as an add-on to risperidone. The trial that used N-acetyl-l-cysteine as a primary treatment noted no significant differences in adverse events between the treatment and placebo groups, although both groups demonstrated a high rate of gastrointestinal symptoms and one participant in the active treatment phase required termination due to increased agitation (72). In the add-on study, one patient in the active treatment group withdrew due to severe sedation (73). In this latter study, adverse effects were not compared statistically between groups, but most adverse effects were mild and had a low prevalence. Such adverse effects included constipation, increased appetite, fatigue, nervousness, and daytime drowsiness. Lastly, a small DPBC study using vitamin C did not report any adverse effects from the treatment (67). Thus, there are several relatively safe and well-tolerated treatments for addressing abnormal redox metabolism, but there does appear to be a low rate of adverse effects, reinforcing the notion that a medical professional should guide treatment.
Three DBPC studies, one small (110), one medium (111), and one medium-to-large (109) sized, were conducted using sapropterin as a treatment for ASD. None of these studies have reported a higher prevalence of adverse effects in the treatment group as compared to the placebo group and none of these studies attributed any dropouts to the treatment. Thus, sapropterin appears to be a well-tolerated treatment.
Discussion
One advantage of the treatments outlined above is that the physiological mechanisms that they address are known and biomarkers are available to identify children who may respond to these treatments. Preliminary studies suggest that there are a substantial number of ASD children with these metabolic abnormalities. For example, mitochondrial abnormalities may be seen in 5–80% of children with ASD (31, 43–45, 53, 54) and FRα autoantibodies may be found in 47% (103) to 75% (17) of children with ASD. Clearly, further studies will be required to clarify the percentage of these subgroups.
Further large-scale, multicenter DBPC clinical trials are needed for these promising treatments in order to document the efficacy and define the subgroups that best respond to these treatments. As more treatable disorders are documented and as data accumulates to demonstrate the efficacy of treatments for these disorders, clinical algorithms to approach the work-up for a child with ASD need to be developed by a consensus of experts. Indeed, developing guidelines will be the next step for applying many of these scientific findings. Clearly many children with ASD may be able to benefit from such treatments, which are focused on improving dysfunctional physiology. Given the fact that no approved medical treatment exists which addresses the underlying pathophysiology or core symptoms of ASD, these treatments could make a substantial difference in the lives of children with ASD and their families. With the high prevalence of ASD, treatments that successfully treat even only a fraction of children affected with ASD would translate into substantial benefits for millions of individuals with ASD and their families. In summary, it appears that many of these treatments may provide benefit for a substantial proportion of children with ASD.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. Washington, DC: American Psychiatric Association (1994).
2. Volkmar FR, McPartland JC. From Kanner to DSM-5: autism as an evolving diagnostic concept. Annu Rev Clin Psychol (2013) 10:193–212. doi: 10.1146/annurev-clinpsy-032813-153710
3. Developmental Disabilities Monitoring Network Surveillance 2010 Year Principal Investigators; Centers for Disease Control and Prevention. Prevalence of autism spectrum disorder among children aged 8 years – autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill Summ (2014) 63:1–21.
4. Autism and Developmental Disabilities Monitoring Network Surveillance Year 2002 Principal Investigators; Centers for Disease Control and Prevention. Prevalence of autism spectrum disorders – autism and developmental disabilities monitoring network, 14 sites, United States, 2002. MMWR Surveill Summ (2007) 56:12–28.
