 Zuzana Marincak Vrankova1,2,3
Zuzana Marincak Vrankova1,2,3 Jan Krivanek4Zdenek Danek2,3Jiri Zelinka2Alena Brysova1
Jan Krivanek4Zdenek Danek2,3Jiri Zelinka2Alena Brysova1 Lydie Izakovicova Holla1
Lydie Izakovicova Holla1 James K. Hartsfield Jr5
James K. Hartsfield Jr5 Petra Borilova Linhartova1,2,3*
Petra Borilova Linhartova1,2,3*
- 1Clinic of Stomatology, Institution Shared with St. Anne's University Hospital, Faculty of Medicine, Masaryk University, Brno, Czech Republic
- 2Clinic of Maxillofacial Surgery, Institution Shared with the University Hospital Brno, Faculty of Medicine, Masaryk University, Brno, Czech Republic
- 3RECETOX, Faculty of Science, Masaryk University, Kotlarska 2, Brno, Czech Republic
- 4Department of Histology and Embryology, Faculty of Medicine, Masaryk University, Brno, Czech Republic
- 5E. Preston Hicks Professor of Orthodontics and Oral Health Research, University of Kentucky Center for the Biologic Basis of Oral/Systemic Diseases, Hereditary Genetics/Genomics Core, Lexington, KE, United States
Pediatric obstructive sleep apnea (POSA) is a complex disease with multifactorial etiopathogenesis. The presence of craniofacial dysmorphisms influencing the patency of the upper airway is considered a risk factor for POSA development. The craniofacial features associated with sleep-related breathing disorders (SRBD) – craniosynostosis, retrognathia and micrognathia, midface and maxillary hypoplasia – have high heritability and, in a less severe form, could be also found in non-syndromic children suffering from POSA. As genetic factors play a role in both POSA and craniofacial dysmorphisms, we hypothesize that some genes associated with specific craniofacial features that are involved in the development of the orofacial area may be also considered candidate genes for POSA. The genetic background of POSA in children is less explored than in adults; so far, only one genome-wide association study for POSA has been conducted; however, children with craniofacial disorders were excluded from that study. In this narrative review, we discuss syndromes that are commonly associated with severe craniofacial dysmorphisms and a high prevalence of sleep-related breathing disorders (SRBD), including POSA. We also summarized information about their genetic background and based on this, proposed 30 candidate genes for POSA affecting craniofacial development that may play a role in children with syndromes, and identified seven of these genes that were previously associated with craniofacial features risky for POSA development in non-syndromic children. The evidence-based approach supports the proposition that variants of these candidate genes could lead to POSA phenotype even in these children, and, thus, should be considered in future research in the general pediatric population.
1. Introduction
Both pediatric (POSA) and adult obstructive sleep apnea (OSA) count among sleep-related breathing disorders (SRBD). POSA is considered a multifactorial disease triggered by the combination of genetic predispositions and several risk factors, including obesity, neuromuscular factors, adenotonsillar hypertrophy, and specific craniofacial features (1, 2). In adults, the genetic background leading to the OSA phenotype has been studied more intensively than in children.
So far, several studies on candidate genes, phenome-wide association studies of OSA genomic variation, and genome/phenome-wide association studies (GWAS/PheWAS) on adult patients with OSA have been published (3–5), while only a single GWAS focusing on children has been reported (6). That study included 1,486 subjects, 1 week to 18 years old, 46.3% of whom were European-Americans and 53.7% African-Americans. The study identified genomic loci associated with POSA at 1p36.22, 15q26.1, 18p11.32 (rs114124196), 1q43 (rs12754698), 2p25 (rs72775219). 8q21.11 (rs6472959), 11q24.3 (rs4370952), and 15q21.1 (rs149936782); children with craniofacial disorders were excluded from that study (6).
Moreover, single nucleotide polymorphisms (SNPs) in genes encoding apolipoprotein E, fatty-acid binding protein 4, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and the macrophage migration inhibitory factor were associated with increased or decreased odds of POSA development in children (7–9). These genes are considered to be candidate genes for POSA development (i.e., they are likely to be related to this disease because of their genomic location or known function). All four mentioned genes are associated with lipid metabolism and/or immune system function. It is, therefore, possible that the susceptibility of carriers of these SNPs to POSA is associated with their role in the development of obesity.
However, genetic background is involved, to some extent, in all of the most commonly reported POSA risk factors – besides obesity, body fat distribution, ventilation control mechanisms, upper airway neural control, and soft tissue morphology, genetic background plays a role also in craniofacial dysmorphisms (10–13). In this narrative review, we closely focus on specific genes involved in the development of the orofacial area and of certain craniofacial features, which makes them possible candidate genes for POSA. Thus, we aimed to (i) describe craniofacial anomalies associated with POSA development, (ii) select syndromes characterized by severe craniofacial dysmorphisms associated with OSA and/or high prevalence of pediatric SRBD, (iii) summarize information about the genetic background of these syndromes, and (iv) suggest candidate genes for POSA in non-syndromic patients with craniofacial dysmorphisms.
2. Craniofacial characteristics associated with POSA development
As the upper airway dimensions and morphology of the craniofacial area are closely related, it is no surprise that some abnormalities in its soft and bony structures may contribute to the narrowing and easier collapse of the airway, resulting in OSA, both in children and adults (14–16). Patients suffering from severe skeletal craniofacial malformations could be at a three times higher risk of POSA development than the general pediatric population (17). The importance of craniofacial morphology in OSA development was confirmed also by Kim et al., who reported the presence of craniofacial dysmorphisms, such as the narrow nasomaxillary complex or underdeveloped mandible, in 93.3% of children diagnosed with sleep-disordered breathing (18).
Multiple studies described craniofacial characteristics that are more often present in children suffering from SRBD than in children without these conditions (19–24). These include the size of the maxillo-mandibular complex, their (absolute and mutual) position, and growth pattern, as well as dental occlusion and facial appearance. The craniofacial dysmorphisms associated with the increased risk of POSA development are summarized in Figure 1. Extended facial profile and retrognathia have also been suggested to be more common in children with OSA; however, a recent systematic review by Fagundes et al. did not confirm this association (25).
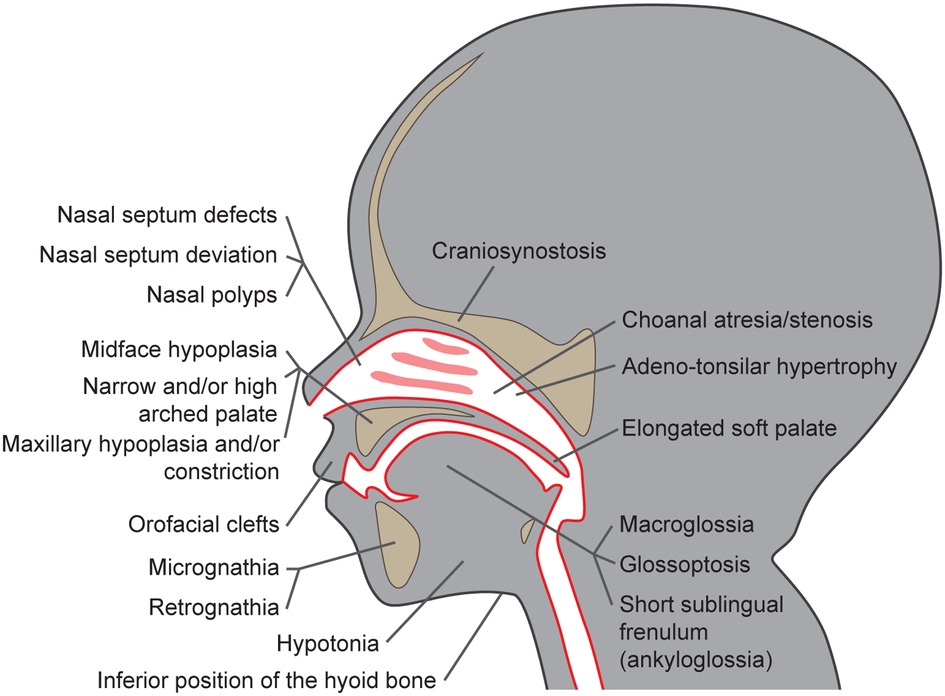
Figure 1. Craniofacial dysmorphisms as risk factors for pediatric obstructive sleep apnea development.
2.1. Skeletal anomalies in the orofacial area risky for POSA development
Premature bone fusion, craniosynostosis, is one of the key features playing role in the narrowing and easier collapse of the airways. It is often diagnosed together with midface hypoplasia, i.e., a combination of the underdevelopment of the maxilla, cheekbones, and eye sockets (although both these features may occur also independently). Even though these features are well-recognized factors in POSA development, the etiology is usually multifactorial and many children suffer from multilevel airway obstruction (24). Underdevelopment of the upper jaw, i.e., maxillary hypoplasia or, in the case of more pronounced narrowing, maxillary constriction, which are often associated also with narrow and/or high arched palate and lateral crossbite, are other characteristics often present in children suffering from POSA (19, 23, 24, 26). Severe reduction of the naso- and oro-pharyngeal airway space may be present in children with craniosynostosis, in patients with clefts originating from prenatal incomplete tissue fusion (20, 21, 27), or in those with anomalies of the mandible, especially if the mandible is undersized, (i.e., micrognathia; (19, 23, 24, 26). However, the underdevelopment of the maxillo-mandibular complex is not the only factor decreasing the airway patency. According to cephalometric studies, sagittal and vertical maxillo-mandibular complex discrepancies, such as mandibular retrognathia, often diagnosed as skeletal class II malocclusion, and increased overjet or open bite, which may appear due to increased mandibular plane angle, are overrepresented in children diagnosed with POSA (22, 26, 28, 29). This hyperdivergent skeletal pattern may lead to the development of the long-face syndrome, which is another facial appearance typical of patients with SRBD (28, 30, 31). Negative anterior overjet and skeletal class III malocclusion are not as often associated with POSA; still, the association is possible, especially if they are caused by severe maxillary deficiency (32). The lower position of the hyoid bone is another skeletal risk factor that can be diagnosed in a cephalogram. As some lingual muscles insert on that bone, their pull in a downward direction can also cause the narrowing of the airway space and, in effect, apnea (14, 33–35).
2.2. Soft tissue anomalies in the orofacial area risky for POSA development
The morphology of soft tissues plays an important role, too. Adeno-tonsillar hypertrophy is a well-described etiological factor of POSA. The deviation or deformity of the nasal septum, hypertrophy of nasal turbinates, or nasal polyps may also increase nasal resistance (36–38) and contribute towards mouth breathing, often accompanied by unphysiological head posture, insufficient lip seal or open bite, all of which are characteristics often present in patients with POSA (16). The lack of nasal breathing accompanied by an imbalance in muscle activity, often associated with the hypotony of orofacial muscles, have a huge impact on the development and growth of the maxillo-mandibular complex and may contribute to its abnormal shape and size (16, 39, 40).
