 Federica Piani1,2,3
Federica Piani1,2,3 Trenton Reinicke1
Trenton Reinicke1 Claudio Borghi3
Claudio Borghi3 Kalie L. Tommerdahl1,2
Kalie L. Tommerdahl1,2 Gabriel Cara-Fuentes1,4Richard J. Johnson1
Gabriel Cara-Fuentes1,4Richard J. Johnson1 Petter Bjornstad1,2*
Petter Bjornstad1,2*- 1Division of Renal Diseases and Hypertension, Department of Medicine, University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 2Section of Pediatric Endocrinology, Department of Pediatrics, Children's Hospital Colorado and University of Colorado Anschutz Medical Campus, Aurora, CO, United States
- 3Department of Medicine and Surgery Sciences, Alma Mater Studiorum–University of Bologna, Bologna, Italy
- 4Section of Pediatric Nephrology, Department of Pediatrics, Children's Hospital Colorado, Aurora, CO, United States
Diabetic kidney disease (DKD) is a common complication of type 1 and 2 diabetes and often presents during adolescence and young adulthood. Given the growing incidence of both type 1 and type 2 diabetes in children and adolescents, DKD represents a significant public health problem. Acute kidney injury (AKI) in youth with diabetes is strongly associated with risk of DKD development. This review will summarize the epidemiology and pathophysiology of AKI in children with diabetes, the relationship between AKI and DKD, and the potential therapeutic interventions. Finally, we will appraise the impact of the recent COVID-19 infection pandemic on AKI in children with diabetes.
Introduction
Type 1 diabetes (T1D) has consistently remained one of the most common chronic diseases affecting children and adolescents (1). In the past few decades, the incidence of youth-onset type 2 diabetes (T2D) has also progressively increased, likely due to rising rates of childhood obesity (2). Both types of diabetes are associated with long term complications that result in increased morbidity and mortality (1). Among these complications, diabetic kidney disease (DKD) is associated with the highest rates of excess mortality observed in young persons with diabetes (1). Indeed, DKD represents the leading cause of end-stage kidney disease (ESKD) and dialysis in the developed world (3). The pathophysiology of DKD is multifaceted and is characterized by progressive chronic kidney disease (CKD) (1, 4). In children and adolescents with diabetes, AKI can magnify the risk for CKD development and progression later in life (5–9).
Several mechanisms have been proposed to explain the accentuated risk of acute kidney injury (AKI) in youth with diabetes, including diabetic ketoacidosis (DKA), acute hyperglycemic events, and chronic poor glycemic control (10–12). Hyperglycemia has been shown to directly induce kidney inflammation and tubulopathy (12), while poor glycemic control can lead to polyuria with resultant volume contraction and hypovolemia, which is subsequently associated with the development of pre-renal AKI (11). In this review we seek to appraise the evolving mechanisms, risk-factors, and management strategies for diabetes-induced AKI in the pediatric population.
Epidemiology and Pathophysiology of AKI in Diabetes
Epidemiology and Definition of AKI in the Pediatric Population
AKI in youth represents a significant and growing challenge for clinicians, as AKI has been demonstrated in 3.9 out of every 1,000 pediatric hospitalizations at-risk in the U.S. (13), including up to 64% of hospitalizations for DKA in youth with T1D (14). AKI is currently defined by the Kidney Disease Improving Global Outcomes (KDIGO) consensus classification based on conventional serum creatinine and urine output (UO) criteria (15). Previous widely used classification criteria have also shown an excellent accuracy in screening for AKI in the pediatric population. The pediatric Risk, Injury, Failure, Loss and End-stage Kidney (pRIFLE) criteria, which include a decrease in estimated creatinine clearance (eCCl) per the Schwartz formula over 8 to 24 h and anuria for 12 h (15), and the Acute Kidney Injury Network (16) criteria, which include an increase in serum creatinine over 6 to 24 h and anuria for 12 h (15). The pRIFLE criteria have been shown to have high sensitivity in detecting AKI (16), and the AKIN criteria have demonstrated high specificity (15).
AKI is typically classified into three main categories: pre-renal, intrinsic/renal, and post-renal (17). The most common form of pediatric AKI is pre-renal, a usually reversible form of kidney dysfunction caused by kidney hypoperfusion (18, 19). In the setting of diabetes, the extracellular volume depletion leading to pre-renal AKI is commonly induced by glycosuria because of poorly controlled diabetes (14, 20). The combination of poor glycemic control with pre-renal AKI can eventually lead to intrinsic renal AKI, characterized by structural damage to the renal parenchyma and the occurrence of tubular necrosis (18, 21). Although DKD has historically been considered a glomerular disease, a growing body of evidence suggests that tubular-interstitial injury may be the first alteration in DKD (18, 21). AKI is also divided by severity into stage 1, 2, or 3 with different definitions according to the applied diagnostic criteria for AKI (e.g., pRIFLE AKI stage 1 is defined as a 25% decrease of estimated GFR (eGFR), stage 2 as a decrease of 50%, and stage 3 as a decrease of 75%) (15). Despite a growing body of literature evaluating the incidence and etiologies of AKI in adults, large epidemiologic studies involving pediatric populations with AKI, with or without diabetes, are lacking, as many studies are either limited to a single center or are focused on specific subpopulations (13, 18) (Table 1).
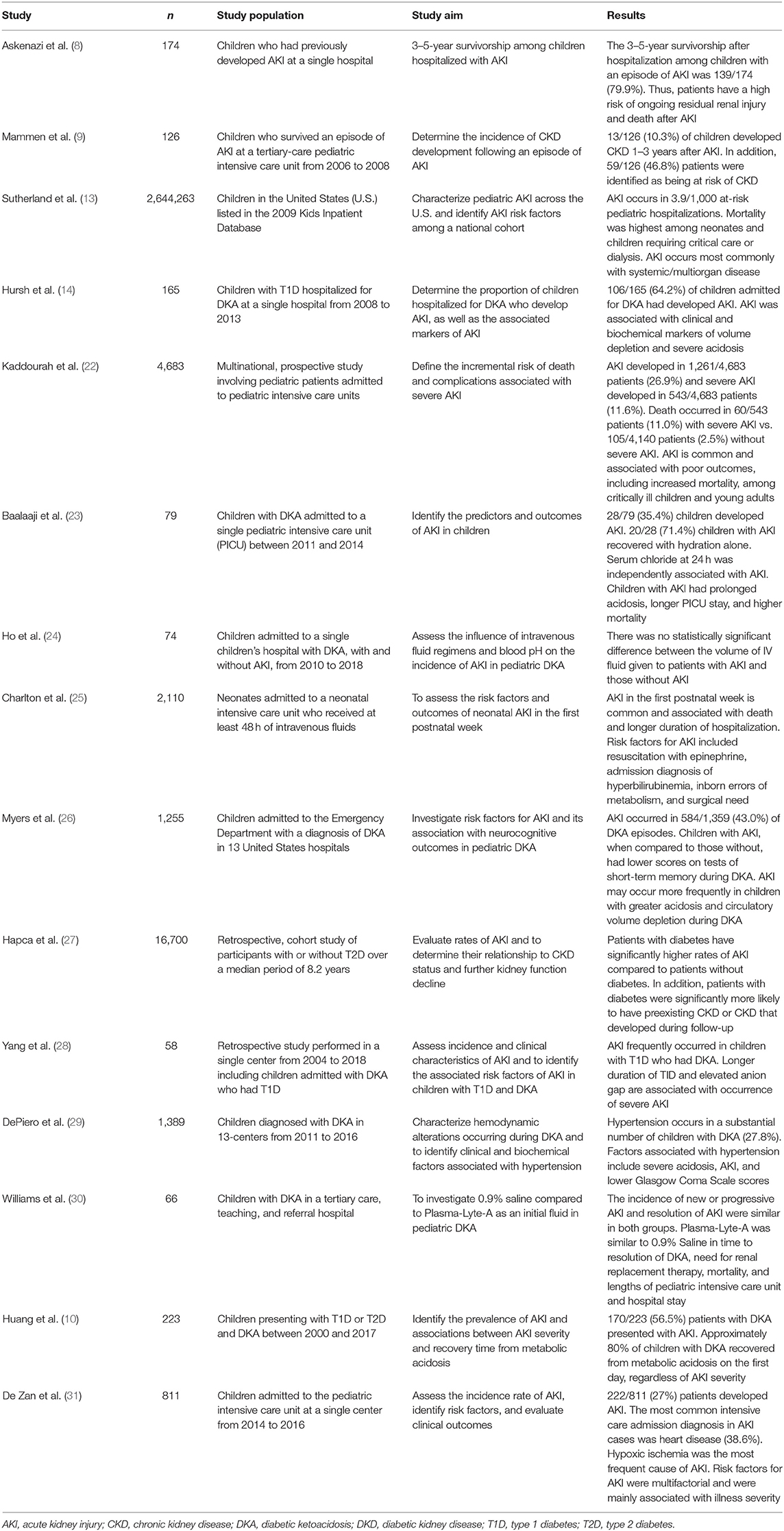
Table 1. Studies on AKI in pediatric populations with and without diabetes.