5. Reichow B, Barton EE, Boyd BA, Hume K. Early intensive behavioral intervention (EIBI) for young children with autism spectrum disorders (ASD). Cochrane Database Syst Rev (2012) 10:CD009260. doi:10.1002/14651858.CD009260.pub2
6. Correll CU, Manu P, Olshanskiy V, Napolitano B, Kane JM, Malhotra AK. Cardiometabolic risk of second-generation antipsychotic medications during first-time use in children and adolescents. JAMA (2009) 302:1765–73. doi:10.1001/jama.2009.1549
7. Bobo WV, Cooper WO, Stein CM, Olfson M, Graham D, Daugherty J, et al. Antipsychotics and the risk of type 2 diabetes mellitus in children and youth. JAMA Psychiatry (2013) 70:1067–75. doi:10.1001/jamapsychiatry.2013.2053
8. Boon-Yasidhi V, Jearnarongrit P, Tulayapichitchock P, Tarugsa J. Adverse effects of risperidone in children with autism spectrum disorders in a naturalistic clinical setting at Siriraj Hospital, Thailand. Psychiatry J (2014) 2014:136158. doi:10.1155/2014/136158
9. Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry (2012) 17:389–401. doi:10.1038/mp.2011.165
10. Schaefer GB, Mendelsohn NJ. Genetics evaluation for the etiologic diagnosis of autism spectrum disorders. Genet Med (2008) 10:4–12. doi:10.1097/GIM.0b013e31815efdd7
11. Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature (2012) 485:242–5. doi:10.1038/nature11011
12. Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature (2012) 485:237–41. doi:10.1038/nature10945
13. Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry (2011) 68:1095–102. doi:10.1001/archgenpsychiatry.2011.76
14. Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. JAMA (2014) 311:1770–7. doi:10.1001/jama.2014.4144
15. Rossignol DA, Genuis SJ, Frye RE. Environmental toxicants and autism spectrum disorders: a systematic review. Transl Psychiatry (2014) 4:e360. doi:10.1038/tp.2014.4
16. Frye RE, James SJ. Metabolic pathology of autism in relation to redox metabolism. Biomark Med (2014) 8:321–30. doi:10.2217/bmm.13.158
17. Frye RE, Sequeira JM, Quadros EV, James SJ, Rossignol DA. Cerebral folate receptor autoantibodies in autism spectrum disorder. Mol Psychiatry (2013) 18:369–81. doi:10.1038/mp.2011.175
18. Frye RE. Central tetrahydrobiopterin concentration in neurodevelopmental disorders. Front Neurosci (2010) 4:52. doi:10.3389/fnins.2010.00052
19. Frye RE, Huffman LC, Elliott GR. Tetrahydrobiopterin as a novel therapeutic intervention for autism. Neurotherapeutics (2010) 7:241–9. doi:10.1016/j.nurt.2010.05.004
20. Frye RE, Delatorre R, Taylor HB, Slattery J, Melnyk S, Chowdhury N, et al. Metabolic effects of sapropterin treatment in autism spectrum disorder: a preliminary study. Transl Psychiatry (2013) 3:e237. doi:10.1038/tp.2013.14
21. Evangeliou A, Vlachonikolis I, Mihailidou H, Spilioti M, Skarpalezou A, Makaronas N, et al. Application of a ketogenic diet in children with autistic behavior: pilot study. J Child Neurol (2003) 18:113–8. doi:10.1177/08830738030180020501
22. James SJ, Melnyk S, Jernigan S, Cleves MA, Halsted CH, Wong DH, et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. Am J Med Genet B Neuropsychiatr Genet (2006) 141B:947–56. doi:10.1002/ajmg.b.30366
23. Rose S, Melnyk S, Pavliv O, Bai S, Nick TG, Frye RE, et al. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl Psychiatry (2012) 2:e134. doi:10.1038/tp.2012.61
24. Tierney E, Bukelis I, Thompson RE, Ahmed K, Aneja A, Kratz L, et al. Abnormalities of cholesterol metabolism in autism spectrum disorders. Am J Med Genet B Neuropsychiatr Genet (2006) 141B:666–8. doi:10.1002/ajmg.b.30368
25. Gargus JJ, Lerner MA. Familial autism with primary carnitine deficiency, sudden death, hypotonia and hypochromic anemia. Am J Human Gen (1997) 61:A98.