The tongue is another factor playing a key role in the narrowing and collapse of the upper airway. The short sublingual frenulum (or ankyloglossia) in its most severe form leads to a low tongue position and disrupted tongue movement and has been already associated with the POSA phenotype (16, 41–43). An insufficient stimulation of the palatal suture, caused by this unphysiological tongue position, may result in the formation of a narrow palate and decreased volume of nasal cavities, which, again, contributes to the preference for mouth breathing and airway narrowing (16, 33). POSA has also a high prevalence in patients with glossoptosis, which is a down- and backward position of the base of the tongue (44). The combination of glossoptosis with micrognathia or retrognathia leads to a high risk of tongue-based airway obstruction (24, 45). In addition, macroglossia and/or an elongated soft palate could reduce airway volume and contribute to airway obstruction (14, 34).
These craniofacial characteristics are associated with several syndromes; however, they could be also found in non-syndromic children (14, 15, 19). Even though they are usually present in less severe forms, they could still contribute to airway obstruction. Craniofacial features associated with POSA could be easily diagnosed and their heritability is estimated to be high. This is especially true for the size of the maxillo-mandibular complex and the timing of its growth (11, 13, 46).
3. Craniofacial syndromes associated with a high prevalence of pediatric SRBD
The prevalence of SRBD, including POSA, may be very high in syndromic children with a severe form of craniofacial dysmorphism. In a population-based case-control study, an OSA diagnosis was associated with the presence of craniofacial anomalies, in particular with orofacial clefting and Down syndrome (46). To better understand the role of genetic factors in both POSA and craniofacial anomalies associated with this diagnosis, we have reviewed the current body of literature and selected syndromes, which: (1) are characterized by severe craniofacial abnormalities associated with POSA, (2) have a high prevalence, or have been already related to the co-incidence of SRBD and POSA in children, and (3) have a known genetic background.
Based on these criteria, 26 syndromes and disorders were selected, namely achondroplasia, Antley-Bixler, Apert, Auriculocondylar, Beare-Stevenson, Cohen, and Collins syndromes, congenital central hypoventilation, craniofacial microsomia (Goldenhar syndrome, oculo-auriculo-vertebral spectrum), craniofrontonasal dysplasia, Crouzon, Down, Ehlers-Danlos, Ellis-van Creveld, Jackson-Weiss, Marfan, and Marshall-Stickler syndromes, mucopolysaccharidosis IV and VI, Muenke, Noonan, abd Pfeiffer syndromes, Pierre Robin sequence, Prader-Willi, Saethre-Chotzen and Treacher-Collins syndromes. From the craniofacial dysmorphisms associated with OSA, craniosynostosis, oral clefts, midface and maxillary hypoplasia, narrow high-arched palate, micrognathia, retrognathia, choanal atresia, macroglossia, and glossoptosis were the features found most frequently in these syndromes (21, 4–70). It is necessary to mention that in syndromes associated with high POSA prevalence, a combination of several of these features is often present. For example, the Pierre Robin sequence associated with high POSA prevalence consists of the following: micrognathia, glossoptosis, narrow and/or high-arched palate, and cleft palate (45).
The information about the genetic background and prevalence of SRBD in these syndromes, including POSA, is summarized in Supplementary Table S1 in the Supplement. The prevalence of pediatric SRBD in children suffering from the mentioned syndromes ranges between 10%–87.5%, which is much higher than in the common pediatric population (2%–4%) (21, 45, 53, 54, 56, 58, 63, 69–87). High prevalences of SRBD were found particularly in populations of children with Treacher-Collins syndrome, mucopolysaccharidosis IV and VI, Apert, and Prader-Willi syndrome, in which limited midfacial development is a characteristic feature (21, 53, 54, 69, 70, 72). Despite their shared relationship to craniofacial dysmorphisms and high SRBD prevalence, these syndromic phenotypes are associated with different genes. In total, aneuploidy in Down syndrome and variations in 30 genes in the other 25 mentioned syndromes (see Supplementary Table S1 in the Supplement) are considered causative or risk factors for SRBD development.
4. Possible candidate genes for POSA development in children with craniofacial dysmorphisms
We prepared an overview of possible candidate genes and loci for pediatric SRBD. Figure 2 depicts genes and loci associated both with POSA in children without craniofacial features, and those associated with syndromes manifested by craniofacial features risky for SRBD in children (6–9).
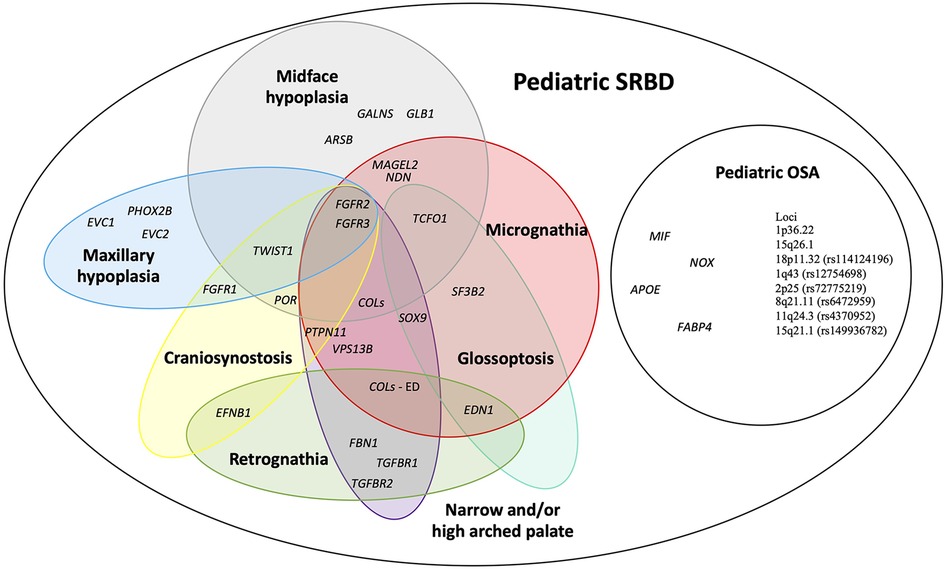
Figure 2. An overview of possible candidate genes for pediatric sleep-related breathing disorders (SRBD), especially pediatric obstructive sleep apnea (POSA) in children with craniofacial dysmorphisms. APOE, apolipoprotein E; ARSB, N-acetylgalactosamine-4 sulfatase; COLs (- ED), collagens gene family (genes associated with Ehlers-Danlos syndrome); EFNB1, ephrin-B1; EDN1, endothelin 1; EVC1, EvC ciliary complex subunit 1; EVC2, EvC ciliary complex subunit 2; FABP4, fatty acid-binding protein 4; FBN1, fibrillin 1; FGFR1, fibroblast growth factor receptor 1; FGFR2, fibroblast growth factor receptor 2; FGFR3, fibroblast growth factor receptor 3; GALNS, galactosamine-6-sulfatase; GLB1, b-D-galactosidase; MAGEL2, MAGE-like protein 2; MIF, macrophage migration inhibitory factor; NDN, necidin; NOX, NADPH oxidase 1; PHOX2B, paired like homeobox 2B; POR, cytochrome P450 oxidoreductase; PTPN11, protein tyrosine phosphatase non-receptor type 11; SF3B2, splicing factor 3B subunit 2; SOX9, SRY-box 9; SNORD116, CD box 116; TCOF1, treacle ribosome biogenesis factor 1; TGFBR1, transforming growth factor-β receptor 1; TGFBR2, transforming growth factor-β receptor 2; TWIST1, twist family bHLH transcription factor 1; VPS13B, vacuolar protein sorting 13 homolog B.
Although these syndromes do not share the same genetic background, some of the associated genes affect similar processes, such as the skeletal system development (including the cranial area), organ growth, or embryonic organ morphogenesis. Figure 3 demonstrates both known and predicted interactions and similarities among 30 considered genes; their functions and importance are described below.
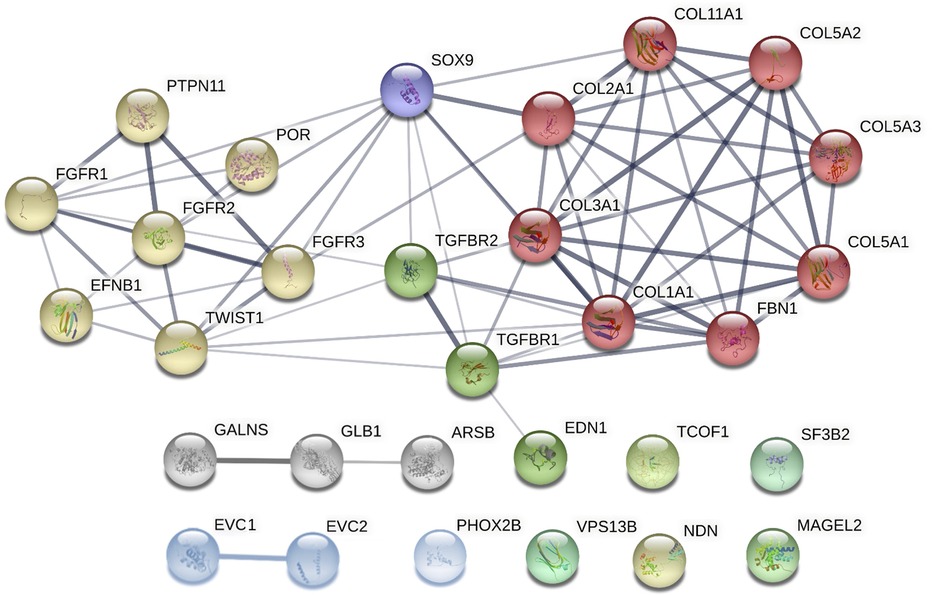
Figure 3. Clusters of candidate genes for pediatric obstructive sleep apnea (POSA) development in children with craniofacial dysmorphisms and their interactions (created in string, https://string-db.org/cgi/network?taskId = bF5B2GAcAUOE&sessionId = bSUqeCIqj6OL). Proteins encoded by these genes are clustered into 3 main groups using the Markov Clustering Algorithm. yellow dots: genes associated with non-/syndromic craniosynostosis. red, purple, and green dots: other genes associated with non-/syndromic retrognathia and/or micrognathia. grey and blue dots: other genes associated with non-/syndromic midface or maxillary hypoplasia. ARSB, N-acetylgalactosamine-4 sulfatase; COL1A1, collagen type I alpha 1 chain; COL2A1, collagen type II alpha 1 chain; COL3A1, collagen type III alpha 1 chain; COL5A1, collagen type V alpha 1 chain; COL5A2, collagen type V alpha 2 chain; COL5A3, collagen type V alpha 3 chain; COL11A1, collagen type XI alpha 1 chain; EFNB1, ephrin-B1; EDN1, endothelin 1; EVC1, EvC ciliary complex subunit 1; EVC2, EvC ciliary complex subunit 2; FBN1, fibrillin 1; FGFR1, fibroblast growth factor receptor 1; FGFR2, fibroblast growth factor receptor 2; FGFR3, fibroblast growth factor receptor 3; GALNS, galactosamine-6-sulfatase; GLB1, b-D-galactosidase; MAGEL2, MAGE-like protein 2; NDN, necidin; PHOX2B, paired like homeobox 2B; POR, cytochrome P450 oxidoreductase; PTPN11, protein tyrosine phosphatase non-receptor type 11; SF3B2, splicing factor 3B subunit 2; SOX9, SRY-box 9; SNORD116, CD box 116; TCOF1, treacle ribosome biogenesis factor 1; TGFBR1, transforming growth factor-β receptor 1; TGFBR2, transforming growth factor-β receptor 2; TWIST1, twist family bHLH transcription factor 1; VPS13B, vacuolar protein sorting 13 homolog B.
