 Daniel R. Knight1
Daniel R. Knight1 Thomas V. Riley1,2,3,4*
Thomas V. Riley1,2,3,4*- 1Medical, Molecular, and Forensic Sciences, Murdoch University, Perth, WA, Australia
- 2School of Medical and Health Sciences, Edith Cowan University, Joondalup, WA, Australia
- 3School of Biomedical Sciences, The University of Western Australia, Nedlands, WA, Australia
- 4PathWest Laboratory Medicine, Department of Microbiology, Nedlands, WA, Australia
Clostridium difficile is toxin-producing antimicrobial resistant (AMR) enteropathogen historically associated with diarrhea and pseudomembranous colitis in hospitalized patients. In recent years, there have been dramatic increases in the incidence and severity of C. difficile infection (CDI), and associated morbidity and mortality, in both healthcare and community settings. C. difficile is an ancient and diverse species that displays a sympatric lifestyle, establishing itself in a range of ecological niches external to the healthcare system. These sources/reservoirs include food, water, soil, and over a dozen animal species, in particular, livestock such as pigs and cattle. In a manner analogous to human infection, excessive antimicrobial exposure, particularly to cephalosporins, is driving the expansion of C. difficile in livestock populations worldwide. Subsequent spore contamination of meat, vegetables grown in soil containing animal feces, agricultural by-products such as compost and manure, and the environment in general (households, lawns, and public spaces) is contributing to a persistent community source/reservoir of C. difficile and the insidious rise of CDI in the community. The whole-genome sequencing era continues to redefine our view of this complex pathogen. The application of high-resolution microbial genomics in a One Health framework (encompassing clinical, veterinary, and environment derived datasets) is the optimal paradigm for advancing our understanding of CDI in humans and animals. This approach has begun to yield critical insights into the genetic diversity, evolution, AMR, and zoonotic potential of C. difficile. In Europe, North America, and Australia, microevolutionary analysis of the C. difficile core genome shows strains common to humans and animals (livestock or companion animals) do not form distinct populations but share a recent evolutionary history. Moreover, for C. difficile sequence type 11 and PCR ribotypes 078 and 014, major lineages of One Health importance, this approach has substantiated inter-species clonal transmission between animals and humans. These findings indicate either a zoonosis or anthroponosis. Moreover, they challenge the existing paradigm and the long-held misconception that CDI is primarily a healthcare-associated infection. In this article, evolutionary, and zoonotic aspects of CDI are discussed, including the anthropomorphic factors that contribute to the spread of C. difficile from the farm to the community.
Introduction
Last year was the 40th anniversary of the publication in 1978 of a series of papers from several research groups that provided proof that Clostridium difficile caused pseudomembranous colitis (1–4). While the spectrum of gastrointestinal disease caused by C. difficile has broadened significantly since then, for much of those 40 years C. difficile was thought of as causing disease almost exclusively within high-risk hospitalized patient populations (5). In evolutionary terms, 40 years is a negligible length of time. The Clostridia are an ancient prokaryotic lineage, estimated to have diverged from the bacterial domain 2.34 Ga (billion years) ago around the time when concentrations of molecular oxygen in the atmosphere began to increase (6). With the advances of next-generation sequencing, the taxonomy of the Clostridia is currently undergoing a major revision. Indeed, given the significant differences between C. difficile and some other pathogenic clostridia, it has been proposed that it be renamed Clostridioides difficile (7). While this has caused some angst in the C. difficile community, both names are currently viewed as being “validly published” and therefore acceptable (8).
In recent years, the vast majority of emerging or re-emerging infections have been vector-borne or zoonoses—animal diseases that are transmissible to humans (9). Most attention has focused on viral infections because of highly publicized outbreaks; SARS, avian influenza, and Ebola. However, disease associated with C. difficile infection (CDI) has killed more people worldwide in the last 15 years than all these viral infections combined, around 30,000 per year in the USA alone according to the CDC (10). CDI should always have been considered a zoonosis, either direct or indirect. In some definitions of zoonoses, non-human animal hosts play an essential role in maintaining the infection in nature and humans are only incidental hosts. In CDI, all animals (human and non-human) are likely hosts; the wide variety of animals from which C. difficile has already been isolated suggests this (11).
What then is the natural history of CDI following exposure to C. difficile? C. difficile is ubiquitous in the environment. C. difficile colonizes the gastrointestinal tracts of all animals during the neonatal period, multiplies, and is excreted, but cannot/does not compete well when other bacterial species start to colonize. The exact timing of this change is not clear, but it is probably linked to changes in diet in babies, i.e., weaning. Through a process known as colonization resistance, a well-developed microbiota provides protection against overgrowth of C. difficile by inhibiting germination, vegetative growth, and toxin production (12). In human and non-human animals, antimicrobial exposure creates an environment that could be thought of as mimicking the neonatal gut—characterized by an underdeveloped microbiota and consequently reduced or absent colonization resistance. In such a compromised host gut, C. difficile spores rapidly germinate and begin to produce potent cytotoxins (toxin A and toxin B) which cause extensive colonic inflammation and epithelial tissue damage, the net effect being a rapid fluid loss into the intestinal lumen which manifests as diarrhea (13). Some strains also produce a binary toxin, an ADP-ribosyltransferase that causes actin cytoskeletal disruption, and is associated with more severe CDI, a higher case-fatality rate and refractory disease (14).
When those antimicrobials were cephalosporins in the 1980s and 90s, antimicrobials to which C. difficile is intrinsically resistant, there was an expansion of CDI in hospitals that continues today. Since the 1990s in North America, cephalosporins have been licensed for use in food animals. There has been an amplification of C. difficile in food animals since then, with subsequent contamination of meat, and vegetables grown in soil containing animal feces. In some animals such as piglets, there is overt disease with significant impact on industry. “Animal” strains of C. difficile are now infecting humans. C. difficile ribotype (RT) 027 was found in animals in North America in the early 2000s (15) but probably moved from animals to humans a decade earlier around the time that RT027 developed resistance to fluoroquinolone antimicrobials (16). This strain was likely to have initially caused infections in the community at a time when community-acquired (CA) CDI [defined as cases with symptom onset in the community or ≤ 48 h after admission to a healthcare facility (17)] was thought infrequent, and diarrhea in the community was rarely investigated. The mutation to fluoroquinolone resistance and high use of fluoroquinolones drove RT027 spread, in North America and later Europe, once it entered the hospital system (16). A similar process now appears to be occurring with C. difficile RT078, another animal strain that has increased significantly as a cause of CA-CDI in Europe over the last 10 years (18, 19). C. difficile continues to expand in food animal populations, driven by cephalosporin use, and animal strains of C. difficile are driving the worldwide increase in CA-CDI.
The whole-genome sequencing era continues to redefine our view of this complex pathogen. The application of high-resolution microbial genomics in a One Health framework (encompassing clinical, veterinary, and environment derived datasets) is the optimal paradigm for advancing our understanding of CDI in humans and animals. This approach has begun to yield critical insights into the genetic diversity, evolution, AMR, and zoonotic potential of C. difficile. In this review, evolutionary and zoonotic aspects of CDI are discussed, including the anthropomorphic factors that contribute to the spread of C. difficile from the farm to the community.
Community-Acquired CDI
Surveillance data indicate that CA-CDI comprises a significant fraction of total CDI cases and that the incidence of CA-CDI has been increasing globally (20). In the United States, CA-CDI accounts for around a third of all CDI cases and increased 4-fold during the period 1991–2005 (18, 21–24). In another US study, comparable incidence rates for CA-CDI and hospital-associated CDI (HA-CDI) were reported (11.2 cases/100,000 person-years and 12.1 cases/100,000 person-years, respectively) (18). A recent European multi-center study (97 hospitals in 34 European countries) found 14% of 506 cases were classified CA-CDI (25). In Australia, data from 2011 to 2012 showed CA-CDI accounted for up to a quarter of all cases (26% of 5,109 CDI cases) and has been increasing in recent years (26–28). More recent studies from the USA report higher proportions of CA-CDI around 40% (24). Many studies have noted that individuals with CA-CDI often do not have the “classical” risk factors for CDI acquisition and are generally younger, healthy, and female, without contact with hospitalized patients nor prior antimicrobial exposure (5, 20, 29). In up to 40% of CA-CDI cases, infection is more severe and there are adverse outcomes (hospitalization, treatment failure, complications, colectomy, and recurrence) (19, 30). Notably, C. difficile strains acquired in the community can differ in genotype from predominant hospital strains (31), however, C. difficile RT078 (see below) has emerged as a significant pathogen associated with both HA- and CA-CDI in the Northern Hemisphere (21, 24, 32–35).
Zoonotic and Environmental Sources of C. difficile
C. difficile shows remarkable adaption to life within a diverse array of natural and host environments, including its primary habitat the mammalian gastrointestinal tract (as a commensal and/or pathogen), and several secondary habitats such as water, soil, and compost. We have previously reviewed aspects of C. difficile prevalence, pathogenicity and antimicrobial resistance (AMR) in non-human reservoirs (36), as have others including excellent reviews by Rodriguez et al. (11) and Candel-Pérez et al. (37). Here we will briefly summarize the key prevalence and molecular data that suggest a zoonotic origin for CDI. Figure 1 summarizes C. difficile prevalence data in farm animals, food and the environment taken from 86 studies in 23 countries worldwide (15, 38–122). In many of these studies, differences in C. difficile prevalence, strain lineage, toxigenic status, and AMR were identified. These were influenced by a variety of factors including the age of the animal, geographic region, methods used for isolation (e.g., sample type, spore selection, enrichment vs. no enrichment) and veterinary and agricultural practices [see recent reviews (11, 37)].
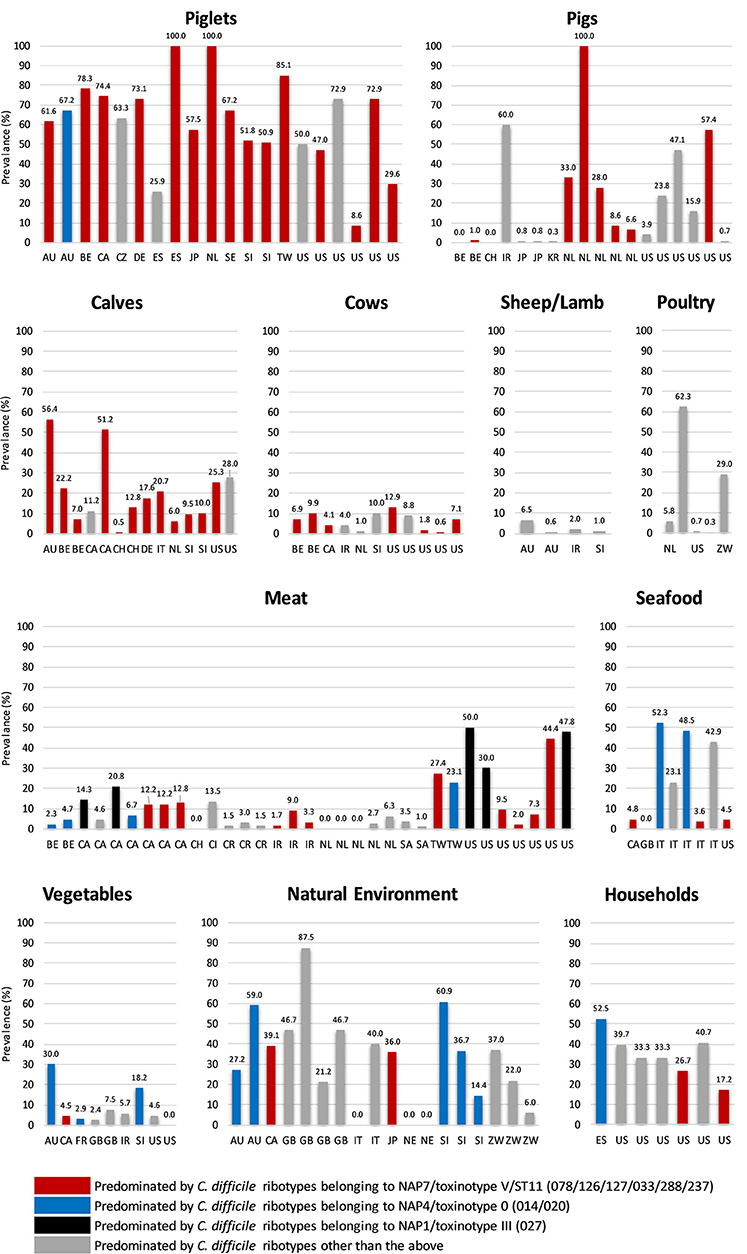
Figure 1. Global prevalence of C. difficile in farm animals, food and the environment. Data were taken from 86 studies in 23 countries worldwide (15, 38–122). Categories: Poultry (hens and broilers, Seafood (salmon, perch, clams, shrimp, and mussels), Meat (veal, beef, pork, lamb, chicken, goat, buffalo, and turkey), Vegetables (salads, lettuce, pea sprouts, ginger, carrots, beetroot, potatoes, onions, and spinach), Household (sandbox, shoes, toilet, vacuum, sink, floor), and Natural Environment (compost, lawn, soil, sediment, lake, and river). Two-letter country codes (International Organization for Standardization, ISO): AU, Australia; BE, Belgium; CA, Canada; CH, Switzerland; CI, Ivory Coast; CR, Costa Rica; CZ, Czech Republic; DE, Germany; ES, Spain; FR, France; GB, Great Britain and Northern Ireland; IR, Iran; IT, Italy; JP, Japan; KR, Korea; NE, Nigeria; NL, The Netherlands; SA, Saudi Arabia; SE, Sweden; SI, Slovenia; TW, Taiwan; US, United States of America; ZW, Zimbabwe. NAP, North American Pulse Type. RT027 and all ST11 RTs listed are binary toxin-positive.