26. Filipek PA, Juranek J, Nguyen MT, Cummings C, Gargus JJ. Relative carnitine deficiency in autism. J Autism Dev Disord (2004) 34:615–23. doi:10.1007/s10803-004-5283-1
27. Celestino-Soper PB, Violante S, Crawford EL, Luo R, Lionel AC, Delaby E, et al. A common X-linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc Natl Acad Sci U S A (2012) 109:7974–81. doi:10.1073/pnas.1120210109
28. Frye RE, Melnyk S, Macfabe DF. Unique acyl-carnitine profiles are potential biomarkers for acquired mitochondrial disease in autism spectrum disorder. Transl Psychiatry (2013) 3:e220. doi:10.1038/tp.2012.143
29. Novarino G, El-Fishawy P, Kayserili H, Meguid NA, Scott EM, Schroth J, et al. Mutations in BCKD-kinase lead to a potentially treatable form of autism with epilepsy. Science (2012) 338:394–7. doi:10.1126/science.1224631
30. Rossignol DA, Frye RE. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front Physiol (2014) 5:150. doi:10.3389/fphys.2014.00150
31. Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry (2012) 17:290–314. doi:10.1038/mp.2010.136
32. Condie J, Goldstein J, Wainwright MS. Acquired microcephaly, regression of milestones, mitochondrial dysfunction, and episodic rigidity in a 46,XY male with a de novo MECP2 gene mutation. J Child Neurol (2010) 25:633–6. doi:10.1177/0883073809342004
33. Gibson JH, Slobedman B, Kaipananickal H, Williamson SL, Minchenko D, El-Osta A, et al. Downstream targets of methyl CpG binding protein 2 and their abnormal expression in the frontal cortex of the human Rett syndrome brain. BMC Neurosci (2010) 11:53. doi:10.1186/1471-2202-11-53
34. Grosser E, Hirt U, Janc OA, Menzfeld C, Fischer M, Kempkes B, et al. Oxidative burden and mitochondrial dysfunction in a mouse model of Rett syndrome. Neurobiol Dis (2012) 48:102–14. doi:10.1016/j.nbd.2012.06.007
35. Napoli E, Ross-Inta C, Wong S, Hung C, Fujisawa Y, Sakaguchi D, et al. Mitochondrial dysfunction in Pten haplo-insufficient mice with social deficits and repetitive behavior: interplay between Pten and p53. PLoS One (2012) 7:e42504. doi:10.1371/journal.pone.0042504
36. Frye RE. Mitochondrial disease in 22q13 duplication syndrome. J Child Neurol (2012) 27:942–9. doi:10.1177/0883073811429858
37. Filipek PA, Juranek J, Smith M, Mays LZ, Ramos ER, Bocian M, et al. Mitochondrial dysfunction in autistic patients with 15q inverted duplication. Ann Neurol (2003) 53:801–4. doi:10.1002/ana.10596
38. Frye RE. 15q11.2-13 Duplication, mitochondrial dysfunction, and developmental disorders. J Child Neurol (2009) 24:1316–20. doi:10.1177/0883073809333531
39. Su H, Fan W, Coskun PE, Vesa J, Gold JA, Jiang YH, et al. Mitochondrial dysfunction in CA1 hippocampal neurons of the UBE3A deficient mouse model for Angelman syndrome. Neurosci Lett (2011) 487:129–33. doi:10.1016/j.neulet.2009.06.079
40. Schuelke M, Krude H, Finckh B, Mayatepek E, Janssen A, Schmelz M, et al. Septo-optic dysplasia associated with a new mitochondrial cytochrome b mutation. Ann Neurol (2002) 51:388–92. doi:10.1002/ana.10151
41. Pallardo FV, Lloret A, Lebel M, D’Ischia M, Cogger VC, Le Couteur DG, et al. Mitochondrial dysfunction in some oxidative stress-related genetic diseases: ataxia-Telangiectasia, Down syndrome, Fanconi anaemia and Werner syndrome. Biogerontology (2010) 11:401–19. doi:10.1007/s10522-010-9269-4
42. Pagano G, Castello G. Oxidative stress and mitochondrial dysfunction in Down syndrome. Adv Exp Med Biol (2012) 724:291–9. doi:10.1007/978-1-4614-0653-2_22
43. Frye RE. Biomarker of abnormal energy metabolism in children with autism spectrum disorder. N A J Med Sci (2012) 5:141–7.
