 Andrew T. Schnaubelt1†
Andrew T. Schnaubelt1† David M. Brett-Major2†Janet Williamson1Bailey Barcal1
David M. Brett-Major2†Janet Williamson1Bailey Barcal1 Julie Carstens1
Julie Carstens1 Ashley Peer1
Ashley Peer1 Michael Wiley1
Michael Wiley1 M. Jana Broadhurst1*
M. Jana Broadhurst1*- 1Department of Pathology, Microbiology, and Immunology, College of Medicine, University of Nebraska Medical Center, Omaha, NE, United States
- 2Department of Epidemiology, College of Public Health, University of Nebraska Medical Center, Omaha, NE, United States
Continually emerging SARS-CoV-2 variants pose challenges to clinical and public health interventions, necessitating sustainable approaches to real-time variant monitoring. This case study describes an innovative SARS-CoV-2 screening and surveillance program that demonstrates the utility of sequencing-based variant monitoring using self-collected saliva specimens. We conducted saliva-based SARS-CoV-2 screening in occupational settings in Omaha, Nebraska from December 2021 through November 2022. 8,372 saliva specimens collected from 1,480 participants were tested for SARS-CoV-2 RNA by extraction-free PCR, with 334 positive samples referred for whole-genome sequencing analysis. Program utilization, quality metrics, and sequencing outputs were compared across sites. Specimen quality was high across program settings, demonstrating the suitability of self-collected saliva specimens for PCR and genomic surveillance testing. Virus RNA sequencing successfully determined the variant strain in 83 and 67% of SARS-CoV-2-positive saliva samples collected in two program settings, demonstrating the successful integration of SARS-CoV-2 sequencing for variant determination into screening programs in occupational settings using self-collected saliva with an extraction-free qRT-PCR testing method. We further demonstrate that the sensitivity and efficiency of variant analysis is dependent on the PCR cycle threshold (Ct) cutoff of the diagnostic assay virus gene target. Use of an optimized Ct value cutoff for sequencing referral is recommended. Community-based saliva testing programs can be utilized to enhance variant monitoring, and could be considered in the risk identification of other respiratory infections. This approach offers the advantages of a non-invasive specimen collection, no need for supervised collection by a healthcare worker, supply chain resiliency, distributable access, and scalability.
1 Introduction
SARS-CoV-2 variants of concern continue to emerge globally (1–3) and surveillance testing to detect and monitor variant strains remains a public health priority (4, 5). Surveillance testing for SARS-CoV-2 variant strains primarily relies on virus whole-genome sequencing, typically performed on nasal or nasopharyngeal swab specimens referred to clinical or public health laboratories for polymerase chain reaction (PCR)-based diagnostic testing. Gaps in access to and/or utilization of PCR-based diagnostic testing for SARS-CoV-2 pose a risk to adequate representation of underserved communities in genomic surveillance data. There is a critical need to develop and evaluate community-based testing strategies that increase the representation of high-risk populations in SARS-CoV-2 screening and genomic surveillance programs.
Community-based testing programs increase testing access through facilitated specimen collection in locations such as schools and workplaces, offering an important opportunity to expand representation for SARS-CoV-2 variant surveillance. Saliva has been increasingly utilized for such programs due to the ease of collection, greater acceptability due to a less invasive sampling method compared to nasopharyngeal swabs, specimen stability, and diagnostic reliability (6–9). Genomic sequencing of SARS-CoV-2 from saliva specimens has shown comparable results to nasopharyngeal swabs with specimens collected in a healthcare setting (10). However, the performance of integrated SARS-CoV-2 genomic sequencing during community-based saliva testing programs has not been described. Viral load distribution and specimen quality are major factors impacting the utility of sequencing strategies.
In Fall 2020, we developed and implemented an extraction-free PCR test for SARS-CoV-2 diagnosis using self-collected saliva to address regional gaps in testing access and to meet the need for community-based testing in Nebraska (11). During subsequent waves of the COVID-19 pandemic driven by variant strains of the virus, we developed an innovative community-based testing approach using self-collected saliva specimens for SARS-CoV-2 detection and variant determination by whole-genome sequencing to enhance public health surveillance of emerging SARS-CoV-2 variants. Here we describe program utilization, quality metrics, and outcomes of SARS-CoV-2 genomic sequencing performed on saliva specimens generated from occupational settings in Omaha, Nebraska and tested at the University of Nebraska Medical Center (UNMC). Genomic sequencing is resource intensive and warrants a strategic approach to optimize specimen referral. We examine the sensitivity and efficiency of genomic sequencing for variant analysis in the context of a specimen selection strategy based on the PCR cycle threshold (Ct) value as a measure of viral RNA quantity. This community case study describes an innovative approach to SARS-CoV-2 screening and genomic surveillance, and presents a data-driven strategy to monitor specimen quality and optimize specimen referral for sequencing, key factors in overall program efficacy. This work demonstrates the utility of self-collected saliva to expand SARS-CoV-2 genomic surveillance for variant monitoring enabling successful program implementation and expanded surveillance in high-risk occupational settings.