Markers of AKI in the Pediatric Population
The diagnosis of AKI, per the Kidney Disease Improving Global Outcomes (KDIGO) guidelines, is currently defined by factors including elevated serum creatinine concentration, decreased UO, and renal replacement therapy requirement (32, 33). UO is the oldest known indicator of AKI, yet it retains multiple theoretical advantages over biomarkers such as serum creatinine concentration due to a predefined cut-off value and rapid response to treatment. In contrast, serum creatinine concentration requires comparison to a baseline value which is often either unknown or estimated (34). However, the sole use of UO to diagnose AKI also has several constrains, notably the significant influence of hydration status and blood pressure, the dependence on hemodynamic stability, and the artificial effect of medications such as diuretics and vasopressors (34). Severe glycosuria from uncontrolled diabetes or use of SGLT2 inhibitors can also lead to prerenal or true AKI despite continued UO. Another issue includes the difficulty in accurately measuring UO, particularly in children without Foley catheters in place (34). UO is also not an infallible indicator of AKI as it has been shown that some individuals with severe AKI can still have a preserved UO (34). Serum creatinine concentration is also an imperfect indicator of AKI as it can be delayed and unreliable in the setting of concurrent infections, sepsis, malnutrition, and obesity, which can ultimately lead to an inaccurate diagnosis (34–36). Laboratory and clinical studies are currently targeting the identification of biomarkers that predict the development of early AKI. Some of the most promising AKI biomarkers include neutrophil gelatinase-associated lipocalin (NGAL), kidney injury molecule 1 (KIM-1), interleukin 18 (lL-18), liver-type fatty-acid-binding protein (L-FABP, also called FABP1), insulin-like-growth-factor-binding protein 7 (IGFBP7), and tissue inhibitor of metalloproteinase 2 (TIMP-2) (37, 38).
NGAL is a protease-resistant polypeptide of the lipocalin superfamily identified in human neutrophils and is expressed by the tubular epithelia of the kidneys in response to inflammation (39–41). NGAL is a promising marker for AKI because it is easily detected in urine or plasma as soon as 1 h following kidney damage and prior to changes in serum creatinine, and it correlates with the severity and duration of AKI (37). Additionally, urine NGAL has been extensively studied in pediatrics and has been shown to be an early and sensitive biomarker for both AKI and DKD (42).
KIM-1, a type-1 transmembrane protein expressed in the renal proximal tubular cells, is significantly upregulated following ischemia (43, 44). In a 2019 study by Assadi and Sharbaf urinary KIM-1 demonstrated the strongest performance for the early detection of AKI among critically ill children with circulatory collapse when compared to NGAL, IL-18, and sCr (45). However, while current evidence supports the use of KIM-1 as a promising new avenue for the detection of AKI, concentrations may be affected by a variety of additional factors including the type of assay, timing and clinical setting of the sample collection, and patient age (46). Similar to NGAL, KIM-1 has also been shown to be elevated in children with diabetes and higher concentrations may associated with the development of early DKD (47).
IL-18 is a cytokine in the IL-1 superfamily that is synthesized by monocytes, macrophages, and proximal tubular epithelial cells of the kidney. Urine IL-18 is currently a promising predictor of early AKI as concentrations increase rapidly after ischemic kidney injury, nearly 12 h before clinical AKI is diagnosed by other means (37). Yet, identification of additional biomarkers for AKI is still necessary as IL-18's accuracy in predicting AKI among children and adolescents is highest when combined with other biomarkers (37).
L-FABP is a cytoplasmic protein involved in intracellular lipid trafficking and endogenous cytoprotection against oxidative stress that is expressed in the proximal epithelial tubular cells of the kidneys (34, 48). Urine L-FABP has been shown to predict both AKI and adverse clinical outcomes in pediatrics, reflecting the negative impact of oxidative stress on the kidneys and the associated resultant proximal tubule cellular injury (38). However, urine L-FABP concentrations may be elevated by factors including obesity, insulin resistance, and high blood pressure, even without concurrent kidney injury (49).
IGFBP7 and TIMP-2 are cell-cycle arrest proteins expressed in higher concentrations from the renal tubular cells during cellular stress (50). Evaluations of the urinary excretion of TIMP-2 and IGFBP7 have been proven useful in the detection of AKI and the urinary concentration of the product of TIMP-2 and IGFBP7 [(TIMP-2) × (IGFBP7)] was the first urinary AKI biomarker approved by the United States Food and Drug Administration (51). Notably, this biomarker combination has also been validated in post-operative youth following cardiopulmonary bypass, demonstrating accuracy in predicting AKI 6 h after major surgery as well (38).
Other novel biomarkers are currently being investigated for the diagnosis and prognostic stratification of AKI in youth, including microRNAs, urinary low-molecular-weight proteins, and urinary tubular enzymes (37). The validation of these emerging biomarkers in large epidemiological studies is still necessary, but the biomarkers previously discussed hold significant clinical promise as supplements to our current tools for the identification of AKI in youth, namely serum creatinine concentration and UO.
Pathophysiologic Mechanisms of AKI in Youth With Diabetes
The pathophysiologic mechanisms leading to diabetes-induced kidney injury are complex and oftentimes multifactorial (Figure 1). Structural and functional changes in the endothelial cells of the vasculature and the epithelial cells of the kidney tubules have been postulated to promote the production of cytokines and chemokines, which induce inflammation, ischemic tubular epithelial and endothelial injury, and isolated proximal tubulopathy (19, 52, 53).
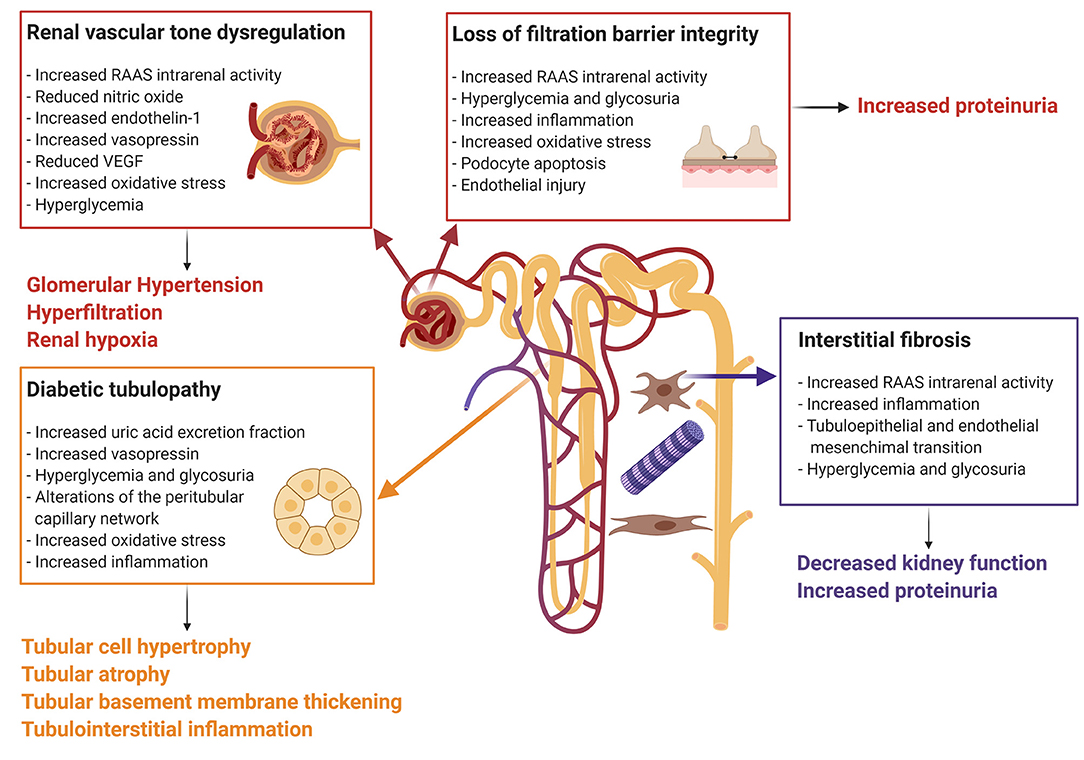
Figure 1. Mechanisms of diabetes-induced kidney injury. Every box represents one mechanism of diabetes-associated kidney injury with the respective causes. Connected to each box the clinical effect of the pathophysiologic pathway. The Figure was created with BioRender.com. RAAS, renin-angiotensin-aldosterone system; VEGF, vascular endothelial growth factor.
Risk factors for the development of AKI in children and adolescents with diabetes are numerous and include, but are not limited to, episodes of DKA as well as the presence of acute and chronic hyperglycemia. DKA is a severe risk factor for the development of AKI, particularly in youth with T1D, and is characterized by a combination of hyperglycemia, metabolic acidosis, and the production of ketone bodies (54, 55). DKA is currently the leading cause of hospitalization, morbidity, and mortality in youth with T1D (14, 56, 57) and in a study by Hursh et al. up to 64% of youth with T1D hospitalized for DKA developed AKI (14). This percentage increased to 85% if the individual required admission to the Intensive Care Unit (14). Severe hyperglycemia associated with DKA leads to osmotic diuresis, dehydration, and significant pre-renal AKI (54, 55).