44. Giulivi C, Zhang YF, Omanska-Klusek A, Ross-Inta C, Wong S, Hertz-Picciotto I, et al. Mitochondrial dysfunction in autism. JAMA (2010) 304:2389–96. doi:10.1001/jama.2010.1706
45. Napoli E, Wong S, Hertz-Picciotto I, Giulivi C. Deficits in bioenergetics and impaired immune response in granulocytes from children with autism. Pediatrics (2014) 133:e1405–10. doi:10.1542/peds.2013-1545
46. Chauhan A, Gu F, Essa MM, Wegiel J, Kaur K, Brown WT, et al. Brain region-specific deficit in mitochondrial electron transport chain complexes in children with autism. J Neurochem (2011) 117:209–20. doi:10.1111/j.1471-4159.2011.07189.x
47. Anitha A, Nakamura K, Thanseem I, Yamada K, Iwayama Y, Toyota T, et al. Brain region-specific altered expression and association of mitochondria-related genes in autism. Mol Autism (2012) 3:12. doi:10.1186/2040-2392-3-12
48. Anitha A, Nakamura K, Thanseem I, Matsuzaki H, Miyachi T, Tsujii M, et al. Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain Pathol (2013) 23:294–302. doi:10.1111/bpa.12002
49. Tang G, Gutierrez Rios P, Kuo SH, Akman HO, Rosoklija G, Tanji K, et al. Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol Dis (2013) 54:349–61. doi:10.1016/j.nbd.2013.01.006
50. Kriaucionis S, Paterson A, Curtis J, Guy J, Macleod N, Bird A. Gene expression analysis exposes mitochondrial abnormalities in a mouse model of Rett syndrome. Mol Cell Biol (2006) 26:5033–42. doi:10.1128/MCB.01665-05
51. Graf WD, Marin-Garcia J, Gao HG, Pizzo S, Naviaux RK, Markusic D, et al. Autism associated with the mitochondrial DNA G8363A transfer RNA(Lys) mutation. J Child Neurol (2000) 15:357–61. doi:10.1177/088307380001500601
52. Frye RE, Naviaux RK. Autistic disorder with complex IV overactivity: a new mitochondrial syndrome. J Pediatr Neurol (2011) 9:427–34. doi:10.3233/JPN-2001-0507
53. Rose S, Frye RE, Slattery J, Wynne R, Tippett M, Melnyk S, et al. Oxidative stress induces mitochondrial dysfunction in a subset of autistic lymphoblastoid cell lines. Transl Psychiatry (2014) 4:e377. doi:10.1038/tp.2014.15
54. Rose S, Frye RE, Slattery J, Wynne R, Tippett M, Pavliv O, et al. Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS One (2014) 9:e85436. doi:10.1371/journal.pone.0085436
55. Minshew NJ, Goldstein G, Dombrowski SM, Panchalingam K, Pettegrew JW. A preliminary 31P MRS study of autism: evidence for under synthesis and increased degradation of brain membranes. Biol Psychiatry (1993) 33:762–73. doi:10.1016/0006-3223(93)90017-8
56. Mostafa GA, El-Gamal HA, El-Wakkad ASE, El-Shorbagy OE, Hamza MM. Polyunsaturated fatty acids, carnitine and lactate as biological markers of brain energy in autistic children. Int J Child Neuropsychiatry (2005) 2:179–88.
