
 K. Scott Beard1,2
K. Scott Beard1,2
- 1Denver Veterans Affairs Medical Center, Denver, CO, USA
- 2University of Colorado Anschutz Medical Campus, Aurora, CO, USA
Nosocomial pneumonia (NP) is the third most common hospital-acquired infection and the leading cause of death due to hospital-acquired infection in the US. During pneumonia and non-pneumonia severe illness, respiratory tract secretions become enriched with the serine protease neutrophil elastase (NE). Several NE activities promote onset and severity of NP. NE in the airways causes proteolytic tissue damage, augments inflammation, may promote invasion of respiratory epithelium by bacteria, and disrupts respiratory epithelial barrier function. These NE activities culminate in enhanced bacterial replication, impaired gas exchange, fluid intrusion into the airways, and loss of bacterial containment that can result in bacteremia. Therefore, neutralizing NE activity may reduce the frequency and severity of NP. We evaluated human alpha-1 antitrypsin (AAT), the prototype endogenous NE inhibitor, as a suppressor of bacterial pneumonia and pneumonia-related pathogenesis. In AAT+/+ transgenic mice that express human AAT in lungs, mortality due to Pseudomonas aeruginosa (P.aer) pneumonia was reduced 90% compared to non-transgenic control animals. Exogenous human AAT given to non-transgenic mice also significantly reduced P.aer pneumonia mortality. P.aer-infected AAT+/+ mice demonstrated reduced lung tissue damage, decreased bacterial concentrations in lungs and blood, and diminished circulating cytokine concentrations compared to infected non-transgenic mice. In vitro, AAT suppressed P.aer internalization into respiratory epithelial cells and inhibited NE or P.aer-induced disruption of epithelial cell barrier function. The beneficial effects of human AAT in murine P.aer pneumonia raise the possibility of AAT use as a prophylactic treatment for NP in humans, and suggest a role for AAT as an innate immune mediator.
Introduction
Pneumonia is often categorized by location of acquisition. Community-acquired pneumonia (CAP) is contracted in the non-health care setting, and nosocomial pneumonia (NP) is acquired in the hospital. NP is of special interest due to a high prevalence in hospitalized patients, a poor prognosis, and a large expenditure of resources. NP is the third most common hospital-acquired infection in the US (1–3), with about 250,000 cases of NP in 2002 (3). NP is the leading cause of death due to hospital-acquired infection (2, 3), and in 2002 there were nearly 36,000 NP-related deaths in the US (crude mortality of about 15%) (3). NP is caused predominantly by Gram-negative rods (nearly 42% of isolates), with Pseudomonas aeruginosa (P.aer) accounting for about 20% of isolates (2, 4, 5).
Alarmingly, mortality due to lower respiratory tract infection was not diminished over 25 years of observation in the US (6). Attributable mortality is especially high for P.aer NP that is acquired while receiving mechanical ventilation, reported at 43% (1). Bacteria that cause NP have demonstrated escalating resistance to antibiotics over time (5). Unfortunately, the current antibiotic pipeline has slowed and emergence of more potent antimicrobial drugs is unlikely to address NP-related therapeutic challenges (7). NP carries substantial resource burdens that include annual total treatment costs in the US nearing $5.4 billion (3, 8). These considerations indicate that NP is a substantial clinical problem, and recent treatments and preventative (prophylactic) measures have shown little clinical impact (9).
The pathogenesis of pneumonia usually involves bacterial colonization of the upper airways followed by aspiration of these bacteria into the lower respiratory tract (4, 10, 11). Pneumonia develops when bacteria and secretions aspirated into the lower airways is sufficient to overcome lower respiratory tract host defenses. Lower levels of aspirated bacteria may cause pneumonia during illness that weakens host defenses. Bacterial components induce inflammation by stimulating alveolar macrophages and respiratory epithelial cells to produce pro-inflammatory cytokines such as interleukin (IL)-1, tumor necrosis factor (TNF)α, and the chemokine IL-8 (12, 13). IL-8 recruits and activates neutrophils, which secrete neutrophil elastase (NE) into the respiratory tract. Clinical studies have demonstrated substantial NE levels in bronchoalveolar lavage fluid (BALF) in patients with P.aer NP (14, 15).
Neutrophil elastase activities in the airways promote pneumonia severity (12). Since NE is an omnivorous protease, NE can directly damage lung epithelial cells and supporting tissues. This results in reduced capacity to eliminate bacteria, defective gas exchange, and exudation of fluid into lung airspaces (12, 16, 17). NE induces production of pro-inflammatory cytokines in the lungs that augment inflammation (16, 18, 19), and NE inactivates several extracellular immune mediators such as immunoglobulins, complement components, and cathelicidin (16, 20, 21). NE also induces expression of MUC1 (a cell-surface mucin) that can serve as a receptor for P.aer and perhaps other bacteria (22, 23). Since bacteria can spontaneously invade respiratory epithelium (22, 24 –26), NE-enhanced binding of bacteria to respiratory epithelial cells likely initiates invasion. Translocation of bacteria into the cell interior permits evasion from extracellular antimicrobial substances such as antibodies, complement, lysozyme, lactoferrin, cathelicidin-related molecules, and defensins (13, 27). Intracellular translocation also sequesters bacteria from the antibacterial activities of macrophages and neutrophils. Since NE can also increase vascular permeability, NE may enhance translocation of bacteria across the endothelial cell barrier and initiate bacteremia (12). During established pneumonia, these NE activities in the lower respiratory tract amplify lung inflammation and tissue damage (12). NE may also promote bacterial proliferation and bacteremia during pneumonia. Elevated lower respiratory tract NE has also been demonstrated during non-pneumonia systemic illness. This may result in NE-induced defects in host defense that link underlying systemic disease to increased NP risk (28 –30).
Since excessive NE activity in the respiratory tract participates in pneumonia pathogenesis, NE inhibition is a target for therapeutic intervention. Alpha-1 antitrypsin (AAT) is the prototype endogenous inhibitor of serine proteases such as NE. AAT is a 394 amino acid, 52 kDa glycoprotein produced primarily by the liver and secreted into the circulation. AAT is the most abundant endogenous serine protease inhibitor in the circulation, with serum concentrations reported as 1.0–2.7 mg/mL in healthy adults (31). AAT is an acute phase protein (32), and circulating concentrations can increase two to fourfold during systemic inflammation (33, 34). AAT concentrations in lung tissues are substantially less than concentrations in the circulation, with extracellular lung fluid levels of about 10% serum concentrations (35). The function of AAT is classically described as neutralization of NE in the lung in order to limit NE-induced tissue damage (36). There are over 100 AAT variants in humans, and clinical interest in AAT has focused on genetic AAT deficiency caused by inheritance of two copies of the abnormal Z-type AAT gene. Z-type AAT is characterized by defective export from the liver into the circulation, and serum AAT levels are reduced to 10–15% of normal (37). The best-described clinical consequences of AAT deficiency include pulmonary emphysema and liver disease (37). Pulmonary emphysema is thought to originate from an imbalance between protease (NE) and antiprotease (AAT) that favors protease destruction of lung tissue (37). The only specific treatment for AAT deficiency is replacement therapy with intravenous AAT purified from healthy donor plasma.
In a prior report, AAT was used as a treatment for chronic bacterial pneumonia in rats (38). In that report, 7 days of aerosolized AAT was administered as a post-infection treatment in rats with Pseudomonas lung infection. AAT-treated rats demonstrated a delayed reduction in bacterial levels in lungs and reduced lung inflammation. We assessed a different approach to AAT use in pneumonia. In our studies, we tested the hypothesis that human AAT would provide preventative (prophylactic) protection against acute pneumonia, since AAT clinical use is likely optimal as a preventative treatment. Since human AAT neutralizes murine NE, human AAT can be used to examine NE suppression in mouse models (39). Therefore, we conducted mouse studies to examine the protective effect of AAT present before bacterial infection. Mortality, lung histopathology, bacterial quantification in the lungs and peripheral blood, and serum cytokine levels were determined. We also assessed mechanisms of AAT protection in vitro. AAT was examined for direct antibacterial activity, for effect on P.aer cell-surface binding and internalization into A549 human lung epithelial cells, and for effect on NE- or P.aer-induced disruption of A549 monolayer barrier integrity.
Materials and Methods
Cells and Reagents
The gentamicin-sensitive PA01 P.aer bacterium was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in Luria–Bertani broth (LB, Fisher Scientific, Fair Lawn, NJ, USA) as suspension cultures or on LB-agar plates at 37°C. A549, a human lung alveolar epithelial cell line, was also obtained from the ATCC. A549 culture medium comprised RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich, St. Louis, MO, USA), and 2 mM L-glutamine without or with 100 U/mL penicillin and 100 μg/mL streptomycin (all from Mediatech, Inc., Herndon, VA, USA). All A549 cell culture incubations were conducted in a 37°C and 5% CO2 atmosphere incubator. Clinical-grade AAT was obtained from Baxter (Aralast NP, Westlake Village, CA, USA). Human NE was obtained from Innovative Research (Novi, MI, USA). All cultures were analyzed for toxicity using a lactate dehydrogenase (LDH) release assay (Promega, Madison, WI, USA).
Mouse Studies
All procedures and care complied with the Institutional Animal Care and Use Committee at the Denver Veterans Affairs Medical Center. Female C57Bl/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). A transgenic AAT+/+ mouse strain containing a human AAT gene under control of the lung Surfactant Protein C promoter (40), was backcrossed onto a C57Bl/6 background and obtained from Eli Lewis, University of Colorado Denver (41).
Mouse Infection with P.aer
For all mouse infection experiments, 8- to 12-week-old female AAT+/+ transgenic mice or non-transgenic C57Bl/6 control mice were anesthetized with isoflurane (Vedco, Inc., St. Joseph, MO, USA) and infected by nasal aspiration of a 50 μL suspension containing 1 × 106 colony forming units (cfu) of viable P.aer. Body weights were measured twice daily, and moribund mice (body weight loss>20%) were euthanized. In experiments examining the effect of exogenous AAT on P.aer pneumonia, C57Bl/6 non-transgenic mice received three 30 min treatments of aerosolized AAT or sterile saline (control) using a nebulizer (nebulizer obtained from Mabis Healthcare, Waukegan, IL, USA) at 48, 24, and 2 h before P.aer nasal aspiration. The nebulization rate was 0.2 mL/min and the total AAT dose was 120 mg per treatment. In addition, mice were given three intraperitoneal (ip) injections with either 200 μL (2 mg) AAT or 200 μL saline (control) at 48, 24, and 2 h before infection with P.aer.