4.1. Genes associated with non-/syndromic craniosynostosis
The etiology of craniosynostosis may involve genetic, epigenetic, and/or environmental factors (88). Craniosynostosis is associated with a high prevalence of POSA. It is a common feature in patients with Antley-Bixler, Apert, Beare-Stevenson, Crouzon, Pfeiffer, Muenke, Jackson-Weiss, Craniofrontonasal, and Saethre-Chotzen syndromes (89–92). SRDB was present also in 50% of children suffering from non-syndromic craniosynostosis (NSC) (89).
Deviations in the development of the craniofacial area are also associated with a variability in the fibroblast growth factor receptor (FGFR) genes, which are important for cell specialization as well as for bone growth and modeling, especially in the process of ossification and bone fusion (48, 93, 94). Severe mutations in FGFR genes are associated with premature cranial bone fusion and craniosynostosis. These mutations were found in several craniofacial syndromes with a high prevalence of SRBD in children, such as achondroplasia, Antley-Bixler, Beare-Stevenson, Jackson-Weiss, Apert, Crouzon, Pfeiffer, and Saethre-Chotzen syndromes (21, 51, 92, 95–99). These FGFR-related craniosynostosis syndromes are autosomal-dominantly inherited.
Moreover, variants in FGFR genes could also lead to NSC (95, 100, 101). Genes most commonly mutated in familial craniosynostosis include, besides FGFR2 and FGFR3, the twist family bHLH transcription factor 1 (TWIST1) and ephrin-B1 (EFNB1) (102). More than 100 mutations in the EFNB1 gene have been found to cause the craniofrontonasal syndrome, which was confirmed in a study with knockout mice models (103). This rare x-linked disorder shows paradoxically greater severity in heterozygous females than in hemizygous males. TWIST1 acts through Eph–ephrin interactions to regulate the development of the boundary that forms the coronal suture (104). The TWIST1 gene associated with the Saethre-Chotzen syndrome is believed to regulate bone formation through other genes, such as FGFR and RUNX2 (63, 64). Genetic testing of FGFR1, FGFR2, FGFR3, and TWIST1 was even suggested as a first-line test for patients with NSC (101).
Interestingly, not only rare mutations of these genes but also SNPs of these genes are associated with craniofacial dysmorphia. For example, Da Fontoura et al. found an association between SNPs rs11200014 and rs2162540 in FGFR2 and sagittal maxilla-mandibular discrepancy, so-called skeletal malocclusion (both skeletal class II and III). They also found an association between the SNP rs2189000 in TWIST1 and a larger body and shorter ramus of the mandible (62). Although FGFR3 gene variants are associated with Muenke and Crouzon syndromes manifested by craniosynostosis, this feature, surprisingly, was not exhibited in the FGFR3 A385E/+ mice model (105–107). Thus, FGFR2 seems to be more important for craniosynostosis development than FGFR3. On the other hand, a mutation in FGFR3 causes achondroplasia, which, according to a recent study by Legare et al., has craniosynostosis as a co-occurring feature (108).
A missense mutation in the Protein Tyrosine Phosphatase Non-Receptor Type 11 (PTPN11) gene was found in almost 50% of patients diagnosed with Noonan syndrome (109). This gene encodes tyrosine phosphatase Shp-2, an enzyme involved in multiple signal transduction cascades including receptors for growth factors involved in the developmental processes, such as FGFR (110). The Noonan syndrome is manifested by micrognathia, maxillomandibular discrepancy, narrow and/or high-arched palate, and long face syndrome (hyperdivergence) (111, 112). Also, craniosynostosis was described in some patients suffering from this syndrome. Mutations in the PTPN11, KRAS, or Leucine-Rich Repeat Scaffold Protein (SHOC2) gene are causally involved in craniosynostosis (113–115). In patients with the Antley-Bixler syndrome, characterized by craniosynostosis, brachycephaly, midface hypoplasia, and choanal atresia and/or stenosis, variants have been found not only in FGFR2, but also in the gene encoding cytochrome p450 oxidoreductase (POR) (116–119). This enzyme transfers electrons from NADPH to all microsomal cytochrome P450 enzymes. While individuals with an ABS-like phenotype and normal steroidogenesis are carriers of FGFR2 mutations, those with genital anomalies and disordered steroidogenesis should be recognized as having a POR deficiency (116).
4.2. Genes associated with non-/syndromic retrognathia and/or micrognathia
The SRY-box 9 transcription factor (SOX9) gene plays an important regulatory role during craniofacial development (120). In a rat model with upper airway obstruction, SOX9 level was found to be downregulated, which explains the bone architecture abnormalities (121). This gene is also associated with the Pierre Robin sequence (122). Repressed SOX9 expression leads to changes in the expression of genes essential for normal development of the mandible, causing micrognathia, and, consequently, glossoptosis, airway obstruction, and often, cleft palate (123). The expression of SOX9 is influenced, among others, by FGFR3. Therefore, dysregulation of SOX9 levels, a major regulator of chondrogenesis, is an important underlying mechanism in skeletal diseases caused by mutations in FGFR3 (124–126). Interestingly, the SNP rs12941170 of SOX9 was associated with non-syndromic orofacial clefting. However, its role in these non-syndromic clefts remains unclear (124).
Similarly to SOX9, variants in endothelin 1 (EDN1), the Splicing factor 3B subunit (SF3B2), and Treacle ribosome biogenesis factor 1 (TCOF1) were associated with syndromes manifesting in children by both micrognathia and glossoptosis. EDN1 encodes a vasoactive peptide belonging to the family of endothelins and is associated with the auriculocondylar syndrome (127), a rare syndrome that usually affects facial features. It is characterized by micrognathia, microstomia, and anomalies in the temporomandibular joint and the condyle (127). Also, studies using mice models with the EDN1 gene knocked out or deficient have shown several craniofacial dysmorphisms, mandibular dysfunction, and severe retrognathism (62, 127). SF3B2 may be, according to a study by Timberlake et al., an important factor in the development of craniofacial microsomia, which was also confirmed by a recent review covering this congenital facial anomaly (128, 129). TCOF1 presents an important factor for the undisrupted formation and development of the craniofacial area, cartilage, and skeleton (55, 130, 131). Mutations in these genes were found in patients with Treacher-Collins syndrome (130, 131).
Ehlers-Danlos syndrome, manifesting through retrognathia, micrognathia, and maxillary constriction, has been previously proposed as a genetic model for pediatric OSA (60, 61, 132). Variants in genes encoding and/or influencing the expression of collagens (COL gene family) and others (see Supplementary Table S1 in the Supplement) were associated with this rare connective tissue disorder (60, 133). The minor allele of SNP rs2249492 in the Collagen type I alpha 1 chain (COL1A1) has been previously associated with the increased risk of a sagittal maxilla-mandibular discrepancy (skeletal class III malocclusion) in non-syndromic children (62). The results of the study by Topârcean et al. showed a tendency towards a class II skeletal malocclusion pattern determined by mandibular retrognathism rather than maxillary prognathism among the individuals possessing the mutant allele of this SNP (134).
Other genes for collagens, COL2A1 and COL11A1, are associated with Marshall-Stickler syndrome (86). Collagens II and XI are present throughout the Meckel's cartilage, which provides mechanical support for the developing mandible. The characteristic craniofacial features of Marshall-Stickler syndrome are midface hypoplasia, micrognathia, cleft palate, and Pierre Robin anomaly (50). Variants in COL2A1 and COL11A1 were also associated with the Robin sequence in nonsyndromic patients (135).
Besides collagens, fibrillin and elastin are also present in the architectural scaffolds that impart specific mechanical properties to tissues and organs. The FBN1 gene is essential for the production of fibrillin, and its mutation could cause Marfan syndrome (57, 136).
Fibrillin is crucial for bone and muscle rigidity; hence, its disruption can increase the laxity of airway connective tissues and predispose them to easier collapsibility (56). At the same time, patients often have their maxillo-mandibular complex in a retrognathic position, with a narrow maxilla and palate, and a “long face” appearance (56–58, 137).
Besides FBN1, mutations in the transforming growth factor-β receptor 1 (TGFBR1) and transforming growth factor-β receptor 2 (TGFBR2) may also be found in Marfan syndrome (138). TGFBR2 protein forms a complex with TGFBR1, and both are involved in a signaling pathway responsible for the proliferation, differentiation, and apoptosis of cells throughout the body (139). They are extremely important for bone growth and extracellular matrix formation; moreover, they play a role in the fusion of craniofacial sutures (140). The development of micrognathia and retrognathism was observed in mice with an impaired TGFB2 gene, giving evidence to its importance in craniofacial morphology (141).
The gene for vacuolar protein sorting-associated protein 13B (VPS13B), also called the COH1 gene, encodes a protein forming a part of the Golgi apparatus membrane. Its disruption may be involved through various cellular mechanisms, in several clinical features of Cohen syndrome (142, 143), including micrognathia, constricted hard palate, insufficient lip seal, and truncal obesity. All of these issues increase the risk of the collapse of the upper airway and the development of POSA (65, 66, 142, 143).
Mutations in necidin (NDN) and the melanoma antigen family member L2 (MAGEL2), both localized on chromosome 15, were found in the Prader-Willi syndrome, a complex genetic disorder characterized by several features, such as midface hypoplasia and micrognathia (144). The phenotype of this syndrome includes hypoplastic midface area, hypotonia, and a changed viscosity in secretions. All these factors facilitate the collapse of upper airways and apnea (52, 53, 144). Some polymorphisms in NDN were determined in extremely obese German children and adolescents as well as in neonates examined by polysomnography. However, there was a lack of association with juvenile-onset human obesity or sleep and respiratory parameters (145, 146).