2 Methods
2.1 Program settings
The specimens and testing included in this analysis are from saliva-based SARS-CoV-2 diagnostic testing programs supported by the UNMC Emerging Pathogens Laboratory in two program settings between December 2021 and November 2022: (1) UNMC campus (Omaha, Nebraska). Voluntary saliva testing was made available to employees and students four days per week. Specimens were self-collected at an on-campus collection site with supervision by trained staff and hand-carried to the laboratory. (2) Omaha workplaces (various locations in Omaha, Nebraska). Mandatory employee saliva testing occurred prior to aggregated work activities, according to industry and organizational requirements. Specimens were self-collected at home or at temporary workplace collection sites with supervision by trained community members and delivered to the laboratory by ground couriers. All individuals who registered for these testing programs and submitted at least one specimen for testing were included in this study. This study received a non-human subjects research determination from the University of Nebraska Medical Center Institutional Review Board, as the work constituted quality assurance within a public health surveillance activity.
2.2 Program site preparation and training, test ordering and reporting
Each site managed their own saliva collection schedules and programs, with training and supplies provided by the testing laboratory. SARS-CoV-2 testing was provided at no cost to the individuals who sought testing. Representatives from each program site were trained on test ordering, specimen collection and packaging, and specimen transport or shipping to the laboratory. Registration and consent for saliva testing and electronic reporting was provided through the web-based Nebraska University Laboratory Information Reporting Tool (NULirt) system (12). NULirt reports individual test results by email. NULirt also provides electronic results reporting to the Department of Health and Human Services disease surveillance system, Nebraska electronic disease surveillance system, and the Centers for Disease Control and Prevention health information exchange.
2.3 Saliva specimen collection
Saliva self-collection was performed by the passive drool method (13), using the clinically validated Nebraska COVID-19 Saliva PCR Test Saliva Collection Kit under the supervision of trained staff or volunteers, or unsupervised using the Nebraska COVID-19 Saliva PCR Test Home Collection Kit. Half-length plastic straws (S.P. Richards Co) were used to collect saliva into dry 1.5 mL collection tubes (Eppendorf Co). Following surface decontamination with ethanol, each sample was placed in an individual biohazard bag and then packaged as a batch into a transport or shipping box. The packaged saliva specimens were transported or shipped under ambient conditions in accordance with International Air Transport Association regulations. Packaged SARS-CoV-2-positive saliva specimens were tested for the stability of RNA target detection following up to 96 h of exposure to summer or winter shipping conditions in accordance with FDA guidance (14). No significant drift in cycle threshold (Ct) values for the SARS-CoV-2 RNA target or the human RNA internal control target were observed in the stability study.
2.4 Saliva SARS-CoV-2 PCR testing and sequencing analysis
SARS-CoV-2 detection in saliva by quantitative reverse-transcription (qRT)-PCR was conducted by the CLIA-certified EPL at UNMC using the Nebraska COVID-19 Saliva PCR Test, as previously described (11). This diagnostic test is a dual-plex, extraction-free qRT-PCR assay adapted from the SalivaDirect protocol (15, 16) that detects SARS-CoV-2 N1gene RNA in saliva. In brief, 50 μL of well-mixed saliva is added to 6.3 μL of proteinase K (New England Biolabs), then shaken and heated at 2,200 RPM and 95°C for 5 min. Five microliters of the sample preparation are added to 15 μL of PCR master mix (TaqPath 1-Step RT-1PCR Master Mix, ThermoFisher Scientific) containing primers and probes for detection of SARS-CoV-2 N1 gene RNA and human ribonuclease P (RNase P) RNA (Integrated DNA Technologies). RT-PCR is performed on QuantStudio7 pro thermocyclers (ThermoFisher Scientific). A positive test is defined by N1 gene target amplification with a Ct value <40; higher levels of gene target yield lower Ct values. Human RNase P RNA is measured as an internal control for specimen adequacy and amplification efficiency. A test was considered valid if the RNase P gene target amplification was adequate with a Ct value <35.
SARS-CoV-2 sequencing supported a national initiative funded by the Centers for Disease Control and Prevention to expand surveillance for emerging variant strains. Saliva specimens that tested positive for the presence of SARS-CoV-2 were stored frozen at −20°C until they were processed for genomic sequencing. Nucleic acid was extracted from 400 μL of saliva using the MagMAX™ Viral/Pathogen II (MVP II) Nucleic Acid Isolation Kit (Applied Biosystems, A48383) on the KingFisher Flex instrument following the manufacturer’s protocol and resuspended in 55 μL of elution buffer. Both Illumina and Oxford Nanopore Technology (ONT) sequencing platforms were used for supply chain resiliency. RNA was reverse transcribed using the SuperScript™ IV First-Strand Synthesis System (ThermoFisher, 1,809,150). For the Illumina based sequencing, SARS-CoV-2 was amplified and converted to sequencing libraries with the xGen SARS-CoV-2 Amplicon Panel (IDT,10009827, 10,009,832, 10,009,845). Libraries were normalized to 4 nM using Normalase and loaded on a 2 × 150 MiniSeq High Output cartridge. Consensus genomes were generated using the TAYLOR pipeline with default settings (17, 18). For the ONT based sequencing, SARS-CoV-2 was amplified and libraries were prepared according to the nCoV-2019 sequencing protocol (GunIT) v2 developed by the ARTIC Network (19), and loaded onto a R9.4.1 MinION flow cell. Consensus genomes were generated using the ARTIC network pipeline with default settings (20). SARS-CoV-2 consensus genomes with at least 70% coverage were submitted to Nextclade (21) for variant identification.