57. Frye RE, Delatorre R, Taylor H, Slattery J, Melnyk S, Chowdhury N, et al. Redox metabolism abnormalities in autistic children associated with mitochondrial disease. Transl Psychiatry (2013) 3:e273. doi:10.1038/tp.2013.51
58. Geier DA, Kern JK, Davis G, King PG, Adams JB, Young JL, et al. A prospective double-blind, randomized clinical trial of levocarnitine to treat autism spectrum disorders. Med Sci Monit (2011) 17:I15–23. doi:10.12659/MSM.881792
59. Fahmy SF, El-Hamamsy MH, Zaki OK, Badary OA. L-carnitine supplementation improves the behavioral symptoms in autistic children. Res Autism Spectr Disord (2013) 7:159–66. doi:10.1016/j.rasd.2012.07.006
60. Poling JS, Frye RE, Shoffner J, Zimmerman AW. Developmental regression and mitochondrial dysfunction in a child with autism. J Child Neurol (2006) 21:170–2. doi:10.1177/08830738060210021401
61. Gargus JJ, Imtiaz F. Mitochondrial energy-deficient endophenotype in autism. Am J Biochem Biotech (2008) 4:198–207. doi:10.3844/ajbbsp.2008.198.207
62. Pastural E, Ritchie S, Lu Y, Jin W, Kavianpour A, Khine Su-Myat K, et al. Novel plasma phospholipid biomarkers of autism: mitochondrial dysfunction as a putative causative mechanism. Prostaglandins Leukot Essent Fatty Acids (2009) 81:253–64. doi:10.1016/j.plefa.2009.06.003
63. Ezugha H, Goldenthal M, Valencia I, Anderson CE, Legido A, Marks H. 5q14.3 Deletion manifesting as mitochondrial disease and autism: case report. J Child Neurol (2010) 25:1232–5. doi:10.1177/0883073809361165
64. Adams JB, Holloway C. Pilot study of a moderate dose multivitamin/mineral supplement for children with autistic spectrum disorder. J Altern Complement Med (2004) 10:1033–9. doi:10.1089/acm.2004.10.1033
65. Adams JB, Audhya T, McDonough-Means S, Rubin RA, Quig D, Geis E, et al. Effect of a vitamin/mineral supplement on children and adults with autism. BMC Pediatr (2011) 11:111. doi:10.1186/1471-2431-11-111
66. Rossignol DA. Novel and emerging treatments for autism spectrum disorders: a systematic review. Ann Clin Psychiatry (2009) 21:213–36.
67. Dolske MC, Spollen J, McKay S, Lancashire E, Tolbert L. A preliminary trial of ascorbic acid as supplemental therapy for autism. Prog Neuropsychopharmacol Biol Psychiatry (1993) 17:765–74. doi:10.1016/0278-5846(93)90058-Z
68. Nakano K, Noda N, Tachikawa E, Urano MAN, Takazawa M, Nakayama T, et al. A preliminary study of methylcobalamin therapy in autism. J Tokyo Womens Med Univ (2005) 75:64–9. doi:10.1016/j.chc.2013.03.002
69. James SJ, Melnyk S, Fuchs G, Reid T, Jernigan S, Pavliv O, et al. Efficacy of methylcobalamin and folinic acid treatment on glutathione redox status in children with autism. Am J Clin Nutr (2009) 89:425–30. doi:10.3945/ajcn.2008.26615
70. Frye RE, Melnyk S, Fuchs G, Reid T, Jernigan S, Pavliv O, et al. Effectiveness of methylcobalamin and folinic acid treatment on adaptive behavior in children with autistic disorder is related to glutathione redox status. Autism Res Treat (2013) 2013:609705. doi:10.1155/2013/609705