4.3. Genes associated with non-/syndromic midface or maxillary hypoplasia
Midface or maxillary hypoplasia are typical features of several syndromes, including mucopolysaccharidosis, Ellis-van Creveld, or congenital central hypoventilation syndrome, the genetic backgrounds of which are described below.
Mucopolysaccharidosis (MPS) is a metabolic disorder characterized by the deficiency or total absence of enzymes responsible for the degradation of glycosaminoglycans. It can be classified into 7 types based on the specific malfunctioning enzyme and clinical manifestations (147). The Morquio syndrome (MPS IV) can be caused by a mutation either in the N-acetylgalactosamine-6-sulfatase (GALNS) gene (MPS type IVA), or in the gene for galactosidase beta 1 (GLB1; MPS type IVB). Among other clinical manifestations, the Morquio syndrome includes also craniofacial dysmorphisms such as mid-facial hypoplasia, condylar deformities, open bite, macroglossia, or abnormal teeth (148, 149).
The mutated gene for arylsulfatase B (ARSB) leads to the reduced function of the enzyme, causing a lysosomal storage disorder – MPS type VI, also known as Maroteaux-Lamy syndrome (117). This syndrome is associated with orofacial manifestations such as macroglossia, malocclusions, or disrupted dental eruption (150).
In syndromic children, the TWIST gene and the genes of the FGFR family, described in detail above, were associated with maxillary hypoplasia. In addition, the EvC ciliary complex subunit 1 (EVC1) and subunit 2 (EVC2) genes were found to be causative for the formation of the Ellis-van Creveld syndrome manifested also by maxillary hypoplasia and mandibular prognathism (151, 152). They encode proteins, the functions of which are not completely understood yet, but appear to be important in the physiological growth and development of bones and teeth (153).
The paired-like homeobox 2B (PHOX2B) transcription factor plays a crucial role in the autonomic nervous system development. Mutations in the PHOX2B gene are known to cause the congenital central hypoventilation syndrome with a specific craniofacial phenotype – maxillary hypoplasia, box-shaped face, and brachycephaly (68). However, a “silent” mutation in this gene was found in children with class III skeletal malocclusion and a history of sleep apnea (63, 154, 155).
5. Discussion
As POSA may cause serious health problems in young, growing patients, it would be highly beneficial to diagnose the increased risk of its development as soon as possible. While much of the POSA etiopathogenesis remains underexplored, craniofacial dysmorphisms leading to the narrowing of the airways undoubtedly play an important role (15, 17). Their severe forms can be found in craniofacial syndromes, which are also associated with a much higher prevalence of SRBD and POSA compared to the general pediatric population (21, 45, 53, 54, 56, 58, 63, 69–87). However, similar craniofacial features may be present also in healthy, non-syndromic patients. These skeletal variations could be mild when compared to syndromic phenotypes, but they could still lead to the collapse of the upper airways and POSA development. This is supported by Kim et al. who reported that the majority of non-syndromic, non-obese children diagnosed with POSA have craniofacial anomalies that are possible risk factors for POSA (18).
Several studies have already explored genes associated with OSA etiopathogenesis in adults, including the genes associated with the craniofacial area and characteristic features (4, 156). The heritability of craniofacial traits varies but is generally estimated to be high and very similar in healthy subjects and in patients suffering from OSA (11, 13, 46, 157). This is supported by several studies reporting an increased incidence of the above-mentioned OSA risk features among relatives (157–160). The first phenome-wide association study of genomic variation in adult OSA was recently published by Veatch et al. (5). None of the three SNPs in the leptin receptor (LEPR), the matrix metallopeptidase 9 (MMP9), and the Gamma-aminobutyric acid type B receptor subunit 1 (GABBR1), the association of which with OSA diagnosis was validated in their study, was associated with other non-OSA clinical traits once they controlled for multiple testing (5).
Cade at al. performed a GWAS investigating genetic associations of OSA in Hispanic/Latino Americans from three cohorts. They identified two loci (rs11691765 in the G protein-coupled receptor 83 gene, GPR83; and rs35424364 in the pseudogene CCDC162P) associated with the AHI and the respiratory event duration, respectively (3). Another GWAS study, focusing on European Caucasians, reported five genes to be associated with facial characteristics, namely the paired-box gene 3 (PAX3), the PR-set domain 16 (PRDM16), the transcription factor TP63, small integral membrane protein 23 (C5orf50), and the Collagen type XVII alpha 1 chain (COL17A1A), the variants of which contribute to the facial morphology in young adults (4). Some variants of these genes and their possible association with craniofacial abnormalities were also explored in another GWAS study focusing on young adults of European-ancestry from the Avon Longitudinal Study of Parents and Children (161) as well as in mice models (162, 163).
Unfortunately, most publications focus on the genetic background of OSA in adult patients, not the pediatric population. To this date, only one GWAS has been performed in relation to POSA, including European American and African American children without craniofacial disorders (6). The study identified several genomic loci (see the Introduction). However, only one genetic marker, located at 18p11.32, was shared by groups of both ancestries. Their study, therefore, emphasizes the importance of study populations with diverse ethnic backgrounds to identify unique and shared genetic markers that contribute to the heterogeneity of POSA (6).
It follows that specific genes involved in the development of the orofacial area and associated with craniofacial OSA features should be also considered as candidate genes for POSA. Here, we provide an overview of genes that are known to be involved in the development of craniofacial syndromes in children with high SRBD prevalence, including POSA, see Supplementary Table S1 in the Supplement. All these genes are, to some extent, involved in the formation of tissues of the orofacial area. The candidate genes for POSA can be classified into three major groups based on their involvement in the development of specific craniofacial features. These groups would consist of genes associated with non-/syndromic (i) craniosynostosis, (ii) retrognathia and/or micrognathia, and (iii) midface or maxillary hypoplasia. While certain mutations cause various rare syndromes, other variants in these same genes were suggested to be associated with non-syndromic skeletal variations in the orofacial area (62, 63, 101, 154, 164). So far, variants in FGFR1, FGFR2, FGFR3, TWIST, SOX9, COL1A1, and PHOX2B are known to play a role in syndrome development as well as in the development of skeletal malocclusions (sagittal maxillo-mandibular complex discrepancies in non-syndromic patients). These genes, therefore, can be considered promising candidate genes for testing of genetic susceptibility to POSA development in various populations.
Although the inheritance pattern of POSA as well as OSA is unclear, most cases with these diseases do not adhere to classical models of inheritance, suggesting that multiple genes could be involved in their development. We believe that besides the GWAS approach, strategies based on candidate genes are also necessary for further research of both these multifactorial diseases. Considering the results of the mentioned genetic association studies (3–9) it appears that there is not much overlap between candidate variants/genes for the POSA and OSA development. In addition, these studies also revealed a high interpopulation variability that should be taken into account in the further research of these disorders. The low match in candidate genes for OSA between children and adults is to be expected since those diseases differ in their etiopathogenesis, clinical presentation as well as polysomnographic characteristics; there are also major differences in therapy approaches and possible consequences if left untreated (165).
Recently, Yoon et al. proposed a clinical guideline for application of multidisciplinary care in children with SRBD, emphasizing the importance of dentofacial interventions that target variable growth patterns (166). In the last years, craniofacial modification by orthodontic techniques is increasingly incorporated into the multidisciplinary management of SRBD in children and adolescents. In view of the multifactorial etiology of POSA, a better understanding of the risk factors contributing to its development may be useful not only for predicting the risk of POSA development but, even more importantly, for selecting the best therapeutic approach. Research of genetic predispositons to OSA in children as well as in adults may improve our understanding of the underlying biological mechanisms of susceptibility to these diseases.
6. Conclusion
Genetic background plays an important role in both POSA and craniofacial dysmorphisms. Therefore, genes associated with specific craniofacial features more common in patients suffering from POSA may be also considered candidate genes for this disease. We have reviewed a large body of literature and focused on the genes known to be involved in the development of cranio-facial syndromes with a high POSA prevalence. Based on the review, we chose 30 candidate genes for pediatric SRBD. Variants in seven of them (FGFR1, FGFR2, FGFR3, TWIST, SOX9, COL1A1, and PHOX2B) are known to play a role not only in syndrome development but also in skeletal malocclusions that are typical of pediatric orthodontic patients. Considering this, these seven genes appear to have the highest potential for targeted analysis of POSA risk in non-syndromic children.