Histopathological Assessment of Lung Involvement in Mice Infected with P.aer
Alpha-1 antitrypsin+/+ transgenic mice and C57Bl/6 non-transgenic control mice were infected by nasal inhalation of 1 × 106 cfu of viable P.aer. Twenty-four hours after infection, mice were anesthetized with Nembutal (Oak Pharmaceuticals, Lake Forest, IL, USA) and the lungs were inflated in situ with 4% paraformaldehyde in phosphate buffered saline (PBS) (pH 7.4) to fix the tissues. The lungs were excised and stored at 4°C overnight in 4% paraformaldehyde. After 24 h, the lungs were washed in 5.0 mL PBS, embedded in paraffin, and sectioned. Hematoxylin and eosin (H&E) staining was performed at National Jewish Health Core Laboratory (Denver, CO, USA). Whole mount sections of both lungs from each mouse were evaluated by a single examiner (DTM) blinded to experimental condition. Pneumonia was classified as lobar pneumonia or bronchopneumonia in each lung lobe according to the extent of inflammation in each of the five lobes. If the large majority of a lobe (>80%) showed evidence of inflammation, involvement was defined as lobar. Bronchopneumonia in a lobe was defined by the presence of distinct peri-bronchial inflammation that involved<80% of the lung tissue. All lung sections were also examined for alveolar hemorrhage and the number of lobes with hemorrhage was counted for each mouse (usually five lung lobes per mouse). In a second analysis, the percent of each lobe occupied by inflammatory infiltrate was estimated. An overall percent of lung inflammation for each mouse was calculated as the mean percent involvement in all lobes in the two lungs. The presence and severity of necrosis was also examined and subjectively reported as early or advanced.
Quantification of P.aer in Mouse Lungs and Peripheral Blood
C57Bl/6 control and AAT+/+ mice were infected by nasal inhalation of 1 × 106 cfu of P.aer. Twenty-four hours after infection, mice were euthanized and both lungs excised and weighed. Lungs were then homogenized in 1.0 mL PBS using a Tissue Tearor homogenizer (BioSpec Products, Bartlesville, OK, USA). Lung homogenates were diluted 1:100 into 1.0 mL LB medium, 200 μL aliquots of this dilution spread onto duplicate LB-agar plates, and the plates incubated at 37°C for 18 h. After counting the number of colonies, the number of bacteria per gram of lung tissue was calculated by multiplying the average number of colonies per agar plate by 500 and then dividing this value by the total lung weight to obtain the number of colonies per gram lung tissue.
For quantification of P.aer in blood, peripheral blood was obtained via heart puncture under sterile conditions from the same mice used for lung bacterial cultures. One milliliter of cardiac blood was transferred into a sterile tube containing 1.0 mL of BacT/ALERT SA blood culture medium (bioMérieux, Inc., Durham, NC, USA). Two hundred microliter aliquots of this dilution were spread onto duplicate LB-agar plates, and the plates were incubated at 37°C for 18 h. The bacterial colonies on the plates were counted, and the average number of colonies per plate was multiplied by 10 to obtain the number of P.aer bacteria in 1.0 mL blood.
Cytokine Measurements in Mouse Serum
Alpha-1 antitrypsin+/+ transgenic mice and C57Bl/6 non-transgenic control mice were infected by nasal inhalation of 1 × 106 cfu of P.aer. Twenty-four hours after infection, the mice were euthanized and peripheral blood obtained via heart puncture. The blood was collected in BD Microtainer tubes containing lithium heparin (BD), and centrifuged at 2000 × g for 3 min. The supernatant plasma was transferred to a fresh tube and stored at −70°C until cytokine quantification. Mouse IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-10, IL-12, IL-17, MCP-1, IFNγ, TNFα, MIP-1α, GMCSF, and RANTES were measured using a multiplex array chemiluminescence device (Quansys Biosciences, Logan, UT, USA).
P.aer Bacterial Growth in Liquid Suspension Cultures
Separate 2.0 mL aliquots of LB broth were inoculated with 2 × 106 cfu of P.aer, one without and one with 5 mg/mL AAT, and the cultures were placed in a shaking incubator at 37°C. Culture aliquots were obtained during each hour of incubation for 8 h. The culture optical density (OD) at 600 nm was obtained as a measure of bacterial content (38).
P.aer Adherence and Internalization into A549 Cells
For bacterial adherence experiments, 1 × 106 A549 cells were added into wells in a 24-well polystyrene tissue culture plate and incubated for 24 h in antibiotic-free culture medium. After 24 h, A549 cells adhered to culture well bottom surfaces and the culture medium was replaced with 1.0 mL fresh antibiotic-free culture medium without or with 5 mg/mL AAT. For cells exposed to human NE, human NE (2.0 μg/mL final concentration) was added to wells 1 h after AAT addition. Following an additional 1 h incubation, 0.2 × 106 cfu (0.2 multiplicity of infection, MOI) of viable log-phase P.aer were added to all wells. Following addition of P.aer, the cultures were incubated for 1 h at 37°C and 5% CO2. The supernatants were aspirated and the cells washed three times with PBS to remove bacteria that were not cell-associated. The cells were lysed by removing PBS and adding 0.5 mL of 0.25% Triton X-100 into each well for 10 min. The lysates were diluted 1:1000 with LB broth, 200 μL aliquots were spread onto duplicate LB-agar plates using a spinner disk, and the plates incubated at 37°C for 18 h to facilitate bacterial growth. After counting all bacterial colonies on the agar plates, the total number of colonies in culture wells was calculated by multiplying the average number of colonies per plate by 2500.
For bacterial internalization experiments, 1 × 106 A549 cells were added into five wells in 24-well polystyrene tissue culture plates and incubated for 24 h in antibiotic-free culture medium. After 24 h, the A549 cells adhered to culture well bottom surfaces and the culture medium was replaced with 1.0 mL fresh antibiotic-free culture medium without or with 5 mg/mL AAT. For cultured cells exposed to human NE, human NE (2.0 μg/mL final concentration) was added to wells 1 h after AAT addition. Following an additional 1 h incubation, 0.2 × 106 cfu (0.2 MOI) of viable log-phase P.aer were added to all wells. The cultures were incubated for 2 h at 37°C and 5% CO2, and the supernatants aspirated and the cells washed three times with PBS to remove bacteria that were not cell-associated. The PBS was aspirated, 1.0 mL of antibiotic-free culture medium supplemented with 200 μg/mL Gentamicin (Sigma-Aldrich) was added to the wells, and the cells incubated for an additional 2 h to kill non-internalized bacteria. Cells were then washed three times with PBS and lysed by removing the PBS and adding 0.5 mL of 0.25% Triton X-100 into each well for 10 min. The lysates were diluted 1:100 with LB broth, 200 μL aliquots were spread onto duplicate LB-agar plates using a spinner disk, and the plates incubated at 37°C for 18 h to facilitate bacterial growth. After counting all colonies on the agar plates, the total number of colonies in culture wells was calculated by multiplying the average number of colonies per plate by 250.
Transepithelial Electrical Resistance (TEER) Assay
Transepithelial electrical resistance (TEER) is used as a measure of barrier integrity of cell monolayers in culture, where electrical resistance across a cell monolayer is directly proportional to barrier integrity (42). TEER assay experiments were conducted using six-well polystyrene tissue culture plates, with each well containing a cell culture insert with bottom-surface 0.4 μm pores (Fisher Scientific). One million A549 cells were added to the pore-containing inserts (upper chambers) along with 2.5 mL antibiotic-free culture medium. Three milliliters of antibiotic-free culture medium was added to wells in the six-well plates (lower chambers) and the cultures incubated for 24 h. This produced confluent A549 monolayers that formed a barrier between the upper and lower chambers. The medium was aspirated from upper and lower chambers and replaced with fresh antibiotic-free culture medium without (medium control) or with 5 mg/mL AAT. For cells exposed to human NE, 2.0 μg/mL final concentration human NE was added to upper chambers 1 h after AAT addition. A 5.0 MOI (5 × 106 cfu) of viable log-phase P.aer bacteria were added to the upper (insert) chambers of all culture wells except for medium-alone control wells. For TEER experiments conducted in the absence of P.aer, TEER measurements were performed at 1, 3, and 6 h following addition of NE, and for experiments conducted in the presence of P.aer, TEER measurements were performed at 24, 30, and 48 h using an EVOM resistance meter as instructed by the manufacturer (World Precision Instruments, Sarasota, FL, USA). Electrical resistance was also measured in an insert containing well in the absence of A549 cells (defined as electrical resistance = 0). This value for resistance was subtracted from all other measurements. For each experimental condition, a time 0 measurement (TEER measured just prior to addition of P.aer) was set to 100% and subsequent measurements were calculated as a portion of 100%.
Statistical Analysis
For mouse mortality experiments, results were calculated as percent survival and Kaplan–Meir mortality curves were generated. Statistical significance was assessed by the log-rank test (Prism by GraphPad, La Jolla, CA, USA). For bacterial quantification comparisons, the Mann–Whitney U-test was performed. For TEER assay studies and experiments comparing serum cytokine levels, differences between experimental conditions were evaluated using ANOVA repeated measures with Bonferroni’s comparison test. p < 0.05 was defined as statistically significant for all analyses.
Results
P.aer Pneumonia Mortality is Reduced in AAT+/+ Mice and in Mice Given Exogenous Human AAT
Mortality was quantified following P.aer infection in transgenic mice that express human AAT (AAT+/+) and in C57Bl/6 (control) mice. As shown in Figure 1A, 10 AAT+/+ mice demonstrated significantly less pneumonia-related death (p < 0.0001) compared to 9 control mice, with 90% of the AAT+/+ mice surviving infection and 0% of control mice surviving infection. For the first 48 h, weight loss was comparable between the two groups (data not shown). After 48 h, surviving mice began to recover lost weight. In Figure 1B, exogenous human AAT was administered to 16 C57Bl/6 mice by both aerosol delivery using a nebulizer and ip injection. Seventeen control animals received PBS using the same two routes of administration. Forty-four percent of AAT-treated mice compared to 12% of control mice survived infection (p = 0.021).