71. Ghanizadeh A, Derakhshan N. N-acetylcysteine for treatment of autism, a case report. J Res Med Sci (2012) 17:985–7.
72. Hardan AY, Fung LK, Libove RA, Obukhanych TV, Nair S, Herzenberg LA, et al. A randomized controlled pilot trial of oral N-acetylcysteine in children with autism. Biol Psychiatry (2012) 71:956–61. doi:10.1016/j.biopsych.2012.01.014
73. Ghanizadeh A, Moghimi-Sarani E. A randomized double blind placebo controlled clinical trial of N-acetylcysteine added to risperidone for treating autistic disorders. BMC Psychiatry (2013) 13:196. doi:10.1186/1471-244X-13-196
74. Gvozdjakova A, Kucharska J, Ostatnikova D, Babinska K, Nakladal D, Crane FL. Ubiquinol improves symptoms in children with autism. Oxid Med Cell Longev (2014) 2014:798957. doi:10.1155/2014/798957
75. Chez MG, Buchanan CP, Aimonovitch MC, Becker M, Schaefer K, Black C, et al. Double-blind, placebo-controlled study of L-carnosine supplementation in children with autistic spectrum disorders. J Child Neurol (2002) 17:833–7. doi:10.1177/08830738020170111501
76. Avula S, Parikh S, Demarest S, Kurz J, Gropman A. Treatment of mitochondrial disorders. Curr Treat Options Neurol (2014) 16:292. doi:10.1007/s11940-014-0292-7
77. Boris M, Goldblatt A, Galanko J, James SJ. Association of MTHFR gene variants with autism. J Am Phys Surg (2004) 9:106–8.
78. Goin-Kochel RP, Porter AE, Peters SU, Shinawi M, Sahoo T, Beaudet AL. The MTHFR 677C– >T polymorphism and behaviors in children with autism: exploratory genotype-phenotype correlations. Autism Res (2009) 2:98–108. doi:10.1002/aur.70
79. Mohammad NS, Jain JM, Chintakindi KP, Singh RP, Naik U, Akella RR. Aberrations in folate metabolic pathway and altered susceptibility to autism. Psychiatr Genet (2009) 19:171–6. doi:10.1097/YPG.0b013e32832cebd2
80. Pasca SP, Dronca E, Kaucsar T, Craciun EC, Endreffy E, Ferencz BK, et al. One carbon metabolism disturbances and the C677T MTHFR gene polymorphism in children with autism spectrum disorders. J Cell Mol Med (2009) 13:4229–38. doi:10.1111/j.1582-4934.2008.00463.x
81. Liu X, Solehdin F, Cohen IL, Gonzalez MG, Jenkins EC, Lewis ME, et al. Population- and family-based studies associate the MTHFR gene with idiopathic autism in simplex families. J Autism Dev Disord (2011) 41:938–44. doi:10.1007/s10803-010-1120-x
82. Frustaci A, Neri M, Cesario A, Adams JB, Domenici E, Dalla Bernardina B, et al. Oxidative stress-related biomarkers in autism: systematic review and meta-analyses. Free Radic Biol Med (2012) 52:2128–41. doi:10.1016/j.freeradbiomed.2012.03.011
83. Guo T, Chen H, Liu B, Ji W, Yang C. Methylenetetrahydrofolate reductase polymorphisms C677T and risk of autism in the Chinese Han population. Genet Test Mol Biomarkers (2012) 16:968–73. doi:10.1089/gtmb.2012.0091
84. Schmidt RJ, Tancredi DJ, Ozonoff S, Hansen RL, Hartiala J, Allayee H, et al. Maternal periconceptional folic acid intake and risk of autism spectrum disorders and developmental delay in the CHARGE (childhood autism risks from genetics and environment) case-control study. Am J Clin Nutr (2012) 96:80–9. doi:10.3945/ajcn.110.004416
85. Pu D, Shen Y, Wu J. Association between MTHFR gene polymorphisms and the risk of autism spectrum disorders: a meta-analysis. Autism Res (2013) 6:384–92. doi:10.1002/aur.1300
86. Adams M, Lucock M, Stuart J, Fardell S, Baker K, Ng X. Preliminary evidence for involvement of the folate gene polymorphism 19bp deletion-DHFR in occurrence of autism. Neurosci Lett (2007) 422:24–9. doi:10.1016/j.neulet.2007.05.025
87. Wollack JB, Makori B, Ahlawat S, Koneru R, Picinich SC, Smith A, et al. Characterization of folate uptake by choroid plexus epithelial cells in a rat primary culture model. J Neurochem (2008) 104:1494–503. doi:10.1111/j.1471-4159.2007.05095.x