C. difficile is known to colonize numerous food-producing animals including pigs, cattle, sheep, lambs, and poultry. Neonatal animals are viewed as significant reservoirs for C. difficile (Figure 1). Prevalence in domestic pigs and piglets averages around ~43%, ranging from 0% [Belgium and Switzerland (98, 103)] to ~50% [USA and Slovenia (61, 70)] and 100% [Spain and The Netherlands (62, 68)]. In cattle and calves, C. difficile prevalence averages around 14%, ranging from 0.5% [Switzerland (98)] to ~20% [Italy, Belgium and the USA (43, 46, 103)] to ~50% [Australia and Canada (38, 40)]. On average, a lower prevalence has been reported in ovine hosts [sheep and lambs, ~6% (77)] with prevalence in poultry [hens, broiler chickens] varying considerably [0.3% in the USA (82), to 29.0% in Zimbabwe (83) and 62% in Slovenia (80), mean ~19%]. Due to an absence of colonization resistance afforded by a mature intestinal microflora, during the first weeks of life neonatal pigs and calves are susceptible to disease caused by C. difficile. Although data is limited for calves (46) the pathophysiology of CDI in piglets is well-described; diarrhea, dehydration, weight loss, enteritis histologically similar to human lesions, and high mortality (123–125).
Other non-human animal reservoirs of C. difficile include cats and dogs (prevalence 0–100%), horses and foals (3–33%) and numerous wild animal species including rabbits, zebra, kangaroos, birds, shrews, Kodiak bears, racoons, camels, donkeys, feral swine, elephants, ibex, molluscs, tamarin monkeys, chimpanzees and, most recently, polar bears (0–100%) (37, 126, 127). The most common C. difficile lineage identified in many of these animal studies is multilocus sequence type (MLST, ST) 11, predominated by RT078 and its close relatives RTs 033, 045, 066, 126, 127, and 288 (all binary toxin positive, toxinotype V and cause CDI in humans) (Figure 1). Surprisingly, in Australia, the predominant RT found in pig herds is RT014, one of the most common strains causing CDI in humans worldwide (128) (see below).
C. difficile has been recovered from meats and plant-based foods sourced from processing plants, shops, farms and markets throughout Europe, North America and the Middle East (Figure 1). These include retail meat (veal, beef, pork, lamb, chicken, goat, buffalo, and turkey), seafood (salmon, perch, clams, shrimp, and mussels), and salads and vegetables (lettuce, pea sprouts, ginger, carrots, potatoes, onions, and spinach). As is the case with farm animals, the prevalence of C. difficile in food varies widely with food type and geographic origin. A high prevalence of C. difficile in retail pork, beef, and chicken has been reported in the USA (42%) but studies elsewhere report a much lower prevalence (Taiwan, 23%; Cote d'Ivoire, 14%), especially in Europe (~3.0%) (105, 129, 130). The prevalence of C. difficile in seafood varies considerably from ~5.0% in Canada, USA and Wales (99, 108, 118) to ~50.0% in Italy where its presence has been tentatively linked to sewage contamination in local rivers (95). Similarly, the prevalence of C. difficile on vegetables varies from 3 to 8% in North America and Europe [ready to eat salads (85, 101, 107, 109, 111, 118)] to 20–56% in Australia [organic beetroot and potatoes (84)] reflecting, possibly, different methods of processing. The molecular epidemiology of C. difficile recovered from food largely mirrors that of farm animals (ST11 RTs and common healthcare-associated lineages including 014 and 027, Figure 1).
Farm to Fork: Agricultural Practices Presenting a Risk for CA-CDI
In its spore form, C. difficile persists in various different natural ecosystems [soil, rivers, oceans, lakes, and sediments (114–116, 118, 119)], animals and food (11), and many abiotic environments for example toilets, floors, sinks, and soles of shoes (112, 113, 131). The high transmissibility of the spore (132) combined with its inherent resilience to desiccation, extremes of temperature, and disinfection (133) facilitates the transmission of C. difficile between these ecosystems. C. difficile spores could be transmitted from the farm environment to humans through a number of mechanisms including direct contact, airborne dispersal, avian, rodent or arthropod vectors (134–137), contamination of meat with feces during slaughter (53, 138) and via animal effluent or effluent by-products such as compost (139). However, CDI is a complex phenomenon encompassing pathogen, host, anthropomorphic and environmental factors, and our understanding of CDI transmission dynamics between production animals and humans is nowhere near perfect. Within Australia, two agricultural practices have been identified which present a credible risk for transmission of C. difficile causing CA-CDI: (i) slaughtering of neonatal animals destined for human consumption, and (ii) the recycling of effluent for agricultural purposes such as manufacturing compost which is then disseminated into the community setting (140, 141).
The prevalence of C. difficile in Australian veal calves is high although this decreases significantly with increasing age of the animal; 56% from <7-day-old calves, 3.8% in 2–6 month-old calves, and 1.8% in adult cattle (38). The C. difficile population within these cattle was dominated by ST11 RTs that all cause disease in humans. Moreover, at slaughter, the prevalence of C. difficile in calve feces was 60.0% and a significant proportion of calf carcasses (25.3%) was positive (with a spore concentration of 33 CFU/cm2), as a result of spore contamination from gastrointestinal contents during the slaughter process (138). As before, clinically important ST11 RT lineages dominated (138). Australia is one of the very few countries that cull male neonatal dairy calves (veal calves), a practice that exists because they are born male and considered surplus to industry requirements. With C. difficile prevalence highest in this neonatal period (127), the unique slaughter age of these animals presents a significant and perhaps under-appreciated risk for contamination of carcasses during the slaughter process. Further, C. difficile spores contaminating carcases would likely survive chilling, freezing, and cooking processes (142–145) and may compromise the quality of veal for domestic and export markets. To date, C. difficile has not been recovered from retail meat in Australia although only limited surveys have been undertaken mainly on meat from adult animals. Consumer demand for newborn veal in Australia is low and thus there is likely to be limited exposure of consumers to contaminated meats. However, Australia is the third largest beef and veal producer in the world (146), exporting 1.9 million tons of beef and veal per annum to over 100 countries, particularly in Africa, Asia and the Middle East. It is possible that contaminated Australian veal may be contributing to CDI in these regions, however, with the exception of Taiwan where ST11 strains are commonly reported in humans with CDI and farm animals (64, 102, 147), CDI surveillance is lacking in many of these countries. Whatever the level of risk to the domestic and export consumer, it is possible that it can be significantly mitigated by increasing the age that the animal is slaughtered to >3 or more weeks (38).
In the case of Australian piglets and dissemination of the major healthcare-associated lineage RT014, a growing body of evidence points to zoonotic transmission extending from the farrowing shed to the community. First, Australian piglets are major amplification reservoirs for C. difficile (67% prevalence nationwide with RT014 comprising 23% of isolates) (52). Second, whilst suckling age piglets are not slaughtered for meat on a large scale, C. difficile spores are abundant in treated biosolids, effluent, and piggery wastewater (121, 148–150). These by-products of the pig industry are subsequently recycled to pasture and agriculture for composting and direct irrigation/fertilization of crops and lawn. Third, C. difficile has been recovered from 30% of “high-street” retail compost samples in Australia (122), 59% of new roll-on lawn samples in Australia (151) and 20% of various root vegetables from mainstream and organic markets (84). Both lawn and organic vegetables are invariably grown in compost/soil containing animal manure. In these studies, RT014 comprised 7, 39, and 10% of isolates, respectively. Finally, the use of potent, late generation cephalosporins in human and veterinary medicine is a major driver of (i) C. difficile colonization and onset of disease in pigs; (ii) amplification and persistence of C. difficile in piggeries; (iii) spill-over of spores into the environment; and (iv) onset of CDI in the community (135, 140, 141).
Genomic Insights Into The Evolution And Transmission Of C. Difficile In Animals And Humans
Microevolution in the C. difficile Core Genome
The next generation sequencing era has seen the development of exquisitely sensitive, cost-effective, and rapid, benchtop whole-genome sequencing (WGS) technologies. Combined with new WGS-based genotyping tools, these technologies are shaping the future of infectious diseases surveillance. Core genome single nucleotide variant (SNV) analysis is an ultra-fine scale discriminatory method that uses WGS to detect transmission and outbreaks of bacterial pathogens (152, 153). SNV analysis is restricted to the non-repetitive, non-recombinative core genome which contains essential genes common to all isolates under analysis that are often vertically inherited and most likely to have the strongest signal-to-noise ratio for inferring phylogeny (152, 153). For C. difficile, SNV analysis uses a fixed-rate molecular clock derived from serial isolation of strains from clinical cases, estimated to be in the region of 1.47 × 10−7 to 5.33 × 10−7 mutations per site per year, to identify signatures of plausible clonal transmission (154, 155). This equates to 1–2 SNVs per genome per year. For studies of C. difficile transmission, a clonal group is therefore defined as two or more strains differing by <2 SNVs in their core genome, with ≥10 SNVs used as a threshold for genetically distinct isolates (154–157). For longer-term ecological studies, these thresholds may not hold true as the genetically quiescent nature of C. difficile spores may result in underestimating the evolutionary distance between strains (19).
The ultra-fine scale resolution of this technique is superior to conventional C. difficile typing methods including PCR ribotyping, pulsed-field gel electrophoresis (PFGE), MLST, Rep-PCR, toxinotyping, and amplified fragment-length polymorphism (AFLP) fingerprinting (152). It also shows discriminatory power comparable, and in some cases superior, to multilocus variable-number tandem repeat analysis (MLVA) (152, 157) and the recently developed core genome MLST scheme (158). Supplementary Table 1 provides a summary of bioinformatics tools and algorithms involved in a C. difficile SNV pipeline.
For C. difficile, SNV-based typing has been used to study the microevolution of CDI in the hospital setting (154) and to investigate localized transmission and international dissemination of major clinically important lineages such as RTs 027 (16) and 017 (159). But as outlined below, this approach has also been used to delineate cryptic transmission pathways of C. difficile between animals, humans, and their shared environment. In doing so, these genomic studies have redefined our understanding of the ecology and evolution of this complex species.
C. difficile RT078
C. difficile RT078 belongs to evolutionary clade 5 and is the principal ST11 sublineage (Figure 2). Between 2005 and 2008, RT078 rose from 11th to become the 3rd most frequently encountered RT in European hospitals (25), an increase particularly evident in the Netherlands where, from 2005 to 2008, Dutch hospitals would see the total prevalence of RT078-associated cases increase from 3 to 13% (32). These RT078 cases of CDI were in younger patients and with community-onset (32, 33). Comparable rates have been found in North America (21, 24, 35) with one study reporting 46% of all RT078 isolates were community acquired (160). As with many toxigenic C. difficile RTs, RT078 can be carried asymptomatically (161, 162). C. difficile RT078 has established significant reservoirs in North American, European, and Asian pigs and cattle and is often reported as the dominant type irrespective of age, diarrheal status or other farm-specific factors (37, 127). In an important Dutch study of C. difficile spore acquisition, Hopman et al. (68) demonstrated that piglets delivered by cesarean-section were C. difficile-negative yet were rapidly colonized with C. difficile RT078 spores within 48 h.
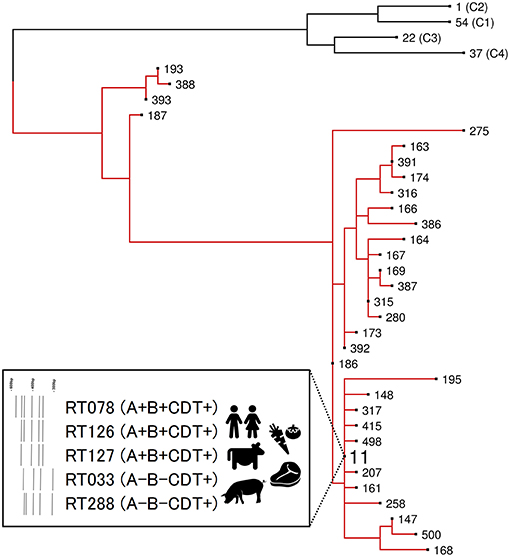
Figure 2. Evolutionary clade 5. Maximum-likelihood phylogeny based on concatenated MLST allele sequences (seven loci, 3,501 bp) for 32 known clade 5 STs. For global phylogenetic context, well-characterized representatives of major MLST clades C1 (ST54, RT012), C2 (ST1, RT027), C3 (ST22, RT023), and C4 (ST37, RT017) are also shown. Branches for clade 5 are shown in red. The inset box highlights major ST11 RT sublineages of clinical and agricultural/livestock importance with associated toxin gene profiles and graphical representation of 16s−23s rRNA PCR ribotyping banding patterns.