Author contributions
ZMV: as an orthodontist specialized in sleep medicine, performed the literature search, co-wrote this review, and prepared the Figure 1. JK: critically reviewed the article from the perspective of a specialist in the morphology of the orofacial area and prepared the Figure 1. ZD and JZ: critically reviewed the article from the perspective of maxillofacial surgeons. AB and LIH: critically reviewed the article from the perspective of an orthodontist and pediatric dentist, respectively. JH: critically reviewed the article from the perspective of a specialist in craniofacial genetics and orthodontic treatment. PBL: as a specialist in molecular genetics and complex diseases, performed the literature search, co-wrote this review, and prepared Figures 2, 3. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Ministry of Health of the Czech Republic (grant no. NV17-30439A). All rights reserved. This work was supported by a project provided by the Faculty of Medicine Masaryk University Brno MUNI/A/1445/2021 and by a project provided by the University Hospital Brno, Ministry of Health of the Czech Republic – RVO (FNBr, 65269705). The study was created as part of an internal grant supported by the St. Anne’s University Hospital Brno. This publication has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under grant agreement No. 857560. This publication reflects only the authoŕs view and the European Commission is not responsible for any use that may be made of the information it contains. Authors also thank the Research Infrastructure RECETOX RI (grant no LM2023069) and the project CETOCOEN EXCELLENCE (grant no CZ.02.1.01/0.0/0.0/17_043/0009632) financed by the Ministry of Education, Youth and Sports for supportive background. The article was supported by a grant from the Czech Orthodontic Society.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2023.1117493/full#supplementary-material
References
1. Xu Z, Wu Y, Tai J, Feng G, Ge W, Zheng L, et al. Risk factors of obstructive sleep apnea syndrome in children. J Otolaryngol Head Neck Surg. (2020) 49:11. doi: 10.1186/s40463-020-0404-1
2. Xiao L, Su S, Liang J, Jiang Y, Shu Y, Ding L. Analysis of the risk factors associated with obstructive sleep apnea syndrome in Chinese children. Front Pediatr. (2022) 10:1032. doi: 10.3389/fped.2022.900216
3. Cade BE, Chen H, Stilp AM, Gleason KJ, Sofer T, Ancoli-Israel S, et al. Genetic associations with obstructive sleep apnea traits in hispanic/latino Americans. Am J Respir Crit Care Med. (2016) 194:886–97. doi: 10.1164/rccm.201512-2431OC
4. Liu F, van der Lijn F, Schurmann C, Zhu G, Chakravarty MM, Hysi PG, et al. A genome-wide association study identifies five loci influencing facial morphology in Europeans. PLoS Genet. (2012) 8:e1002932. doi: 10.1371/journal.pgen.1002932
5. Veatch OJ, Bauer CR, Keenan BT, Josyula NS, Mazzotti DR, Bagai K, et al. Characterization of genetic and phenotypic heterogeneity of obstructive sleep apnea using electronic health records. BMC Med Genomics. (2020) 13:1–14. doi: 10.1186/s12920-020-00755-4
6. Quinlan CM, Chang X, March M, Mentch FD, Qu H-Q, Liu Y, et al. Identification of novel loci in obstructive sleep apnea in European American and African American children. Sleep. (2022). doi: 10.1093/sleep/zsac182
7. Gozal D, Khalyfa A, Capdevila OS, Kheirandish-Gozal L, Khalyfa AA, Kim J. Cognitive function in prepubertal children with obstructive sleep apnea: a modifying role for nadph oxidase P22 subunit gene polymorphisms? Anttioxid Redox Signal. (2012) 16:171–7. doi: 10.1089/ars.2011.4189
8. Bhushan B, Khalyfa A, Spruyt K, Kheirandish-Gozal L, Capdevila OS, Bhattacharjee R, et al. Fatty-acid binding protein 4 gene polymorphisms and plasma levels in children with obstructive sleep apnea. Sleep Med. (2011) 12:666–71. doi: 10.1016/j.sleep.2010.12.014
9. Khalyfa A, Kheirandish-Gozal L, Capdevila OS, Bhattacharjee R, Gozal D. Macrophage migration inhibitory factor gene polymorphisms and plasma levels in children with obstructive sleep apnea. Pediatric Pulmonol. (2012) 47:1001–11. doi: 10.1002/ppul.22560
10. Taheri S, Mignot E. The genetics of sleep disorders. Lancet Neurol. (2002) 1:242–50. doi: 10.1016/s1474-4422(02)00103-5
11. Casale M, Pappacena M, Rinaldi V, Bressi F, Baptista P, Salvinelli F. Obstructive sleep apnea syndrome: from phenotype to genetic basis. Curr Genomics. (2009) 10:119–26. doi: 10.2174/138920209787846998
12. Gaultier C, Guilleminault C. Genetics, control of breathing, and sleep-disordered breathing: a review. Sleep Med. (2001) 2:281. doi: 10.1016/s1389-9457(01)00098-3
13. Redline S, Tishler PV. The genetics of sleep apnea. Sleep Med Rev. (2000) 4:583–602. doi: 10.1053/smrv.2000.0120
14. Johal A, Patel SI, Battagel JM. The relationship between craniofacial anatomy and obstructive sleep apnoea: a case-controlled study. J Sleep Res. (2007) 16:319–26. doi: 10.1111/j.1365-2869.2007.00599.x
15. Capistrano A, Cordeiro A, Capelozza Filho L, Almeida VC, Martinez S, Almeida-Pedrin R. Facial morphology and obstructive sleep apnea. Dental Press J Orthod. (2015) 20:60–7. doi: 10.1590/2177-6709.20.6.060-067.oar
16. Guilleminault C, Huang YS. From oral facial dysfunction to dysmorphism and the onset of pediatric OSA. Sleep Med Rev. (2018) 40:203–14. doi: 10.1016/j.smrv.2017.06.008
17. Canepa I, Elshebiny T, Valiathan M. OSA prevalence in a general pediatric craniofacial population. J Dent Sleep Med. (2019) 6. doi: 10.15331/jdsm.7084
18. Kim JH, Guilleminault C. The nasomaxillary complex, the mandible, and sleep-disordered breathing. Sleep Breath. (2011) 15:185–93. doi: 10.1007/s11325-011-0504-2
19. Hansen C, Markström A, Sonnesen L. Specific dento-craniofacial characteristics in non-syndromic children can predispose to sleep-disordered breathing. Acta Paediatr. (2022) 111:473–7. doi: 10.1111/apa.16202
20. Moore MH. Upper airway obstruction in the syndromal craniosynostoses. Br J Plast Surg. (1993) 46:355–62. doi: 10.1016/0007-1226(93)90039-e
21. Inverso G, Brustowicz K, Katz E, Padwa B. The prevalence of obstructive sleep apnea in symptomatic patients with syndromic craniosynostosis. Int J Orral Maxillofac Surg. (2016) 45:167–9. doi: 10.1016/j.ijom.2015.10.003
22. Lee Y-H, Huang Y-S, Chen I-C, Lin P-Y, Chuang L-C. Craniofacial, dental arch morphology, and characteristics in preschool children with mild obstructive sleep apnea. J Dent Sci. (2020) 15:193–9. doi: 10.1016/j.jds.2019.09.005
23. Katyal V, Pamula Y, Daynes CN, Martin J, Dreyer CW, Kennedy D, et al. Craniofacial and upper airway morphology in pediatric sleep-disordered breathing and changes in quality of life with rapid maxillary expansion. Am J Orthod Dentofacial Orthop. (2013) 144:860–71. doi: 10.1016/j.ajodo.2013.08.015
24. Tan H-L, Kheirandish-Gozal L, Abel F, Gozal D. Craniofacial syndromes and sleep-related breathing disorders. Sleep Med Rev. (2016) 27:74–88. doi: 10.1016/j.smrv.2015.05.010
25. Fagundes NCF, Gianoni-Capenakas S, Heo G, Flores-Mir C. Craniofacial features in children with obstructive sleep apnea: a systematic review and meta-analysis. J Clin Sleep Med. (2022) 18(7):1865–75. doi: 10.5664/jcsm.9904
26. Aroucha Lyra MC, Aguiar D, Paiva M, Arnaud M, Filho AA, Rosenblatt A, et al. Prevalence of sleep-disordered breathing and associations with malocclusion in children. JJ Clin Sleep Med. (2020) 16:1007–12. doi: 10.5664/jcsm.8370
27. Marino A, Malagnino I, Ranieri R, Villa MP, Malagola C. Craniofacial morphology in preschool children with obstructive sleep apnoea syndrome. Eur J Paediatr Dent. (2009) 10:181–4.20073543
28. Caprioglio A, Zucconi M, Calori G, Troiani V. Habitual snoring, OSA and craniofacial modification. Orthodontic clinical and diagnostic aspects in a case control study. Minerva Stomatol. (1999) 48:125–37.10431534
29. Kawashima S, Niikuni N, Chia-hung L, Takahasi Y, Kohno M, Nakajima I, et al. Cephalometric comparisons of craniofacial and upper airway structures in young children with obstructive sleep apnea syndrome. Ear Nose Throat J. (2000) 79:499–506. doi: 10.1177/014556130007900708
30. Zucconi M, Caprioglio A, Calori G, Ferini-Strambi L, Oldani A, Castronovo C, et al. Craniofacial modifications in children with habitual snoring and obstructive sleep apnoea: a case-control study. Eur Respir J. (1999) 13:411–7. doi: 10.1183/09031936.99.13241199
31. Pacheco MC, Fiorott BS, Finck NS, Araújo MT. Craniofacial changes and symptoms of sleep-disordered breathing in healthy children. Dental Press J Orthod. (2015) 20:80–7. doi: 10.1590/2176-9451.20.3.080-087.oar
32. Coban G, Buyuk SK. Sleep disordered breathing and oral health-related quality of life in children with different skeletal malocclusions. Cranio. (2022) 40:1–8. doi: 10.1080/08869634.2022.2080960
33. Huang YS, Guilleminault C. Pediatric obstructive sleep apnea and the critical role of oral-facial growth: evidences. Front Neurol. (2012) 3:184. doi: 10.3389/fneur.2012.00184
34. Lee RW, Sutherland K, Cistulli PA. Craniofacial morphology in obstructive sleep apnea: a review. Clin Pulm Med. (2010) 17:189–95. doi: 10.1097/CPM.0b013e3181e4bea7
35. Pirilä-Parkkinen K, Löppönen H, Nieminen P, Tolonen U, Pirttiniemi P. Cephalometric evaluation of children with nocturnal sleep-disordered breathing. Eur J Orthod. (2010) 32:662–71. doi: 10.1093/ejo/cjp162
36. Marcus CL. Pathophysiology of childhood obstructive sleep apnea: current concepts. Respir Physiol. (2000) 119:143–54. doi: 10.1093/ejo/cjp162
37. Edwards BA, Eckert DJ, Jordan AS. Obstructive sleep apnoea pathogenesis from mild to severe: is it all the same? Respirology. (2017) 22:33–42. doi: 10.1111/resp.12913
38. Yeom SW, Kim MG, Lee EJ, Chung SK, Kim DH, Noh SJ, et al. Association between septal deviation and OSA diagnoses: a nationwide 9-year follow-up cohort study. J Clin Sleep Med. (2021) 17:2099–106. doi: 10.5664/jcsm.9352
39. Pirilä-Parkkinen K, Pirttiniemi P, Nieminen P, Tolonen U, Pelttari U, Löppönen H. Dental arch morphology in children with sleep-disordered breathing. Eur J Orthod. (2009) 31:160–7. doi: 10.1093/ejo/cjn061
40. Guilleminault C, Akhtar F. Pediatric sleep-disordered breathing: new evidence on its development. Sleep Med Rev. (2015) 24:46–56. doi: 10.1016/j.smrv.2014.11.008
41. Guilleminault C, Huseni S, Lo L. A frequent phenotype for paediatric sleep apnoea: short lingual frenulum. ERJ Open Res. (2016) 2:00043-2016. doi: 10.1183/23120541.00043-2016