2.5 Program performance monitoring and evaluation
Program performance was assessed using both process and outcome measures. The diagnostic testing process measures were RNase P Ct value results and turnaround time. The RNase P internal control result represents specimen adequacy. The turnaround time uses the time it takes from specimen collection to results reporting as a surrogate for consistency of the overall process. The outcome measures were sample rejection rate and invalid result rate. The sample rejection rate reflects the proportion of samples that were not able to be processed due to inadequate labeling, insufficient volume, container leakage, receipt in the lab >96 h from collection, or other issues that impact specimen integrity. The invalid result rate reflects the proportion of samples that yielded inconclusive or unreliable results. These measures reflect diagnostic yield returned to both participants and health systems. The process measure for SARS-CoV-2 genomic sequencing was the number of specimens which yielded sequence coverage greater than 70%. The outcome measure was the percentage of specimens that yielded a variant determination that was reportable to public health authorities. Performance measures are reported for two program settings.
N1 Ct values were assessed in two ways to evaluate the relationship between diagnostic testing results and subsequent referral to sequencing activities. First, they were rank-ordered by sensitivity to determine how many of the achieved variant calls would have been captured if the Ct value was used as a threshold for referral to sequencing (sensitivity rank score). Second, they were rank-ordered by efficiency to determine the fraction of referred specimens that would have yielded a variant determination if that Ct value was employed as the threshold for referral (efficiency rank score). For each Ct value threshold, the sensitivity rank score was then multiplied by the efficiency rank score to provide an overall rank score.
2.6 Demographic, test utilization, and statistical analyses