The virulence potential of RT078 has been likened to that of epidemic RT027 with which it shares similar genetic features. These include the major virulence genes tcdA, tcdB, and cdtA/B involved in toxin production, and an aberrant toxin regulator gene tcdC (deletions, nonsense mutations, and premature stop codons) leading to a reduction in log phase repression of toxin expression. The role for the latter in the observed hyper-virulent disease phenotype seen also in RT078 infections i.e., more toxin, increased mortality and morbidity, remains speculative (32, 163–167). C. difficile RT078 strains are often multidrug-resistant (MDR) (161, 168) and, compared to other RTs, including RT027, show remarkable resilience to extremes of temperature (80 to 96°C) and water treatment processes (142, 143, 145). It has also been proposed that the emergence and global dissemination of RT078 in humans is linked to an enhanced ability to metabolize the food additive trehalose (169). These virulence and survival traits may explain the successful dissemination of this lineage in production animals and humans worldwide. Unsurprisingly, it has received major attention as a potentially zoonotic lineage.
Zoonotic Transmission of C. difficile RT078 Between Humans and Animals
Genetic studies using MLST, MLVA, Rep-PCR and AFLP fingerprinting have all provided significantly higher strain resolution of RT078 populations compared to conventional PCR ribotyping. In 2010, Bakker et al. (170) found 85% of RT078 isolates of human and porcine origin in the Netherlands were genetically related and, in many instances, indistinguishable by high-resolution MLVA. In 2012, Stabler et al. (171) used MLST to analyse 385 C. difficile isolates from different geographical locations (Europe, North America, and Australia) and sources (human, food, and animal). Strains of RT078 from humans, food and animals, some from different countries and continents, were indistinguishable (all sharing seven identical housekeeping genes, ST11) (171). More recent work from Taiwan showed RT078 isolated from pig farms shared identical Rep-PCR fingerprints as RT078 strains derived from humans with CDI in hospitals in the same region (64). Similarly, in Spain, RT078 of human and animal origin were clustered together by AFLP (172). Evidence from Japan suggests RT078 has been introduced from Europe. Usai et al. found Japanese pig RT078 strains clustered (by MLVA) with European human and pig RT078 strains (86), and Niwa et al. found a single MLVA cluster of RT078 responsible for five cases of colitis in Japanese racehorses (173). Both pigs and racehorses are internationally traded in Japan; thus, RT078 may have been imported into Japan from Europe via live animals.
Natural and diverse reservoirs of RT078 support the hypothesis that CDI may have a zoonotic origin. To date, a few key WGS-based studies have led to significant advancement in understanding the true zoonotic potential and evolution of the RT078 and its close relatives. In 2013, Knetsch et al. (161) used core genome SNV typing to compare 65 C. difficile RT078 isolates of human and porcine origin sourced over a 10-year period in The Netherlands. Using Bayesian techniques, an RT078 population-specific mutation rate was estimated to be 2.72 × 10−7 substitutions per site per year, equating to around 1 SNV per genome per year—a figure comparable with earlier estimates (154, 155). A core genome phylogeny showed isolates of human and porcine origin clustering together. Notably, the analysis showed a pair of human and pig isolates from the same pig farm in The Netherlands to be indistinguishable (zero SNVs difference in their core genome). Working in pig husbandry or living in (or visiting) areas with a high density of pigs increased the risk of acquiring C. difficile due to exposure to pig feces (161). Whilst the transmission of RT078 between a pig and pig farmer within the confines of a pig-rearing facility might not be that surprising, it was nonetheless the first ever confirmation that interspecies transmission of C. difficile had occurred (161). The exact mode of transmission between these species remains unclear. Whilst these data appear to support the theory that CDI is a zoonosis, a common environmental source, asymptomatic carriage and/or zooanthroponotic (human to animal) transmission cannot be ruled out.
In 2017, the same authors (174) extended these findings. They investigated microevolution in the core genome of 248 C. difficile RT078 strains sourced from humans and animals in 22 countries. This study provided the first estimate of the global RT078 population structure and yielded new insights into the potential and extent for zoonotic spread. Extensive clustering of C. difficile RT078 from human clinical cases and food animals was observed, with clear instances of interspecies clonal transmission, only this time, the significant clustering of clones supported evidence of bidirectional spread of C. difficile RT078 between production animals and humans. Moreover, there was only limited geographic clustering with clones of C. difficile RT078 spread multiple times across multiple towns, countries and continents, in particular between North America and Europe: one example was the transmission of an RT078 clone between an animal in Canada and humans in the United Kingdom. This indicated interspecies transmission of C. difficile RT078 was not restricted to a local population of humans and production animals, as previously shown in the 2014 Dutch study. Together, these data revealed a highly linked intercontinental transmission network of C. difficile RT078 between humans and animals and provided further evidence that CDI has a significant zoonotic component (174). Yet it also showed that, in contrast to another classic enteric pathogen Salmonella enterica which has distinct animal- and human-associated populations, C. difficile RT078 appeared to be a clonal population moving frequently (and likely over long time periods) between production animal and human hosts, with no geographical constraints.
ST11 Is a Heterogeneous Lineage of Major One Health Importance
ST11 is an ancient evolutionary lineage comprising at least a dozen CDT+ ribotypes that cause CDI in humans with significant ecological niches in production animals worldwide (175) (Figure 2). As is apparent, and for good reasons, there has been a strong focus on the ST11 sub-lineage RT078, however, until recently, little was known about the evolutionary history and zoonotic potential of other ST11 RTs. Our recent study (175) addressed this knowledge gap, using WGS to investigate population structure and clonal transmission in over 200 strains of major ST11 RTs 078, 126, 127, 033, and 288 sourced from human and veterinary/environmental origin across Australia, Asia, Europe, and North America. A core genome phylogeny showed the global ST11 population structure largely mirrored RT sub-lineage, with discrete evolutionary clusters congruent with RTs 078/126, 127, 033/288. Core genome SNV analysis found multiple instances of inter- and intra-species clonal transmission in all RT sub-lineages. Interspecies clonal groups comprised C. difficile isolates derived from health care facilities and farm animals spread across different states, countries, and continents, often without any healthcare association. Our findings independently confirm and extend the work of Knetsch et al. (161, 174) revealing a globally-disseminated network of C. difficile ST11 clones with the capability and proclivity for reciprocal zoonotic and/or anthroponotic transmission. Moreover, this study showed for the first time that non-RT078 ST11 strains such as RTs 126, 127, 033, and 288 also display a high zoonotic potential and should also be considered lineages of emerging One Health importance.
Antimicrobial Resistance and ST11 Evolution
Antimicrobials are a crucial component in the pathogenesis of CDI; they play a central role in the establishment of infection and, paradoxically, remain the preferred option for treatment (176).
AMR is, therefore, a key factor driving epidemiological changes in CDI (1). As we have seen with virulent C. difficile RT027 epidemic lineage, outbreaks emerge when the inherent resistance of C. difficile to cephalosporins is combined with acquired resistance to high-risk antimicrobials known to incite CDI, such as fluoroquinolones (16). In all the above WGS-based studies of RT078 and ST11, substantial AMR repertoires were identified. In the Dutch study (161), interspersed throughout the RT078 phylogeny were clones common to humans and livestock harboring identical mobile genetic elements (MGEs) conferring resistance to streptomycin (Tn6235, aphA1+) and tetracycline (Tn6190, tetM+) (161). In the later study by Knetsch et al. (174), the global population of RT078 contained a broad array of AMR genes encoding resistance to aminoglycosides and streptothricin (aph3′-III, ant6′-Ib, Sat4A), erythromycin (ermB+), and tetracycline (tetM, tetO, tet32, tet40, tet44). The gene cdeA encoding a multidrug efflux transporter was found in all isolates (174).
In our ST11 study (175), half of all strains showed phenotypic resistance to one or more of tetracycline, moxifloxacin, erythromycin, and clindamycin, of which a quarter, predominantly RTs 126/078, were resistant to ≥3 of these agents. Underscoring this resistance was an array of AMR genetic loci including chromosomal mutations in gyrA/B (fluoroquinolone resistance) and MGEs conferring resistance to macrolides and lincosamides (Tn6194; ermB+), and tetracycline (Tn6190; tetM+ and Tn6164; tet44+), the latter a 106 kb genetic island apparently specific to RT078 (177). This was the first such report of Tn6194 from animals in the world. This element is the most common ermB-containing element found in human clinical isolates in Europe and is a defining genetic feature of epidemic RT027 (16, 178, 179). A phenotypically silent vanB2 transposon (likely from Enterococcus faecalis) was also found in a C. difficile RT033 strain isolated from an Australian veal calf at slaughter (180). Another common ruminant species Erysipelothrix rhusiopathiae appeared to be the origin of the numerous aminoglycoside resistance gene clusters present in all ST11 sub-lineages.
In a compelling new study, Dingle et al. (181) present a strong case for antimicrobial selection influencing the recent evolutionary history of C. difficile RT078. A time-scaled phylogeny built from the core genome of over 400 international C. difficile RT078 strains revealed three major clonal expansions (a rapid, recent international spread of RT078 clones). Two-thirds of all RT078 were tetracycline resistant. Remarkably, a common ancestor of each clonal expansion had independently evolved tetracycline resistance via the acquisition of distinct tetM alleles carried on closely related Tn916-like elements, an analogous situation to the emergence of fluoroquinolone resistance in RT027 (16). The parallel tetM associated clonal expansions were estimated to have occurred sometime around the year 2000, at a time when the number of RT078-associated clinical cases (at least in Europe) started to increase. Moreover, the three tetM alleles show significant homology (97–100% sequence identity) with tetM genes belonging to established zoonotic species such as E. faecalis, Escherichia coli, and Streptococcus suis—further supporting an agricultural origin for RT078. The authors note that S. suis has striking parallels with C. difficile RT078—it is a globally disseminated human pathogen which has established substantial reservoirs in pigs and has displayed recent increases in tetracycline resistance (182, 183). In summary, these phylogenetic data are consistent with an evolutionary response to tetracycline selective pressure. The inappropriate and overuse of tetracycline in animal husbandry is well-recognized (184). This selective pressure, combined with the rapid, international spread of C. difficile RT078 via the food chain and other agricultural vectors provides a plausible explanation for the clinical prominence of this lineage in humans.
Interspecies Transfer of C. difficile RT014 Between Humans and Animals
C. difficile RT014 is a toxigenic (A+B+CDT−) and highly successful lineage of C. difficile belonging to MLST clade 1. RT014 is consistently among the most common RTs causing CDI in European healthcare systems, and in Australia it has been the most prevalent RT causing human infection for many years, accounting for ~25% of all CDI cases (10, 185–188). The zoonotic potential of this RT was initially thought to be quite low as its prevalence in production animals in Europe was low and it was absent from livestock in Asia. In Australia, there was a completely different and intriguing story. In 2013, a nationwide cross-sectional study of C. difficile in 21 pig farms in Australia found RT014 to be the most prevalent RT in neonatal pigs aged <14 days, accounting for 23% (n = 26/154) of isolates (52). With rates of CDI in Australia increasing markedly in recent years (24% in 2011–2012 alone) and a significant rise in CA-CDI (26), the establishment of significant RT014 reservoirs in porcine populations in Australia suggests zoonotic transmission as a plausible source of human infection.
To examine the true extent of genetic relatedness, a collection of 40 contemporaneous isolates of RT014 of human and porcine origin in Australia were subjected to WGS (128). A total of three distinct STs were identified in this RT014 collection (STs 2, 13, and 49), and in each, human and porcine populations were intermingled, signaling a very recent shared ancestry. A phylogeny based on evolution in 1,260 core orthologous genes (1,019,160 bp, ~25% of bases in an average C. difficile genome) showed geographically and temporally unconstrained clustering of human and animal C. difficile RT014 strains in all three STs again supporting a close genetic relationship. Finally, a phylogeny-based on evolution in non-recombinant 1,287 core genome SNVs provided ultra-fine scale resolution of the RT014 population, identifying multiple instances of plausible interspecies clonal transmission. In total, 42% of C. difficile RT014 strains from humans with CDI showed a clonal relationship (differing by no more than two SNVs in their core genome) with one or more RT014 strains derived from pigs. Remarkably, many RT014 clones originated from pigs and humans in states separated by thousands of kilometers, collected many months apart, and half of the human isolates in these clonal groups originated from cases classified as CA-CDI, representing the acquisition of CDI outside of the hospital system (Figure 3). Long range transmission of C. difficile RT014 clones suggests direct contact between humans and colonized livestock is perhaps unlikely, and there was no evidence here. Given what we know of the C. difficile colonization-transmission cycle in the farrowing environment and wider livestock industry, it is conceivable that over an extended period there has been frequent long-range indirect interspecies transmission through human exposure to contaminated retail meat but more likely contaminated piggery by-products such as manure and compost in the community setting (Figure 3). Indeed, genomic studies from the USA and Europe have shown that the household environment and pet dogs are colonized with C. difficile RT014/ST2 representing reservoirs of RT014 in the community (124, 125).