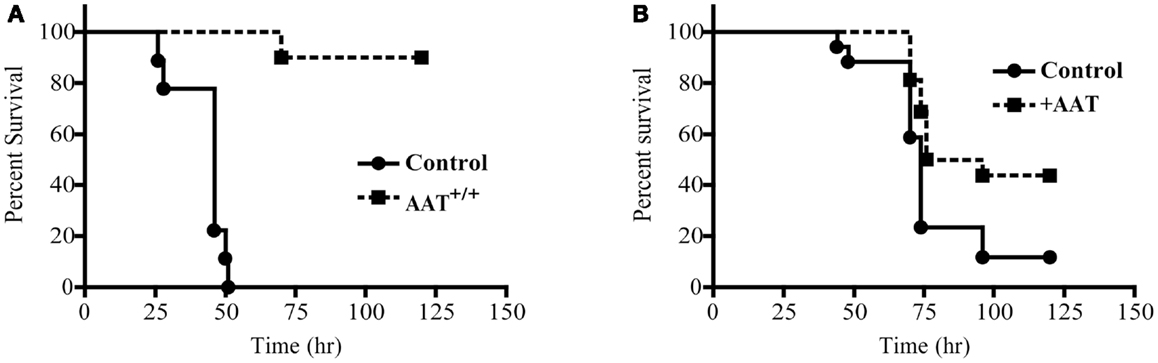
Figure 1. Reduced mortality following P.aer pneumonia in AAT transgenic mice and in mice given exogenous AAT. In (A) 10 transgenic mice that express human AAT in lung tissue (AAT+/+) and 9 C57Bl/6 (Control) mice were infected with inhaled P.aer. Kaplan–Meier survival curves are shown for 120 h following infection. Reduced mortality in AAT+/+ mice was statistically significant (p < 0.0001). In (B) C57Bl/6 mice were treated with exogenous AAT (16 mice, +AAT) or with saline (17 mice, Control). AAT was administered by nebulizer and intraperitoneal injection prior to infection with inhaled P.aer. Kaplan–Meier survival curves are shown for 120 h following infection. Reduced mortality in +AAT mice was statistically significant (p = 0.021).
AAT+/+ Mice Demonstrate Reduced Lung Damage Following P.aer Pneumonia
Six AAT+/+ mice and five C57Bl/6 non-transgenic (control) mice were infected with inhaled P.aer. Twenty-four hours after infection, lungs were fixed, sectioned, stained, and analyzed for pathological effects. One control animal had only four identifiable lung lobes. Table 1 displays blinded histological assessments of the number of lung lobes demonstrating lobar pneumonia, bronchopneumonia, or alveolar hemorrhage. The percent total lung involvement analysis describes the estimated percent of total lung tissue involved with pneumonia.
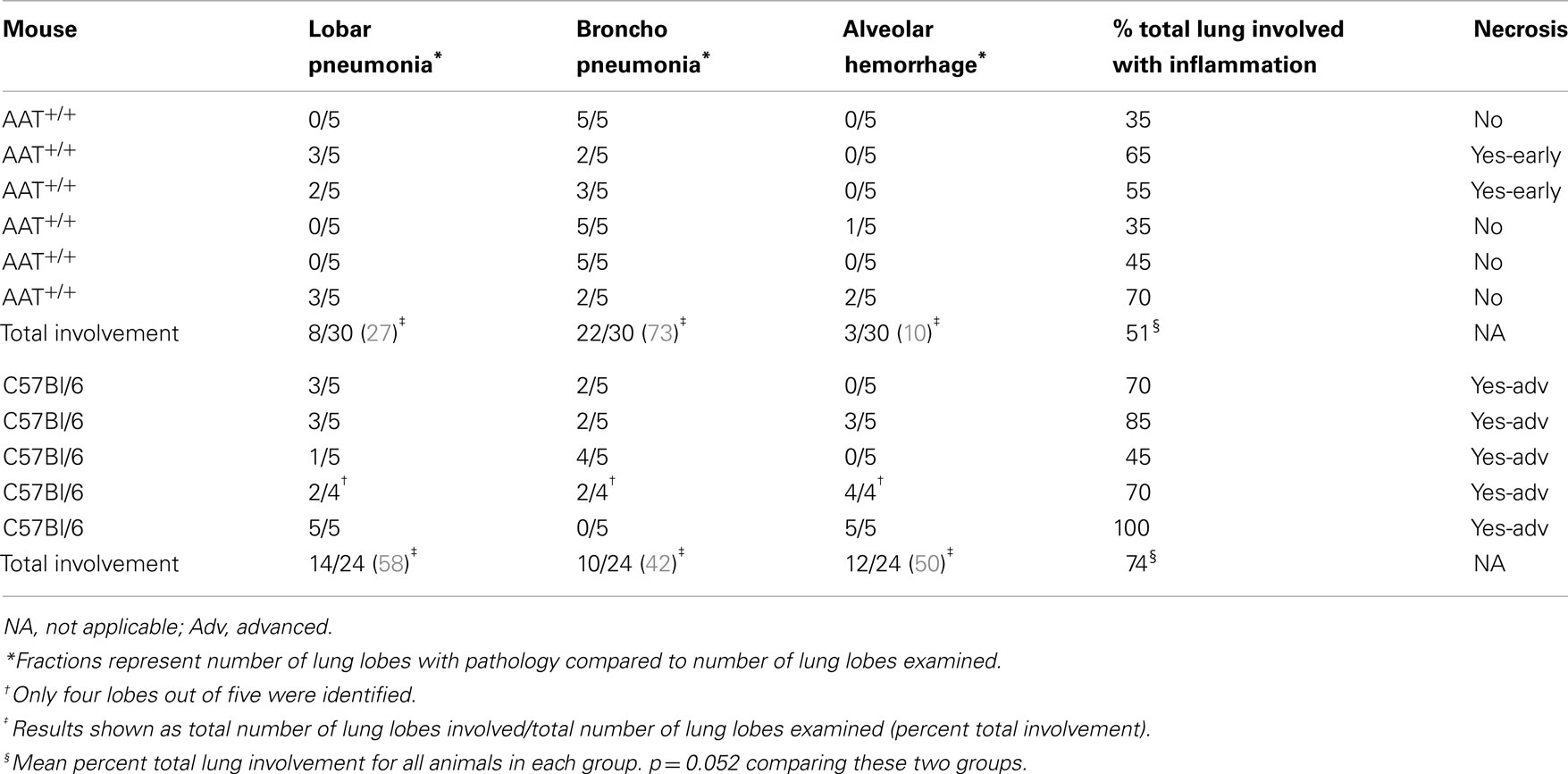
Table 1. Lung histopathology in six AAT+/ + and five C57Bl/6 control mice infected with P.aer.
Reduced severity of pneumonia-associated inflammation was observed in AAT+/+ mice. A lobar pneumonia pattern was observed in 27% of AAT+/+ mouse lobes and in 58% of control mouse lobes. A bronchopneumonia pattern was observed in 73% of AAT+/+ lung lobes and in 42% of control lung lobes. These observations indicate reduced involvement in AAT+/+ mice, since bronchopneumonia is defined as less severe disease involvement than lobar pneumonia (see Materials and Methods). Alveolar hemorrhage was described in 10% of AAT+/+ mice and in 50% of control animals. The summary percent of total lung involved with inflammation showed less involvement in AAT+/+ mice (51%) compared to control animals (74%). Comparison of percent of total lung involvement in these two groups approached (but did not achieve) statistical significance (p = 0.052). Necrosis was observed in two of six AAT+/+ mice, with early stages of necrosis present in both. In contrast, necrosis was observed in five of five non-transgenic control animals, and in every case the necrosis was in advanced stages.
Infected lung sections from representative control and AAT+/+ mice are presented in Figure 2. There is less pneumonia-associated inflammation in the AAT+/+ mouse lung compared to the control mouse lung, and decreased alveolar fluid extravasation and inflammatory cell infiltration in AAT+/+ lung (compare control lung tissue in Figures 2A,B to AAT+/+ lung tissue in Figures 2C,D).
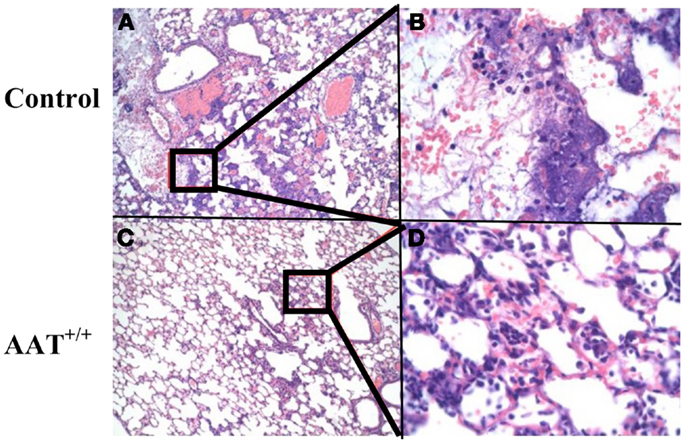
Figure 2. Less inflammation in lungs of AAT transgenic mice infected with P.aer. Six transgenic (AAT+/+) mice and five C57Bl/6 non-transgenic control (Control) mice were infected with inhaled P.aer. Twenty-four hours after infection the mice were euthanized and lungs excised, fixed, sectioned, and stained with H&E. Representative photomicrographs of lung tissues are shown, with control lung sections in (A,B), and AAT+/+ lung sections in (C,D). Inset regions (B,D) are magnified views of the areas indicated in black rectangles in (A,C), respectively. (B,D) show perivascular cuffing with predominant mononuclear infiltration that is common in bacterial pneumonia. Magnification is 20× for (A,C) and 200× for(B,D).
Reduced P.aer Bacterial Concentrations in Lungs and Blood in AAT+/+ Mice
Six AAT+/+ mice and six non-transgenic control mice were infected with inhaled P.aer. Twenty-four hours following infection, lungs were removed and homogenized and peripheral blood was obtained via heart puncture. Compared to controls, AAT+/+ mice demonstrated significantly reduced bacterial levels in lungs (Figure 3A, approximate 77% mean reduction, p = 0.015) and in blood (Figure 3B, approximate 99% mean reduction, p = 0.030).