42. Villa MP, Evangelisti M, Barreto M, Cecili M, Kaditis A. Short lingual frenulum as a risk factor for sleep-disordered breathing in school-age children. Sleep Med. (2020) 66:119–22. doi: 10.1016/j.sleep.2019.09.019
43. Huang Y, Quo S, Berkowski J, Guilleminault C. Short lingual frenulum and obstructive sleep apnea in children. Int J Pediatr Res. (2015) 1:273. doi: 10.23937/2469-5769/1510003
44. Schweiger C, Manica D, Kuhl G. Glossoptosis. Semin Pediatr Surg. (2016) 25(3):123–7. doi: 10.1053/j.sempedsurg.2016.02.002
45. Cielo CM, Marcus CL. Obstructive sleep apnoea in children with craniofacial syndromes. Paediatr Respir Rev. (2015) 16:189–96. doi: 10.1016/j.prrv.2014.11.003
46. Lam DJ, Jensen CC, Mueller BA, Starr JR, Cunningham ML, Weaver EM. Pediatric sleep apnea and craniofacial anomalies: a population-based case–control study. Laryngoscope. (2010) 120:2098–105. doi: 10.1002/lary.21093
47. Onodera K, Niikuni N, Chigono T, Nakajima I, Sakata H, Motizuki H. Sleep disordered breathing in children with achondroplasia. Part 2. relationship with craniofacial and airway morphology. Int J Pediatr Otorhinolaryngol. (2006) 70:453–61. doi: 10.1016/j.ijporl.2005.07.016
48. Vajo Z, Francomano CA, Wilkin DJ. The molecular and genetic basis of fibroblast growth factor receptor 3 disorders: the achondroplasia family of skeletal dysplasias, muenke craniosynostosis, and crouzon syndrome with acanthosis Nigricans. Endocr Rev. (2000) 21:23–39. doi: 10.1210/edrv.21.1.0387
49. Zaffanello M, Antoniazzi F, Tenero L, Nosetti L, Piazza M, Piacentini G. Sleep-disordered breathing in paediatric setting: existing and upcoming of the genetic disorders. Ann Transl Med. (2018) 6:343. doi: 10.21037/atm.2018.07.13
50. Carinci F, Pezzetti F, Locci P, Becchetti E, Carls F, Avantaggiato A, et al. Apert and crouzon syndromes: clinical findings, genes and extracellular matrix. J Craniofac Surg. (2005) 16:361–8. doi: 10.1097/01.scs.0000157078.53871.11
51. Lajeunie E, Heuertz S, El Ghouzzi V, Martinovic J, Renier D, Le Merrer M, et al. Mutation screening in patients with syndromic craniosynostoses indicates that a limited number of recurrent Fgfr2 mutations accounts for severe forms of pfeiffer syndrome. Eur J Hum Genet. (2006) 14:289–98. doi: 10.1038/sj.ejhg.5201558
52. Lin HY, Lin SP, Lin CC, Tsai LP, Chen MR, Chuang CK, et al. Polysomnographic characteristics in patients with prader–willi syndrome. Pediatr Pulmonol. (2007) 42:881–7. doi: 10.1002/ppul.20673
53. Sedky K, Bennett DS, Pumariega A. Prader willi syndrome and obstructive sleep apnea: co-occurrence in the pediatric population. J Clin Sleep Med. (2014) 10:403–9. doi: 10.5664/jcsm.3616
54. Akre H, Øverland B, Åsten P, Skogedal N, Heimdal K. Obstructive sleep apnea in treacher collins syndrome. Eur Arch Otorhinolaryngol. (2012) 269:331–7. doi: 10.1007/s00405-011-1649-0
55. Ma X, Forte AJ, Persing JA, Alonso N, Berlin NL, Steinbacher DM. Reduced three-dimensional airway volume is a function of skeletal dysmorphology in treacher collins syndrome. Plast Reconstr Surg. (2015) 135:382e–92e. doi: 10.1097/PRS.0000000000000993
56. Cistulli PA, Sullivan CE. Sleep apnea in marfan’s syndrome: increased upper airway collapsibility during sleep. Chest. (1995) 108:631–5. doi: 10.1378/chest.108.3.631
57. De Coster P, Pauw GD, Martens L, De Paepe A. Craniofacial structure in marfan syndrome: a cephalometric study. Am J Med Genet A. (2004) 131:240–8. doi: 10.1002/ajmg.a.30393
58. Sedky K, Gaisl T, Bennett DS. Prevalence of obstructive sleep apnea in joint hypermobility syndrome: a systematic review and meta-analysis. J Clin Sleep Med. (2019) 15:293–9. doi: 10.5664/jcsm.7636
59. Robin P. Glossoptosis due to atresia and hypotrophy of the mandible. Am J Dis Child. (1934) 48:541–7.
60. Guilleminault C, Primeau M, Chiu HY, Yuen KM, Leger D, Metlaine A. Sleep-disordered breathing in Ehlers-Danlos syndrome: a genetic model of OSA. Chest. (2013) 144:1503–11. doi: 10.1378/chest.13-0174
61. Malfait F, Wenstrup RJ, De Paepe A. Clinical and genetic aspects of Ehlers-Danlos syndrome, classic type. Genet Med. (2010) 12:597–605. doi: 10.1097/GIM.0b013e3181eed412
62. da Fontoura CS, Miller SF, Wehby GL, Amendt BA, Holton NE, Southard TE, et al. Candidate gene analyses of skeletal variation in malocclusion. J Dent Res. (2015) 94:913–20. doi: 10.1177/0022034515581643
63. Hartsfield JK Jr, Morford LA, Jacob GJ, Kluemper GT. Genetic factors affecting facial morphology associated with sleep apnea.
64. Zhang Y, Blackwell EL, McKnight MT, Knutsen GR, Vu WT, Ruest LB. Specific inactivation of twist1 in the mandibular arch neural crest cells affects the development of the ramus and reveals interactions with Hand2. Dev Dyn. (2012) 241:924–40. doi: 10.1002/dvdy.23776
65. Cohen MM Jr. A new syndrome with hypotonia, obesity, mental deficiency, and facial, oral, ocular, and limb anomalies. J Pediatr. (1973) 83:280–4. doi: 10.1016/s0022-3476(73)80493-7
66. Chandler K, Kidd A, Al-Gazali L, Kolehmainen J, Lehesjoki A-E, Black GC, et al. Diagnostic criteria, clinical characteristics, and natural history of cohen syndrome. J Med Genet. (2003) 40:233–41. doi: 10.1136/jmg.40.4.233
67. Kurihara Y, Kurihara H, Suzuki H, Kodama T, Maemura K, Nagai R, et al. Elevated blood pressure and craniofaclal abnormalities in mice deficient in endothelin-1. Nature. (1994) 368:703–10. doi: 10.1038/368703a0
68. Todd ES, Weinberg SM, Berry-Kravis EM, Silvestri JM, Kenny AS, Rand CM, et al. Facial phenotype in children and young adults with Phox2b-determined congenital central hypoventilation syndrome: quantitative pattern of dysmorphology. Pediatr Res. (2006) 59:39–45. doi: 10.1203/01.pdr.0000191814.73340.1d
69. Berger KI, Fagondes SC, Giugliani R, Hardy KA, Lee KS, McArdle C, et al. Respiratory and sleep disorders in mucopolysaccharidosis. J Inherit Metab Dis. (2013) 36:201–10. doi: 10.1007/s10545-012-9555-1
70. Pal AR, Brown N, Jones SA, Bigger BW, Bruce IA. Obstructive sleep apnea in mps: a systematic review of pretreatment and posttreatment prevalence and severity. J Inborn Errors Metab Screen. (2019) 3:1–10. doi: 10.1177/2326409815616392
71. Afsharpaiman S, Saburi A, Waters KA. Respiratory difficulties and breathing disorders in achondroplasia. Paediatr Respir Rev. (2013) 14:250–5. doi: 10.1016/j.prrv.2013.02.009
72. Plomp RG, Bredero-Boelhouwer HH, Joosten KF, Wolvius EB, Hoeve HL, Poublon RM, et al. Obstructive sleep apnoea in treacher collins syndrome: prevalence, severity and cause. Int J Oral Maxillofac Surg. (2012) 41:696–701. doi: 10.1016/j.ijom.2012.01.018
73. Dahlqvist Å, Rask E, Rosenqvist C-J, Sahlin C, Franklin KA. Sleep apnea and down’s syndrome. Acta Otolaryngol. (2003) 123:1094–7. doi: 10.1080/00016480310015362
74. Ng DK, Chan C-H. Obesity is an important risk factor for sleep disordered breathing in children with down syndrome. Sleep. (2004) 27:1023–4; author reply 5.15453564
75. Churchill SS, Kieckhefer GM, Landis CA, Ward TM. Sleep measurement and monitoring in children with down syndrome: a review of the literature, 1960–2010. Sleep Med Rev. (2012) 16:477–88. doi: 10.1016/j.smrv.2011.10.003
76. Khayat A, Bin-Hassan S, Al-Saleh S. Polysomnographic findings in infants with pierre robin sequence. Ann Thor Med. (2017) 12:25–9. doi: 10.4103/1817-1737.197770
77. Domany KA, Hantragool S, Smith DF, Xu Y, Hossain M, Simakajornboon N. Sleep disorders and their management in children with Ehlers-Danlos syndrome referred to sleep clinics. J Clin Sleep Med. (2018) 14:623–9. doi: 10.5664/jcsm.7058
78. Stöberl AS, Gaisl T, Giunta C, Sievi NA, Singer F, Möller A, et al. Obstructive sleep apnoea in children and adolescents with Ehlers-Danlos syndrome. Respiration. (2019) 97:284–91. doi: 10.1159/000494328
79. Perkins JA, Sie KC, Milczuk H, Richardson MA. Airway management in children with craniofacial anomalies. Cleft Palate- Craniofac J. (1997) 34:135–40. doi: 10.1597/1545-1569_1997_034_0135_amicwc_2.3.co_2
80. Storm AL, Johnson JM, Lammer E, Green GE, Cunniff C. Auriculo-condylar syndrome is associated with highly Variable ear and mandibular defects in multiple kindreds. Am J Med Genet A. (2005) 138:141–5. doi: 10.1002/ajmg.a.30883
81. Wang A, Kun S, Diep B, Davidson Ward SL, Keens TG, Perez IA. Obstructive sleep apnea in patients with congenital central hypoventilation syndrome ventilated by diaphragm pacing without tracheostomy. J Clin Sleep Med. (2018) 14:261–4. doi: 10.5664/jcsm.6948
82. Abraham C, Virbalas J, DelRosso LM. Severe obstructive sleep apnea in a child with goldenhar syndrome and nasal obstruction. J Clin Sleep Med. (2017) 13:825–7. doi: 10.5664/jcsm.6626
83. Baugh A, Wooten W, Chapman B, Drake A, Vaughn B. Sleep characteristics in goldenhar syndrome. Int J Pediatf Otorhinolaryngol. (2015) 79:356–8. doi: 10.1016/j.ijporl.2014.12.024
84. Chan J, Edman JC, Koltai PJ. Obstructive sleep apnea in children. Am Fam Physician. (2004) 69:1147–54.15023015
85. Kourelis K, Gouma P, Naxakis S, Kalogeropoulou C, Goumas P. Oculoauriculovertebral Complex with an atypical cause of obstructive sleep apnea. Int J Pediatr Otorhinolaryngol. (2009) 73:481–5. doi: 10.1016/j.ijporl.2008.11.004
86. Snead MP, Yates JR. Clinical and molecular genetics of stickler syndrome. J Med Genet. (1999) 36:353–9. doi: 10.1136/jmg.36.5.353
87. Miloro M. Mandibular distraction osteogenesis for pediatric airway management. J Oral Maxillofac Surg. (2010) 68:1512–23. doi: 10.1016/j.joms.2009.09.099
88. Kutkowska-Kaźmierczak A, Gos M, Obersztyn E. Craniosynostosis as a clinical and diagnostic problem: molecular pathology and genetic counseling. J Appl Genet. (2018) 59:133–47. doi: 10.1007/s13353-017-0423-4
89. Alsaadi MM, Iqbal SM, Elgamal EA, Salih MA, Gozal D. Sleep-disordered breathing in children with craniosynostosis. Sleep Breath. (2013) 17:389–93. doi: 10.1007/s11325-012-0706-2
90. Driessen C, Joosten KF, Bannink N, Bredero-Boelhouwer HH, Hoeve HL, Wolvius EB, et al. How does obstructive sleep apnoea evolve in syndromic craniosynostosis? A prospective cohort study. Arch Dis Child. (2013) 98:538–43. doi: 10.1136/archdischild-2012-302745
91. Wenger T, Miller D, Evans K. Fgfr craniosynostosis syndromes overview. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, editors. Genereviews®. Seattle, WA: University of Washington (1993).