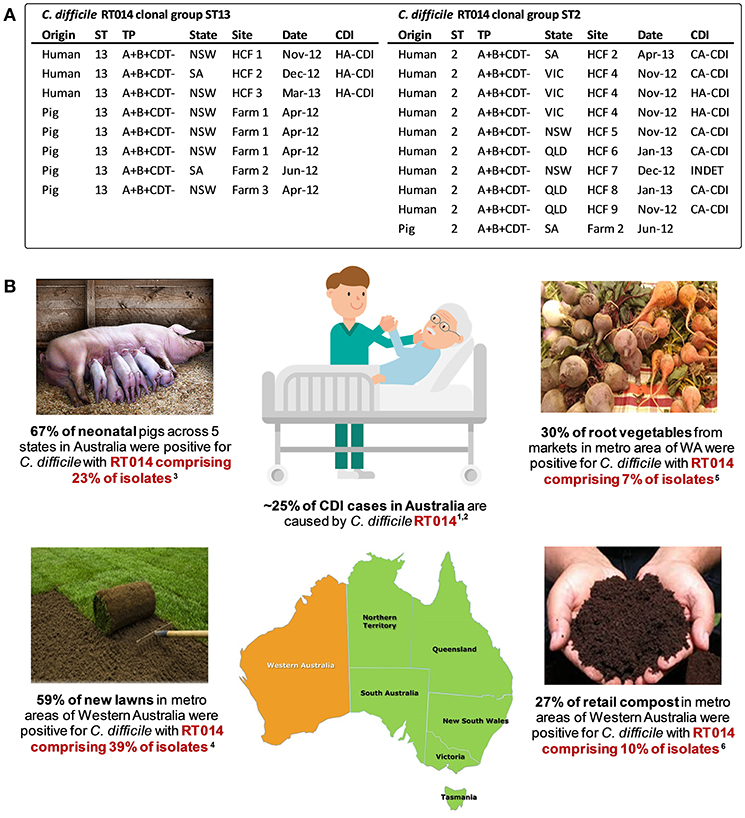
Figure 3. Transmission networks and community reservoirs of C. difficile RT014 in Australia. (A) summary of ST13 (n = 8) and ST2 (n = 10) RT014 clonal groups found in pigs and humans with CDI in Australia, adapted from Knight et al. (128). A clonal group is defined as two or more strains differing by <2 SNVs in their core genome. HCF, healthcare facility; NSW, New South Wales; SA, South Australia; VIC, Victoria; QLD, Queensland; INDET, indeterminate. (B) summary of RT014 ecological niches in Australia. 1Knight et al. (186); 2Collins et al. (188); 3Knight et al. (52); 4Moono et al. (151); 5Lim et al. (84); 6Lim et al. (122).
The C. difficile Pan-Genome: Insights Into the Ecology of a Complex Pathogen
A bacterial pan-genome describes the full complement of genes in a species or individual phylogenetic lineage. It comprises a core component (those genes present in all strains) and an accessory or adaptive component (genes absent from one or more strain or unique to a particular strain) (189). Early microarray-based studies estimated the C. difficile pan-genome to be comprised of 9,640 coding sequences (CDS) with a core genome component many orders of magnitude lower at 600–3,000 CDS (190–192). More recent WGS based studies of RT014 (128), RT078 (174), and ST11 (175) from humans and animals have provided further insights into the genetic diversity, plasticity and ecology of zoonotic C. difficile lineages.
Analysis of 44 Australian RT014 genomes (STs 2, 13, and 49) revealed a large pangenome (7,587 genes) comprising a core genome of 2,296 genes (30.3% of the total gene repertoire) and an accessory genome of 5,291 genes (128). Moreover, the human and porcine populations shared near identical proteomes (128). The global RT078 population (248 genomes from four continents) possessed a large pangenome of 6,239 genes with a core genome of 3,368 genes (53.9% of the total gene repertoire) and an accessory genome of 2,871 genes (174). Finally, the global ST11 population (207 genomes from four continents including RTs 078, 126, 127, 033, and 288) was defined by a massive pan-genome (10,378 genes), a remarkably small core genome of 2,058 genes (only 19.8% of the gene pool) and an accessory genome of 8,320 genes (175). In the case of RT014 and ST11, power-law regression analysis determined the pangenomes to be “open,” that is, size increases indefinitely when adding new genomes. For example, in the ST11 analysis, after sequencing over 200 genomes there is an average of 16 new genes contributed to the gene pool with each additional sequenced strain (175).
The size and openness of a pan-genome is also a very useful proxy for characterizing the lifestyle of a bacterial species (193). The pan-genome data derived from these zoonotic and agricultural-associated C. difficile lineages predict a species with a sympatric lifestyle, occupying niches in extremely diverse environments that are enriched with mixed microbial communities of prokarya and archea (193). This is true of C. difficile, a versatile species which shows extraordinary adaption to multiple ecosystems including the gastrointestinal tract of multiple mammalian hosts, and several secondary habitats such as water, soil, and composts and invertebrate species (179). In contrast to allopatric and intracellular species such as Rickettsia rickettsii and Chlamydia trachomatis, which have small closed pan-genomes and live in isolated niches with limited exchange with the global microbial gene pool, sympatric species like C. difficile (and C. botulinum) have larger, more complex open pan-genomes. Sympatry also means a higher frequency of gene acquisition events and a higher probability of acquiring parasitic DNA i.e., transposons and bacteriophages, both contributing to an increase in pan-genome size (193, 194). Indeed, underscoring the substantial genetic diversity in these zoonotic C. difficile lineages were large and diverse collections of clinically important prophages of the Siphoviridae and Myoviridae (128, 175) and AMR genetic elements (128, 174, 175). As corroborated by Dingle et al. in RT078 (181), many of these underlying AMR elements show evolutionary origins in commensal species residing within the gut of farm animals. Examples being macrolide resistance genes from Campylobacter coli (cryptic), aminoglycoside, and streptothricin genes cassettes from E. rhusiopathiae, and a plethora of tetracycline resistance genes from S. suis, E. faecalis, Megasphaera elsdenii, C. jejuni, and C. perfringens (128, 161, 174, 175). Moreover, AMR elements Tn6194 (ermB+) and Tn5397 (tetM+) are capable of intra-species transfer to different C. difficile RTs and even inter-species transfer to other genera (16, 191, 195).
Together, the phylogenetic, pangenome, and AMR data show that these zoonotic C. difficile lineages have the capability and propensity to move between humans, production animals, and their shared environment. By occupying niches within multiple host species, these C. difficile lineages are able to access and exchange DNA with an enormously diverse metagenome, particularly the ruminant gut and soil microbiota. Such promiscuous behavior provides C. difficile with a potential selective advantage over taxa inhabiting the same gut ecosystem, be it the pig, cow or human intestinal tract, therefore greatly enhancing their ability to adapt to fluctuating environmental factors and their likelihood of success.
Finally, in the case of ST11, it is remarkable that even after sampling >200 ST11 strains from over a dozen unique RT sub-lineages spread over four different continents; the complete gene complement of this lineage was not captured (175). With over 420 STs and >600 RTs currently recognized, it is likely that the complete species pan-genome for C. difficile could be astonishingly high. Such enormous diversity is more typical for phylogenetic distances between genera within a family, rather than strains within a species (179). In light of recent calls for taxonomic revisions (196–199), it is possible that C. difficile may, in fact, be a complex of sub-species divided along the major evolutionary clades.
Future Directions And Challenges
The One Health paradigm is a philosophical approach to improving and safeguarding the health of humans, animals and the environment and, importantly, recognizes that these three areas are inter-related (200). Specifically, improved treatment of disease common to humans and animals can be achieved through the application of interdisciplinary approaches between human and veterinary medicine, and the analysis of environment-derived isolate datasets. In this regard, CDI is the quintessential One Health issue (141). As we have highlighted here, the application of high-resolution microbial genomics in a One Health framework is the optimal paradigm for advancing our understanding of CDI in humans and animals. Together, this body of evidence challenges the existing paradigm and long-held conception that CDI is primarily a healthcare-associated infection and provides compelling evidence that CDI has a significant zoonotic component. More important, these findings should stimulate new discussions about One Health focused interventions for CDI.
Collaboration between human and veterinary medicine will be essential if we are to safeguard the health of humans and production animals (141). First and foremost, measures which reduce the levels of C. difficile spores in the piggery environment are of paramount importance, not only for mitigating the risk of community acquisition but also for improving animal health (141). In human medicine, these measures comprise stringent infection control policies such as case isolation, reduced use of late-generation cephalosporins, hand hygiene and deep environmental cleaning (201, 202). Analogous interventions have been employed in the veterinary hospital setting with a significant reduction in CDI cases (203); however, the vast scale of modern production animal systems may hinder successful implementation. Also, the frequent disagreement between clinicians, veterinarians and the livestock industry regarding appropriate risk management of C. difficile in animal populations remains an additional, significant hurdle to overcome (141, 204).
With several candidate C. difficile vaccines in development (205), immunization of livestock could be a highly effective way to reduce the overall prevalence of C. difficile and is a good example of an integrative One Health approach to tackling CDI (141). Finally, continued genetic and phenotypic surveillance of C. difficile is critical to an enhanced understanding of epidemiological and genetic factors contributing to the emergence, evolution, and spread of CDI (152, 179). Crucially, if we are to identify improved infection prevention and control strategies, and public health interventions designed to mitigate the risk of C. difficile transmission, it is imperative that such studies should have a strong One Health focus by including analysis of C. difficile strains derived from humans, animals and food, and their shared environment. As much of the focus to date has been on the ST11 group and RT014, future studies should examine the potential for clonal relationships between other lineages circulating in clinical and animal/environmental settings. As illustrated by the studies highlighted in this review, WGS will play a central role in this, providing a level of discrimination far beyond that achievable by conventional molecular typing methodologies.
Author Contributions
All authors listed have made equal intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2019.00164/full#supplementary-material
References
1. Larson HE, Price AB, Honour P, Borriello SP. Clostridium difficile and the aetiology of pseudomembranous colitis. Lancet. (1978) 311:1063–6. doi: 10.1016/S0140-6736(78)90912-1
2. George RH, Symonds JM, Dimock F, Brown JD, Arabi Y, Shinagawa N, et al. Identification of Clostridium difficile as a cause of pseudomembranous colitis. Br Med J. (1978) 1:695. doi: 10.1136/bmj.1.6114.695
3. Chang T-W, Bartlett JG, Gorbach SL, Onderdonk AB. Clindamycin-induced enterocolitis in hamsters as a model of pseudomembranous colitis in patients. Infect Immun. (1978) 20:526–9.