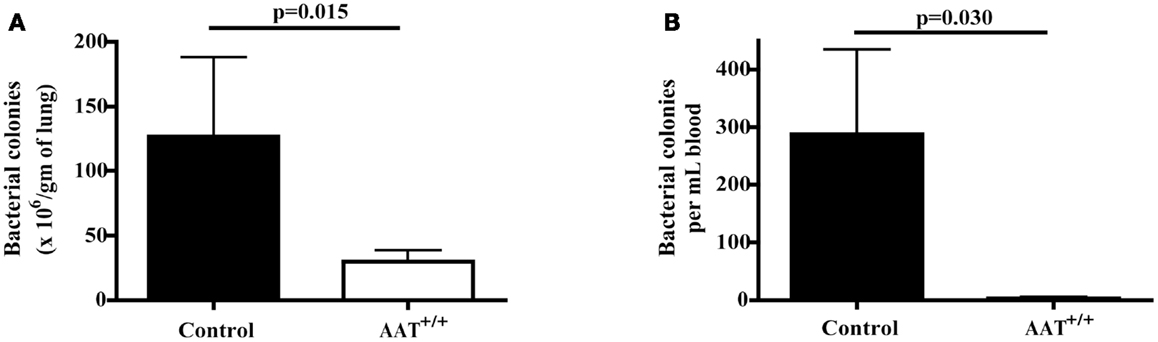
Figure 3. Reduced bacterial concentrations in lungs and blood in AAT transgenic mice following P.aer pneumonia. Six transgenic (AAT+/+) mice and six C57Bl/6 non-transgenic control (Control) mice were infected with inhaled P.aer. After 24 h, mice were euthanized and lung tissues and blood samples collected and processed as described in Section “Materials and Methods.” Bacterial concentrations were quantified in lungs (A) and in blood (B). Graphs depict means + SEM and p-values are indicated in the graphs.
Peripheral Blood Cytokine Concentrations are Reduced in P.aer-Infected AAT+/+ Mice
Excessive inflammation following bacterial infection may contribute to lung tissue damage and death (15, 43 –46), and AAT possesses anti-inflammatory properties (47–53). Therefore, an AAT inhibitory effect on systemic inflammation (indicated by circulating cytokine production) may participate in the decreased mortality in AAT+/+ mice. We examined serum cytokine levels in six AAT+/+ mice and in four non-transgenic C57Bl/6 (control) mice 24 h after inhalation of P.aer. As depicted in Table 2 (shown under the line indicating mice infected with P.aer), infected AAT+/+ mice produced substantially lower blood cytokine levels than infected control animals. Cytokine reduction was numerically substantial for all tested cytokines except for IL 4, and reductions were statistically significant for 9 of the 16 cytokines tested (p < 0.05). In separate experiments conducted in the absence of P.aer infection, serum cytokine concentrations were measured in five AAT+/+ transgenic mice and five C57Bl/6 non-transgenic mice. The results are shown in Table 2 under the line indicating uninfected mice. Serum cytokine concentrations were uniformly low in uninfected AAT+/+ transgenic and C57Bl/6 non-transgenic mice.
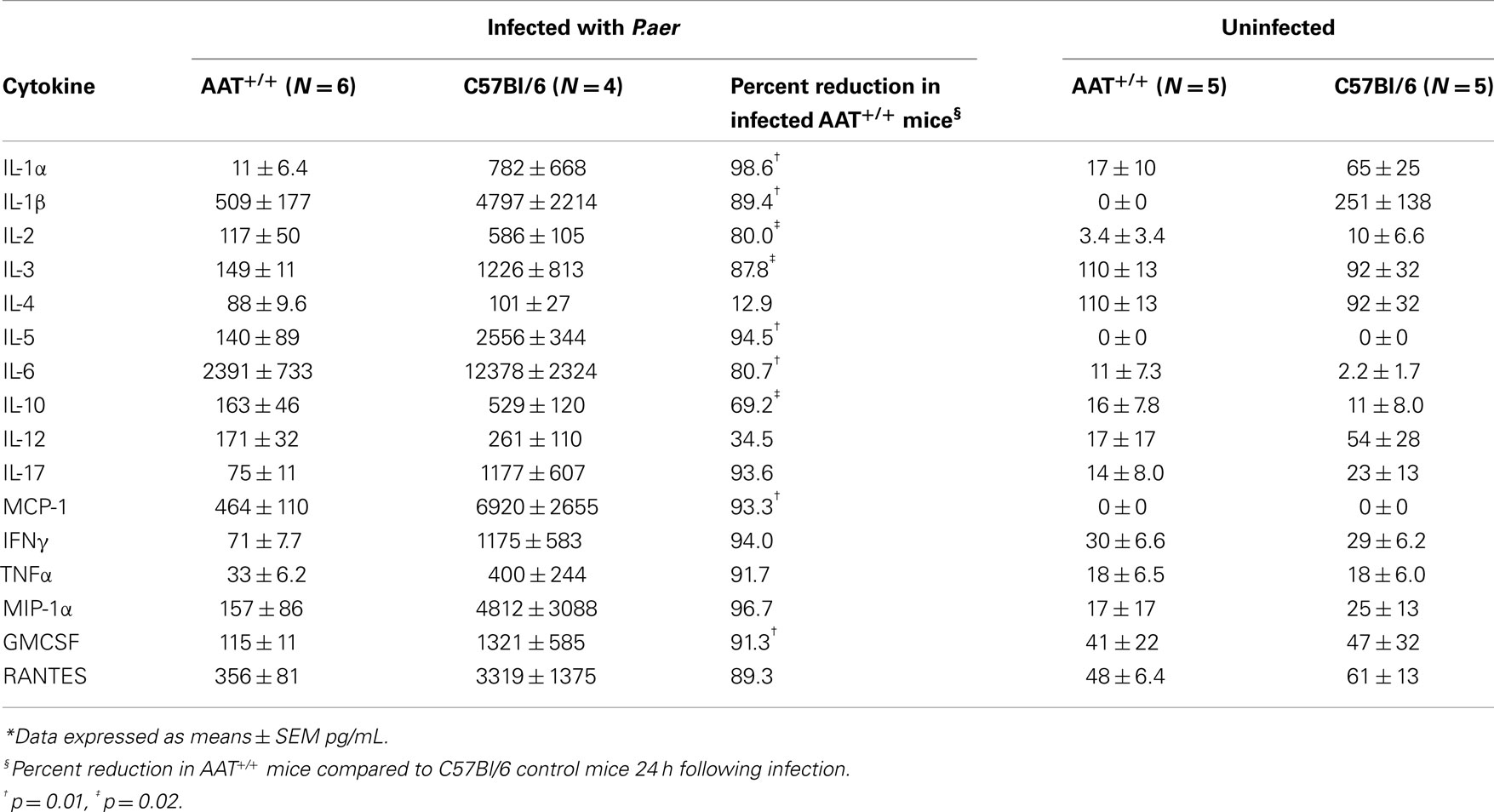
Table 2. Serum cytokine levels in AAT+/ + and C57Bl/6 control mice*.
AAT does not Inhibit P.aer Bacterial Growth
It is possible that AAT protective activity resulted from a direct antibacterial effect. To test this possibility, P.aer was grown in LB broth cultures in the absence or presence of 5 mg/mL AAT. Bacterial concentrations (OD600) were measured hourly to determine bacterial growth. As shown in Figure 4, AAT had no effect on bacterial growth compared to control cultures.
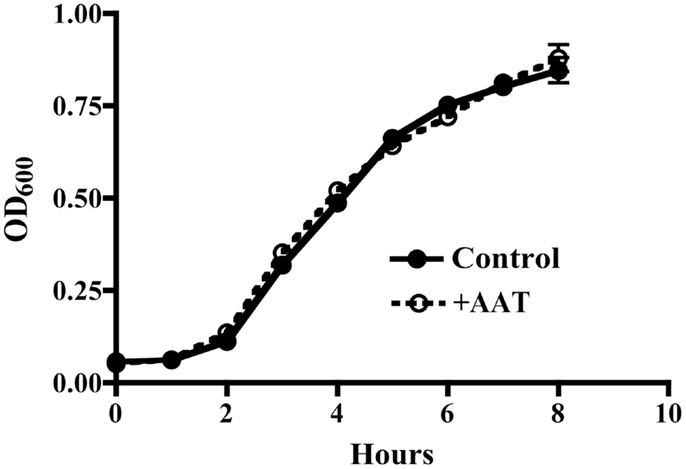
Figure 4. AAT effect on P.aer replication in suspension cultures. P.aer bacteria were inoculated into LB broth cultures in the absence (Control, closed circles with solid line) or presence of 5 mg/mL AAT (+AAT, open circles with dashed line) and incubated in a shaking incubator for 8 h at 37°C. The optical density (OD600) of the cultures was measured hourly. The means ± SEM OD values are shown from three separate experiments.
AAT Inhibits P.aer Internalization into A549 Lung Epithelial Cells, but does not Inhibit Bacterial Adherence
Prior studies showed that P.aer invades primary respiratory epithelial cells and the A549 epithelial cell line (25, 26). A549 cells were exposed to P.aer for 2 h, a period of time that allowed intracellular invasion of bacteria. As shown in Figure 5A, compared to P.aer alone (closed column), AAT significantly inhibited bacterial internalization (second column from left, mean 48% inhibition, p = 0.02). The effect of human NE on P.aer internalization into A549 lung epithelial cells was also assessed. A549 cell cultures exposed to human NE increased P.aer internalization by a mean 19% compared to P.aer alone (compare closed column and third column from left). The presence of AAT (far right column) reduced internalization levels by a mean 58% compared to NE-exposed cells (p = 0.01). All cultures were tested for cytotoxicity using an LDH release assay, and none was detected (not presented).
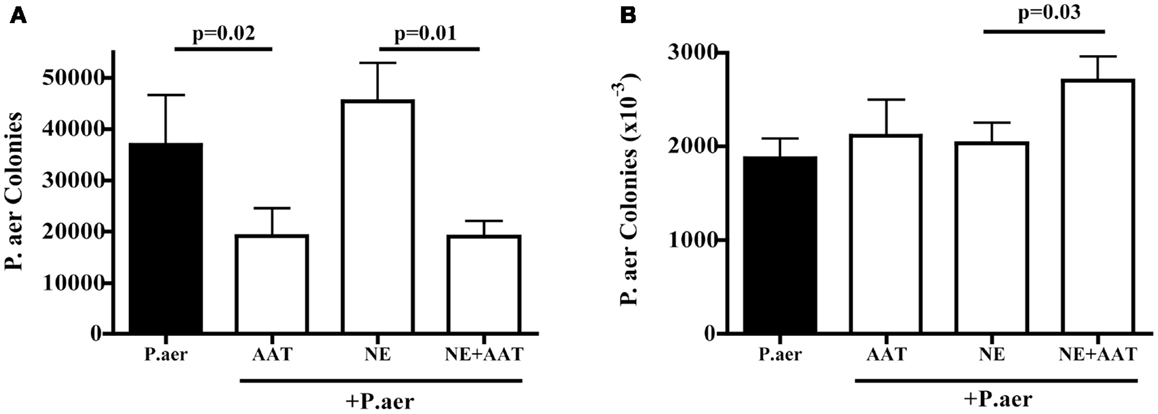
Figure 5. Effect of AAT on P.aer internalization and adhesion in A549 cells. In four separate experiments, A549 cells were exposed to P.aer alone (closed bars) or to P.aer in the presence of 5 mg/mL AAT, 2 μg/mL NE, or NE with AAT. In bacterial internalization experiments (A) and surface adhesion experiments (B), A549 cells were lysed and concentrations of released bacteria calculated after culture on LB-agar plates. p-Values are indicated in the graphs. Data are depicted as means + SEM of bacterial colonies.