Demographic information and test utilization metrics were extracted from test registration and required public health reporting data elements in the NULirt information reporting tool. Comparative data for Douglas County, Nebraska (resident county of Omaha) was downloaded from the CDC COVID-19 Data Tracker (22).
Data management and descriptive analytics were conducted in Microsoft Excel ® and SAS Version 9.4®. Visualization of testing, positive results, and case counts was accomplished with GraphPad Prism 9.5.1.
3 Results
3.1 Utilization of saliva SARS-CoV-2 testing in occupation settings in Omaha, Nebraska
Demographic and individual-level test utilization metrics are provided in Table 1. Figure 1 shows the weekly counts of tests performed and positive test results for both program settings over the study period. At UNMC, the availability of unrestricted, voluntary testing four days per week resulted in a higher number of tests performed and more repeated testing among individual participants compared to participants in other occupational settings (Table 1). Test utilization rates at UNMC peaked during the community case surge related to the emergence of the Omicron variant (Figure 1A). Mandatory screening testing in workplace settings in Omaha also peaked during the surge in community cases related to the emergence of the Omicron variant (Figure 1B) while a sharp reduction in test utilization occurred as organizations de-escalated COVID-19 risk mitigation requirements. In general, as community transmission of COVID-19 increased, more individuals participated in saliva testing programs and a higher proportion of those individuals tested positive for the virus. At UNMC, women and those of Hispanic ethnicity appeared to have lower test positivity rates than men or those not self-identifying as Hispanic (Table 1).
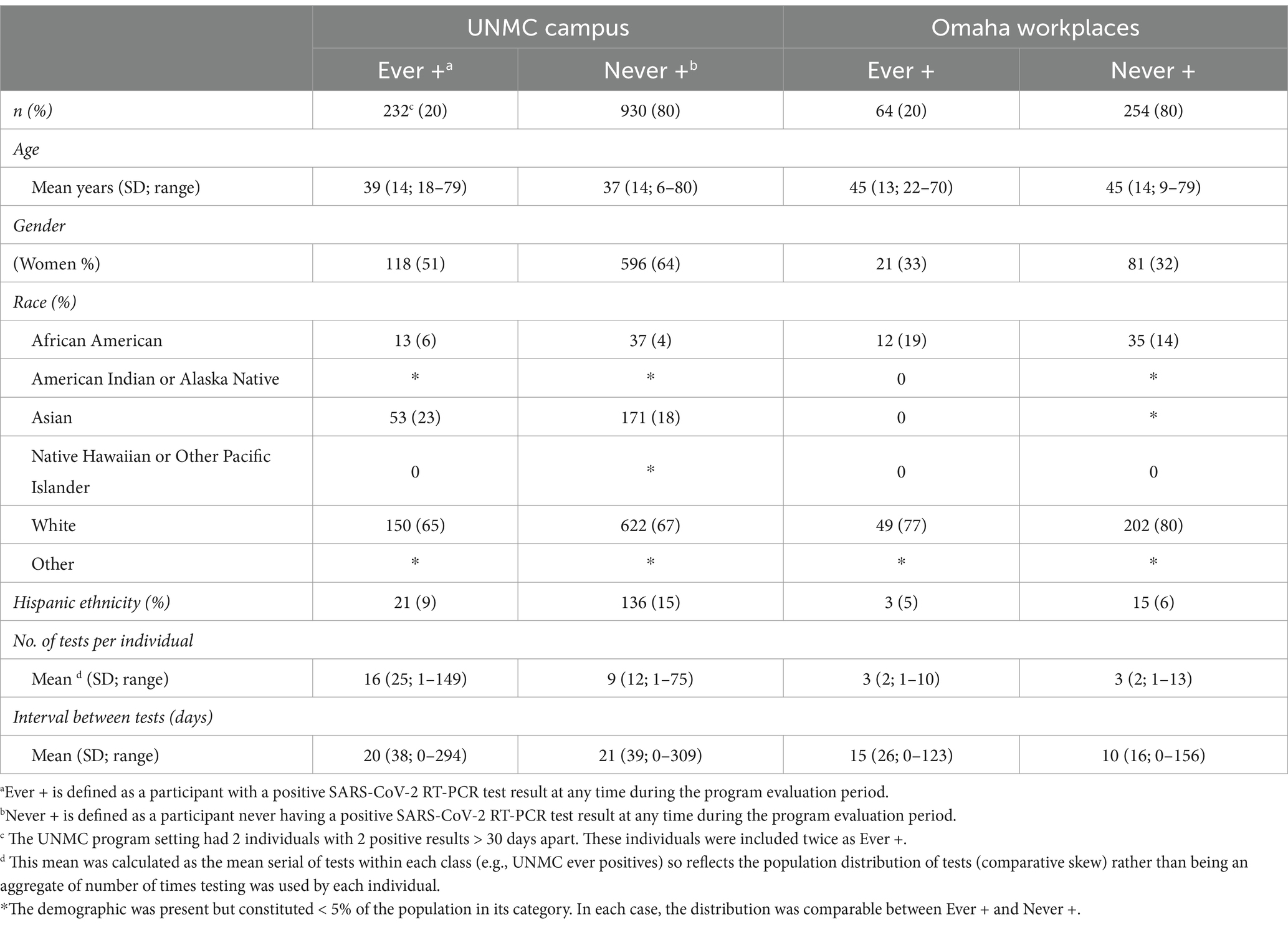
Table 1. Participant demographics and test utilization by program setting, December 2021–November 2022.
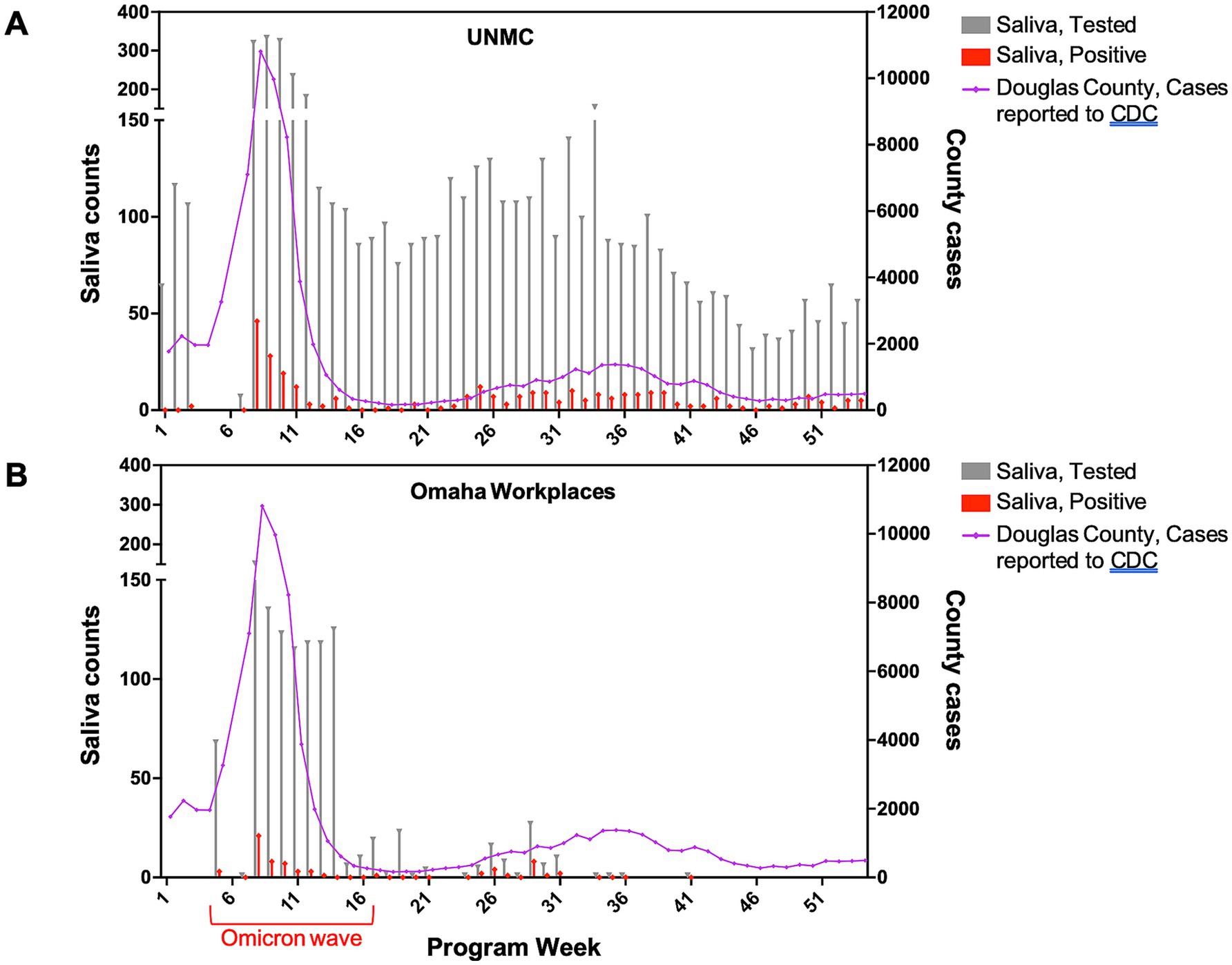
Figure 1. Weekly saliva tests performed and positive tests by program setting, December 2021–November 2022.