4. George WL, Goldstein EC, Sutter V, Ludwig S, Finegold S. Aetiology of antimicrobial-agent-associated colitis. Lancet. (1978) 311:802–3. doi: 10.1016/S0140-6736(78)93001-5
5. Leffler DA, Lamont JT. Editorial: not so nosocomial anymore: the growing threat of community-acquired Clostridium difficile. Am J Gastroenterol. (2012) 107:96–8. doi: 10.1038/ajg.2011.404
6. Sheridan PP, Freeman KH, Brenchley JE. Estimated minimal divergence times of the major bacterial and archaeal phyla. Geomicrobiol J. (2003) 20:1–14. doi: 10.1080/01490450303891
7. Lawson PA, Citron DM, Tyrrell KL, Finegold SM. Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O'Toole 1935) Prevot 1938. Anaerobe. (2016) 40:95–9. doi: 10.1016/j.anaerobe.2016.06.008
8. Oren A, Rupnik M. Clostridium difficile and Clostridioides difficile: two validly published and correct names. Anaerobe. (2018) 52:125–6. doi: 10.1016/j.anaerobe.2018.07.005
9. Heymann DL, Dar OA. Prevention is better than cure for emerging infectious diseases. BMJ. (2014) 348:g1499. doi: 10.1136/bmj.g1499
10. Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, et al. Burden of Clostridium difficile infection in the United States. N Engl J Med. (2015) 372:825–34. doi: 10.1056/NEJMoa1408913
11. Rodriguez Diaz C, Seyboldt C, Rupnik M. Non-human C. difficile reservoirs and sources: animals, food, environment. Adv Exp Med Biol. (2018) 1050:227–43. doi: 10.1007/978-3-319-72799-8_13
12. Britton RA, Young VB. Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterol. (2014) 146:1547–53. doi: 10.1053/j.gastro.2014.01.059
13. Carter GP, Rood JI, Lyras D. The role of toxin A and toxin B in the virulence of Clostridium difficile. Trends Microbiol. (2012) 20:21–9. doi: 10.1016/j.tim.2011.11.003
14. Chandrasekaran R, Lacy DB. The role of toxins in Clostridium difficile infection. FEMS Microbiol Rev. (2017) 41:723–50. doi: 10.1093/femsre/fux048
15. Rodriguez-Palacios A, Stampfli HR, Duffield T, Peregrine AS, Trotz-Williams LA, Arroyo LG, et al. Clostridium difficile PCR ribotypes in calves, Canada. Emerg Infect Dis. (2006) 12:1730–6. doi: 10.3201/eid1211.051581
16. He M, Miyajima F, Roberts P, Ellison L, Pickard DJ, Martin MJ, et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet. (2013) 45:109–13. doi: 10.1038/ng.2478
17. McDonald LC, Coignard B, Dubberke E, Song X, Horan T, Kutty PK. Recommendations for surveillance of Clostridium difficile–associated disease. Infect Control Hosp Epidemiol. (2007) 28:140–5. doi: 10.1086/511798
18. Kuntz JL, Chrischilles EA, Pendergast JF, Herwaldt LA, Polgreen PM. Incidence of and risk factors for community-associated Clostridium difficile infection: a nested case-control study. BMC Infect Dis. (2011) 11:194. doi: 10.1186/1471-2334-11-194
19. Khanna S, Pardi DS, Aronson SL, Kammer PP, Baddour LM. Outcomes in community-acquired Clostridium difficile infection. Aliment Pharmacol Ther. (2012) 35:613–8. doi: 10.1111/j.1365-2036.2011.04984.x
20. Bloomfield LE, Riley TV. Epidemiology and risk factors for community-associated Clostridium difficile infection: a narrative review. Infect Dis Ther. (2016) 5:231–51. doi: 10.1007/s40121-016-0117-y
21. Khanna S, Pardi DS. Community-acquired Clostridium difficile infection: an emerging entity. Clin Infect Dis. (2012) 55:1741–2. doi: 10.1093/cid/cis722
22. Naggie S, Frederick J, Pien BC, Miller BA, Provenzale DT, Goldberg KC, et al. Community-associated Clostridium difficile infection: experience of a veteran affairs medical center in southeastern USA. Infection. (2010) 38:297–300. doi: 10.1007/s15010-010-0025-0
23. Kutty PK, Woods CW, Sena AC, Benoit SR, Naggie S, Frederick J, et al. Risk factors for and estimated incidence of community-associated Clostridium difficile infection, North Carolina, USA. Emerg Infect Dis. (2010) 16:197–204. doi: 10.3201/eid1602.090953
24. Khanna S, Pardi DS, Aronson SL, Kammer PP, Orenstein R, St Sauver JL, et al. The epidemiology of community-acquired Clostridium difficile infection: a population-based study. Am J Gastroenterol. (2012) 107:89–95. doi: 10.1038/ajg.2011.398
25. Bauer MP, Notermans DW, van Benthem BH, Brazier JS, Wilcox MH, Rupnik M, et al. Clostridium difficile infection in Europe: a hospital-based survey. Lancet. (2011) 377:63–73. doi: 10.1016/S0140-6736(10)61266-4
26. Slimings C, Armstrong P, Beckingham WD, Bull AL, Hall L, Kennedy KJ, et al. Increasing incidence of Clostridium difficile infection, Australia, 2011–2012. Med J Aust. (2014) 200:272–6. doi: 10.5694/mja13.11153
27. Mitchell B, Wilson F, McGregor A. An increase in community onset Clostridium difficile infection: a population based study. Healthcare Infect. (2012) 17:127–32. doi: 10.1071/HI12029
28. Eyre DW, Tracey L, Elliott B, Slimings C, Huntington PG, Stuart RL, et al. Emergence and spread of predominantly community-onset Clostridium difficile PCR ribotype 244 infection in Australia, 2010 to 2012. Euro Surveill. (2015) 20:21059. doi: 10.2807/1560-7917.ES2015.20.10.21059
29. Furuya-Kanamori L, Stone JC, Clark J, McKenzie SJ, Yakob L, Paterson DL, et al. Comorbidities, exposure to medications, and the risk of community-acquired Clostridium difficile infection: a systematic review and meta-analysis. Infect Control Hosp Epidemiol. (2015) 36:132–41. doi: 10.1017/ice.2014.39
30. Sandora TJ, Fung M, Flaherty K, Helsing L, Scanlon P, Potter-Bynoe G, et al. Epidemiology and risk factors for Clostridium difficile infection in children. Pediatr Infect Dis J. (2011) 30:580–4. doi: 10.1097/INF.0b013e31820bfb29
31. Bignardi GE, Settle C. Different ribotypes in community-acquired Clostridium difficile. J Hosp Infect. (2008) 70:96–8. doi: 10.1016/j.jhin.2008.04.003
32. Goorhuis A, Bakker D, Corver J, Debast SB, Harmanus C, Notermans DW, et al. Emergence of Clostridium difficile infection due to a new hypervirulent strain, polymerase chain reaction ribotype 078. Clin Infect Dis. (2008) 47:1162–70. doi: 10.1086/592257
33. Patterson L, Wilcox MH, Fawley WN, Verlander NQ, Geoghegan L, Patel BC, et al. Morbidity and mortality associated with Clostridium difficile ribotype 078: a case–case study. J Hosp Infect. (2012) 82:125–8. doi: 10.1016/j.jhin.2012.07.011
34. Dumyati G, Stevens V, Hannett GE, Thompson AD, Long C, Maccannell D, et al. Community-associated Clostridium difficile infections, Monroe County, New York, USA. Clin Infect Dis. (2012) 18:392–400. doi: 10.3201/eid1803.102023
35. Gerding DN, Lessa FC. The epidemiology of Clostridium difficile infection inside and outside health care institutions. Infect Dis Clin North Am. (2015) 29:37–50. doi: 10.1016/j.idc.2014.11.004
36. Moono P, Foster NF, Hampson DJ, Knight DR, Bloomfield LE, Riley TV. Clostridium difficile infection in production animals and avian species: a review. Foodborne Pathog Dis. (2016) 13:647–55. doi: 10.1089/fpd.2016.2181
37. Candel-Perez C, Ros-Berruezo G, Martinez-Gracia C. A review of Clostridioides [Clostridium] difficile occurrence through the food chain. Food Microbiol. (2019) 77:118–29. doi: 10.1016/j.fm.2018.08.012
38. Knight DR, Thean S, Putsathit P, Fenwick S, Riley TV. Cross-sectional study reveals high prevalence of Clostridium difficile non-PCR ribotype 078 strains in Australian veal calves at slaughter. Appl Environ Microbiol. (2013) 79:2630–5. doi: 10.1128/AEM.03951-12
39. Zidaric V, Pardon B, Dos Vultos T, Deprez P, Brouwer MS, Roberts AP, et al. Multiclonal presence of Clostridium difficile PCR ribotypes 078, 126 and 033 within a single calf farm is associated with differences in antibiotic resistance and sporulation properties. Appl Environ Microbiol. (2012) 78:8515–22. doi: 10.1128/AEM.02185-12
40. Costa MC, Stampfli HR, Arroyo LG, Pearl DL, Weese JS. Epidemiology of Clostridium difficile on a veal farm: prevalence, molecular characterization and tetracycline resistance. Vet Microbiol. (2011) 152:379–84. doi: 10.1016/j.vetmic.2011.05.014
41. Costa MC, Reid-Smith R, Gow S, Hannon SJ, Booker C, Rousseau J, et al. Prevalence and molecular characterization of Clostridium difficile isolated from feedlot beef cattle upon arrival and mid-feeding period. BMC Vet Res. (2012) 8:38. doi: 10.1186/1746-6148-8-38
42. Schneeberg A, Neubauer H, Schmoock G, Grossmann E, Seyboldt C. Presence of Clostridium difficile PCR ribotype clusters related to 033, 078 and 045 in diarrhoeic calves in Germany. J Med Microbiol. (2013) 62:1190–8. doi: 10.1099/jmm.0.056473-0
43. Magistrali CF, Maresca C, Cucco L, Bano L, Drigo I, Filippini G, et al. Prevalence and risk factors associated with Clostridium difficile shedding in veal calves in Italy. Anaerobe. (2015) 33:42–7. doi: 10.1016/j.anaerobe.2015.01.010
44. Bandelj P, Blagus R, Briski F, Frlic O, Vergles Rataj A, Rupnik M, et al. Identification of risk factors influencing Clostridium difficile prevalence in middle-size dairy farms. Vet Res. (2016) 47:41. doi: 10.1186/s13567-016-0326-0
45. Romano V, Albanese F, Dumontet S, Krovacek K, Petrini O, Pasquale V. Prevalence and genotypic characterization of Clostridium difficile from ruminants in Switzerland. Zoonoses Public Health. (2012) 59:545–8. doi: 10.1111/j.1863-2378.2012.01540.x
46. Hammitt MC, Bueschel DM, Keel MK, Glock RD, Cuneo P, DeYoung DW, et al. A possible role for Clostridium difficile in the etiology of calf enteritis. Vet Microbiol. (2008) 127:343–52. doi: 10.1016/j.vetmic.2007.09.002
47. Houser BA, Soehnlen MK, Wolfgang DR, Lysczek HR, Burns CM, Jayarao BM. Prevalence of Clostridium difficile toxin genes in the feces of veal calves and incidence of ground veal contamination. Foodborne Pathog Dis. (2012) 9:32–6. doi: 10.1089/fpd.2011.0955
48. Rodriguez-Palacios A, Pickworth C, Loerch S, LeJeune JT. Transient fecal shedding and limited animal-to-animal transmission of Clostridium difficile by naturally infected finishing feedlot cattle. Appl Environ Microbiol. (2011) 77:3391–7. doi: 10.1128/AEM.02736-10
49. Rodriguez-Palacios A, Koohmaraie M, LeJeune JT. Prevalence, enumeration, and antimicrobial agent resistance of Clostridium difficile in cattle at harvest in the United States. J Food Prot. (2011) 74:1618–24. doi: 10.4315/0362-028X.JFP-11-141
50. Kalchayanand N, Arthur TM, Bosilevac JM, Brichta-Harhay DM, Shackelford SD, Wells JE, et al. Isolation and characterization of Clostridium difficile associated with beef cattle and commercially produced ground beef. J Food Prot. (2013) 76:256–64. doi: 10.4315/0362-028X.JFP-12-261
51. Squire MM, Carter GP, Mackin KE, Chakravorty A, Noren T, Elliott B, et al. Novel molecular type of Clostridium difficile in neonatal pigs, Western Australia. Emerg Infect Dis. (2013) 19:790–2. doi: 10.3201/eid1905.121062
52. Knight DR, Squire MM, Riley TV. Nationwide surveillance study of Clostridium difficile in Australian neonatal pigs shows high prevalence and heterogeneity of PCR ribotypes. Appl Environ Microbiol. (2014) 81:119–23. doi: 10.1128/AEM.03032-14
53. Rodriguez C, Avesani V, Van Broeck J, Taminiau B, Delmee M, Daube G. Presence of Clostridium difficile in pigs and cattle intestinal contents and carcass contamination at the slaughterhouse in Belgium. Int J Food Microbiol. (2013) 166:256–62. doi: 10.1016/j.ijfoodmicro.2013.07.017
54. Weese JS, Wakeford T, Reid-Smith R, Rousseau J, Friendship R. Longitudinal investigation of Clostridium difficile shedding in piglets. Anaerobe. (2010) 16:501–4. doi: 10.1016/j.anaerobe.2010.08.001
55. Goldova J, Malinova A, Indra A, Vitek L, Branny P, Jiraskova A. Clostridium difficile in piglets in the Czech Republic. Folia Microbiol. (2012) 57:159–61. doi: 10.1007/s12223-012-0102-0
56. Schneeberg A, Neubauer H, Schmoock G, Baier S, Harlizius J, Nienhoff H, et al. Clostridium difficile genotypes in German piglet populations. J Clin Microbiol. (2013) 51:3796–803. doi: 10.1128/JCM.01440-13
57. Doosti A, Mokhtari-Farsani A. Study of the frequency of Clostridium difficile tcdA, tcdB, cdtA and cdtB genes in feces of calves in south west of Iran. Ann Clin Microbiol Antimicrob. (2014) 13:21. doi: 10.1186/1476-0711-13-21
58. Asai T, Usui M, Hiki M, Kawanishi M, Nagai H, Sasaki Y. Clostridium difficile isolated from the fecal contents of swine in Japan. J Vet Med Sci. (2013) 75:539–41. doi: 10.1292/jvms.12-0353
59. Cho A, Byun JW, Kim JW, Oh SI, Lee MH, Kim HY. Low prevalence of Clostridium difficile in Slaughter pigs in Korea. J Food Prot. (2015) 78:1034–6. doi: 10.4315/0362-028X.JFP-14-493
60. Pirs T, Ocepek M, Rupnik M. Isolation of Clostridium difficile from food animals in Slovenia. J Med Microbiol. (2008) 57:790–2. doi: 10.1099/jmm.0.47669-0
61. Avbersek J, Janezic S, Pate M, Rupnik M, Zidaric V, Logar K, et al. Diversity of Clostridium difficile in pigs and other animals in Slovenia. Anaerobe. (2009) 15:252–5. doi: 10.1016/j.anaerobe.2009.07.004
62. Pelaez T, Alcala L, Blanco JL, Alvarez-Perez S, Marin M, Martin-Lopez A, et al. Characterization of swine isolates of Clostridium difficile in Spain: a potential source of epidemic multidrug resistant strains? Anaerobe. (2013) 22:45–9. doi: 10.1016/j.anaerobe.2013.05.009
63. Norén T, Johansson K, Unemo M. Clostridium difficile PCR ribotype 046 is common among neonatal pigs and humans in Sweden. Clin Microbiol Infect. (2014) 20:O2–O6. doi: 10.1111/1469-0691.12296
64. Tsai BY, Ko WC, Chen TH, Wu YC, Lan PH, Chen YH, et al. Zoonotic potential of the Clostridium difficile RT078 family in Taiwan. Anaerobe. (2016) 41:125–30. doi: 10.1016/j.anaerobe.2016.06.002
65. Debast SB, van Leengoed LA, Goorhuis A, Harmanus C, Kuijper EJ, Bergwerff AA. Clostridium difficile PCR ribotype 078 toxinotype V found in diarrhoeal pigs identical to isolates from affected humans. Environ Microbiol. (2009) 11:505–11. doi: 10.1111/j.1462-2920.2008.01790.x
66. Keessen EC, Leengoed LA, Bakker D, van den Brink KM, Kuijper EJ, Lipman LJA. Prevalence of Clostridium difficile in swine thought to have Clostridium difficile infections (CDI) in eleven swine operations in the Netherlands. Tijdschr Diergeneeskd. (2010) 135:134–7.