To determine if AAT inhibited bacterial association with A549 cell surfaces, A549 cells were exposed to P.aer in the absence or presence of AAT. Incubation for 1 h following addition of P.aer permitted binding of bacteria to the cell surface while minimizing intracellular invasion. As shown in Figure 5B (two bars on left), AAT did not significantly alter P.aer binding to the surface of A549 cells compared to cells exposed to P.aer alone (closed bar). We also assessed the effect of AAT on P.aer adherence in the presence of human NE. Figure 5B shows (third and fourth bars from left) that AAT did not reduce bacterial association with A549 cells. In fact, AAT enhanced P.aer association with cells incubated with human NE (p = 0.03). All cultures were analyzed for toxicity using an LDH release assay, and no cytotoxicity was detected (not shown).
AAT Inhibits NE- or P.aer-Induced Disruption of A549 Cell Monolayers
The pathogenesis of severe pneumonia includes defects in the barrier function of respiratory epithelium. We examined the effect of human NE or viable P.aer on A549 cell monolayer integrity using the TEER assay, which measures the barrier integrity of cell monolayers (42). Figure 6A shows that compared to medium alone, addition of human NE to A549 cells reduced monolayer integrity by a maximum of 20% at 6 h (p < 0.001). Addition of 5 mg/mL AAT almost completely reversed the NE effect (p < 0.001, p < 0.05, or p < 0.01 comparing NE to NE + AAT at 1, 3, or 6 h, respectively).
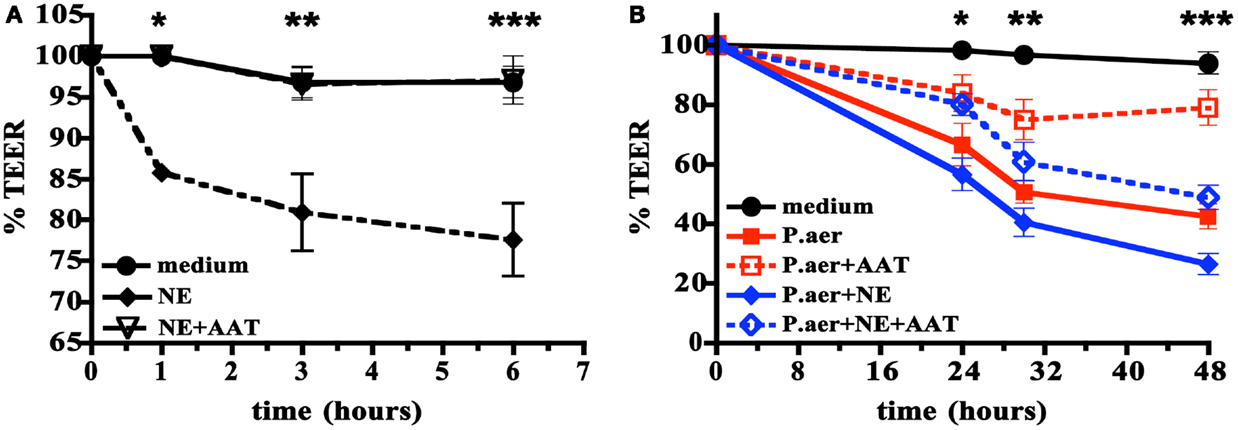
Figure 6. Effect of AAT on human NE- or P.aer-induced disruption of A549 cell monolayers. In (A) A549 cell monolayers were incubated with medium alone (medium, closed circles with solid line), human NE alone (NE, closed diamonds with dashed line), or with human NE in the presence of 5 mg/mL AAT (NE + AAT, open triangles with dashed line). Data are expressed as percent of TEER determined just prior to addition of NE (defined as T = 0 and set to 100%). The means ± SEM are shown for four separate experiments. *For T = 1 h, significant differences were noted for NE compared to medium (p < 0.001) and for NE compared to NE + AAT (p < 0.001). **For T = 3 h, significant differences were noted for NE compared to medium (p < 0.05) and for NE compared to NE + AAT (p < 0.05). ***For T = 6 h, significant differences were noted for NE compared to medium (p < 0.001) and for NE compared to NE + AAT (p < 0.001). In (B) A549 cell monolayers were exposed to medium alone (medium, black closed circles with solid line), P.aer alone (P.aer, red closed squares with solid line), P.aer with AAT (P.aer + AAT, red open squares with dashed line), P.aer and human NE (P.aer + NE, blue closed diamonds with solid line), or with P.aer and human NE with AAT (P.aer + NE + AAT, blue open diamonds with dashed line). Data are expressed as percent of TEER measurements in each condition just prior to addition of P.aer (defined as T = 0 and set to 100%). The means ± SEM are shown for three separate experiments. *For T = 24 h, significant differences were noted for medium compared to P.aer (p < 0.01), medium compared to P.aer + NE (p < 0.001), and for P.aer compared to P.aer + AAT (p < 0.05). **For T = 30 h, significant differences were noted for medium compared to P.aer (p < 0.001), medium compared to P.aer + NE (p < 0.001), P.aer compared to P.aer + AAT (p < 0.01), and for P.aer + NE compared to P.aer + NE + AAT (p < 0.05). ***For T = 48 h, significant differences were noted for medium compared to P.aer (p < 0.001), medium compared to P.aer + NE (p < 0.001), P.aer compared to P.aer + AAT (p < 0.001), and for P.aer + NE compared to P.aer + NE + AAT (p < 0.01).
In Figure 6B, we quantified the effect of viable P.aer alone or P.aer with human NE on A549 monolayer integrity. Cells exposed only to P.aer (P.aer) demonstrated decreased electrical resistance by a maximum 55% at 48 h compared to medium (p < 0.001). The presence of AAT significantly inhibited P.aer-induced monolayer disruption (p < 0.05, p < 0.01, or p < 0.001 comparing P.aer to P.aer + AAT at 24, 32, or 48 h, respectively). We also tested AAT effect in the presence of both P.aer and human NE. As shown in Figure 6B, the combination of P.aer and human NE (P.aer + NE) decreased monolayer resistance by 72% compared to medium at 48 h. AAT significantly reduced monolayer disruption induced by combined P.aer and human NE (p < 0.05, p < 0.05, or p < 0.01 comparing P.aer + NE to P.aer + NE + AAT at 24, 32, or 48 h, respectively). All cultures were analyzed for toxicity using an LDH release assay, and no cytotoxicity was detected (not shown).
Discussion
New approaches are needed to reduce the clinical and economic burdens of NP. More effective NP prophylaxis would be especially welcomed, since prevention of disease or reduced disease severity can produce substantial clinical and economic benefits. Therefore, we focused on preventative (prophylactic) models of AAT use in pneumonia therapy. P.aer was used in our experiments since this bacterium causes up to 20% of NP cases and carries an attributable mortality of nearly 43% (2, 4, 5).
Alpha-1 antitrypsin+/+ mice infected with inhaled P.aer demonstrated nearly 100% reduced mortality compared to non-transgenic control mice (Figure 1A). This result was especially striking since the mortality in control animals was 100% and AAT+/+ animals did not receive antimicrobial treatment. The survival benefit in AAT+/+ mice was likely due to increased AAT in lungs, since human AAT transgene expression in these animals is restricted to lung tissues (40). Since transgenic AAT+/+ mice express human AAT constitutively, this is a model of AAT pneumonia prophylaxis.
Exogenous human AAT was tested in non-transgenic C57Bl/6 mice. AAT delivered by aerosol alone in preliminary experiments did not significantly protect mice subjected to P.aer pneumonia (not shown). The nebulizer we used to deliver aerosolized AAT into the airways produces 5–10 μm particles, and aerosolized droplets of 1–3 μm are required for optimal AAT distribution into peripheral airspaces (54). Therefore, we believe insufficient AAT in the peripheral airways accounted for the absence of mortality effect. We also tested AAT administration by ip injection alone in preliminary studies with the goal of increasing serum concentrations to approximately 5 mg/mL. This AAT concentration suppressed P.aer internalization into respiratory epithelial cells in vitro (Figure 5), and reduced P.aer-induced epithelial barrier disruption in TEER experiments (Figure 6). We determined that two ip AAT doses of 2 mg administered 24 h apart followed by a third ip AAT injection 2 h before infection increased AAT serum levels to approximately 5.0 mg/mL (not shown). However, ip-alone AAT did not improve survival in mice compared to untreated controls (not shown). Since only about 10% of the AAT concentration in the circulation is detected in extracellular lung fluid (35), the increase in lung tissue AAT following ip injection alone was insufficient to protect mice. In contrast, exogenous AAT administration using combined aerosol delivery and ip injection resulted in significant protection from P.aer pneumonia (Figure 1B). However, combined aerosol and ip AAT administration did not achieve the same level of protection (44% survival, Figure 1B) as observed in AAT+/+ transgenic mice (90% survival, Figure 1A). In future studies, we will assess P.aer pneumonia in non-transgenic mice using aerosol AAT delivered using a nebulizer that produces 1–3 μm droplets. We believe optimized AAT delivery to peripheral airways will enable mortality benefit using aerosol AAT monotherapy.
Histopathologic assessments of infected mouse lungs showed reduced inflammation and damage in AAT+/+ mice compared to non-transgenic controls (Figure 2; Table 1). Table 1 documents reduced lung pathology in AAT+/+ mice for all measures tested, except for increased prevalence of bronchopneumonia in AAT+/+ mice. Since bronchopneumonia indicates less severe lung involvement than lobar pneumonia (see Materials and Methods), this observation reflected lower pneumonia severity in AAT+/+ mice. Reduced percent total lung involvement was observed in AAT+/+ mice that approached but did not achieve statistical significance (p = 0.052 compared to non-transgenic controls), probably due to insufficient sample size. Reduced lung damage in AAT+/+ mice may represent one mechanism of improved survival in AAT+/+ mice.