92. Huang N, Pandey AV, Agrawal V, Reardon W, Lapunzina PD, Mowat D, et al. Diversity and function of mutations in P450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis. Am J Hum Genet. (2005) 76:729–49. doi: 10.1086/429417
93. L’Hôte CG, Knowles MA. Cell responses to Fgfr3 signalling: growth, differentiation and apoptosis. Exp Cell Res. (2005) 304:417–31. doi: 10.1016/j.yexcr.2004.11.012
94. Horton WA, Lunstrum GP. Fibroblast growth factor receptor 3 mutations in achondroplasia and related forms of dwarfism. Rev Endocr Metab Disord. (2002) 3:381–5. doi: 10.1023/a:1020914026829
95. Azoury SC, Reddy S, Shukla V, Deng CX. Fibroblast growth factor receptor 2 (Fgfr2) mutation related syndromic craniosynostosis. Int J Biol Sci. (2017) 13:1479–88. doi: 10.7150/ijbs.22373
97. Tartaglia M, Di Rocco C, Lajeunie E, Valeri S, Velardi F, Battaglia PA. Jackson-Weiss syndrome: identification of two novel Fgfr2 missense mutations shared with crouzon and pfeiffer craniosynostotic disorders. Hum Genet. (1997) 101:47–50. doi: 10.1007/s004390050584
98. Slavotinek A, Crawford H, Golabi M, Tao C, Perry H, Oberoi S, et al. Novel Fgfr2 deletion in patient with beare-stevenson-like syndrome. Am J Med Genet A. (2009) 149:1814. doi: 10.1002/ajmg.a.32947
99. Bochukova EG, Roscioli T, Hedges DJ, Taylor IB, Johnson D, David DJ, et al. Rare mutations of Fgfr2 causing apert syndrome: identification of the first partial gene deletion, and an alu element insertion from a new subfamily. Hum Mutat. (2009) 30:204–11. doi: 10.1002/humu.20825
100. Boyadjiev SA, Consortium IC. Genetic analysis of non-syndromic craniosynostosis. Orthod Craniofac Res. (2007) 10:129–37. doi: 10.1111/j.1601-6343.2007.00393.x
101. Lattanzi W, Barba M, Di Pietro L, Boyadjiev SA. Genetic advances in craniosynostosis. Am J Med Genet A. (2017) 173:1406–29. doi: 10.1002/ajmg.a.38159
102. Johnson D, Wilkie AO. Craniosynostosis. Eur J Hum Genet. (2011) 19:369–76. doi: 10.1038/ejhg.2010.235
103. Bereza S, Yong R, Gronthos S, Arthur A, Ranjitkar S, Anderson PJ. Craniomaxillofacial morphology in a murine model of Ephrinb1 conditional deletion in osteoprogenitor cells. Arch I Oral Biol. (2022) 137:105389. doi: 10.1016/j.archoralbio.2022.105389
104. Morriss-Kay GM, Wilkie AO. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat. (2005) 207:637–53. doi: 10.1111/j.1469-7580.2005.00475.x.
105. Cornille M, Moriceau S, Khonsari RH, Heuzé Y, Loisay L, Boitez V, et al. Fgfr3 overactivation in the brain is responsible for memory impairments in crouzon syndrome mouse model. J Exp Med. (2022) 219. doi: 10.1084/jem.20201879
106. Bellus GA, McIntosh I, Smith EA, Aylsworth AS, Kaitila I, Horton WA, et al. A recurrent mutation in the tyrosine kinase domain of fibroblast growth factor receptor 3 causes hypochondroplasia. Nat Genet. (1995) 10:357–9. doi: 10.1038/ng0795-357
107. Khominsky A, Yong R, Ranjitkar S, Townsend G, Anderson PJ. Extensive phenotyping of the orofacial and dental complex in crouzon syndrome. Arch Oral Biol. (2018) 86:123–30. doi: 10.1016/j.archoralbio.2017.10.022
108. Legare JM, Pauli RM, Hecht JT, Bober MB, Smid CJ, Modaff P, et al. Clarity: co-occurrences in achondroplasia—craniosynostosis, seizures, and decreased risk of diabetes mellitus. Am J Med Genet A. (2021) 185:1168–74. doi: 10.1002/ajmg.a.62096
110. Neel BG, Gu H, Pao L. The ’Shp’ing news: Sh2 domain-containing tyrosine phosphatases in cell signaling. Trends Bochem Sci. (2003) 28:284–93. doi: 10.1016/S0968-0004(03)00091-4
112. Cardiel Ríos SA. Correction of a severe class ii malocclusion in a patient with noonan syndrome. Am J Orthod Dentofacial Orthop. (2016) 150:511–20. doi: 10.1016/j.ajodo.2015.09.032
113. Ueda K, Yaoita M, Niihori T, Aoki Y, Okamoto N. Craniosynostosis in patients with rasopathies: accumulating clinical evidence for expanding the phenotype. Am J Med Genet A. (2017) 173:2346–52. doi: 10.1002/ajmg.a.38337
114. Takenouchi T, Sakamoto Y, Miwa T, Torii C, Kosaki R, Kishi K, et al. Severe craniosynostosis with noonan syndrome phenotype associated with Shoc2 mutation: clinical evidence of crosslink between Fgfr and Ras signaling pathways. Am J Med Genet A. (2014) 164a:2869–72. doi: 10.1002/ajmg.a.36705
115. Addissie YA, Kotecha U, Hart RA, Martinez AF, Kruszka P, Muenke M. Craniosynostosis and noonan syndrome with kras mutations: expanding the phenotype with a case report and review of the literature. Am J Med Genet A. (2015) 167a:2657–63. doi: 10.1002/ajmg.a.37259
116. Li H, Zhao A, Xie M, Chen L, Wu H, Shen Y, et al. Antley-Bixler syndrome arising from compound heterozygotes in the P450 oxidoreductase gene: a case report. Transl Pediatr. (2021) 10:3309–18. doi: 10.21037/tp-21-499
117. Garrido E, Cormand B, Hopwood JJ, Chabás A, Grinberg D, Vilageliu L. Maroteaux-Lamy syndrome: functional characterization of pathogenic mutations and polymorphisms in the arylsulfatase B gene. Mol Genet Metab. (2008) 94:305–12. doi: 10.1016/j.ymgme.2008.02.012
118. Kwon YS, Jo JK, Lim YH, Yon JH, Kim KM. Anesthetic management of a neonate with Antley-Bixler syndrome: a case report. Anesth Pain Med. (2011) 6:89–92.
119. Antley R. Trapezoidocephaly, midfacial hypoplasia and cartilage abnormalities with multiple synostoses and skeletal fractures. Birth Defects Orig Artic Ser. (1975) 11:397–401.1227559
120. Benko S, Fantes JA, Amiel J, Kleinjan D-J, Thomas S, Ramsay J, et al. Highly conserved non-coding elements on either side of Sox9 associated with pierre robin sequence. Nat Genet. (2009) 41:359–64. doi: 10.1016/j.prrv.2014.11.003
121. Tarasiuk A, Levi A, Assadi MH, Troib A, Segev Y. Orexin plays a role in growth impediment induced by obstructive sleep breathing in rats. Sleep. (2016) 39:887–97. doi: 10.5665/sleep.5648
122. Gordon CT, Attanasio C, Bhatia S, Benko S, Ansari M, Tan TY, et al. Identification of novel craniofacial regulatory domains located far upstream of sox 9 and disrupted in pierre robin sequence. Hum Mutat. (2014) 35:1011–20. doi: 10.1002/humu.22606
123. Dash S, Bhatt S, Falcon K, Sandell L, Trainor P. Med23 regulates Sox9 expression during craniofacial development. J Dent Res. (2021) 100:406–14. doi: 10.1177/0022034520969109
124. Jia Z-L, He S, Jiang S-Y, Zhang B-H, Duan S-J, Shi J-Y, et al. Rs12941170 at Sox9 gene associated with orofacial clefts in Chinese. Arch Oral Biol. (2017) 76:14–9. doi: 10.1016/j.archoralbio.2016.12.010
125. Shung C-Y, Ota S, Zhou Z-Q, Keene DR, Hurlin PJ. Disruption of a Sox9–Β-catenin circuit by mutant Fgfr3 in thanatophoric dysplasia type ii. Hum Mol Genet. (2012) 21:4628–44. doi: 10.1093/hmg/dds305
126. Zhou Z-Q, Ota S, Deng C, Akiyama H, Hurlin PJ. Mutant activated Fgfr3 impairs endochondral bone growth by preventing Sox9 downregulation in differentiating chondrocytes. Hum Mol Genet. (2015) 24:1764–73. doi: 10.1093/hmg/ddu594
127. Gordon CT, Petit F, Kroisel PM, Jakobsen L, Zechi-Ceide RM, Oufadem M, et al. Mutations in endothelin 1 cause recessive auriculocondylar syndrome and dominant isolated question-mark ears. Am J Hum Genet. (2013) 93:1118–25. doi: 10.1016/j.ajhg.2013.10.023
128. Timberlake AT, Griffin C, Heike CL, Hing AV, Cunningham ML, Chitayat D, et al. Haploinsufficiency of Sf3b2 causes craniofacial microsomia. Nat Commun. (2021) 12:1–11. doi: 10.1038/s41467-021-24852-9
129. Tingaud-Sequeira A, Trimouille A, Sagardoy T, Lacombe D, Rooryck C. Oculo-auriculo-vertebral spectrum: new genes and literature review on a complex disease. J Med Genet. (2022) 59:417–27. doi: 10.1136/jmedgenet-2021-108219
130. Sakai D, Trainor PA. Treacher collins syndrome: unmasking the role of Tcof1/treacle. Int J Biochem Cell Biol. (2009) 41:1229–32. doi: 10.1016/j.biocel.2008.10.026
131. Chen Y, Guo L, Li C-L, Shan J, Xu H-S, Li J-Y, et al. Mutation screening of Chinese treacher collins syndrome patients identified novel Tcof1 mutations. Mol Genet Genomics. (2018) 293:569–77. doi: 10.1007/s00438-017-1384-3
132. Van Camp N, Aerden T, Politis C. Problems in the orofacial region associated with Ehlers-Danlos and marfan syndromes: a case series. Br J Oral Maxillofac Surg. (2020) 58:208–13. doi: 10.1016/j.bjoms.2019.11.018
133. Van Damme T, Colman M, Syx D, Malfait F. The Ehlers-Danlos syndromes against the backdrop of inborn errors of metabolism. Genes. (2022) 13:265. doi: 10.3390/genes13020265
134. Topârcean AM, Acatrinei A, Rusu I, Mircea C, Feştilă D, Lucaciu OP, et al. Preliminary data regarding the influence of the collal Rs2249492 polymorphism on the risk of malocclusion in the Romanian population. Studia Universitatis Babes-Bolyai, Biologia. (2021) 66.