67. Hopman NEM, Oorburg D, Sanders I, Kuijper EJ, Lipman LJA. High occurrence of various Clostridium difficile PCR ribotypes in pigs arriving at the slaughterhouse. Vet Q. (2011) 31:179–81. doi: 10.1080/01652176.2011.649370
68. Hopman NE, Keessen EC, Harmanus C, Sanders IM, van Leengoed LAMG, Kuijper EJ, et al. Acquisition of Clostridium difficile by piglets. Vet Microbiol. (2011) 149:186–92. doi: 10.1016/j.vetmic.2010.10.013
69. Keessen EC, van den Berkt AJ, Haasjes NH, Hermanus C, Kuijper EJ, Lipman LJA. The relationship between farm specific factors and prevalence of Clostridium difficile in slaughter pigs. Vet Microbiol. (2011) 154:130–4. doi: 10.1016/j.vetmic.2011.06.032
70. Norman KN, Harvey RB, Scott HM, Hume ME, Andrews K, Brawley AD. Varied prevalence of Clostridium difficile in an integrated swine operation. Anaerobe. (2009) 15:256–60. doi: 10.1016/j.anaerobe.2009.09.006
71. Baker AA, Davis E, Rehberger T, Rosener D. Prevalence and diversity of toxigenic Clostridium perfringens and Clostridium difficile among swine herds in the midwest. Appl Environ Microbiol. (2010) 76:2961–7. doi: 10.1128/AEM.02459-09
72. Thakur S, Putnam M, Fry PR, Abley M, Gebreyes WA. Prevalence of antimicrobial resistance and association with toxin genes in Clostridium difficile in commercial swine. Am J Vet Res. (2010) 71:1189–94. doi: 10.2460/ajvr.71.10.1189
73. Norman KN, Scott HM, Harvey RB, Norby B, Hume ME, Andrews K. Prevalence and genotypic characteristics of Clostridium difficile in a closed and integrated human and swine population. Appl Environ Microbiol. (2011) 77:5755–60. doi: 10.1128/AEM.05007-11
74. Thitaram SN, Frank J, Lyon S, Siragusa G, Bailey J, Lombard J, et al. Clostridium difficile from healthy food animals: optimized isolation and prevalence. J Food Prot. (2011) 74:130–3. doi: 10.4315/0362-028X.JFP-10-229
75. Fry PR, Thakur S, Abley M, Gebreyes WA. Antimicrobial resistance, toxinotype, and genotypic profiling of Clostridium difficile isolates of swine origin. J Clin Microbiol. (2012) 50:2366–72. doi: 10.1128/JCM.06581-11
76. Susick EK, Putnam M, Bermudez DM, Thakur S. Longitudinal study comparing the dynamics of Clostridium difficile in conventional and antimicrobial free pigs at farm and slaughter. Vet Microbiol. (2012) 157:172–8. doi: 10.1016/j.vetmic.2011.12.017
77. Knight DR, Riley TV. Prevalence of gastrointestinal Clostridium difficile carriage in Australian sheep and lambs. Appl Environ Microbiol. (2013) 79:5689–92. doi: 10.1128/AEM.01888-13
78. Esfandiari Z, Weese JS, Ezzatpanah H, Chamani M, Shoaei P, Yaran M, et al. Isolation and characterization of Clostridium difficile in farm animals from slaughterhouse to retail stage in Isfahan, Iran. Foodborne Pathog Dis. (2015) 12:864–6. doi: 10.1089/fpd.2014.1910
79. Avbersek J, Pirs T, Pate M, Rupnik M, Ocepek M. Clostridium difficile in goats and sheep in Slovenia: characterisation of strains and evidence of age-related shedding. Anaerobe. (2014) 28:163–7. doi: 10.1016/j.anaerobe.2014.06.009
80. Zidaric V, Zemljic M, Janezic S, Kocuvan A, Rupnik M. High diversity of Clostridium difficile genotypes isolated from a single poultry farm producing replacement laying hens. Anaerobe. (2008) 14:325–7. doi: 10.1016/j.anaerobe.2008.10.001
81. Koene MGJ, Mevius D, Wagenaar JA, Harmanus C, Hensgens MPM, Meetsma AM, et al. Clostridium difficile in Dutch animals: their presence, characteristics and similarities with human isolates. Clin Microbiol Infect. (2012) 18:778–84. doi: 10.1111/j.1469-0691.2011.03651.x
82. Rodriguez-Palacios A, Barman T, LeJeune JT. Three-week summer period prevalence of Clostridium difficile in farm animals in a temperate region of the United States (Ohio). Can Vet J. (2014) 55:786–9.
83. Simango C, Mwakurudza S. Clostridium difficile in broiler chickens sold at market places in Zimbabwe and their antimicrobial susceptibility. Int J Food Microbiol. (2008) 124:268–70. doi: 10.1016/j.ijfoodmicro.2008.03.020
84. Lim SC, Foster NF, Elliott B, Riley TV. High prevalence of Clostridium difficile on retail root vegetables, Western Australia. J Appl Microbiol. (2017) 124:585–90. doi: 10.1111/jam.13653
85. Tkalec V, Janezic S, Skok B, Simonic T, Mesaric S, Vrabic T, et al. High Clostridium difficile contamination rates of domestic and imported potatoes compared to some other vegetables in Slovenia. Food Microbiol. (2019) 78:194–200. doi: 10.1016/j.fm.2018.10.017
86. Usui M, Nanbu Y, Oka K, Takahashi M, Inamatsu T, Asai T, et al. Genetic relatedness between Japanese and European isolates of Clostridium difficile originating from piglets and their risk associated with human health. Front Microbiol. (2014) 5:513. doi: 10.3389/fmicb.2014.00513
87. Rodriguez-Palacios A, Staempfli HR, Duffield T, Weese JS. Clostridium difficile in retail ground meat, Canada. Emerg Infect Dis. (2007) 13:485–7. doi: 10.3201/eid1303.060988
88. De Boer E, Zwartkruis-Nahuis A, Heuvelink AE, Harmanus C, Kuijper EJ. Prevalence of Clostridium difficile in retailed meat in the Netherlands. Int J Food Microbiol. (2011) 144:561–4. doi: 10.1016/j.ijfoodmicro.2010.11.007
89. Weese JS, Avery BP, Rousseau J, Reid-Smith RJ. Detection and enumeration of Clostridium difficile spores in retail beef and pork. Appl Environ Microbiol. (2009) 75:5009–11. doi: 10.1128/AEM.00480-09
90. Rahimi E, Jalali M, Weese JS. Prevalence of Clostridium difficile in raw beef, cow, sheep, goat, camel and buffalo meat in Iran. BMC Pub Health. (2014) 14:119. doi: 10.1186/1471-2458-14-119
91. Weese JS, Reid-Smith RJ, Avery BP, Rousseau J. Detection and characterization of Clostridium difficile in retail chicken. Lett Appl Microbiol. (2010) 50:362–5. doi: 10.1111/j.1472-765X.2010.02802.x
92. Harvey RB, Norman KN, Andrews K, Hume ME, Scanlan CM, Callaway TR, et al. Clostridium difficile in poultry and poultry meat. Foodborne Pathog Dis. (2011) 8:1321–3. doi: 10.1089/fpd.2011.0936
93. Harvey RB, Norman KN, Andrews K, Norby B, Hume ME, Scanlan CM, et al. Clostridium difficile in retail meat and processing plants in Texas. J Vet Diagn Invest. (2011) 23:807–11. doi: 10.1177/1040638711407893
94. Curry SR, Marsh JW, Schlackman JL, Harrison LH. Clostridium difficile prevalence in uncooked ground meat products from Pittsburgh, PA. Appl Environ Microbiol. (2012) 78:4183–6. doi: 10.1128/AEM.00842-12
95. Pasquale V, Romano V, Rupnik M, Capuano F, Bove D, Aliberti F, et al. Occurrence of toxigenic Clostridium difficile in edible bivalve molluscs. Food Microbiol. (2012) 31:309–12. doi: 10.1016/j.fm.2012.03.001
96. Quesada-Gómez C, Mulvey MR, Vargas P, del Mar Gamboa-Coronado M, Rodríguez C, Rodríguez-Cavillini E. Isolation of a toxigenic and clinical genotype of Clostridium difficile in retail meats in Costa Rica. J Food Prot. (2013) 76:348–51. doi: 10.4315/0362-028X.JFP-12-169
97. Rodriguez-Palacios A, Reid-Smith RJ, Staempfli HR, Daignault D, Janecko N, Avery BP, et al. Possible seasonality of Clostridium difficile in retail meat, Canada. Emerg Infect Dis. (2009) 15:802–5. doi: 10.3201/eid1505.081084
98. Hoffer E, Haechler H, Frei R, Stephan R. Low occurrence of Clostridium difficile in fecal samples of healthy calves and pigs at slaughter and in minced meat in Switzerland. J Food Prot. (2010) 73:973–5. doi: 10.4315/0362-028X-73.5.973
99. Metcalf D, Avery BP, Janecko N, Matic N, Reid-Smith R, Weese JS. Clostridium difficile in seafood and fish. Anaerobe. (2011) 17:85–6. doi: 10.1016/j.anaerobe.2011.02.008
100. Metcalf D, Reid-Smith RJ, Avery BP, Weese JS. Prevalence of Clostridium difficile in retail pork. Can Vet J. (2010) 51:873–6.