Striking reduction in bacterial proliferation in lungs was observed in AAT+/+ mice (Figure 3A). AAT did not directly affect bacterial replication (Figure 4), indicating AAT antibacterial effect was indirect. Since AAT is best described as an inhibitor of the serine protease NE, AAT inhibition of NE likely participated in AAT bacterial suppression. This implies a role for NE in enhancing bacterial proliferation. NE effects that likely assist bacterial survival include proteolytic destruction of respiratory tissues and inactivation of extracellular soluble anti-pathogen substances such as immunoglobulins, complement components, defensins, lysozyme, lactoferrin, and cathelicidins (12, 13, 16, 20, 21, 39). By quenching NE proteolytic activity, AAT can promote clearance of extracellular bacteria by maintaining the structural integrity of the airways and preserving cilia-mediated bacterial evacuation. AAT-induced NE blockade can also protect the function of soluble anti-pathogen substances. A separate NE activity that can interfere with bacterial clearance is increased expression of cell-surface bacterial binding sites (MUC1) that promotes bacterial invasion into epithelial cells. Internalization shields bacteria from extracellular immune effectors such as soluble anti-pathogen substances and phagocytic cells. It is therefore noteworthy that AAT suppressed P.aer internalization into respiratory epithelial cells in vitro (Figure 5). This AAT effect may assist bacterial killing by prolonging exposure of bacteria to soluble extracellular anti-pathogen mediators and phagocytic cells (13). Taken together, we surmise enhanced P.aer clearance in lungs in AAT+/+ mice (Figure 3A) involved counteracting NE-induced airways tissue destruction, reversing NE-inactivation of soluble antibacterial substances, and blocking P.aer access to the interior of respiratory epithelial cells.
As shown in Figure 3B, significant reduction in the magnitude of bacteremia was also observed in AAT+/+ mice (99% reduction compared to non-transgenic control mice). These results suggest unrestrained NE promoted bacteremia. NE-induced proteolytic lung damage can disrupt epithelial and endothelial barrier integrity and allow translocation of bacteria from lungs into blood. NE can also disrupt epithelial barrier integrity in the absence of tissue destruction or cytolysis (Figure 6, see text below). For these reasons, AAT suppression of NE effects on tissue barrier integrity is a possible mechanism by which AAT reduced bacteremia.
Although our results implicate NE as an enhancer of bacterial survival, prior studies showed NE possesses direct antibacterial activity (55). Therefore, the inhibitory effect of AAT on bacterial proliferation (Figure 3) must be reconciled with this NE antibacterial effect. Since the reported NE antibacterial activities occur intracellularly and apply to bacteria phagocytosed by neutrophils, a detrimental AAT effect on NE-induced bacterial killing would have to occur intracellularly. However, AAT has not been shown to inhibit intracellular proteases, and AAT likely does not interfere with beneficial intracellular NE-mediated antimicrobial effects.
Systemic inflammation was profoundly reduced in infected AAT+/+ mice. Serum concentrations of all 16 cytokines tested in AAT+/+ mice were lower compared to control animals (Table 2). We believe lower circulating cytokine levels in AAT+/+ mice reflected attenuated severity of pneumonia in AAT+/+ animals. The reduced systemic inflammatory response in AAT+/+ transgenic animals may have contributed to improved survival following P.aer pneumonia.
In vitro studies explored mechanisms by which AAT may interrupt pneumonia pathogenesis. AAT was not directly bactericidal for P.aer (Figure 4), and this has been reported previously (38). Pseudomonas and other pneumonia-causing bacteria can invade lung epithelial cells, which may be a mechanism of immune evasion (24, 26, 56–59). Therefore, we assessed AAT for effect on bacterial invasion of respiratory epithelial cells. In our studies, AAT significantly reduced P.aer internalization into A549 cells (Figure 5A). Unexpectedly, we did not observe an AAT effect on P.aer adherence to A549 cells (Figure 5B). This suggests AAT suppression of P.aer internalization occurred at a post-binding step during the process of invasion. The small but statistically significant increase in P.aer binding to cells exposed to combined AAT and human NE (compared to infected cultures with human NE alone) is of uncertain significance (Figure 5B). Results shown in Figure 5 suggest AAT assists clearance of bacteria by blocking intracellular invasion and exposing bacteria to extracellular soluble anti-pathogen mediators and phagocytic cells. We previously reported that AAT inhibited spontaneous internalization of Mycobacterium abscessus into human monocyte-derived macrophages (60). These parallel results raise the possibility that an unappreciated AAT function is to deny access of pathogens to the intracellular compartment. It is also noteworthy that AAT reduced spontaneous (absence of human NE) P.aer invasion of A549 cells (second bar from left in Figure 5A), indicating that AAT can block invasion using a mechanism unrelated to NE inhibition. Other reports also suggest AAT possesses activities independent of serine protease inhibition. These include suppression of cytokine production in whole blood or in monocytes, and inhibition of HIV replication (50, 61–66).
Respiratory epithelial barrier dysfunction is a pivotal component of severe pneumonia that can result in impaired gas exchange, seepage of fluid into the respiratory tract, and decreased bacterial containment with ensuing bacteremia (46). In TEER experiments in A549 cell monolayers, human NE, P.aer, or combined NE and P.aer significantly reduced monolayer barrier integrity (Figure 6). AAT reduced monolayer disruption induced by human NE, P.aer, or combined human NE and P.aer (Figures 6A,B). Therefore, one mechanism of AAT protection against pneumonia-related pathology is suppressed disruption of the pulmonary epithelium barrier. No cytotoxicity was observed in the TEER experiments (not shown), suggesting monolayer alterations involved changes in the integrity of intercellular junctions. Prior reports showed P.aer or LPS disrupted respiratory cell barrier function, and altered intercellular junction integrity accompanied these effects (67, 68). We speculate that AAT prevents epithelial barrier disruption in vivo by blocking alterations in intercellular tight junctions induced during inflammation and infection.
Our studies suggest that AAT is an endogenous molecule with pneumonia-suppressive function, which implies that AAT deficit weakens host defense against pneumonia. In a study of deaths in persons with genetic AAT deficiency, Tomashefski et al. (69) noted that mortality due to pneumonia approached 37%, the highest immediate cause of death in these patients. In comparison, pneumonia (including influenza) was the cause of death in 2.2% of the US population in 2009 (70). The extraordinary mortality caused by pneumonia in AAT deficient persons suggests predisposition to severe pneumonia due to AAT deficit. Similarly, studies in patients without known AAT deficiency suggest association between a deficit in AAT function in lung tissues and pneumonia. Braun et al. (71) measured BALF AAT levels in patients with acute pneumonia. Although BALF AAT concentrations were higher in pneumonia patients than in healthy controls, the serine protease inhibitor function of AAT was reduced in pneumonia patients. The authors surmised that proteases and reactive oxygen species (produced by neutrophils and lung epithelia) degraded AAT serine protease inhibitor function in the airways. Taken together, these reports document association between reduced AAT levels or reduced AAT function and pneumonia, and suggest a pneumonia-suppressive role for AAT. A pneumonia-suppressive role for AAT implies AAT augmentation should reduce frequency or severity of pneumonia. In fact, two reports suggest this is the case. Lieberman examined the self-reported incidence of lung infections in patients with genetic AAT deficiency, and noted three to five lung infections per year in AAT deficient patients not receiving AAT replacement therapy (72). In contrast, only zero to one infections per year were described in AAT deficient patients receiving intravenous AAT replacement therapy. In a rat model of chronic P.aer lung infection, aerosol delivery of AAT decreased lung inflammation and accelerated bacterial clearance (38).
Conclusion
Several limitations apply to our studies. We examined AAT effects as pneumonia prophylaxis (AAT present before onset of pneumonia), and we did not evaluate AAT administration as a treatment after infection. Although we believe AAT administration will be beneficial after onset of pneumonia, AAT effect as a non-preventative therapy cannot be confidently inferred from our results. Also, the generalizability of the AAT anti-pneumonia effect to bacteria other than P.aer is uncertain. Technical limitations likely prevented us from obtaining mortality reduction in mice treated with exogenous AAT equivalent to mortality reduction in AAT+/+ mice (Figure 1). Optimal AAT deposition in peripheral lung tissues requires aerosol droplets of 1–3 μm, and the nebulizer used in our studies produced particle sizes of 5–10 μm. Additionally, our in vitro pathogenesis studies were conducted in the A549 respiratory epithelial cell line, and cell lines do not necessarily reflect characteristics of primary cells in vivo. The AAT concentration used in our in vitro studies (5 mg/mL) is consistent with serum levels attained during acute disease. However, AAT concentrations in the lung microenvironment that enable AAT antibacterial effects are unknown. This introduces some uncertainty regarding the relevance of our in vitro mechanistic studies to our in vivo results. On the other hand, since augmented AAT levels in the respiratory tract enhanced survival (Figure 1), increasing AAT biologic activity sufficient to confer benefit in vivo is possible. Finally, although animal pneumonia models are useful, projecting our results to efficacy in humans entails risk.
In this report, we show protective AAT effects in mice with experimentally induced acute P.aer pneumonia. AAT expression in the lungs of AAT+/+ mice was associated with reduced pneumonia mortality, lower severity of lung inflammation, decreased bacterial proliferation in lungs, prevention of bacteremia, and suppressed systemic inflammatory response. In vitro studies demonstrated AAT blockade of P.aer internalization into lung epithelial cells and AAT suppression of NE- or P.aer-induced epithelial cell barrier disruption. AAT did not demonstrate direct antibacterial activity, and AAT protection likely involved both NE neutralization and effects independent of NE neutralization. We believe AAT protection in mouse pneumonia (Figure 1) was due to inhibition of NE-induced proteolytic lung damage, reduction of NE lysis of extracellular anti-pathogen immune mediators, blockade of P.aer invasion of respiratory epithelial cells, and suppression of epithelial barrier dysfunction. Our studies are different from those reported by Cantin and Woods (38). Although our results and those in the Cantin and Woods report show AAT reduced bacterial density in lungs, Cantin and Woods studied chronic Pseudomonas lung infection in rats. Also, aerosol AAT was administered daily for 7 days after infection. The outcomes in Cantin and Woods included reduced lung bacteria and lung inflammation 7 days post-AAT initiation, and reduced airway NE activity 6 h after onset of AAT administration.