Additionally, DKA has also been associated with early tubulopathy that may be caused by dysregulation of uric acid and vasopressin (58–62). Indeed, a recent study by Burckhardt et al. found that copeptin, a surrogate marker for vasopressin, was elevated in children with DKA (63). Elevated copeptin concentrations have also been associated with markers of tubular injury and an increased risk for the development of DKD (62, 64). We have previously demonstrated that adults with T1D and DKD had higher concentrations of copeptin compared to both healthy controls and adults with T1D without DKD (65). Furthermore, elevations in copeptin are more strongly correlated with intrarenal renin-angiotensin-aldosterone system (RAAS) activation in individuals with T1D compared to healthy controls (65). We have also demonstrated in a study of 169 adolescents with T1D that adolescents with T1D in the highest tertile of copeptin concentration had a significantly higher urinary albumin-to-creatinine ratio compared to those in the lowest tertile (66). Furthermore, several studies have shown that chronically elevated vasopressin can exacerbate kidney injury in animal models (67, 68).
Thus, we hypothesize that vasopressin dysregulation may increase the risk of AKI during an episode of DKA and may subsequently accentuate the risk for future DKD. Activation of the intrarenal RAAS has been shown to impact regulation of renal vascular tone and children and adolescents with T1D and T2D have demonstrated greater resistance indexes when compared to their healthy peers without diabetes (69–71). Additionally, nitric oxide (NO), endothelin-1 (ET-1), vasopressin, and vascular endothelial growth factor (VEGF) have also been shown to be impaired as regulators of kidney vascular tone in individuals with diabetes (58, 65, 72–74).
Dysregulation of kidney vascular endothelial tone secondary to poor glycemic control is thought to be primarily responsible for the development of glomerular hyperfiltration, a common early finding in young persons with diabetes (75, 76). Persistent glomerular hyperfiltration leads to the development of intraglomerular hypertension and subsequent glomerulosclerosis which results in a progressive impairment in kidney function and eventual DKD (75, 76). A dysregulation in baseline kidney vascular tone may accelerate the damage due to AKI, particularly in the setting of pre-renal AKI where the body relies on changes in kidney vascular resistance to maintain blood pressure (18, 19). Kidney hypoperfusion secondary to dehydration or shock also can be significantly worsened by the absence of an appropriate counterregulatory vascular response to maintain kidney blood flow (75, 76). Consequently, the incidence of AKI in the setting of sepsis has been found to be significantly higher in people with diabetes compared to people without diabetes (4, 77).
Hyperuricemia may also contribute to the development of diabetes-related acute and chronic kidney disease (60). High uric acid concentrations have been shown to induce both a crystal-mediated and crystal-independent nephropathy, with the additional development of tubulointerstitial fibrosis, proximal tubulopathy, and glomerular hypertension (61, 78). Elevations in serum uric acid concentrations have also been associated with the development of AKI and medications that lower serum uric acid concentrations have demonstrated a potential to reduce the incidence of AKI (79–82). However, it is important to note that elevated uric acid concentrations may be due to dehydration that may directly induce kidney injury. We have previously demonstrated that youth with T1D and hyperfiltration had a higher urinary fractional uric acid excretion compared to both healthy controls and peers with T1D who have normofiltration (81). Additionally, overweight and obese youth with T1D demonstrated a stronger negative correlation between serum uric acid concentrations and eGFR when compared to their normal weight peers with T1D (83). Thus, high serum uric acid concentrations exhibit a clear association with AKI. Additionally, not only do young persons with diabetes have higher baseline concentrations of serum uric acid, they also demonstrate a stronger association between elevations in uric acid concentration and impaired kidney function.
Another pathophysiologic pathway that has been shown to greatly contribute to the development of diabetes-induced AKI is the presence of persistent hyperglycemia, a hallmark of diabetes. The degree of persistent hyperglycemia has been shown to correlate with longer durations of stay in the intensive care unit as well as an increased risk for the development of AKI which can eventually lead to both CKD and ESKD (4, 12, 78). Notably, laboratory studies have demonstrated endothelial cell apoptosis, interstitial vascular rarefaction, mitochondrial dysfunction, proximal tubular cell inflammation, profibrogenic cytokine secretion, and podocyte apoptosis and autophagy in response to hyperglycemia (4).
Among the glomerular endothelial cells, hyperglycemia also induces cellular apoptosis via the Nuclear Factor-κB (NF-κB) and c-Jun NH2-terminal kinase (JNK) pathways (84, 85) and causes interstitial vascular rarefaction which leads to kidney hypoxic injury and can compromise the generation of necessary ATP (86). Overproduction of reactive oxygen species (ROS) has also been shown to induce endothelial cell damage and reduce the amount of nitric oxide available to modulate vascular tone, thereby contributing to dysregulation of sympathetic tone and sodium homeostasis in the kidneys (87).
Mitochondrial dysfunction has also been identified in the proximal tubules of individuals with diabetes during states of kidney hypoxia, further delaying future recovery from kidney injury (88). Furthermore, in diabetic rat models, the enzyme Myo-inositol oxygenase (MIOX) is upregulated in the setting of mitochondrial dysfunction, thereby inducing ROS production, cellular apoptosis, and eventually, DKD (89). MIOX is modulated by specificity protein-1 (Sp-1), a transcription factor which could serve as a potential site for the development of interventions targeting the progression of DKD as inhibition of Sp-1 has been shown to reduce ROS production (90).
Inflammatory cytokines in the proximal tubule have also been shown to play a significant role in the progression of AKI to diabetes-related CKD. Tissue necrosis factor (TNF)-α, interleukin (IL)-1, and IL-6 have been reported to be upregulated in the setting of diabetes-related CKD and are known to stimulate an inflammatory cascade that contributes to the progression of CKD (91–94). Hyperglycemia has also been shown to induce the expression of adhesion molecules and chemokines in proximal tubular cells that result in inflammation, fibrosis, and kidney injury (95, 96). Additionally, a maladaptive compensatory response for this hyperglycemia-induced inflammatory cascade in the proximal tubule leads to a profibrogenic cytokine secretion and subsequent fibrosis that plays a significant role in the transition of AKI to CKD (97). It has been demonstrated that proximal tubular cells cultured in hyperglycemic media secrete transforming growth factor beta (TGF-β)-dependent extracellular matrix (98, 99) and activation of this pathway, which plays a central role in mediating kidney fibrosis, has been shown to be significantly upregulated in both diabetic rat models and biopsies from humans with diabetes (100). Hyperglycemia has also induced podocyte apoptosis in murine models of diabetes and in adults with T2D, leading to kidney injury and future diabetes-related CKD. Podocyte apoptosis in the setting of hyperglycemia occurs through mechanisms that include, but are not limited to, increased ROS, RAAS activation, and upregulation of the mammalian target of rapamycin (mTOR) pathway (4, 101–105). However, unlike hyperglycemia-induced injury to the proximal tubule cells of the kidney, podocyte apoptosis in DKD is likely irreversible and leads to permanent damage (106).
Risk of AKI in the Pediatric Population With Diabetes
Risk factors for AKI are numerous and include concurrent hospitalizations and comorbid conditions such as hypertension and diabetes. Diabetes is an important risk factor for AKI as it has been shown to independently associate with longer hospitalizations, increased severity of concomitant infections, and future development of CKD (5, 107–112). Indeed, there is a strong association between AKI, CKD, and ESKD (4, 6, 113–115), as it has been demonstrated that both single and repetitive episodes of AKI significantly increase the risk for developing CKD in youth and adults with diabetes (5–9). Additionally, the incidence of AKI is further compounded by factors including surgical interventions (116–120), aminoglycoside usage (121), and sepsis (4, 77), and this occurs to a greater degree in individuals with vs. without diabetes. Yet, while AKI and CKD remain strongly interconnected, as evidenced by a robust body of literature detailing the transition from AKI to CKD, the effects of AKI on CKD are less clear, thus resulting in a disproportionate focus on CKD vs. AKI in diabetes research (4, 13, 34).
Therapeutic Strategies and Implications
To date, there is no universal effective therapy for AKI. As AKI is the result of different pathophysiologic mechanisms, the treatment consists of addressing the underlying causes (e.g., dehydration), correcting the hydro-electrolyte imbalances, withdrawing potential nephrotoxins, starting diuretics, and, when needed, renal replacement therapy (122). As previously mentioned, children with diabetes and DKD have a higher risk of developing AKI. In this paragraph we will discuss how DKD therapeutic strategies may impact the risk of AKI in youth with diabetes.
RAAS inhibitors, in addition to strategies that target euglycemia and minimize cardiovascular risk factors, remain the cornerstone of our existing treatment methods for DKD and has shown to attenuate proteinuria (123–125). In the SEARCH for Diabetes in Youth (SEARCH) study, youth with T2D were found to have significantly higher urinary albumin-to-creatinine ratios than youth with T1D (126), and this difference remained significant after multivariable adjustment for obesity, hypertension, and dyslipidemia, all known risk factors for DKD that are more prevalent in youth with T2D vs. T1D (126). Consequently, due to known differences in risk for the development of DKD, treatment of youth with T2D with RAAS inhibitors may be more frequently indicated than treatment of youth with T1D. However, RAAS inhibitors should be withdraw during AKI due to their effects on intrarenal hemodynamic function as well as potential direct nephrotoxic effect, which could magnify the acute kidney insult (123–125).