101. Metcalf DS, Costa MC, Dew WM, Weese JS. Clostridium difficile in vegetables, Canada. Lett Appl Microbiol. (2010) 51:600–2. doi: 10.1111/j.1472-765X.2010.02933.x
102. Wu YC, Chen CM, Kuo CJ, Lee JJ, Chen PC, Chang YC, et al. Prevalence and molecular characterization of Clostridium difficile isolates from a pig slaughterhouse, pork, and humans in Taiwan. Int J Food Microbiol. (2017) 242:37–44. doi: 10.1016/j.ijfoodmicro.2016.11.010
103. Rodriguez C, Taminiau B, Van Broeck J, Avesani V, Delmee M, Daube G. Clostridium difficile in young farm animals and slaughter animals in Belgium. Anaerobe. (2012) 18:621–5. doi: 10.1016/j.anaerobe.2012.09.008
104. Bakri M. Prevalence of Clostridium difficile in raw cow, sheep, and goat meat in Jazan, Saudi Arabia. Saudi J Biol Sci. (2018) 25:783–5. doi: 10.1016/j.sjbs.2016.07.002
105. Kouassi KA, Dadie AT, N'Guessan KF, Dje KM, Loukou YG. Clostridium perfringens and Clostridium difficile in cooked beef sold in Cote d'Ivoire and their antimicrobial susceptibility. Anaerobe. (2014) 28:90–4. doi: 10.1016/j.anaerobe.2014.05.012
106. Troiano T, Harmanus C, Sanders IMJG, Pasquale V, Dumontet S, Capuano F, et al. Toxigenic Clostridium difficile PCR ribotypes in edible marine bivalve molluscs in Italy. Int J Food Microbiol. (2015) 208:30–4. doi: 10.1016/j.ijfoodmicro.2015.05.002
107. Eckert C, Burghoffer B, Barbut F. Contamination of ready-to-eat raw vegetables with Clostridium difficile in France. J Med Microbiol. (2013) 62 (Pt 9):1435–8. doi: 10.1099/jmm.0.056358-0
108. Norman KN, Harvey RB, Andrews K, Hume ME, Callaway TR, Anderson RC, et al. Survey of Clostridium difficile in retail seafood in College Station, Texas. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. (2014) 31:1127–9. doi: 10.1080/19440049.2014.888785
109. Rodriguez-Palacios A, Ilic S, LeJeune JT. Clostridium difficile with moxifloxacin/clindamycin resistance in vegetables in Ohio, USA, and prevalence meta-analysis. J Pathog. (2014) 2014:158601. doi: 10.1155/2014/158601
110. Yamoudy M, Mirlohi M, Isfahani BN, Jalali M, Esfandiari Z, Hosseini NS. Isolation of toxigenic Clostridium difficile from ready-to-eat salads by multiplex polymerase chain reaction in Isfahan, Iran. Adv Biomed Res. (2015) 4:87. doi: 10.4103/2277-9175.156650
111. Bakri MM, Brown DJ, Butcher JP, Sutherland AD. Clostridium difficile in ready-to-eat salads, Scotland. Emerg Infect Dis. (2009) 15:817–8. doi: 10.3201/eid1505.081186
112. Alam MJ, Anu A, Walk ST, Garey KW. Investigation of potentially pathogenic Clostridium difficile contamination in household environs. Anaerobe. (2014) 27:31–3. doi: 10.1016/j.anaerobe.2014.03.002
113. Shaughnessy MK, Bobr A, Kuskowski MA, Johnston BD, Sadowsky MJ, Khoruts A, et al. Environmental contamination in households of patients with recurrent clostridium difficile infection. Appl Environ Microbiol. (2016) 82:2686–92. doi: 10.1128/AEM.03888-15
114. Orden C, Neila C, Blanco JL, Alvarez-Perez S, Harmanus C, Kuijper EJ, et al. Recreational sandboxes for children and dogs can be a source of epidemic ribotypes of Clostridium difficile. Zoonoses Public Health. (2018) 65:88–95. doi: 10.1111/zph.12374
115. Janezic S, Potocnik M, Zidaric V, Rupnik M. Highly divergent Clostridium difficile strains isolated from the environment. PLoS ONE. (2016) 11:e0167101. doi: 10.1371/journal.pone.0167101
116. Zidaric V, Beigot S, Lapajne S, Rupnik M. The occurrence and high diversity of Clostridium difficile genotypes in rivers. Anaerobe. (2010) 16:371–5. doi: 10.1016/j.anaerobe.2010.06.001
117. Chukwu EE, Ogunsola FT, Nwaokorie FO, Coker AO. Characterization of clostridium species from food commodities and faecal specimens in Lagos state, Nigeria. West Afr J Med. (2015) 34:167–73.
118. Al Saif N, Brazier JS. The distribution of Clostridium difficile in the environment of South Wales. J Med Microbiol. (1996) 45:133–7. doi: 10.1099/00222615-45-2-133
119. Simango C. Prevalence of Clostridium difficile in the environment in a rural community in Zimbabwe. Trans R Soc Trop Med Hyg. (2006) 100:1146–50. doi: 10.1016/j.trstmh.2006.01.009
120. Pasquale V, Romano VJ, Rupnik M, Dumontet S, CiŽnár I, Aliberti F, et al. Isolation and characterization of Clostridium difficile from shellfish and marine environments. Folia Microbiol. (2011) 56:431–7. doi: 10.1007/s12223-011-0068-3
121. Xu C, Weese JS, Flemming C, Odumeru J, Warriner K. Fate of Clostridium difficile during wastewater treatment and incidence in Southern Ontario watersheds. J Appl Microbiol. (2014) 117:891–904. doi: 10.1111/jam.12575
122. Lim S-C, Moono P, Riley TV. Clostridium difficile found in gardening products: innocent bystander or the cause of community-acquired C. difficile infection through contamination of foods and environments? In: Proceeding 44nd The Australian Society for Microbiology (ASM). Perth, WA: ASM (2016).
123. Keel MK, Songer JG. The comparative pathology of Clostridium difficile-associated disease. Vet Pathol. (2006) 43:225–40. doi: 10.1354/vp.43-3-225
124. Nagy J, Bilkei G. Neonatal piglet losses associated with Escherichia coli and Clostridium difficile infection in a Slovakian outdoor production unit. Vet J. (2003) 166:98–100. doi: 10.1016/S1090-0233(02)00252-6
125. Songer JG, Anderson MA. Clostridium difficile: an important pathogen of food animals. Anaerobe. (2006) 12:1–4. doi: 10.1016/j.anaerobe.2005.09.001
126. Weese JS, Salgado-Bierman F, Rupnik M, Smith DA, van Coeverden de Groot P. Clostridium (Clostridioides) difficile shedding by polar bears (Ursus maritimus) in the Canadian Arctic. Anaerobe. (2019) 57:35–8. doi: 10.1016/j.anaerobe.2019.03.013
127. Rodriguez C, Taminiau B, Van Broeck J, Delmee M, Daube G. Clostridium difficile in food and animals: a comprehensive review. Adv Exp Med Biol. (2016) 932:65–92. doi: 10.1007/5584_2016_27
128. Knight DR, Squire MM, Collins DA, Riley TV. Genome analysis of Clostridium difficile PCR ribotype 014 lineage in Australian pigs and humans reveals a diverse genetic repertoire and signatures of long-range interspecies transmission. Front Microbiol. (2017) 7:2138. doi: 10.3389/fmicb.2016.02138
129. Weese JS. Clostridium difficile in food-innocent bystander or serious threat? Clin Microbiol Infect. (2010) 16:3–10. doi: 10.1111/j.1469-0691.2009.03108.x
130. Songer JG, Trinh HT, Killgore GE, Thompson AD, McDonald LC, Limbago BM. Clostridium difficile in retail meat products, USA, 2007. Emerg Infect Dis. (2009) 15:819–21. doi: 10.3201/eid1505.081071
131. Janezic S, Mlakar S, Rupnik M. Dissemination of Clostridium difficile spores between environment and households: dog paws and shoes. Zoonoses Public Health. (2018) 65: 669–74 doi: 10.1111/zph.12475
132. Gerding DN, Muto CA, Owens RC Jr. Measures to control and prevent Clostridium difficile infection. Clin Infect Dis. (2008) 46:43–9. doi: 10.1086/521861
133. Edwards AN, Karim ST, Pascual RA, Jowhar LM, Anderson SE, McBride SM. Chemical and stress resistances of Clostridium difficile spores and vegetative cells. Front Microbiol. (2016) 7:1698. doi: 10.3389/fmicb.2016.01698
134. Rupnik M, Songer JG. Clostridium difficile: its potential as a source of foodborne disease. Adv Food Nutr Res. (2010) 60:53–66. doi: 10.1016/S1043-4526(10)60003-4
135. Squire MM, Riley TV. Clostridium difficile infection: the next big thing! Microbiol Aus. (2012) 33:163–4. Available online at: http://microbiology.publish.csiro.au/?paper=MA12163
136. Burt SA, Siemeling L, Kuijper EJ, Lipman LJ. Vermin on pig farms are vectors for Clostridium difficile PCR ribotypes 078 and 045. Vet Microbiol. (2012) 160:256–8. doi: 10.1016/j.vetmic.2012.05.014
137. Keessen EC, Donswijk CJ, Hol SP, Hermanus C, Kuijper EJ, Lipman LJ. Aerial dissemination of Clostridium difficile on a pig farm and its environment. Environ Res. (2011) 111:1027–32. doi: 10.1016/j.envres.2011.09.014
138. Knight DR, Putsathit P, Elliott B, Riley TV. Contamination of Australian newborn calf carcasses at slaughter with Clostridium difficile. Clin Microbiol Infect. (2016) 22:266.e1–7. doi: 10.1016/j.cmi.2015.11.017
139. Xu C, Wang D, Huber A, Weese SJ, Warriner K. Persistence of Clostridium difficile in wastewater treatment-derived biosolids during land application or windrow composting. J Appl Microbiol. (2016) 120:312–20. doi: 10.1111/jam.13018
140. Squire MM, Knight DR, Riley TV. Community-acquired Clostridium difficile infection and Australian food animals. Microbiol Aus. (2015) 36:111–3. doi: 10.1071/MA15040
141. Squire MM, Riley TV. Clostridium difficile infection in humans and piglets: a ‘One Health' opportunity. Curr Top Microbiol Immunol. (2013) 365:299–314. doi: 10.1007/82_2012_237
142. Rodriguez-Palacios A, Ilic S, LeJeune JT. Subboiling moist heat favors the selection of enteric pathogen Clostridium difficile PCR ribotype 078 spores in food. Can J Infect Dis Med Microbiol. (2016) 2016:1462405. doi: 10.1155/2016/1462405
143. Deng K, Plaza-Garrido A, Torres JA, Paredes-Sabja D. Survival of Clostridium difficile spores at low temperatures. Food Microbiol. (2015) 46:218–21. doi: 10.1016/j.fm.2014.07.022
144. Rodriguez-Palacios A, LeJeune JT. Moist heat resistance, spore aging, and superdormancy in Clostridium difficile. Appl Environ Microbiol. (2011) 77:3085–91. doi: 10.1128/AEM.01589-10
145. Rodriguez-Palacios A, Reid-Smith RJ, Staempfli HR, Weese JS. Clostridium difficile survives minimal temperature recommended for cooking ground meats. Anaerobe. (2010) 16:540–2. doi: 10.1016/j.anaerobe.2010.05.004
146. MLA. Market Information & Industry Insights – Australian Cattle Industry Projections 2015. Meat and Livestock Australia (MLA) (2015). Available online at: http://www.mla.com.au/About-MLA/News-and-media/Media-releases/2015-cattle-industry-projections-released (accessed March 1, 2019).
147. Wu YC, Lee JJ, Tsai BY, Liu YF, Chen CM, Tien N, et al. Potentially hypervirulent Clostridium difficile PCR ribotype 078 lineage isolates in pigs and possible implications for humans in Taiwan. Int J Med Microbiol. (2016) 306:115–22. doi: 10.1016/j.ijmm.2016.02.002
148. Viau E, Peccia J. Survey of wastewater indicators and human pathogen genomes in biosolids produced by class A and class B stabilization treatments. Appl Environ Microbiol. (2009) 75:164–74. doi: 10.1128/AEM.01331-08
149. Romano V, Pasquale V, Krovacek K, Mauri F, Demarta A, Dumontet S. Toxigenic Clostridium difficile PCR ribotypes from wastewater treatment plants in southern Switzerland. Appl Environ Microbiol. (2012) 78:6643–6. doi: 10.1128/AEM.01379-12
150. Squire MM, Lim SC, Foster NF, Riley TV. Detection of Clostridium difficile after treatment in a two-stage pond system. In: van Barneveld RJ, editor. Manipulating Pig Production, Vol 8: Proceedings 13th Biannual Conference of the Australian Pig Science Association (APSA). Adelaide, SA. (2011). p. 215.