Alpha-1 antitrypsin appears to possess broad-spectrum anti-pathogen function, since AAT at physiological concentrations demonstrated inhibitory effects in vitro against Mycobacterium abscessus and HIV (64, 73–75). In fact, an AAT-derived synthetic molecule suppressed HIV replication following intravenous infusion into infected patients (66). Given these observations, we speculate that AAT is an innate immune mediator. It appears that AAT blocks activity of host molecules required for pathogen replication (such as NE) and AAT counteracts pathogen evasion of host immune defenses. Since extracellular lung fluid levels of AAT are only about 10% of serum concentrations (35), the lung may represent an “Achilles heel” of AAT immune protection. This observation provides rationale for using inhaled AAT for prophylaxis or treatment of respiratory tract infections. Results in this report suggest that clinical application may include AAT use as an inhaled drug for NP prophylaxis. Combination treatment using inhaled and intravenous AAT delivery may provide additional protection. Since AAT does not target pathogen-specific molecules, AAT antimicrobial effects may be impervious to genetic mutations that alter pathogen components. Furthermore, since AAT has been available for clinical use since 1988 and AAT has an impressive record of safe use in humans, application of AAT for this indication can proceed rapidly to the bedside (37, 76, 77).
Conflict of Interest Statement
Leland Shapiro is a shareholder of Omni Bio Pharmaceutical, Inc. All other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This project was funded by Omni Bio Pharmaceutical, Inc.
References
1. Fagon JY, Chastre J, Hance AJ, Montravers P, Novara A, Gibert C. Nosocomial pneumonia in ventilated patients: a cohort study evaluating attributable mortality and hospital stay. Am J Med (1993) 94:281–8. doi:10.1016/0002-934309390060-3
2. Kollef MH. Epidemiology and risk factors for nosocomial pneumonia. Emphasis on prevention. Clin Chest Med (1999) 20:653–70. doi:10.1016/S0272-52310570242-2
3. Klevens RM, Edwards JR, Richards CL Jr, Horan TC, Gaynes RP, Pollock DA, et al. Estimating health care-associated infections and deaths in U.S. hospitals, 2002. Public Health Rep (2007) 122:160–6.
4. ATS. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med (2005) 171:388–416. doi:10.1164/rccm.200405-644ST
5. Jones RN. Microbial etiologies of hospital-acquired bacterial pneumonia and ventilator-associated bacterial pneumonia. Clin Infect Dis (2010) 51(Suppl 1):S81–7. doi:10.1086/653053
6. Armstrong GL, Conn LA, Pinner RW. Trends in infectious disease mortality in the United States during the 20th century. JAMA (1999) 281:61–6. doi:10.1001/jama.281.1.61
7. Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis (2009) 48:1–12. doi:10.1086/595011
8. Scott RD. The Direct Medical Costs of Healthcare-Associated Infections in US Hospitals and the Benefits of Prevention. Atlanta: Centers for Disease Control and Prevention (2009). Available from: http://www.cdc.gov/hai/pdfs/hai/scott_costpaper.pdf.
9. O’Grady NP, Murray PR, Ames N. Preventing ventilator-associated pneumonia: does the evidence support the practice? JAMA (2012) 307:2534–9. doi:10.1001/jama.2012.6445
10. Kollef MH, Schuster DP. The acute respiratory distress syndrome. N Engl J Med (1995) 332:27–7. doi:10.1056/NEJM199501053320106
11. CDC. Guidelines for prevention of nosocomial pneumonia. Centers for Disease Control and Prevention. MMWR Recomm Rep (1997) 46:1–79.
12. Moraes TJ, Chow CW, Downey GP. Proteases and lung injury. Crit Care Med (2003) 31:S189–94. doi:10.1097/01.CCM.0000057842.90746.1E
13. Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol (2011) 45:189–201. doi:10.1165/rcmb.2011-0011RT
14. El Solh AA, Akinnusi ME, Wiener-Kronish JP, Lynch SV, Pineda LA, Szarpa K. Persistent infection with Pseudomonas aeruginosa in ventilator-associated pneumonia. Am J Respir Crit Care Med (2008) 178:513–9. doi:10.1164/rccm.200802-239OC
15. Craig A, Mai J, Cai S, Jeyaseelan S. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect Immun (2009) 77:568–75. doi:10.1128/IAI.00832-08
16. Stockley RA. Role of inflammation in respiratory tract infections. Am J Med (1995) 99:S8–13. doi:10.1016/S0002-934309980304-0
17. Hermant B, Bibert S, Concord E, Dublet B, Weidenhaupt M, Vernet T, et al. Identification of proteases involved in the proteolysis of vascular endothelium cadherin during neutrophil transmigration. J Biol Chem (2003) 278:14002–12. doi:10.1074/jbc.M300351200
18. Couillin I, Vasseur V, Charron S, Gasse P, Tavernier M, Guillet J, et al. IL-1R1/MyD88 signaling is critical for elastase-induced lung inflammation and emphysema. J Immunol (2009) 183:8195–202. doi:10.4049/jimmunol.0803154
19. Meyer-Hoffert U, Wiedow O. Neutrophil serine proteases: mediators of innate immune responses. Curr Opin Hematol (2011) 18:19–24. doi:10.1097/MOH.0b013e32834115d1
20. Tralau T, Meyer-Hoffert U, Schroder JM, Wiedow O. Human leukocyte elastase and cathepsin G are specific inhibitors of C5a-dependent neutrophil enzyme release and chemotaxis. Exp Dermatol (2004) 13:316–25. doi:10.1111/j.0906-6705.2004.00145.x
21. Stromstedt AA, Pasupuleti M, Schmidtchen A, Malmsten M. Evaluation of strategies for improving proteolytic resistance of antimicrobial peptides by using variants of EFK17, an internal segment of LL-37. Antimicrob Agents Chemother (2009) 53:593–602. doi:10.1128/AAC.00477-08
22. Lillehoj EP, Hyun SW, Kim BT, Zhang XG, Lee DI, Rowland S, et al. Muc1 mucins on the cell surface are adhesion sites for Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol (2001) 280:L181–7.
23. Sando L, Pearson R, Gray C, Parker P, Hawken R, Thomson PC, et al. Bovine Muc1 is a highly polymorphic gene encoding an extensively glycosylated mucin that binds bacteria. J Dairy Sci (2009) 92:5276–91. doi:10.3168/jds.2009-2216
24. Plotkowski MC, Beck G, Tournier JM, Bernardo-Filho M, Marques EA, Puchelle E. Adherence of Pseudomonas aeruginosa to respiratory epithelium and the effect of leucocyte elastase. J Med Microbiol (1989) 30:285–93. doi:10.1099/00222615-30-4-285
25. Plotkowski MC, Saliba AM, Pereira SH, Cervante MP, Bajolet-Laudinat O. Pseudomonas aeruginosa selective adherence to and entry into human endothelial cells. Infect Immun (1994) 62:5456–63.
26. Chi E, Mehl T, Nunn D, Lory S. Interaction of Pseudomonas aeruginosa with A549 pneumocyte cells. Infect Immun (1991) 59:822–8.
27. Lehrer RI, Ganz T. Cathelicidins: a family of endogenous antimicrobial peptides. Curr Opin Hematol (2002) 9:18–22. doi:10.1097/00062752-200201000-00004
28. Lee CT, Fein AM, Lippmann M, Holtzman H, Kimbel P, Weinbaum G. Elastolytic activity in pulmonary lavage fluid from patients with adult respiratory-distress syndrome. N Engl J Med (1981) 304:192–6. doi:10.1056/NEJM198101223040402
29. Tanaka H, Sugimoto H, Yoshioka T, Sugimoto T. Role of granulocyte elastase in tissue injury in patients with septic shock complicated by multiple-organ failure. Ann Surg (1991) 213:81–5. doi:10.1097/00000658-199101000-00014
30. Kieninger AN, Lipsett PA. Hospital-acquired pneumonia: pathophysiology diagnosis, and treatment. Surg Clin North Am (2009) 89:439–61. doi:10.1016/j.suc.2008.11.001
31. Donato LJ, Jenkins SM, Smith C, Katzmann JA, Snyder MR. Reference and interpretive ranges for alpha(1)-antitrypsin quantitation by phenotype in adult and pediatric populations. Am J Clin Pathol (2012) 138:398–405. doi:10.1309/AJCPMEEJK32ACYFP
32. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med (1999) 340:448–54. doi:10.1056/NEJM199902113400607
33. Blank CA, Brantly M. Clinical features and molecular characteristics of alpha 1-antitrypsin deficiency. Ann Allergy (1994) 72:105–20; quiz 120–2.
34. Min WK, Chun S, Hwang SH, Park H. No relationship between serum lipoprotein(a) and albumin concentrations in patients with acute phase response. Ann Clin Biochem (1999) 36(Pt 5):617–21.