In addition to glucose-lowering effects, many anti-hyperglycemic agents available today also showcase significant nephroprotective properties in DKD. To date, anti-hyperglycemic agents that have demonstrated nephroprotective properties include metformin and thiazolidinediones, as well as newer classes of glycemic-lowering agents including dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagon-like peptide-1 (GLP-1) receptor agonists, and sodium/glucose cotransporter 2 (SGLT2) inhibitors (127–129). Insulin, metformin and GLP-1 receptor agonists are the only medications approved for diabetes treatment in children in the US. Despite being widely used in the of the management of patients with diabetes-related CKD, metformin has been shown to accumulate in the setting of an impaired eGFR (e.g., 30–60 mL/min/1.73 m2), thereby causing toxicity that can lead to impairments in mitochondrial function and possible non-hypoxic type B lactic acidosis (130). Thiazolidinediones, such as pioglitazone, are insulin sensitizing agents that target Peroxisome Proliferators Activated Receptors (PPARs) to regulate gene expression and thereby decrease hepatic gluconeogenesis, increase adiponectin concentrations, and increase insulin-dependent glucose uptake in muscle and fat tissues (131). Studies have previously demonstrated a possible role for PPARs in the protection against AKI (132–134) and a large meta-analysis reported an association between treatment with thiazolidinediones and a significant decrease in urinary albumin excretion (129). Yet, no randomized controlled trials have been completed to thoroughly explore the acute and chronic kidney protective effects of thiazolidinediones (135). Next, some studies have also suggested beneficial nephroprotective effects for both DPP-4 inhibitors and GLP-1 receptor agonists (127, 136). However, a recent Cochrane review was less definitive and concluded that the known effects of DPP-4 inhibitors and GLP-1 receptor agonists on eGFR are uncertain (136). Additionally, SGLT2 inhibitors have been shown to be beneficial in the prevention of AKI in two different randomized, placebo-controlled trials: Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients (EMPA-REG OUTCOME) and Dapagliflozin Effect on Cardiovascular Events-Thrombolysis in Myocardial Infarction 58 (DECLARE-TIMI 58) (137, 138). However, similarly to DPP-4 inhibitors and GLP-1 receptor agonists, a recent Cochrane review found that SGLT2 inhibitors had little to no effect on the risk for AKI (136).
Several ongoing or recently terminated trials have also analyzed the effects of other classes of drugs, particularly those targeting the kidney vasculature and the complex homeostatic mechanisms of vasoconstriction and vasodilation, in the prevention and treatment of DKD (74). Several compounds have shown promise with possible nephroprotective effects against both AKI and CKD; however, many of the human trials are still ongoing and most exclude pediatric participants. In animal models, endothelin receptor antagonists, PPAR agonists, phosphodiesterase inhibitors, and novel mineralocorticoid receptor blockers have demonstrated possible nephroprotective effects against AKI and may represent future therapeutic strategies for the prevention and treatment of AKI and CKD (123, 139–142). In particular, finerenone, a non-steroidal selective mineralocorticoid receptor antagonist, has recently been shown to reduce the risk of CKD progression in adults with T2D in the Efficacy and Safety of Finerenone in Subjects with Type 2 Diabetes Mellitus and Diabetic Kidney Disease (FIDELIO-DKD) study (143). Notably, finerenone was also able to prevent the transition from AKI to CKD in animal models and could represent an early therapeutic intervention for individuals demonstrating AKI to prevent long term complications (144).
In summary, in addition to known anti-hyperglycemic effects, many glucose-lowering agents have also demonstrated evidence of nephroprotection against both diabetes-associated AKI and CKD, two comorbidities that significantly increase the risk of morbidity and mortality in youth with diabetes. Additionally, newer classes of drugs that target intrarenal hemodynamic dysfunction in children and adolescents with diabetes are a promising new avenue for the treatment of both AKI and CKD. Consequently, well-designed pediatric trials are warranted to address the steadily increasing burden of AKI in children and adolescents with diabetes and subsequently prevent the development and progression of chronic DKD.
Covid-19 Impact
Epidemiologic studies have demonstrated evidence of AKI in more than 35% of adults who are hospitalized for the novel coronavirus disease 2019 (COVID-19) (145, 146). The prevalence of diabetes in patients with COVID-19 varies from 5 to 58% (147), yet patients with diabetes are far more likely to develop severe COVID-19 and consequent AKI than their counterparts without diabetes, with a prevalence of critical disease varying from 14 to 32% in adults with diabetes and COVID-19 (147). Acute proximal tubular damage, collapsing glomerulopathy, and podocyte injury have been found to be pathologic hallmarks of kidney injury in adults with confirmed COVID-19 (148–150).
In contrast, AKI in youth with COVID-19 has mostly been described as a consequence of dehydration secondary to gastrointestinal side effects (151, 152). One possible explanation that has been proposed is the presence of immature angiotensin converting enzyme 2 (ACE2) receptors in children and adolescents (151). ACE2 receptors are expressed in both the proximal tubular cells and podocytes and thus may be a site of cytokine-mediated effects as a result of a heightened immune system response in the setting of an active COVID-19 infection (149, 153). However, COVID-19-induced effects on the ACE2 receptor may not fully describe the AKI seen in children and adolescents with COVID-19 as other factors including the presence of a cytokine storm and/or a pro-coagulative state have also been shown to be independently associated with AKI in the setting of COVID-19 (145, 151). This finding has been corroborated by evidence of a close temporal relationship between the development of AKI and impending respiratory failure in patients with COVID-19 (145).
Thus, while AKI remains a somewhat rare complication of COVID-19 infection in pediatrics, it is notable that a concurrent diagnosis of diabetes increases the risk of associated kidney injury. Methods to assist with rapid diagnosis and treatment of AKI in youth, particularly in those with diabetes, could prevent further AKI progression and help mitigate the risk of future CKD development.
Author Contributions
FP, TR, and PB contributed to the conception and design of the review paper. All authors contributed to the manuscript revision, read, and approved the submitted version.
Conflict of Interest
PB has acted as a consultant for AstraZeneca, Bayer, Bristol-Myers Squibb, Boehringer Ingelheim, Sanofi, Novo Nordisk, Lilly USA, and Horizon Pharma. PB serves on the advisory board of AstraZeneca, NovoNordisk, XORTX, and Boehringer Ingelheim. KT receives salary and research support from the NIDDK (5T32DK007135-43), ISPAD-JDRF Research Fellowship, Center for Women's Health Research at the University of Colorado, and the Department of Pediatrics, Section of Endocrinology at the University of Colorado School of Medicine. RJ has consulted for Horizon Pharma and also has equity with XORTX Therapeutics and with Colorado Research Partners, LLC.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Afkarian M. Diabetic kidney disease in children and adolescents. Pediatr Nephrol. (2015) 30:65–74; quiz 70–1. doi: 10.1007/s00467-014-2796-5
2. Pulgaron ER, Delamater AM. Obesity and type 2 diabetes in children: epidemiology and treatment. Curr Diab Rep. (2014) 14:508. doi: 10.1007/s11892-014-0508-y
3. Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease: challenges, progress, and possibilities. Clin J Am Soc Nephrol. (2017) 12:2032–45. doi: 10.2215/CJN.11491116
4. Yu SM, Bonventre JV. Acute kidney injury and progression of diabetic kidney disease. Adv Chronic Kidney Dis. (2018) 25:166–80. doi: 10.1053/j.ackd.2017.12.005
5. Thakar CV, Christianson A, Himmelfarb J, Leonard AC. Acute kidney injury episodes and chronic kidney disease risk in diabetes mellitus. Clin J Am Soc Nephrol. (2011) 6:2567–72. doi: 10.2215/CJN.01120211
6. Ishani A, Nelson D, Clothier B, Schult T, Nugent S, Greer N, et al. The magnitude of acute serum creatinine increase after cardiac surgery and the risk of chronic kidney disease, progression of kidney disease, and death. Arch Intern Med. (2011) 171:226–33. doi: 10.1001/archinternmed.2010.514
7. Garg AX, Suri RS, Barrowman N, Rehman F, Matsell D, Rosas-Arellano MP, et al. Long-term renal prognosis of diarrhea-associated hemolytic uremic syndrome: a systematic review, meta-analysis, and meta-regression. JAMA. (2003) 290:1360–70. doi: 10.1001/jama.290.10.1360
8. Askenazi DJ, Feig DI, Graham NM, Hui-Stickle S, Goldstein SL. 3-5 year longitudinal follow-up of pediatric patients after acute renal failure. Kidney Int. (2006) 69:184–9. doi: 10.1038/sj.ki.5000032
9. Mammen C, Al Abbas A, Skippen P, Nadel H, Levine D, Collet JP, et al. Long-term risk of CKD in children surviving episodes of acute kidney injury in the intensive care unit: a prospective cohort study. Am J Kidney Dis. (2012) 59:523–30. doi: 10.1053/j.ajkd.2011.10.048
10. Huang SK, Huang CY, Lin CH, Cheng BW, Chiang YT, Lee YC, et al. Acute kidney injury is a common complication in children and adolescents hospitalized for diabetic ketoacidosis. PLoS ONE. (2020) 15:e0239160. doi: 10.1371/journal.pone.0239160
11. Vallon V. Do tubular changes in the diabetic kidney affect the susceptibility to acute kidney injury? Nephron Clin Pract. (2014) 127:133–8. doi: 10.1159/000363554
12. Gordillo R, Ahluwalia T, Woroniecki R. Hyperglycemia and acute kidney injury in critically ill children. Int J Nephrol Renovasc Dis. (2016) 9:201–4. doi: 10.2147/IJNRD.S115096
13. Sutherland SM, Ji J, Sheikhi FH, Widen E, Tian L, Alexander SR, et al. AKI in hospitalized children: epidemiology and clinical associations in a national cohort. Clin J Am Soc Nephrol. (2013) 8:1661–9. doi: 10.2215/CJN.00270113
14. Hursh BE, Ronsley R, Islam N, Mammen C, Panagiotopoulos C. Acute kidney injury in children with type 1 diabetes hospitalized for diabetic ketoacidosis. JAMA Pediatr. (2017) 171:e170020. doi: 10.1001/jamapediatrics.2017.0020
15. Ricci Z, Romagnoli S. Acute kidney injury: diagnosis and classification in adults and children. Contrib Nephrol. (2018) 193:1–12. doi: 10.1159/000484956
16. Hasegawa K, Wakino S, Simic P, Sakamaki Y, Minakuchi H, Fujimura K, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. (2013) 19:1496–504. doi: 10.1038/nm.3363
17. Koza Y. Acute kidney injury: current concepts and new insights. J Inj Violence Res. (2016) 8:58–62. doi: 10.5249/jivr.v8i1.610
18. Makris K, Spanou L. Acute kidney injury: definition, pathophysiology and clinical phenotypes. Clin Biochem Rev. (2016) 37:85–98.
19. Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol. (2012) 2:1303–53. doi: 10.1002/cphy.c110041
21. Devarajan P. Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol. (2006) 17:1503–20. doi: 10.1681/ASN.2006010017
22. Kaddourah A, Basu RK, Bagshaw SM, Goldstein SL. Epidemiology of acute kidney injury in critically ill children and young adults. N Engl J Med. (2017) 376:11–20. doi: 10.1056/NEJMoa1611391
23. Baalaaji M, Jayashree M, Nallasamy K, Singhi S, Bansal A. Predictors and outcome of acute kidney injury in children with diabetic ketoacidosis. Indian Pediatr. (2018) 55:311–4. doi: 10.1007/s13312-018-1274-8
24. Ho C, Wong C, Randell T. G575(P) acute kidney injury and diabetic ketoacidosis; intravenous fluids or ph the culprit? Arch Dis Childh. (2019) 104 (Suppl. 2):A232. doi: 10.1136/archdischild-2019-rcpch.556
25. Charlton JR, Boohaker L, Askenazi D, Brophy PD, D'Angio C, Fuloria M, et al. Incidence and risk factors of early onset neonatal AKI. Clin J Am Soc Nephrol. (2019) 14:184–95. doi: 10.2215/CJN.03670318
26. Myers SR, Glaser NS, Trainor JL, Nigrovic LE, Garro A, Tzimenatos L, et al. Frequency and risk factors of acute kidney injury during diabetic ketoacidosis in children and association with neurocognitive outcomes. JAMA Netw Open. (2020) 3:e2025481. doi: 10.1001/jamanetworkopen.2020.25481
27. Hapca S, Siddiqui MK, Kwan RSY, Lim M, Matthew S, Doney ASF, et al. The relationship between AKI and CKD in patients with type 2 diabetes: an observational cohort study. J Am Soc Nephrol. (2020) 32:138–50. doi: 10.1681/ASN.2020030323
28. Yang EM, Lee HG, Oh KY, Kim CJ. Acute kidney injury in pediatric diabetic ketoacidosis. Indian J Pediatr. (2020) 88:568–73. doi: 10.1007/s12098-020-03549-9
29. DePiero A, Kuppermann N, Brown KM, Schunk JE, McManemy JK, Rewers A, et al. Hypertension during diabetic ketoacidosis in children. J Pediatr. (2020) 223:156–63.e5. doi: 10.1016/j.jpeds.2020.04.066
30. Williams V, Jayashree M, Nallasamy K, Dayal D, Rawat A. 0.9% saline versus plasma-lyte as initial fluid in children with diabetic ketoacidosis (SPinK trial): a double-blind randomized controlled trial. Crit Care. (2020) 24:1. doi: 10.1186/s13054-019-2683-3
31. De Zan F, Amigoni A, Pozzato R, Pettenazzo A, Murer L, Vidal E. Acute kidney injury in critically ill children: a retrospective analysis of risk factors. Blood Purif. (2020) 49:1–7. doi: 10.1159/000502081
32. Rahman M, Shad F, Smith MC. Acute kidney injury: a guide to diagnosis and management. Am Fam Physician. (2012) 86:631–9. Available online at: https://www.aafp.org/afp/2012/1001/p631.html
33. Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract. (2012) 120:c179–84. doi: 10.1159/000339789
34. Makris K, Spanou L. Acute kidney injury: diagnostic approaches and controversies. Clin Biochem Rev. (2016) 37:153–75.
35. Marenzi G, Cosentino N, Bartorelli AL. Acute kidney injury in patients with acute coronary syndromes. Heart. (2015) 101:1778–85. doi: 10.1136/heartjnl-2015-307773
36. Aggarwal N, Porter AC, Tang IY, Becker BN, Akkina SK. Creatinine-based estimations of kidney function are unreliable in obese kidney donors. J Transplant. (2012) 2012:872894. doi: 10.1155/2012/872894
37. Rizvi MS, Kashani KB. Biomarkers for early detection of acute kidney injury. J Appl Lab Med. (2019) 2:386–99. doi: 10.1373/jalm.2017.023325
38. Sandokji I, Greenberg JH. Novel biomarkers of acute kidney injury in children: an update on recent findings. Curr Opin Pediatr. (2020) 32:354–9. doi: 10.1097/MOP.0000000000000891
39. Kjeldsen L, Johnsen AH, Sengeløv H, Borregaard N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem. (1993) 268:10425–32. doi: 10.1016/S0021-9258(18)82217-7
40. Cowland JB, Borregaard N. Molecular characterization and pattern of tissue expression of the gene for neutrophil gelatinase-associated lipocalin from humans. Genomics. (1997) 45:17–23. doi: 10.1006/geno.1997.4896
41. Sanjeevani S, Pruthi S, Kalra S, Goel A, Kalra OP. Role of neutrophil gelatinase-associated lipocalin for early detection of acute kidney injury. Int J Crit Illn Inj Sci. (2014) 4:223–8. doi: 10.4103/2229-5151.141420
42. Tang XY, Zhou JB, Luo FQ, Han YP, Zhao W, Diao ZL, et al. Urine NGAL as an early biomarker for diabetic kidney disease: accumulated evidence from observational studies. Ren Fail. (2019) 41:446–54. doi: 10.1080/0886022X.2019.1617736
43. Ichimura T, Bonventre JV, Bailly V, Wei H, Hession CA, Cate RL, et al. Kidney injury molecule-1 (KIM-1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up-regulated in renal cells after injury. J Biol Chem. (1998) 273:4135–42. doi: 10.1074/jbc.273.7.4135
44. Han WK, Bailly V, Abichandani R, Thadhani R, Bonventre JV. Kidney injury molecule-1 (KIM-1): a novel biomarker for human renal proximal tubule injury. Kidney Int. (2002) 62:237–44. doi: 10.1046/j.1523-1755.2002.00433.x
45. Assadi F, Sharbaf FG. Urine KIM-1 as a potential biomarker of acute renal injury after circulatory collapse in children. Pediatr Emerg Care. (2019) 35:104–7. doi: 10.1097/PEC.0000000000000886
46. Shao X, Tian L, Xu W, Zhang Z, Wang C, Qi C, et al. Diagnostic value of urinary kidney injury molecule 1 for acute kidney injury: a meta-analysis. PLoS ONE. (2014) 9:e84131. doi: 10.1371/journal.pone.0084131
47. Ucakturk A, Avci B, Genc G, Ozkaya O, Aydin M. Kidney injury molecule-1 and neutrophil gelatinase associated lipocalin in normoalbuminuric diabetic children. J Pediatr Endocrinol Metab. (2016) 29:145–51. doi: 10.1515/jpem-2015-0138
48. Wang G, Bonkovsky HL, de Lemos A, Burczynski FJ. Recent insights into the biological functions of liver fatty acid binding protein 1. J Lipid Res. (2015) 56:2238–47. doi: 10.1194/jlr.R056705
49. Shi J, Zhang Y, Gu W, Cui B, Xu M, Yan Q, et al. Serum liver fatty acid binding protein levels correlate positively with obesity and insulin resistance in Chinese young adults. PLoS ONE. (2012) 7:e48777. doi: 10.1371/journal.pone.0048777
50. Vijayan A, Faubel S, Askenazi DJ, Cerda J, Fissell WH, Heung M, et al. Clinical use of the urine biomarker [TIMP-2] × [IGFBP7] for acute kidney injury risk assessment. Am J Kidney Dis. (2016) 68:19–28. doi: 10.1053/j.ajkd.2015.12.033
51. Guzzi LM, Bergler T, Binnall B, Engelman DT, Forni L, Germain MJ, et al. Clinical use of [TIMP-2]∙[IGFBP7] biomarker testing to assess risk of acute kidney injury in critical care: guidance from an expert panel. Crit Care. (2019) 23:225. doi: 10.1186/s13054-019-2504-8
52. Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int. (2004) 66:480–5. doi: 10.1111/j.1523-1755.2004.761_2.x
53. Friedewald JJ, Rabb H. Inflammatory cells in ischemic acute renal failure. Kidney Int. (2004) 66:486–91. doi: 10.1111/j.1523-1755.2004.761_3.x
54. Castellanos L, Tuffaha M, Koren D, Levitsky LL. Management of diabetic ketoacidosis in children and adolescents with type 1 diabetes mellitus. Paediatr Drugs. (2020) 22:357–67. doi: 10.1007/s40272-020-00397-0
55. Gilbert RE. Proximal tubulopathy: prime mover and key therapeutic target in diabetic kidney disease. Diabetes. (2017) 66:791–800. doi: 10.2337/db16-0796
56. Realsen J, Goettle H, Chase HP. Morbidity and mortality of diabetic ketoacidosis with and without insulin pump care. Diabetes Technol Ther. (2012) 14:1149–54. doi: 10.1089/dia.2012.0161