135. Melkoniemi M, Koillinen H, Männikkö M, Warman ML, Pihlajamaa T, Kääriäinen H, et al. Collagen Xi sequence variations in nonsyndromic cleft palate, robin sequence and micrognathia. Eur J Hum Genet. (2003) 11:265–70. doi: 10.1038/sj.ejhg.5200950
136. Loeys B, De Backer J, Van Acker P, Wettinck K, Pals G, Nuytinck L, et al. Comprehensive molecular screening of the Fbn1 gene favors locus homogeneity of classical marfan syndrome. Hum Mutat. (2004) 24:140–6. doi: 10.1002/humu.20070
137. Docimo R, Maturo P, D’Auria F, Grego S, Costacurta M, Perugia C, et al. Association between oro-facial defects and systemic alterations in children affected by marfan syndrome. J Clin Diagn Res. (2013) 7:700–3. doi: 10.7860/JCDR/2013/5656.2885
138. Du Q, Zhang D, Zhuang Y, Xia Q, Wen T, Jia H. The molecular genetics of marfan syndrome. Int J Med Sci. (2021) 18:2752–66. doi: 10.7150/ijms.60685
139. Heldin CH, Landström M, Moustakas A. Mechanism of Tgf-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. (2009) 21:166–76. doi: 10.1016/j.ceb.2009.01.021
140. Frazier BC, Mooney MP, Losken HW, Barbano T, Moursi A, Siegel MI, et al. Comparison of craniofacial phenotype in craniosynostotic rabbits treated with anti–Tgf-Β2 at suturectomy site. Cleft Palate Craniofac J. (2008) 45:571–82. doi: 10.1597/07-095.1
141. Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, et al. Tgfbeta2 knockout mice have multiple developmental defects that are non-overlapping with other Tgfbeta knockout phenotypes. Development. (1997) 124:2659–70. doi: 10.1242/dev.124.13.2659
142. Limoge F, Faivre L, Gautier T, Petit JM, Gautier E, Masson D, et al. Insulin response dysregulation explains abnormal fat storage and increased risk of diabetes Mellitus type 2 in cohen syndrome. Hum Mol Genet. (2015) 24:6603–13. doi: 10.1093/hmg/ddv366
143. Duplomb L, Duvet S, Picot D, Jego G, El Chehadeh-Djebbar S, Marle N, et al. Cohen syndrome is associated with major glycosylation defects. Hum Mol Genet. (2014) 23:2391–9. doi: 10.1093/hmg/ddt630
144. Irizarry KA, Miller M, Freemark M, Haqq AM. Prader willi syndrome: genetics, metabolomics, hormonal function, and new approaches to therapy. Adv Pediatr. (2016) 63:47. doi: 10.1016/j.yapd.2016.04.005
145. Oeffner F, Korn T, Roth H, Ziegler A, Hinney A, Goldschmidt H, et al. Systematic screening for mutations in the human necdin gene (Ndn): identification of two naturally occurring polymorphisms and association analysis in body weight regulation. Int J Obes Relat Metab Disord. (2001) 25:767–9. doi: 10.1038/sj.ijo.0801626
146. Wong SB, Zhao LL, Chuang SH, Tsai WH, Yu CH, Tsai LP. Is prone sleeping dangerous for neonates? Polysomnographic characteristics and Ndn gene analysis. Ci Ji Yi Xue Za Zhi. (2019) 31:113–7. doi: 10.4103/tcmj.tcmj_29_18
147. Gómez-González A, Rosales-Berber MÁ, Ávila-Rojas D, Pozos-Guillén A, Garrocho-Rangel A. Pediatric dental management of an uncommon case of mucopolysaccharidosis type iv a (morquio a syndrome): a case report of a three-year follow-up. Case Rep Dent. (2020) 2020. doi: 10.1155/2020/2565486
148. de Almeida-Barros RQ, de Medeiros PFV, de Almeida Azevedo MQ, de Oliveira Lira Ortega A, Yamamoto ATA, Dornelas SKL, et al. Evaluation of oral manifestations of patients with mucopolysaccharidosis iv and vi: clinical and imaging study. Clin Oral Investig. (2018) 22:201–8. doi: 10.1007/s00784-017-2100-8
150. Guimarães M, Farias S, Costa A, Amorim R. Maroteaux-Lamy syndrome: orofacial features after treatment by bone marrow transplant. Oral Health Prev Dent. (2010) 8:139–42.
151. Galdzicka M, Patnala S, Hirshman M, Cai J-F, Nitowsky H, Egeland J, et al. A new gene, Evc2, is mutated in Ellis–Van creveld syndrome. Mol Genet Metab. (2002) 77:291–5. doi: 10.1016/s1096-7192(02)00178-6
152. Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, et al. Mutations in a new gene in Ellis-Van creveld syndrome and weyers acrodental dysostosis. Nat Genet. (2000) 24:283–6. doi: 10.1038/73508
153. Valencia M, Lapunzina P, Lim D, Zannolli R, Bartholdi D, Wollnik B, et al. Widening the mutation spectrum of Evc and Evc2: ectopic expression of weyer variants in Nih 3t3 fibroblasts disrupts hedgehog signaling. Hum Mutat. (2009) 30:1667–75. doi: 10.1002/humu.21117
154. Lavezzi AM, Casale V, Oneda R, Gioventù S, Matturri L, Farronato G. Obstructive sleep apnea syndrome (osas) in children with class iii malocclusion: involvement of the Phox2b gene. Sleep Breath. (2013) 17:1275–80. doi: 10.1007/s11325-013-0833-4
155. Mainieri G, Montini A, Nicotera A, Di Rosa G, Provini F, Loddo G. The genetics of sleep disorders in children: a narrative review. Brain Sci. (2021) 11:1259. doi: 10.3390/brainsci11101259
156. Larkin EK, Patel SR, Goodloe RJ, Li Y, Zhu X, Gray-McGuire C, et al. A candidate gene study of obstructive sleep apnea in European Americans and African Americans. Am J Respir Crit Care Med. (2010) 182:947–53. doi: 10.1164/rccm.201002-0192OC
157. Chi L, Comyn F-L, Keenan BT, Cater J, Maislin G, Pack AI, et al. Heritability of craniofacial structures in normal subjects and patients with sleep apnea. Sleep. (2014) 37:1689–98. doi: 10.5665/sleep.4082
158. Tiro A, Dzemidzic V, Salaga-Nefic S, Redzic I, Nakas E. Heritability of craniofacial characteristics in twins-cephalometric study. Med Arch. (2019) 73:205. doi: 10.5455/medarh.2019.73.205-208
159. Šešelj M, Duren DL, Sherwood RJ. Heritability of the human craniofacial Complex. Anat Rec. (2015) 298:1535–47. doi: 10.1002/ar.23186
160. Mathur R, Douglas NJ. Family studies in patients with the sleep apnea-hypopnea syndrome. Ann Intern Med. (1995) 122:174–8. doi: 10.7326/0003-4819-122-3-199502010-00003
161. Paternoster L, Zhurov AI, Toma AM, Kemp JP, St Pourcain B, Timpson NJ, et al. Genome-wide association study of three-dimensional facial morphology identifies a variant in Pax3 associated with nasion position. Am J Hum Genet. (2012) 90:478–85. doi: 10.1016/j.ajhg.2011.12.021
162. Bjork BC, Turbe-Doan A, Prysak M, Herron BJ, Beier DR. Prdm16 is required for normal palatogenesis in mice. Hum Mol Genet. (2010) 19:774–89. doi: 10.1093/hmg/ddp543
163. Warner DR, Horn KH, Mudd L, Webb CL, Greene RM, Pisano MM. Prdm16/Mel1: a novel smad binding protein expressed in murine embryonic orofacial tissue. Biochim Biophys Acta. (2007) 1773:814–20. doi: 10.1016/j.bbamcr.2007.03.016
164. Robinson PN, Arteaga-Solis E, Baldock C, Collod-Béroud G, Booms P, De Paepe A, et al. The molecular genetics of marfan syndrome and related disorders. J Med Genet. (2006) 43:769–87. doi: 10.1136/jmg.2005.039669
165. Katz ES, Marcus CL. Obstructive sleep apnea: children versus adults. In: Pack AI, editor. Sleep apnea. Boca Raton: CRC Press (2016). p. 382–410.
Keywords: pediatric obstructive sleep apnea, syndrome, craniofacial dysmorphism, candidate gene, skeletal anomaly
Citation: Marincak Vrankova Z, Krivanek J, Danek Z, Zelinka J, Brysova A, Izakovicova Holla L, Hartsfield Jr JK and Borilova Linhartova P (2023) Candidate genes for obstructive sleep apnea in non-syndromic children with craniofacial dysmorphisms – a narrative review. Front. Pediatr. 11:1117493. doi: 10.3389/fped.2023.1117493
Received: 6 December 2022; Accepted: 6 June 2023;
Published: 27 June 2023.
Edited by:
Long Guo, RIKEN Center for Integrative Medical Sciences, JapanReviewed by:
Nathalia Carolina Fernandes Fagundes, University of Alberta, CanadaNuntigar Sonsuwan, Chiang Mai University, Thailand
© 2023 Marincak Vrankova, Krivanek, Danek, Zelinka, Brysova, Izakovicova Holla, Hartsfield and Borilova Linhartova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Petra Borilova Linhartova cGV0cmEubGluaGFydG92YUByZWNldG94Lm11bmkuY3o=