35. Wewers MD, Casolaro MA, Sellers SE, Swayze SC, McPhaul KM, Wittes JT, et al. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med (1987) 316:1055–62. doi:10.1056/NEJM198704233161704
36. Sandhaus RA, Turino G. Neutrophil elastase-mediated lung disease. COPD (2013) 10(Suppl 1):60–3. doi:10.3109/15412555.2013.764403
37. ATS. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med (2003) 168:818–900. doi:10.1164/rccm.168.7.818
38. Cantin AM, Woods DE. Aerosolized prolastin suppresses bacterial proliferation in a model of chronic Pseudomonas aeruginosa lung infection. Am J Respir Crit Care Med (1999) 160:1130–5. doi:10.1164/ajrccm.160.4.9807166
39. Wiesner O, Litwiller RD, Hummel AM, Viss MA, McDonald CJ, Jenne DE, et al. Differences between human proteinase 3 and neutrophil elastase and their murine homologues are relevant for murine model experiments. FEBS Lett (2005) 579:5305–12. doi:10.1016/j.febslet.2005.08.056
40. Dhami R, Zay K, Gilks B, Porter S, Wright JL, Churg A. Pulmonary epithelial expression of human alpha1-antitrypsin in transgenic mice results in delivery of alpha1-antitrypsin protein to the interstitium. J Mol Med (1999) 77:377–85. doi:10.1007/s001090050364
41. Lewis EC, Mizrahi M, Toledano M, Defelice N, Wright JL, Churg A, et al. alpha1-Antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci USA (2008) 105:16236–41. doi:10.1073/pnas.0807627105
42. Warfel JM, Steele AD, D’agnillo F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am J Pathol (2005) 166:1871–81. doi:10.1016/S0002-94401062496-0
43. Behnia M, Robertson KA, Martin WJ 2nd. Lung infections: role of apoptosis in host defense and pathogenesis of disease. Chest (2000) 117:1771–7. doi:10.1378/chest.117.6.1771
44. Schaaf B, Wieghorst A, Aries SP, Dalhoff K, Braun J. Neutrophil inflammation and activation in bronchiectasis: comparison with pneumonia and idiopathic pulmonary fibrosis. Respiration (2000) 67:52–9. doi:10.1159/000029463
45. Hotchkiss RS, Dunne WM, Swanson PE, Davis CG, Tinsley KW, Chang KC, et al. Role of apoptosis in Pseudomonas aeruginosa pneumonia. Science (2001) 294::1783. doi:10.1126/science.294.5548.1783a
46. Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med (2008) 358:716–27. doi:10.1056/NEJMra074111
47. Petrache I, Fijalkowska I, Medler TR, Skirball J, Cruz P, Zhen L, et al. Alpha-1 antitrypsin inhibits caspase-3 activity, preventing lung endothelial cell apoptosis. Am J Pathol (2006a) 169:1155–66. doi:10.2353/ajpath.2006.060058
48. Petrache I, Fijalkowska I, Zhen L, Medler TR, Brown E, Cruz P, et al. A novel antiapoptotic role for alpha1-antitrypsin in the prevention of pulmonary emphysema. Am J Respir Crit Care Med (2006b) 173:1222–8. doi:10.1164/rccm.200512-1842OC
49. Zhang B, Lu Y, Campbell-Thompson M, Spencer T, Wasserfall C, Atkinson M, et al. Alpha1-antitrypsin protects beta-cells from apoptosis. Diabetes (2007) 56:1316–23. doi:10.2337/db06-1273
50. Pott GB, Chan ED, Dinarello CA, Shapiro L. Alpha-1-antitrypsin is an endogenous inhibitor of proinflammatory cytokine production in whole blood. J Leukoc Biol (2009) 85:886–95. doi:10.1189/jlb.0208145
51. Seki H, Fukunaga K, Arita M, Arai H, Nakanishi H, Taguchi R, et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J Immunol (2009) 184:836–43. doi:10.4049/jimmunol.0901809
52. Janciauskiene SM, Bals R, Koczulla R, Vogelmeier C, Kohnlein T, Welte T. The discovery of alpha1-antitrypsin and its role in health and disease. Respir Med (2011) 105:1129–39. doi:10.1016/j.rmed.2011.02.002
53. Chan ED, Pott GB, Silkoff PE, Ralston AH, Bryan CL, Shapiro L. Alpha-1-antitrypsin inhibits nitric oxide production. J Leukoc Biol (2012) 92:1251–60. doi:10.1189/jlb.0212071
54. Usmani OS, Barnes PJ. Assessing and treating small airways disease in asthma and chronic obstructive pulmonary disease. Ann Med (2011) 44:146–56. doi:10.3109/07853890.2011.585656
55. Belaaouaj A, McCarthy R, Baumann M, Gao Z, Ley TJ, Abraham SN, et al. Mice lacking neutrophil elastase reveal impaired host defense against gram negative bacterial sepsis. Nat Med (1998) 4:615–8. doi:10.1038/nm0598-615
56. de Lima Pimenta A, Di Martino P, Le Bouder E, Hulen C, Blight MA. In vitro identification of two adherence factors required for in vivo virulence of Pseudomonas fluorescens. Microbes Infect (2003) 5:1177–87. doi:10.1016/j.micinf.2003.09.002
57. Knappstein S, Ide T, Schmidt MA, Heusipp G. Alpha 1-antitrypsin binds to and interferes with functionality of EspB from atypical and typical enteropathogenic Escherichia coli strains. Infect Immun (2004) 72:4344–50. doi:10.1128/IAI.72.8.4344-4350.2004
58. Leroy-Dudal J, Gagniere H, Cossard E, Carreiras F, Di Martino P. Role of alphavbeta5 integrins and vitronectin in Pseudomonas aeruginosa PAK interaction with A549 respiratory cells. Microbes Infect (2004) 6:875–81. doi:10.1016/j.micinf.2004.05.004
59. Angus AA, Lee AA, Augustin DK, Lee EJ, Evans DJ, Fleiszig SM. Pseudomonas aeruginosa induces membrane blebs in epithelial cells, which are utilized as a niche for intracellular replication and motility. Infect Immun (2008) 76:1992–2001. doi:10.1128/IAI.01221-07
60. Chan ED, Kaminska AM, Gill W, Chmura K, Feldman NE, Bai X, et al. Alpha-1-antitrypsin (AAT) anomalies are associated with lung disease due to rapidly growing mycobacteria and AAT inhibits Mycobacterium abscessus infection of macrophages. Scand J Infect Dis (2007a) 39:690–6. doi:10.1080/00365540701225744
61. Tilg H, Vannier E, Vachino G, Dinarello CA, Mier JW. Antiinflammatory properties of hepatic acute phase proteins: preferential induction of interleukin 1 (IL-1) receptor antagonist over IL-1 beta synthesis by human peripheral blood mononuclear cells. J Exp Med (1993) 178:1629–36. doi:10.1084/jem.178.5.16297693853
62. Janciauskiene S, Moraga F, Lindgren S. C-terminal fragment of alpha1-antitrypsin activates human monocytes to a pro-inflammatory state through interactions with the CD36 scavenger receptor and LDL receptor. Atherosclerosis (2001) 158:41–51. doi:10.1016/S0021-91500000767-X
63. Janciauskiene SM, Nita IM, Stevens T. Alpha1-antitrypsin, old dog, new tricks. Alpha1-antitrypsin exerts in vitro anti-inflammatory activity in human monocytes by elevating cAMP. J Biol Chem (2007) 282:8573–82. doi:10.1074/jbc.M607976200
64. Munch J, Standker L, Adermann K, Schulz A, Schindler M, Chinnadurai R, et al. Discovery and optimization of a natural HIV-1 entry inhibitor targeting the gp41 fusion peptide. Cell (2007) 129:263–75. doi:10.1016/j.cell.2007.02.042
65. Subramaniyam D, Virtala R, Pawlowski K, Clausen IG, Warkentin S, Stevens T, et al. TNF-alpha-induced self expression in human lung endothelial cells is inhibited by native and oxidized alpha1-antitrypsin. Int J Biochem Cell Biol (2008) 40:258–71. doi:10.1016/j.biocel.2007.07.016
66. Forssmann WG, The YH, Stoll M, Adermann K, Albrecht U, Tillmann HC, et al. Short-term monotherapy in HIV-infected patients with a virus entry inhibitor against the gp41 fusion peptide. Sci Transl Med (2010) 2:63re63. doi:10.1126/scitranslmed.3001697
67. Halldorsson S, Gudjonsson T, Gottfredsson M, Singh PK, Gudmundsson GH, Baldursson O. Azithromycin maintains airway epithelial integrity during Pseudomonas aeruginosa infection. Am J Respir Cell Mol Biol (2009) 42:62–8. doi:10.1165/rcmb.2008-0357OC
68. He D, Su Y, Usatyuk PV, Spannhake EW, Kogut P, Solway J, et al. Lysophosphatidic acid enhances pulmonary epithelial barrier integrity and protects endotoxin-induced epithelial barrier disruption and lung injury. J Biol Chem (2009) 284:24123–32. doi:10.1074/jbc.M109.007393
69. Tomashefski JF Jr, Crystal RG, Wiedemann HP, Mascha E, Stoller JK. The bronchopulmonary pathology of alpha-1 antitrypsin (AAT) deficiency: findings of the Death Review Committee of the national registry for individuals with Severe Deficiency of Alpha-1 Antitrypsin. Hum Pathol (2004) 35:1452–61. doi:10.1016/j.humpath.2004.08.013
70. Kochanek KD, Xu J, Murphy SL, Minino AM, Kung H. Deaths: preliminary data for 2009. Natl Vital Stat Rep (2011) 59:1–54.
71. Braun J, Dalhoff K, Schaaf B, Wood WG, Wiessmann KJ. Characterization of protein-antiproteinase imbalance in bronchoalveolar lavage from patients with pneumonia. Eur Respir J (1994) 7:127–33. doi:10.1183/09031936.94.07010127
72. Lieberman J. Augmentation therapy reduces frequency of lung infections in antitrypsin deficiency: a new hypothesis with supporting data. Chest (2000) 118:1480–5. doi:10.1378/chest.118.5.1480
73. Shapiro L, Pott GB, Ralston AH. Alpha-1-antitrypsin inhibits human immunodeficiency virus type 1. FASEB J (2001) 15:115–22. doi:10.1096/fj.00-0311com
74. Chan WM,Andrews C, Dragan L, Fredrick D, Armstrong L, Lyons C, et al. Three novel mutations in KIF21A highlight the importance of the third coiled-coil stalk domain in the etiology of CFEOM1. BMC Genet (2007b) 8::26. doi:10.1186/1471-2156-8-26
75. Zhou X, Shapiro L, Fellingham G, Willardson BM, Burton GF. HIV replication in CD4+ T lymphocytes in the presence and absence of follicular dendritic cells: inhibition of replication mediated by alpha-1-antitrypsin through altered IkappaBalpha ubiquitination. J Immunol (2011) 186:3148–55. doi:10.4049/jimmunol.1001358
76. Hubbard RC, Sellers S, Czerski D, Stephens L, Crystal RG. Biochemical efficacy and safety of monthly augmentation therapy for alpha 1-antitrypsin deficiency. JAMA (1988) 260:1259–64. doi:10.1001/jama.260.9.1259
Keywords: pneumonia, alpha-1 antitrypsin, cytokines, sepsis, Pseudomonas, neutrophil elastase
Citation: Pott GB, Beard KS, Bryan CL, Merrick DT and Shapiro L (2013) Alpha-1 antitrypsin reduces severity of Pseudomonas pneumonia in mice and inhibits epithelial barrier disruption and Pseudomonas invasion of respiratory epithelial cells. Front. Public Health 1:19. doi: 10.3389/fpubh.2013.00019
Received: 02 May 2013; Accepted: 07 June 2013;
Published online: 21 June 2013.
Edited by:
Anna Norrby-Teglund, Karolinska Institutet, SwedenReviewed by:
Eva Medina, Helmholtz Centre for Infection Research, GermanyPeter Bergman, Karolinska Institutet, Sweden
Copyright: © 2013 Pott, Beard, Bryan, Merrick and Shapiro. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Leland Shapiro, Department of Medicine, Division of Infectious Diseases, Veterans Affairs Medical Center, University of Colorado Denver, 12700 East 19th Avenue, Box B168, Aurora, CO 80045, USA e-mail:bGVsYW5kLnNoYXBpcm9AdWNkZW52ZXIuZWR1