88. Ramaekers VT, Blau N. Cerebral folate deficiency. Dev Med Child Neurol (2004) 46:843–51. doi:10.1111/j.1469-8749.2004.tb00451.x
89. Moretti P, Sahoo T, Hyland K, Bottiglieri T, Peters S, Del Gaudio D, et al. Cerebral folate deficiency with developmental delay, autism, and response to folinic acid. Neurology (2005) 64:1088–90. doi:10.1212/01.WNL.0000154641.08211.B7
90. Ramaekers VT, Rothenberg SP, Sequeira JM, Opladen T, Blau N, Quadros EV, et al. Autoantibodies to folate receptors in the cerebral folate deficiency syndrome. N Engl J Med (2005) 352:1985–91. doi:10.1056/NEJMoa043160
91. Ramaekers VT, Blau N, Sequeira JM, Nassogne MC, Quadros EV. Folate receptor autoimmunity and cerebral folate deficiency in low-functioning autism with neurological deficits. Neuropediatrics (2007) 38:276–81. doi:10.1055/s-2008-1065354
92. Moretti P, Peters SU, Del Gaudio D, Sahoo T, Hyland K, Bottiglieri T, et al. Brief report: autistic symptoms, developmental regression, mental retardation, epilepsy, and dyskinesias in CNS folate deficiency. J Autism Dev Disord (2008) 38:1170–7. doi:10.1007/s10803-007-0492-z
93. Ramaekers VT, Sequeira JM, Blau N, Quadros EV. A milk-free diet downregulates folate receptor autoimmunity in cerebral folate deficiency syndrome. Dev Med Child Neurol (2008) 50:346–52. doi:10.1111/j.1469-8749.2008.02053.x
94. Shoffner J, Hyams L, Langley GN, Cossette S, Mylacraine L, Dale J, et al. Fever plus mitochondrial disease could be risk factors for autistic regression. J Child Neurol (2010) 25:429–34. doi:10.1177/0883073809342128
95. Ramaekers VT, Hansen SI, Holm J, Opladen T, Senderek J, Hausler M, et al. Reduced folate transport to the CNS in female Rett patients. Neurology (2003) 61:506–15. doi:10.1212/01.WNL.0000078939.64774.1B
96. Ramaekers VT, Sequeira JM, Artuch R, Blau N, Temudo T, Ormazabal A, et al. Folate receptor autoantibodies and spinal fluid 5-methyltetrahydrofolate deficiency in Rett syndrome. Neuropediatrics (2007) 38:179–83. doi:10.1055/s-2007-991148
97. Perez-Duenas B, Ormazabal A, Toma C, Torrico B, Cormand B, Serrano M, et al. Cerebral folate deficiency syndromes in childhood: clinical, analytical, and etiologic aspects. Arch Neurol (2011) 68:615–21. doi:10.1001/archneurol.2011.80
98. Allen RJ, Dimauro S, Coulter DL, Papadimitriou A, Rothenberg SP. Kearns-Sayre syndrome with reduced plasma and cerebrospinal fluid folate. Ann Neurol (1983) 13:679–82. doi:10.1002/ana.410130620
99. Pineda M, Ormazabal A, Lopez-Gallardo E, Nascimento A, Solano A, Herrero MD, et al. Cerebral folate deficiency and leukoencephalopathy caused by a mitochondrial DNA deletion. Ann Neurol (2006) 59:394–8. doi:10.1002/ana.20746
100. Garcia-Cazorla A, Quadros EV, Nascimento A, Garcia-Silva MT, Briones P, Montoya J, et al. Mitochondrial diseases associated with cerebral folate deficiency. Neurology (2008) 70:1360–2. doi:10.1212/01.wnl.0000309223.98616.e4
101. Hasselmann O, Blau N, Ramaekers VT, Quadros EV, Sequeira JM, Weissert M. Cerebral folate deficiency and CNS inflammatory markers in Alpers disease. Mol Genet Metab (2010) 99:58–61. doi:10.1016/j.ymgme.2009.08.005
102. Frye RE, Rossignol DA. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatr Res (2011) 69:41R–7R. doi:10.1203/PDR.0b013e318212f16b
103. Ramaekers VT, Quadros EV, Sequeira JM. Role of folate receptor autoantibodies in infantile autism. Mol Psychiatry (2013) 18:270–1. doi:10.1038/mp.2012.22
104. Suren P, Roth C, Bresnahan M, Haugen M, Hornig M, Hirtz D, et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. JAMA (2013) 309:570–7. doi:10.1001/jama.2012.155925
105. James SJ, Cutler P, Melnyk S, Jernigan S, Janak L, Gaylor DW, et al. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am J Clin Nutr (2004) 80:1611–7.