57. Razavi Z. Frequency of ketoacidosis in newly diagnosed type 1 diabetic children. Oman Med J. (2010) 25:114–7. doi: 10.5001/omj.2010.31
58. Roussel R, Velho G, Bankir L. Vasopressin and diabetic nephropathy. Curr Opin Nephrol Hypertens. (2017) 26:311–8. doi: 10.1097/MNH.0000000000000335
59. Bjornstad P, Cherney D, Maahs DM. Early diabetic nephropathy in type 1 diabetes: new insights. Curr Opin Endocrinol Diabetes Obes. (2014) 21:279–86. doi: 10.1097/MED.0000000000000074
60. Bjornstad P, Lanaspa MA, Ishimoto T, Kosugi T, Kume S, Jalal D, et al. Fructose and uric acid in diabetic nephropathy. Diabetologia. (2015) 58:1993–2002. doi: 10.1007/s00125-015-3650-4
61. Verzola D, Ratto E, Villaggio B, Parodi EL, Pontremoli R, Garibotto G, et al. Uric acid promotes apoptosis in human proximal tubule cells by oxidative stress and the activation of NADPH oxidase NOX 4. PLoS ONE. (2014) 9:e115210. doi: 10.1371/journal.pone.0115210
62. Bjornstad P, Maahs DM, Jensen T, Lanaspa MA, Johnson RJ, Rewers M, et al. Elevated copeptin is associated with atherosclerosis and diabetic kidney disease in adults with type 1 diabetes. J Diabetes Complications. (2016) 30:1093–6. doi: 10.1016/j.jdiacomp.2016.04.012
63. Burckhardt MA, Gotta V, Beglinger S, Renggli L, Bachmann S, Hess M, et al. Copeptin kinetics and its relationship to osmolality during rehydration for diabetic ketoacidosis in children. J Clin Endocrinol Metab. (2020) 105:dgaa568. doi: 10.1210/clinem/dgaa568
64. Velho G, El Boustany R, Lefèvre G, Mohammedi K, Fumeron F, Potier L, et al. Plasma copeptin, kidney outcomes, ischemic heart disease, and all-cause mortality in people with long-standing type 1 diabetes. Diabetes Care. (2016) 39:2288–95. doi: 10.2337/dc16-1003
65. Piani F, Reinicke T, Lytvyn Y, Melena I, Lovblom LE, Lai V, et al. Vasopressin associated with renal vascular resistance in adults with longstanding type 1 diabetes with and without diabetic kidney disease. J Diabetes Complications. (2020) 35:107807. doi: 10.1016/j.jdiacomp.2020.107807
66. Wiromrat P, Bjornstad P, Vinovskis C, Chung LT, Roncal C, Pyle L, et al. Elevated copeptin, arterial stiffness, and elevated albumin excretion in adolescents with type 1 diabetes. Pediatr Diabetes. (2019) 20:1110–7. doi: 10.1111/pedi.12909
67. Roncal-Jimenez CA, Milagres T, Andres-Hernando A, Kuwabara M, Jensen T, Song Z, et al. Effects of exogenous desmopressin on a model of heat stress nephropathy in mice. Am J Physiol Renal Physiol. (2017) 312:F418–26. doi: 10.1152/ajprenal.00495.2016
68. El Boustany R, Taveau C, Chollet C, Velho G, Bankir L, Alhenc-Gelas F, et al. Antagonism of vasopressin V2 receptor improves albuminuria at the early stage of diabetic nephropathy in a mouse model of type 2 diabetes. J Diabetes Complications. (2017) 31:929–32. doi: 10.1016/j.jdiacomp.2017.04.005
69. Abd El Ghaffar S, El Kaffas K, Hegazy R, Mostafa M. Renal doppler indices in diabetic children with insulin resistance syndrome. Pediatr Diabetes. (2010) 11:479–86. doi: 10.1111/j.1399-5448.2009.00628.x
70. Raes A, Matthys D, Donckerwolcke R, Craen M, Van Aken S, Vande Walle J. Renal functional changes in relation to hemodynamic parameters during exercise test in normoalbuminuric insulin-dependent children. Acta Paediatr. (2007) 96:548–51. doi: 10.1111/j.1651-2227.2006.00157.x
71. Youssef DM, Fawzy FM. Value of renal resistive index as an early marker of diabetic nephropathy in children with type-1 diabetes mellitus. Saudi J Kidney Dis Transpl. (2012) 23:985–92. doi: 10.4103/1319-2442.100880
72. Anguiano L, Riera M, Pascual J, Soler MJ. Endothelin blockade in diabetic kidney disease. J Clin Med. (2015) 4:1171–92. doi: 10.3390/jcm4061171
73. Fu J, Lee K, Chuang PY, Liu Z, He JC. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. Am J Physiol Renal Physiol. (2015) 308:F287–97. doi: 10.1152/ajprenal.00533.2014
74. Leung WK, Gao L, Siu PM, Lai CW. Diabetic nephropathy and endothelial dysfunction: current and future therapies, and emerging of vascular imaging for preclinical renal-kinetic study. Life Sci. (2016) 166:121–30. doi: 10.1016/j.lfs.2016.10.015
75. Bjornstad P, Cherney DZ. Renal hyperfiltration in adolescents with type 2 diabetes: physiology, sex differences, and implications for diabetic kidney disease. Curr Diab Rep. (2018) 18:22. doi: 10.1007/s11892-018-0996-2
76. Premaratne E, Verma S, Ekinci EI, Theverkalam G, Jerums G, MacIsaac RJ. The impact of hyperfiltration on the diabetic kidney. Diabetes Metab. (2015) 41:5–17. doi: 10.1016/j.diabet.2014.10.003
77. Venot M, Weis L, Clec'h C, Darmon M, Allaouchiche B, Goldgran-Tolédano D, et al. Acute kidney injury in severe sepsis and septic shock in patients with and without diabetes mellitus: a multicenter study. PLoS ONE. (2015) 10:e0127411. doi: 10.1371/journal.pone.0127411
78. Piani F, Johnson RJ. Does gouty nephropathy exist, and is it more common than we think? Kidney Int. (2021) 99:31–3. doi: 10.1016/j.kint.2020.10.015
79. Ejaz AA, Johnson RJ, Shimada M, Mohandas R, Alquadan KF, Beaver TM, et al. The role of uric acid in acute kidney injury. Nephron. (2019) 142:275–83. doi: 10.1159/000499939
80. Fathallah-Shaykh SA, Cramer MT. Uric acid and the kidney. Pediatr Nephrol. (2014) 29:999–1008. doi: 10.1007/s00467-013-2549-x
81. Bjornstad P, Roncal C, Milagres T, Pyle L, Lanaspa MA, Bishop FK, et al. Hyperfiltration and uricosuria in adolescents with type 1 diabetes. Pediatr Nephrol. (2016) 31:787–93. doi: 10.1007/s00467-015-3299-8
82. Lytvyn Y, Bjornstad P, Lovshin JA, Singh SK, Boulet G, Farooqi MA, et al. Association between uric acid, renal haemodynamics and arterial stiffness over the natural history of type 1 diabetes. Diabetes Obes Metab. (2019) 21:1388–98. doi: 10.1111/dom.13665
83. Santucci MP, Muzzio ML, Peredo MS, Brovarone L, Scricciolo R, Diez C, et al. Different alterations of glomerular filtration rate and their association with uric acid in children and adolescents with type 1 diabetes or with overweight/obesity. Pediatr Diabetes. (2020) 21:657–63. doi: 10.1111/pedi.13008
84. Ho FM, Liu SH, Liau CS, Huang PJ, Lin-Shiau SY. High glucose-induced apoptosis in human endothelial cells is mediated by sequential activations of c-Jun NH(2)-terminal kinase and caspase-3. Circulation. (2000) 101:2618–24. doi: 10.1161/01.CIR.101.22.2618
85. Ho FM, Lin WW, Chen BC, Chao CM, Yang CR, Lin LY, et al. High glucose-induced apoptosis in human vascular endothelial cells is mediated through NF-kappaB and c-Jun NH2-terminal kinase pathway and prevented by PI3K/Akt/eNOS pathway. Cell Signal. (2006) 18:391–9. doi: 10.1016/j.cellsig.2005.05.009
86. Lindenmeyer MT, Kretzler M, Boucherot A, Berra S, Yasuda Y, Henger A, et al. Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J Am Soc Nephrol. (2007) 18:1765–76. doi: 10.1681/ASN.2006121304
87. Han HJ, Lee YJ, Park SH, Lee JH, Taub M. High glucose-induced oxidative stress inhibits Na+/glucose cotransporter activity in renal proximal tubule cells. Am J Physiol Renal Physiol. (2005) 288:F988–96. doi: 10.1152/ajprenal.00327.2004
88. Zuk A, Bonventre JV. Acute kidney injury. Annu Rev Med. (2016) 67:293–307. doi: 10.1146/annurev-med-050214-013407