3.2 Quality metrics for saliva SARS-CoV-2 PCR testing and genomic sequencing
Self-collected saliva specimens demonstrated reliable quality for PCR testing in both program settings, despite variable collection venues, specimen transport times, and environmental conditions over four seasons. Low rates of specimen rejection (<0.3%) and invalid results (<0.2%) were observed (Table 2). The time from specimen collection to receipt in the laboratory varied between program settings (average 3.4 and 11.4 h for UNMC and Omaha workplaces, respectively; Table 2). However, Ct values for the RNase P internal control gene target were comparable for specimen sets from both program sites (average 22.0 and 21.7 for UNMC and Omaha workplaces, respectively; Table 2) indicating stability of RNA for PCR analysis.
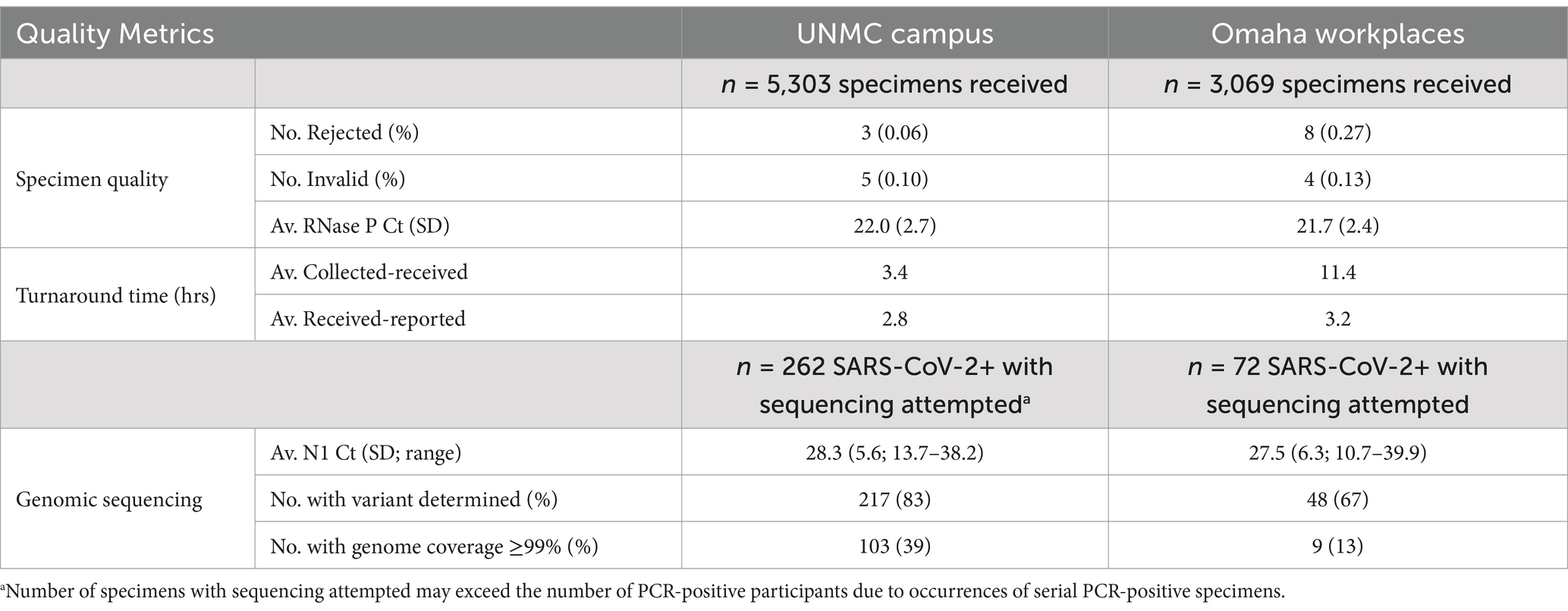
Table 2. Quality metrics for saliva SARS-CoV-2 PCR testing and genomic sequencing by program setting, December 2021–November 2022.