106. Rose S, Melnyk S, Trusty TA, Pavliv O, Seidel L, Li J, et al. Intracellular and extracellular redox status and free radical generation in primary immune cells from children with autism. Autism Res Treat (2012) 2012:986519. doi:10.1155/2012/986519
107. Sajdel-Sulkowska EM, Xu M, McGinnis W, Koibuchi N. Brain region-specific changes in oxidative stress and neurotrophin levels in autism spectrum disorders (ASD). Cerebellum (2011) 10:43–8. doi:10.1007/s12311-010-0223-4
108. Adams JB, Audhya T, McDonough-Means S. Nutritional and metabolic status of children with autism vs. neuro-typical children, and the association with autism severity. Nutr Metab (2011) 8:34. doi:10.1186/1743-7075-8-34
109. Nareuse H, Hayash TI, Takesada M, Nakane A, Yamazaki K, Noguchi T, et al. Therapeutic effect of tetrahydrobiopterin in infantile autism. Proc Jpn Acad (1987) 63:231–3. doi:10.2183/pjab.63.231
110. Danfors T, von Knorring AL, Hartvig P, Langstrom B, Moulder R, Stromberg B, et al. Tetrahydrobiopterin in the treatment of children with autistic disorder: a double-blind placebo-controlled crossover study. J Clin Psychopharmacol (2005) 25:485–9. doi:10.1097/01.jcp.0000177667.35016.e9
111. Klaiman C, Huffman L, Masaki L, Elliott GR. Tetrahydrobiopterin as a treatment for autism spectrum disorders: a double-blind, placebo-controlled trial. J Child Adolesc Psychopharmacol (2013) 23:320–8. doi:10.1089/cap.2012.0127
112. Kim HW, Cho SC, Kim JW, Cho IH, Kim SA, Park M, et al. Family-based association study between NOS-I and -IIA polymorphisms and autism spectrum disorders in Korean trios. Am J Med Genet B Neuropsychiatr Genet (2009) 150B:300–6. doi:10.1002/ajmg.b.30798
113. Delorme R, Betancur C, Scheid I, Anckarsater H, Chaste P, Jamain S, et al. Mutation screening of NOS1AP gene in a large sample of psychiatric patients and controls. BMC Med Genet (2010) 11:108. doi:10.1186/1471-2350-11-108
114. Lakshmi Priya MD, Geetha A. A biochemical study on the level of proteins and their percentage of nitration in the hair and nail of autistic children. Clin Chim Acta (2011) 412:1036–42. doi:10.1016/j.cca.2011.02.021
Keywords: autism spectrum disorders, mitochondria, folate receptor alpha, folinic acid, folate metabolism, redox regulation, oxidative stress, tetrahydrobiopterin
Citation: Frye RE and Rossignol DA (2014) Treatments for biomedical abnormalities associated with autism spectrum disorder. Front. Pediatr. 2:66. doi: 10.3389/fped.2014.00066
Received: 09 May 2014; Accepted: 09 June 2014;
Published online: 27 June 2014.
Edited by:
Roberto Canitano, University Hospital of Siena, ItalyReviewed by:
Andrew Walter Zimmerman, Harvard Medical School, USARobert Hendren, University of California San Francisco, USA
Copyright: © 2014 Frye and Rossignol. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard Eugene Frye, Arkansas Children’s Hospital Research Institute, Slot 512-41B, 13 Children’s Way, Little Rock, AR 72202, USA e-mail:cmVmcnllQHVhbXMuZWR1