89. Zhan M, Usman IM, Sun L, Kanwar YS. Disruption of renal tubular mitochondrial quality control by Myo-inositol oxygenase in diabetic kidney disease. J Am Soc Nephrol. (2015) 26:1304–21. doi: 10.1681/ASN.2014050457
90. Sharma I, Dutta RK, Singh NK, Kanwar YS. High glucose-induced hypomethylation promotes binding of Sp-1 to myo-inositol oxygenase: implication in the pathobiology of diabetic tubulopathy. Am J Pathol. (2017) 187:724–39. doi: 10.1016/j.ajpath.2016.12.011
91. Navarro JF, Milena FJ, Mora C, León C, García J. Renal pro-inflammatory cytokine gene expression in diabetic nephropathy: effect of angiotensin-converting enzyme inhibition and pentoxifylline administration. Am J Nephrol. (2006) 26:562–70. doi: 10.1159/000098004
92. Suzuki D, Miyazaki M, Naka R, Koji T, Yagame M, Jinde K, et al. In situ hybridization of interleukin 6 in diabetic nephropathy. Diabetes. (1995) 44:1233–8. doi: 10.2337/diabetes.44.10.1233
93. Sekizuka K, Tomino Y, Sei C, Kurusu A, Tashiro K, Yamaguchi Y, et al. Detection of serum IL-6 in patients with diabetic nephropathy. Nephron. (1994) 68:284–5. doi: 10.1159/000188281
94. Navarro-González JF, Mora-Fernández C, Muros de Fuentes M, García-Pérez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. (2011) 7:327–40. doi: 10.1038/nrneph.2011.51
95. Tang SC, Leung JC, Chan LY, Tsang AW, Lai KN. Activation of tubular epithelial cells in diabetic nephropathy and the role of the peroxisome proliferator-activated receptor-gamma agonist. J Am Soc Nephrol. (2006) 17:1633–43. doi: 10.1681/ASN.2005101113
96. Tang SCW, Leung JCK, Lai KN. Diabetic tubulopathy: an emerging entity. Contrib Nephrol. (2011) 170:124–34. doi: 10.1159/000325647
97. Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol. (2015) 26:1765–76. doi: 10.1681/ASN.2015010006
98. Ziyadeh FN, Snipes ER, Watanabe M, Alvarez RJ, Goldfarb S, Haverty TP. High glucose induces cell hypertrophy and stimulates collagen gene transcription in proximal tubule. Am J Physiol. (1990) 259 (4 Pt. 2):F704–14. doi: 10.1152/ajprenal.1990.259.4.F704
99. Rocco MV, Chen Y, Goldfarb S, Ziyadeh FN. Elevated glucose stimulates TGF-beta gene expression and bioactivity in proximal tubule. Kidney Int. (1992) 41:107–14. doi: 10.1038/ki.1992.14
100. Yamamoto T, Nakamura T, Noble NA, Ruoslahti E, Border WA. Expression of transforming growth factor beta is elevated in human and experimental diabetic nephropathy. Proc Natl Acad Sci USA. (1993) 90:1814–8. doi: 10.1073/pnas.90.5.1814
101. Anil Kumar P, Welsh GI, Saleem MA, Menon RK. Molecular and cellular events mediating glomerular podocyte dysfunction and depletion in diabetes mellitus. Front Endocrinol. (2014) 5:151. doi: 10.3389/fendo.2014.00151
102. Chuang PY, Yu Q, Fang W, Uribarri J, He JC. Advanced glycation endproducts induce podocyte apoptosis by activation of the FOXO4 transcription factor. Kidney Int. (2007) 72:965–76. doi: 10.1038/sj.ki.5002456
103. Zhou LL, Cao W, Xie C, Tian J, Zhou Z, Zhou Q, et al. The receptor of advanced glycation end products plays a central role in advanced oxidation protein products-induced podocyte apoptosis. Kidney Int. (2012) 82:759–70. doi: 10.1038/ki.2012.184
104. Verzola D, Gandolfo MT, Ferrario F, Rastaldi MP, Villaggio B, Gianiorio F, et al. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int. (2007) 72:1262–72. doi: 10.1038/sj.ki.5002531
105. Susztak K, Raff AC, Schiffer M, Böttinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. (2006) 55:225–33. doi: 10.2337/diabetes.55.01.06.db05-0894
106. Anders HJ. Immune system modulation of kidney regeneration–mechanisms and implications. Nat Rev Nephrol. (2014) 10:347–58. doi: 10.1038/nrneph.2014.68
107. Chen J, Zeng H, Ouyang X, Zhu M, Huang Q, Yu W, et al. The incidence, risk factors, and long-term outcomes of acute kidney injury in hospitalized diabetic ketoacidosis patients. BMC Nephrol. (2020) 21:48. doi: 10.1186/s12882-020-1709-z
108. Jain A, McDonald HI, Nitsch D, Tomlinson L, Thomas SL. Risk factors for developing acute kidney injury in older people with diabetes and community-acquired pneumonia: a population-based UK cohort study. BMC Nephrol. (2017) 18:142. doi: 10.1186/s12882-017-0566-x
109. Heung M, Chawla LS. Acute kidney injury: gateway to chronic kidney disease. Nephron Clin Pract. (2014) 127:30–4. doi: 10.1159/000363675
110. Saran R, Robinson B, Abbott KC, Agodoa LY, Albertus P, Ayanian J, et al. US renal data system 2016 annual data report: epidemiology of kidney disease in the United States. Am J Kidney Dis. (2017) 69 (3 Suppl. 1):A7–8. doi: 10.1053/j.ajkd.2017.01.036
111. Waikar SS, Liu KD, Chertow GM. Diagnosis, epidemiology and outcomes of acute kidney injury. Clin J Am Soc Nephrol. (2008) 3:844–61. doi: 10.2215/CJN.05191107
112. James MT, Grams ME, Woodward M, Elley CR, Green JA, Wheeler DC, et al. A meta-analysis of the association of estimated GFR, albuminuria, diabetes mellitus, and hypertension with acute kidney injury. Am J Kidney Dis. (2015) 66:602–12. doi: 10.1053/j.ajkd.2015.02.338
113. Southern DA, James MT, Wilton SB, DeKoning L, Quan H, Knudtson ML, et al. Expanding the impact of a longstanding Canadian cardiac registry through data linkage: challenges and opportunities. Int J Popul Data Sci. (2018) 3:441. doi: 10.23889/ijpds.v3i3.441
114. Chew ST, Ng RR, Liu W, Chow KY, Ti LK. Acute kidney injury increases the risk of end-stage renal disease after cardiac surgery in an Asian population: a prospective cohort study. BMC Nephrol. (2017) 18:60. doi: 10.1186/s12882-017-0476-y
115. Hsu CY, Chertow GM, McCulloch CE, Fan D, Ordoñez JD, Go AS. Nonrecovery of kidney function and death after acute on chronic renal failure. Clin J Am Soc Nephrol. (2009) 4:891–8. doi: 10.2215/CJN.05571008
116. Mehta RH, Grab JD, O'Brien SM, Bridges CR, Gammie JS, Haan CK, et al. Bedside tool for predicting the risk of postoperative dialysis in patients undergoing cardiac surgery. Circulation. (2006) 114:2208–16; quiz 2208. doi: 10.1161/CIRCULATIONAHA.106.635573
117. Kheterpal S, Tremper KK, Heung M, Rosenberg AL, Englesbe M, Shanks AM, et al. Development and validation of an acute kidney injury risk index for patients undergoing general surgery: results from a national data set. Anesthesiology. (2009) 110:505–15. doi: 10.1097/ALN.0b013e3181979440
118. Parolari A, Pesce LL, Pacini D, Mazzanti V, Salis S, Sciacovelli C, et al. Risk factors for perioperative acute kidney injury after adult cardiac surgery: role of perioperative management. Ann Thorac Surg. (2012) 93:584–91. doi: 10.1016/j.athoracsur.2011.09.073
119. Ko B, Garcia S, Mithani S, Tholakanahalli V, Adabag S. Risk of acute kidney injury in patients who undergo coronary angiography and cardiac surgery in close succession. Eur Heart J. (2012) 33:2065–70. doi: 10.1093/eurheartj/ehr493
120. Hertzberg D, Sartipy U, Holzmann MJ. Type 1 and type 2 diabetes mellitus and risk of acute kidney injury after coronary artery bypass grafting. Am Heart J. (2015) 170:895–902. doi: 10.1016/j.ahj.2015.08.013
121. Oliveira JF, Silva CA, Barbieri CD, Oliveira GM, Zanetta DM, Burdmann EA. Prevalence and risk factors for aminoglycoside nephrotoxicity in intensive care units. Antimicrob Agents Chemother. (2009) 53:2887–91. doi: 10.1128/AAC.01430-08
122. Basu RK. Acute kidney injury in hospitalized pediatric patients. Pediatr Ann. (2018) 47:e286–91. doi: 10.3928/19382359-20180619-02