SARS-CoV-2 whole-genome sequencing was attempted for all PCR-positive saliva specimens when resources permitted. During a reagent shortage in January 2022 (coinciding with a surge in cases due to the emergence of the Omicron variant) specimens with N1 Ct values >32 were de-prioritized for sequencing referral, resulting in the exclusion of 8/342 (2.3%) PCR-positive specimens from sequencing analysis. N1 gene target Ct values in PCR-positive specimens referred for sequencing showed similar averages (28.3 and 27.5 for UNMC and Omaha workplaces, respectively; Table 2) and distributions (Supplementary Figure S1) in both program settings, suggesting comparable viral load experiences in the participant populations. SARS-CoV-2 variant determination (requiring at least 70% genomic sequence coverage) was achieved in 217/262 (83%) and 48/72 (67%) of PCR-positive saliva specimens collected from UNMC and Omaha workplaces, respectively (Table 2). Variants detected across program periods included B.1.617.2, B.1.1.529, BA.1 and sublineages, BA.2 and sublineages, BA. 4 and sublineages, and BA.5 and sublineages (Supplementary Figure S2). SARS-CoV-2 sequence coverage of ≥99% (i.e., adequate for phylogenetic analysis for outbreak investigation) was achieved in fewer specimens (Table 2).
3.3 Examination of optimal Ct value to trigger referral for sequencing analysis
The ability to generate adequate SARS-CoV-2 genomic sequence coverage for variant determination is dependent on the amount of virus in the specimen, which is inversely related to the Ct value of the virus PCR target. SARS-CoV-2 genome coverage by sequencing analysis showed a robust inverse correlation with N1 target Ct value (Figure 2A). In linear regression analyses of percent genome coverage, including negative natural logarithmic and logarithmic transforms, the relationship with N1 Ct value remained significant when adjusted for location (UNMC, Omaha Workplaces; p < 0.0001). Mantel–Haenszel chi square analysis showed a high likelihood of higher percent genome coverage at a N1 Ct value of less than 35 (p < 0.0001).
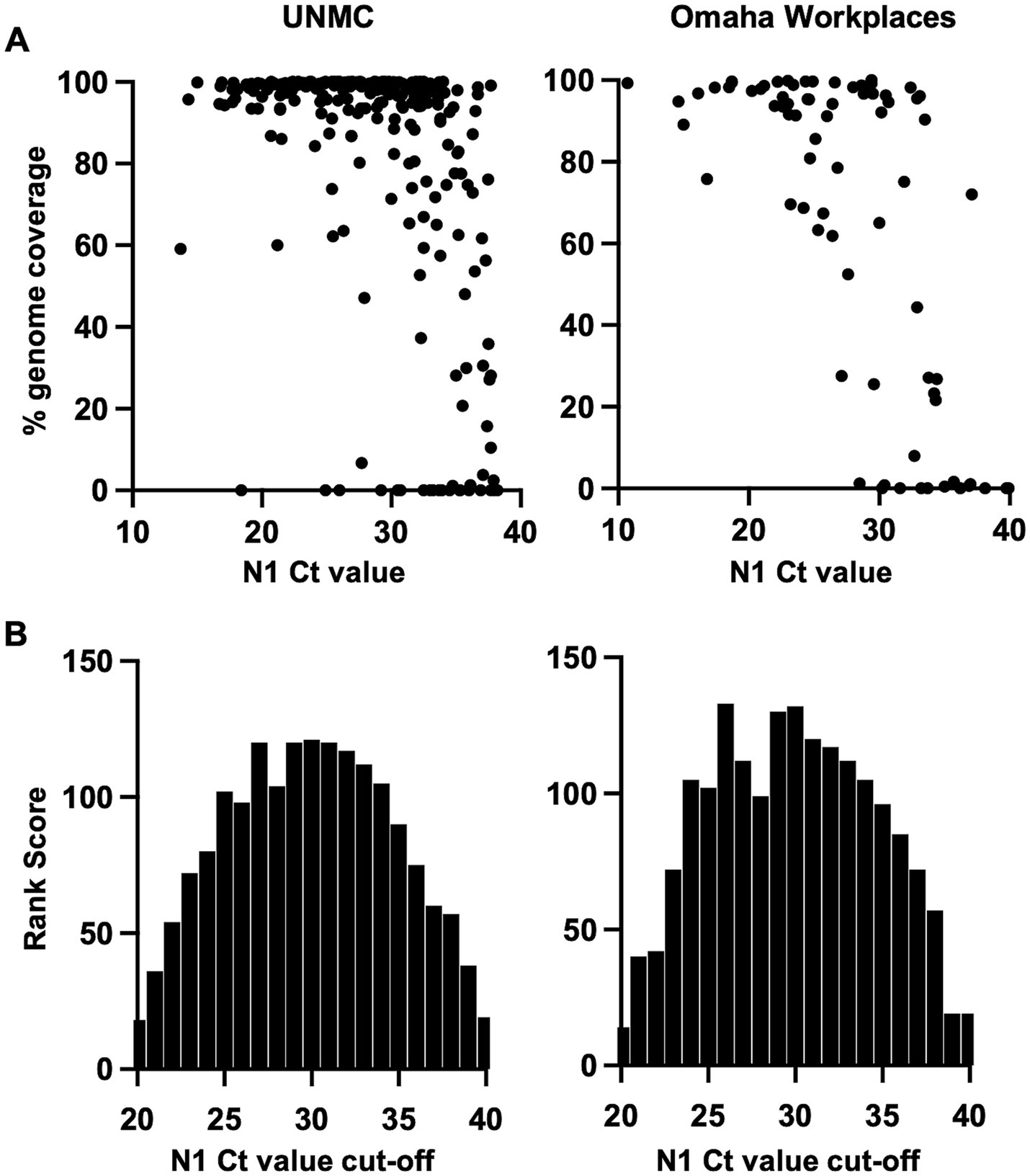
Figure 2. (A) SARS-CoV-2 genome coverage achieved by sequencing analysis across N1 cycle threshold (Ct) values of PCR-positive saliva specimens. (B) Analysis of optimal N1 Ct value cutoff for sequencing referral of PCR-positive saliva specimens based on the rank score for sensitivity and efficiency of SARS-CoV-2 variant determination.