151. Moono P, Lim SC, Riley TV. High prevalence of toxigenic Clostridium difficile in public space lawns in Western Australia. Sci Rep. (2017) 7:41196. doi: 10.1038/srep41196
152. Eyre DW, Walker AS. Clostridium difficile surveillance: harnessing new technologies to control transmission. Expert Rev Anti Infect Ther. (2013) 11:1193–205. doi: 10.1586/14787210.2013.845987
153. Olson ND, Lund SP, Colman RE, Foster JT, Sahl JW, Schupp JM, et al. Best practices for evaluating single nucleotide variant calling methods for microbial genomics. Front Genet. (2015) 6:235. doi: 10.3389/fgene.2015.00235
154. Eyre DW, Cule ML, Wilson DJ, Griffiths D, Vaughan A, O'Connor L, et al. Diverse sources of Clostridium difficile infection identified on whole-genome sequencing. N Engl J Med. (2013) 369:1195–205. doi: 10.1056/NEJMoa1216064
155. Didelot X, Eyre DW, Cule M, Ip CL, Ansari MA, Griffiths D, et al. Microevolutionary analysis of Clostridium difficile genomes to investigate transmission. Genome Biol. (2012) 13:R118. doi: 10.1186/gb-2012-13-12-r118
156. Eyre DW, Fawley WN, Rajgopal A, Settle C, Mortimer K, Goldenberg SD, et al. Comparison of control of Clostridium difficile infection in six english hospitals using whole-genome sequencing. Clin Infect Dis. (2017) 65:433–41. doi: 10.1093/cid/cix338
157. Eyre DW, Fawley WN, Best EL, Griffiths D, Stoesser NE, Crook DW, et al. Comparison of multilocus variable-number tandem-repeat analysis and whole-genome sequencing for investigation of Clostridium difficile transmission. J Clin Microbiol. (2013) 51:4141–9. doi: 10.1128/JCM.01095-13
158. Bletz S, Janezic S, Harmsen D, Rupnik M, Mellmann A. Defining and evaluating a core genome multilocus sequence typing scheme for genome-wide typing of Clostridium difficile. J Clin Microbiol. (2018) 56:e01987–17. doi: 10.1128/JCM.01987-17
159. Cairns MD, Preston MD, Hall CL, Gerding DN, Hawkey PM, Kato H, et al. Comparative genome analysis and global phylogeny of the toxin variant Clostridium difficile PCR ribotype 017 reveals the evolution of two independent sub-lineages. J Clin Microbiol. (2016) 55:865–76. doi: 10.1128/JCM.00487-17
160. Jhung MA, Thompson AD, Killgore GE, Zukowski WE, Songer G, Warny M, et al. Toxinotype V Clostridium difficile in humans and food animals. Emerg Infect Dis. (2008) 14:1039–45. doi: 10.3201/eid1407.071641
161. Knetsch CW, Connor TR, Mutreja A, van Dorp SM, Sanders IM, Browne HP, et al. Whole genome sequencing reveals potential spread of Clostridium difficile between humans and farm animals in the Netherlands, 2002 to 2011. Euro Surveill. (2014) 19:30–41. doi: 10.2807/1560-7917.ES2014.19.45.20954
162. Stoesser N, Eyre DW, Quan TP, Godwin H, Pill G, Mbuvi E, et al. Epidemiology of Clostridium difficile in infants in Oxfordshire, UK: risk factors for colonization and carriage, and genetic overlap with regional C. difficile infection strains. PLoS ONE. (2017) 12:e0182307. doi: 10.1371/journal.pone.0182307
163. Barbut F, Decre D, Lalande V, Burghoffer A, Noussair L, Gigandon A, et al. Clinical features of Clostridium difficile-associated diarrhoea due to binary toxin (actin-specific ADP-ribosyltransferase)-producing strains. J Med Microbiol. (2005) 54:181–5. doi: 10.1099/jmm.0.45804-0
164. Stewart DB, Berg AS, Hegarty JP. Single nucleotide polymorphisms of the tcdC gene and presence of the binary toxin gene predict recurrent episodes of Clostridium difficile infection. Ann Surg. (2013) 260:299–304. doi: 10.1097/SLA.0000000000000469
165. Knetsch CW, Hensgens MPM, Harmanus C, van der Bijl MW, Savelkoul PH, Kuijper EJ, et al. Genetic markers for Clostridium difficile lineages linked to hypervirulence. Microbiol. (2011) 157:3113–23. doi: 10.1099/mic.0.051953-0
166. Carter GP, Douce GR, Govind R, Howarth PM, Mackin KE, Spencer J, et al. The anti-sigma factor TcdC modulates hypervirulence in an epidemic BI/NAP1/027 clinical isolate of Clostridium difficile. PLoS Pathog. (2011) 7:e1002317. doi: 10.1371/journal.ppat.1002317
167. Curry SR, Marsh JW, Muto CA, O'Leary MM, Pasculle AW, Harrison LH. tcdC genotypes associated with severe TcdC truncation in an epidemic clone and other strains of Clostridium difficile. J Clin Microbiol. (2007) 45:215–21. doi: 10.1128/JCM.01599-06
168. Spigaglia P, Barbanti F, Mastrantonio P. Multidrug resistance in European Clostridium difficile clinical isolates. J Antimicrob Chemother. (2011) 66:2227–34. doi: 10.1093/jac/dkr292
169. Collins J, Danhof H, Britton RA. The role of trehalose in the global spread of epidemic Clostridium difficile. Gut Microbes. (2018) 10:204–9. doi: 10.1080/19490976.2018.1491266
170. Bakker D, Corver J, Harmanus C, Goorhuis A, Keessen EC, Fawley WN, et al. Relatedness of human and animal Clostridium difficile PCR ribotype 078 isolates determined on the basis of multilocus variable-number tandem-repeat analysis and tetracycline resistance. J Clin Microbiol. (2010) 48:3744–9. doi: 10.1128/JCM.01171-10
171. Stabler RA, Dawson LF, Valiente E, Cairns MD, Martin MJ, Donahue EH, et al. Macro and micro diversity of Clostridium difficile isolates from diverse sources and geographical locations. PLoS ONE. (2012) 7:e31559. doi: 10.1371/journal.pone.0031559
172. Álvarez-Pérez S, Blanco J, Harmanus C, Kuijper E, García M. Subtyping and antimicrobial susceptibility of Clostridium difficile PCR ribotype 078/126 isolates of human and animal origin. Vet Microbiol. (2017) 199:15–22. doi: 10.1016/j.vetmic.2016.12.001
173. Niwa H, Kato H, Hobo S, Kinoshita Y, Ueno T, Katayama Y, et al. Postoperative Clostridium difficile infection with PCR ribotype 078 strain identified at necropsy in five Thoroughbred racehorses. Vet Record. (2013) 173:607. doi: 10.1136/vr.101960
174. Knetsch CW, Kumar N, Forster SC, Connor TR, Browne HP, Harmanus C, et al. Zoonotic transfer of Clostridium difficile harboring antimicrobial resistance between farm animals and humans. J Clin Microbiol. (2018) 56:e01384–17. doi: 10.1128/JCM.01384-17
175. Knight DR, Kullin B, Androga GO, Barbut F, Eckert C, Johnson S, et al. Evolutionary and genomic insights into Clostridioides difficile sequence type 11: a diverse, zoonotic and antimicrobial resistant lineage of global One Health importance. MBio. (2019) 10:e00446–19. doi: 10.1128/mBio.00446-19
176. Baines SD, Wilcox MH. Antimicrobial resistance and reduced susceptibility in Clostridium difficile: potential consequences for induction, treatment, and recurrence of Clostridium difficile infection. Antibiotics. (2015) 4:267–98. doi: 10.3390/antibiotics4030267
177. Corver J, Bakker D, Brouwer MS, Harmanus C, Hensgens MP, Roberts AP, et al. Analysis of a Clostridium difficile PCR ribotype 078 100 kilobase island reveals the presence of a novel transposon, Tn6164. BMC Microbiol. (2012) 12:130. doi: 10.1186/1471-2180-12-130
178. Spigaglia P. Recent advances in the understanding of antibiotic resistance in Clostridium difficile infection. Ther Adv Infect Dis. (2016) 3:23–42. doi: 10.1177/2049936115622891
179. Knight DR, Elliott B, Chang BJ, Perkins TT, Riley TV. Diversity and evolution in the genome of Clostridium difficile. Clin Microbiol Rev. (2015) 28:721–41. doi: 10.1128/CMR.00127-14
180. Knight DR, Androga GO, Ballard SA, Howden BP, Riley TV. A phenotypically silent vanB2 operon carried on a Tn1549-like element in Clostridium difficile. mSphere. (2016) 1:e00177–16. doi: 10.1128/mSphere.00177-16
181. Dingle K, Didelot X, Quan P, Eyre DW, Stoesser N, Marwick C, et al. A role for tetracycline selection in recent evolution of the agriculture-associated Clostridium difficile PCR-ribotype 078. mBIO. (2019) 10:e02790–18. doi: 10.1128/mBio.02790-18
182. Weinert LA, Chaudhuri RR, Wang J, Peters SE, Corander J, Jombart T, et al. Genomic signatures of human and animal disease in the zoonotic pathogen Streptococcus suis. Nat Commun. (2015) 6:6740. doi: 10.1038/ncomms7740
183. Wertheim HF, Nghia HD, Taylor W, Schultsz C. Streptococcus suis: an emerging human pathogen. Clin Infect Dis. (2009) 48:617–25. doi: 10.1086/596763
184. Robinson TP, Wertheim HF, Kakkar M, Kariuki S, Bu D, Price LB. Animal production and antimicrobial resistance in the clinic. Lancet. (2016) 387:e1–3. doi: 10.1016/S0140-6736(15)00730-8
185. Freeman J, Vernon J, Morris K, Nicholson S, Todhunter S, Longshaw C, et al. Pan-European longitudinal surveillance of antibiotic resistance among prevalent Clostridium difficile ribotypes. Clin Microbiol Infect. (2014) 21:248 e9–e16. doi: 10.1016/j.cmi.2014.09.017
186. Knight DR, Giglio S, Huntington PG, Korman TM, Kotsanas D, Moore CV, et al. Surveillance for antimicrobial resistance in Australian isolates of Clostridium difficile, 2013–14. J Antimicrob Chemother. (2015) 70:2992–9. doi: 10.1093/jac/dkv220
187. Foster NF, Collins DA, Ditchburn SL, Duncan CN, van Schalkwyk JW, Golledge CL, et al. Epidemiology of Clostridium difficile infection in two tertiary-care hospitals in Perth, Western Australia: a cross-sectional study. N Microb N Infect. (2014) 2:64–71. doi: 10.1002/nmi2.43
188. Collins DA, Putsathit P, Elliott B, Riley TV. Laboratory-based surveillance of Clostridium difficile strains circulating in the Australian healthcare setting in 2012. Pathology. (2017) 49:309–13. doi: 10.1016/j.pathol.2016.10.013
189. Tettelin H, Riley D, Cattuto C, Medini D. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol. (2008) 11:472–7. doi: 10.1016/j.mib.2008.09.006
190. Scaria J, Ponnala L, Janvilisri T, Yan W, Mueller LA, Chang YF. Analysis of ultra low genome conservation in Clostridium difficile. PLoS ONE. (2010) 5:e15147. doi: 10.1371/journal.pone.0015147
191. He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci USA. (2010) 107:7527–32. doi: 10.1073/pnas.0914322107
192. Forgetta V, Oughton MT, Marquis P, Brukner I, Blanchette R, Haub K, et al. Fourteen-genome comparison identifies DNA markers for severe-disease-associated strains of Clostridium difficile. J Clin Microbiol. (2011) 49:2230–8. doi: 10.1128/JCM.00391-11
193. Rouli L, Merhej V, Fournier P-E, Raoult D. The bacterial pangenome as a new tool for analysing pathogenic bacteria. N Microb New Infect. (2015) 7:72–85. doi: 10.1016/j.nmni.2015.06.005
194. Georgiades K, Raoult D. Defining pathogenic bacterial species in the genomic era. Front Microbiol. (2010) 1:151. doi: 10.3389/fmicb.2010.00151
195. Wasels F, Monot M, Spigaglia P, Barbanti F, Ma L, Bouchier C, et al. Inter- and intraspecies transfer of a Clostridium difficile conjugative transposon conferring resistance to MLSB. Microb Drug Resist. (2014) 20:555–60. doi: 10.1089/mdr.2014.0015
196. Kunin V, Ahren D, Goldovsky L, Janssen P, Ouzounis CA. Measuring genome conservation across taxa: divided strains and united kingdoms. Nucleic Acids Res. (2005) 33:616–21. doi: 10.1093/nar/gki181
197. Yutin N, Galperin MY. A genomic update on clostridial phylogeny: gram-negative spore formers and other misplaced Clostridia. Environ Microbiol. (2013) 15:2631–41. doi: 10.1111/1462-2920.12173
198. Collins MD, Lawson PA, Willems A, Cordoba JJ, Fernandez-Garayzabal J, Garcia P, et al. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int J Syst Bacteriol. (1994) 44:812–26. doi: 10.1099/00207713-44-4-812
199. Ludwig W, Schleifer KH, Whitman WB. Revised road map to the phylum Firmicutes. In: De Vos P, Garrity GM, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer K-H and Whitman WB, editors. Bergey's Manual of Systematic Bacteriology, 2nd ed, Vol 3: The Firmicutes. New York, NY: Springer (2009). p. 1–14. doi: 10.1007/978-0-387-68489-5_1
200. Lerner H, Berg C. The concept of health in One Health and some practical implications for research and education: what is One Health? Infect Ecol Epidemiol. (2015) 5:25300. doi: 10.3402/iee.v5.25300
201. Thomas C, Stevenson M, Williamson DJ, Riley TV. Clostridium difficile-associated diarrhea: epidemiological data from Western Australia associated with a modified antibiotic policy. Clin Infect Dis. (2002) 35:1457–62. doi: 10.1086/342691
202. Price J, Cheek E, Lippett S, Cubbon M, Gerding DN, Sambol SP, et al. Impact of an intervention to control Clostridium difficile infection on hospital-and community-onset disease; an interrupted time series analysis. Clin Microbiol Infect. (2010) 16:1297–302. doi: 10.1111/j.1469-0691.2009.03077.x
203. Weese JS, Armstrong J. Outbreak of Clostridium difficile-associated disease in a small animal veterinary teaching hospital. J Vet Intern Med. (2003) 17:813–6. doi: 10.1111/j.1939-1676.2003.tb02519.x
204. Riley TV. Is Clostridium difficile a threat to Australia's biosecurity? Med J Aust. (2009) 190:661–2. doi: 10.5694/j.1326-5377.2009.tb02630.x
Keywords: evolution, transmission, Clostridium difficile, one health, livestock, zoonosis
Citation: Knight DR and Riley TV (2019) Genomic Delineation of Zoonotic Origins of Clostridium difficile. Front. Public Health 7:164. doi: 10.3389/fpubh.2019.00164
Received: 11 April 2019; Accepted: 03 June 2019;
Published: 20 June 2019.
Edited by:
Marc Jean Struelens, European Centre for Disease Prevention and Control, SwedenReviewed by:
David Eyre, University of Oxford, United KingdomSergio Alvarez-Perez, KU Leuven, Belgium
Copyright © 2019 Knight and Riley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas V. Riley, VC5SaWxleUBtdXJkb2NoLmVkdS5hdQ==