123. Thomas MC, Brownlee M, Susztak K, Sharma K, Jandeleit-Dahm KA, Zoungas S, et al. Diabetic kidney disease. Nat Rev Dis Primers. (2015) 1:15018. doi: 10.1038/nrdp.2015.70
124. National Kidney Foundation. KDOQI clinical practice guideline for diabetes and CKD: 2012 update. Am J Kidney Dis. (2012) 60:850–86. doi: 10.1053/j.ajkd.2012.07.005
125. Perkins BA, Ficociello LH, Silva KH, Finkelstein DM, Warram JH, Krolewski AS. Regression of microalbuminuria in type 1 diabetes. N Engl J Med. (2003) 348:2285–93. doi: 10.1056/NEJMoa021835
126. Maahs DM, Snively BM, Bell RA, Dolan L, Hirsch I, Imperatore G, et al. Higher prevalence of elevated albumin excretion in youth with type 2 than type 1 diabetes: the SEARCH for diabetes in youth study. Diabetes Care. (2007) 30:2593–8. doi: 10.2337/dc07-0450
127. Schernthaner G, Mogensen CE, Schernthaner GH. The effects of GLP-1 analogues, DPP-4 inhibitors and SGLT2 inhibitors on the renal system. Diab Vasc Dis Res. (2014) 11:306–23. doi: 10.1177/1479164114542802
128. Ravindran S, Kuruvilla V, Wilbur K, Munusamy S. Nephroprotective effects of metformin in diabetic nephropathy. J Cell Physiol. (2017) 232:731–42. doi: 10.1002/jcp.25598
129. Sarafidis PA, Stafylas PC, Georgianos PI, Saratzis AN, Lasaridis AN. Effect of thiazolidinediones on albuminuria and proteinuria in diabetes: a meta-analysis. Am J Kidney Dis. (2010) 55:835–47. doi: 10.1053/j.ajkd.2009.11.013
130. Rhee CM, Kalantar-Zadeh K. Diabetes mellitus: Complex interplay between metformin, AKI and lactic acidosis. Nat Rev Nephrol. (2017) 13:521–2. doi: 10.1038/nrneph.2017.105
131. Nanjan MJ, Mohammed M, Prashantha Kumar BR, Chandrasekar MJN. Thiazolidinediones as antidiabetic agents: a critical review. Bioorg Chem. (2018) 77:548–67. doi: 10.1016/j.bioorg.2018.02.009
132. Iwaki T, Bennion BG, Stenson EK, Lynn JC, Otinga C, Djukovic D, et al. PPARα contributes to protection against metabolic and inflammatory derangements associated with acute kidney injury in experimental sepsis. Physiol Rep. (2019) 7:e14078. doi: 10.14814/phy2.14078
133. Singh AP, Singh N, Pathak D, Bedi PMS. Estradiol attenuates ischemia reperfusion-induced acute kidney injury through PPAR-γ stimulated eNOS activation in rats. Mol Cell Biochem. (2019) 453:1–9. doi: 10.1007/s11010-018-3427-4
134. Kaur T, Singh D, Singh AP, Pathak D, Arora S, Singh B, et al. Stevioside protects against rhabdomyolysis-induced acute kidney injury through PPAR-γ agonism in rats. Drug Dev Res. (2020) doi: 10.1002/ddr.21722
135. Sarafidis PA, Bakris GL. Protection of the kidney by thiazolidinediones: an assessment from bench to bedside. Kidney Int. (2006) 70:1223–33. doi: 10.1038/sj.ki.5001620
136. Lo C, Toyama T, Wang Y, Lin J, Hirakawa Y, Jun M, et al. Insulin and glucose-lowering agents for treating people with diabetes and chronic kidney disease. Cochrane Database Syst Rev. (2018) 9:CD011798. doi: 10.1002/14651858.CD011798.pub2
137. Chu C, Lu YP, Yin L, Hocher B. The SGLT2 inhibitor empagliflozin might be a new approach for the prevention of acute kidney injury. Kidney Blood Press Res. (2019) 44:149–57. doi: 10.1159/000498963
138. Menne J, Dumann E, Haller H, Schmidt BMW. Acute kidney injury and adverse renal events in patients receiving SGLT2-inhibitors: a systematic review and meta-analysis. PLoS Med. (2019) 16:e1002983. doi: 10.1371/journal.pmed.1002983
139. Afyouni NE, Halili H, Moslemi F, Nematbakhsh M, Talebi A, Shirdavani S, et al. Preventive role of endothelin antagonist on kidney ischemia: reperfusion injury in male and female rats. Int J Prev Med. (2015) 6:128. doi: 10.4103/2008-7802.172549
140. Mohandas R, Dass B, Ejaz AA. Kinetics of vascular endothelial growth factor and endothelin 1 levels in acute kidney injury. Am J Kidney Dis. (2019) 74:712–3. doi: 10.1053/j.ajkd.2019.08.004
141. Bates CM, Lin F. Future strategies in the treatment of acute renal failure: growth factors, stem cells, and other novel therapies. Curr Opin Pediatr. (2005) 17:215–20. doi: 10.1097/01.mop.0000156269.48510.4e
142. Georgiadis G, Zisis IE, Docea AO, Tsarouhas K, Fragkiadoulaki I, Mavridis C, et al. Current concepts on the reno-protective effects of phosphodiesterase 5 inhibitors in acute kidney injury: systematic search and review. J Clin Med. (2020) 9:1284. doi: 10.3390/jcm9051284
143. Bakris GL, Agarwal R, Anker SD, Pitt B, Ruilope LM, Rossing P, et al. Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes. N Engl J Med. (2020) 383:2219–29. doi: 10.1056/NEJMoa2025845
144. Lattenist L, Lechner SM, Messaoudi S, Le Mercier A, El Moghrabi S, Prince S, et al. Nonsteroidal mineralocorticoid receptor antagonist finerenone protects against acute kidney injury-mediated chronic kidney disease: role of oxidative stress. Hypertension. (2017) 69:870–8. doi: 10.1161/HYPERTENSIONAHA.116.08526
145. Hirsch JS, Ng JH, Ross DW, Sharma P, Shah HH, Barnett RL, et al. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. (2020) 98:209–18. doi: 10.1016/j.kint.2020.05.006
146. Chan L, Chaudhary K, Saha A, Chauhan K, Vaid A, Zhao S, et al. AKI in hospitalized patients with COVID-19. J Am Soc Nephrol. (2020) 32:151–60. doi: 10.1681/ASN.2020050615
147. Singh AK, Gupta R, Ghosh A, Misra A. Diabetes in COVID-19: prevalence, pathophysiology, prognosis and practical considerations. Diabetes Metab Syndr. (2020) 14:303–10. doi: 10.1016/j.dsx.2020.04.004
148. Gagliardi I, Patella G, Michael A, Serra R, Provenzano M, Andreucci M. COVID-19 and the kidney: from epidemiology to clinical practice. J Clin Med. (2020) 9:2506. doi: 10.3390/jcm9082506
149. Kudose S, Batal I, Santoriello D, Xu K, Barasch J, Peleg Y, et al. Kidney biopsy findings in patients with COVID-19. J Am Soc Nephrol. (2020) 31:1959–68. doi: 10.1681/ASN.2020060802
150. Su H, Yang M, Wan C, Yi LX, Tang F, Zhu HY, et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. (2020) 98:219–227. doi: 10.1016/j.kint.2020.04.003
151. Wang X, Chen X, Tang F, Luo W, Fang J, Qi C, et al. Be aware of acute kidney injury in critically ill children with COVID-19, Pediatr Nephrol. (2021) 36:163–9. doi: 10.1007/s00467-020-04715-z
152. Deep A, Bansal M, Ricci Z. Acute kidney injury and special considerations during renal replacement therapy in children with coronavirus disease-19: perspective from the critical care nephrology section of the European society of paediatric and neonatal intensive care. Blood Purif . (2020) 50:150–60. doi: 10.1159/000509677
Keywords: acute kidney injury, diabetes, diabetic kidney disease, pediatric, COVID-19
Citation: Piani F, Reinicke T, Borghi C, Tommerdahl KL, Cara-Fuentes G, Johnson RJ and Bjornstad P (2021) Acute Kidney Injury in Pediatric Diabetic Kidney Disease. Front. Pediatr. 9:668033. doi: 10.3389/fped.2021.668033
Received: 15 February 2021; Accepted: 17 May 2021;
Published: 15 June 2021.
Edited by:
Michael Zappitelli, Hospital for Sick Children, CanadaReviewed by:
Emily Lauren Joyce, Rainbow Babies & Children's Hospital, United StatesNicole Hayde, Children's Hospital at Montefiore, United States
Copyright © 2021 Piani, Reinicke, Borghi, Tommerdahl, Cara-Fuentes, Johnson and Bjornstad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Petter Bjornstad, UGV0dGVyLk0uQmpvcm5zdGFkQENVQW5zY2h1dHouZWR1