As sequencing analysis is resource intensive, we sought to determine optimal N1 Ct value cutoffs for sequencing referral in our specimen sets. The approach of referring all PCR-positive specimens (i.e., N1 Ct <40) for sequencing yields the highest sensitivity for variant determination. However, the efficiency of sequencing is expected to improve if specimens with a low level of virus (i.e., high Ct value) are excluded. The sensitivity and efficiency of sequencing for SARS-CoV-2 variant determination was calculated across a range of positive N1 Ct value cut-offs from 20 to 40, and rank scores were applied (Supplementary Table S1). The product of rank scores for sensitivity and efficiency (“Rank Score,” Supplementary Table S1) was plotted across N1 Ct cut-off values, demonstrating a similar distribution in both program settings with the highest scores occurring at Ct cut-off values of 26–33 (Figure 2B).
4 Discussion
Innovative and sustainable strategies are needed to support community-based SARS-CoV-2 genomic surveillance, particularly in populations with limited healthcare access (23–25). We demonstrate successful integration of SARS-CoV-2 sequencing for variant determination into screening programs in occupational settings using self-collected saliva with an extraction-free qRT-PCR testing method. This approach offers the advantages of a non-invasive specimen collection, no need for supervised collection by a healthcare worker, supply chain resiliency, distributable access, and scalability. High specimen quality across program settings demonstrated the suitability of self-collected saliva specimens for PCR and genomic surveillance testing.
Despite similar distributions of SARS-CoV-2 N1 gene target Ct values, positive specimens collected at the UNMC program site showed a higher rate of successful variant determination by genomic sequencing than those collected off campus and transported to the laboratory. We also observed a lower rate of obtaining ≥99% genome coverage (which is typically required for investigating transmission chains) in specimens collected off campus. This may reflect RNA degradation during transport that, while not impacting amplification of the PCR target, diminishes whole-genome sequencing coverage. Biases inherent in self-selection for testing and readily available testing may have contributed to this finding (e.g., self-perceived risk and testing early in the course of infection). Work is ongoing to evaluate sources of variation in sequence recovery between program settings. Strategies to mitigate RNA degradation during specimen transport include the use of RNA preservative reagents and maintenance of cold chain. However, these strategies add cost and complexity and must be carefully evaluated to determine the added value to testing program quality.
To the best of our knowledge, we present the first generalizable approach to optimize specimen referral for variant determination by sequencing analysis. Applying rank scores to a range of virus gene target Ct value cut-offs for sensitivity and efficiency of variant determination can be used by laboratories to assess their processes employing local data. A universal Ct value cutoff cannot be applied, as the sensitivity of variant determination is dependent on sequencing method and RNA integrity, and efficiency is dependent on viral load (Ct value) distribution in the testing population. During periods of low community transmission, prioritizing sensitivity for variant determination is advisable, whereas efficiency becomes most relevant during periods of high community transmission and resource scarcity.
Each testing program was unique with respect to the participating populations and recruitment design. Consequently, performance differences could be influenced by non-delineated factors. The participant population described in this study was predominantly white adults (majority women for the UNMC campus and majority men for the Omaha workplaces). Factors such as vaccination status, age, and immune states may impact viral load (and therefore Ct values); however, we did not collect vaccination status or store individual demographic or clinical data linked to Ct values. The bulk of observations were among Omicron-lineage SARS-CoV-2 viruses. Findings in programs using extraction-based PCR may differ, reinforcing the importance of pursuing such approaches with local data. Future work will include (1) the application of saliva-based genomic surveillance to additional SARS-CoV-2 variants and other respiratory pathogens including seasonal and avian influenza viruses, (2) implementation of saliva-based genomic surveillance in additional community settings to assess the generalizability of our findings across diverse demographic groups, (3) exploration of pathogen and population characteristics that influence program quality metrics, and (3) optimization of specimen preservation for downstream molecular detection and genomic characterization.
Representation of underserved communities in genomic surveillance of respiratory pathogens can be improved through increased community access to PCR-based testing. Improving the effectiveness and sustainability of sequencing-based variant monitoring relies upon optimizing laboratory-anchored surveillance programming and specimen referral patterns. The utilization of self-collected saliva for respiratory pathogen surveillance and genomic monitoring may facilitate broader community engagement and representation, supporting public health policy and risk communication driven by locally relevant data. This approach may also be implemented for focused surveillance of high-risk groups, such as individuals with occupational exposures to emerging respiratory pathogen threats including avian influenza viruses.
Saliva has been successfully utilized in SARS-CoV-2 screening and surveillance programs in healthcare settings, college campuses, and schools (6, 8–11). Our study further demonstrates successful implementation of saliva-based testing and genomic surveillance in occupational settings during periods of high and low community case rates. However, community-based testing programs using self-collected saliva specimens may face challenges spanning quality assurance, information technology including test registration and reporting, community acceptability, privacy, regulatory compliance, and cost. Additional studies performed in diverse settings and populations should be pursued to further establish successful strategies to meet these challenges. As highlighted by our study, defining quality metrics to track key program outputs is critical for iterative program optimization informed by local data.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements.
Author contributions
AS: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Validation, Writing – original draft, Writing – review & editing. DB-M: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. JW: Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – review & editing. BB: Data curation, Formal analysis, Investigation, Methodology, Project administration, Supervision, Writing – review & editing. AP: Formal analysis, Investigation, Methodology, Writing – review & editing. MW: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing. MB: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by a CDC award under BAA 75D301-21-R-71738, intramural funds, and contract reimbursement for testing services.
Acknowledgments
We are grateful for training and specimen collection support provided by Tori Winter, Kathleen Angell, Julia Bai, Olivia Brett-Major, Sara Donovan, Lauren Longacre, and Louis Walker.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpubh.2025.1360862/full#supplementary-material
References
1. van Dorp, L, Acman, M, Richard, D, Shaw, LP, Ford, CE, Ormond, L, et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. J Mol Epidemiol Evol Genet Infect Dis. (2020) 83:104351. doi: 10.1016/j.meegid
2. Goswami, C, Sheldon, M, Bixby, C, Keddache, M, Bogdanowicz, A, Wang, Y, et al. Identification of SARS-CoV-2 variants using viral sequencing for the Centers for Disease Control and Prevention genomic surveillance program. BMC Infect Dis. (2022) 22:404. doi: 10.1186/s12879-022-07374-7
3. Worobey, M, Pekar, J, Larsen, BB, Nelson, MI, Hill, V, Joy, JB, et al. The emergence of SARS-CoV-2 in Europe and North America. Science. (2020) 370:564–70. doi: 10.1126/science.abc8169
4. Genomic sequencing of SARS-CoV-2: a guide to implementation for maximum impact on public health. (2021). Available online at: https://www.who.int/publications-detail-redirect/9789240018440 (Accessed March 28, 2023).
5. Gangavarapu, K, Latif, AA, Mullen, JL, Alkuzweny, M, Hufbauer, E, Tsueng, G, et al. Outbreak.info genomic reports: scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Nat Methods. (2023) 20:512–22. doi: 10.1038/s41592-023-01769-3
6. Wyllie, AL, Fournier, J, Casanovas-Massana, A, Campbell, M, Tokuyama, M, Vijayakumar, P, et al. Saliva or nasopharyngeal swab specimens for detection of SARS-CoV-2. N Engl J Med. (2020) 383:1283–6. doi: 10.1056/NEJMc2016359
7. Rayack, EJ, Askari, HM, Zirinsky, E, Lapidus, S, Sheikha, H, Peno, C, et al. Routine saliva testing for SARS-CoV-2 in children: methods for partnering with community childcare centers. Front Public Health. (2023) 11:1003158. doi: 10.3389/fpubh.2023.1003158
8. Bordi, L, Sberna, G, Lalle, E, Fabeni, L, Mazzotta, V, Lanini, S, et al. Comparison of SARS-CoV-2 detection in nasopharyngeal swab and saliva samples from patients infected with omicron variant. Int J Mol Sci. (2023) 24:4847. doi: 10.3390/ijms24054847
9. Chopoorian, A, Banada, P, Reiss, R, Elson, D, Desind, S, Park, C, et al. Persistence of SARS-CoV-2 in saliva: implications for late-stage diagnosis and infectious duration. PLoS One. (2023) 18:e0282708. doi: 10.1371/journal.pone.0282708
10. Alpert, T, Vogels, CBF, Breban, MI, Petrone, ME, Wyllie, AL, Grubaugh, ND, et al. Sequencing SARS-CoV-2 genomes from saliva. Virus Evol. (2022) 8:veab098. doi: 10.1093/ve/veab098
11. Crowe, J, Schnaubelt, AT, SchmidtBonne, S, Angell, K, Bai, J, Eske, T, et al. Assessment of a program for SARS-CoV-2 screening and environmental monitoring in an urban public School District. JAMA Netw Open. (2021) 4:e2126447. doi: 10.1001/jamanetworkopen.2021.26447
12. Campbell, WS, Donahue, M, Williams, RM, McCutchen, E, Broadhurst, J, Schnaubelt, A, et al. A public health laboratory information system in support of health emergencies: the Nebraska public health laboratory experience. Public Health Rep. (2023) 138:602–9. doi: 10.1177/00333549231168459
13. Ceron, JJ, Lamy, E, Martinez-Subiela, S, Lopez-Jornet, P, Capela-Silva, F, Eckersall, P, et al. Use of saliva for diagnosis and monitoring the SARS-CoV-2: a general perspective. J Clin Med. (2020) 9:1491. doi: 10.3390/jcm9051491
14. Policy for Coronavirus Disease-2019 Tests. (2023). Food and Drug Administration. Available online at: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-coronavirus-disease-2019-tests-revised (Accessed November 15, 2021).
15. Vogels, C, Brackney, DE, Chaney, CK, Ott, IM, Grubaugh, N, and Wyllie, A. SalivaDirect: RNA extraction-free SARS-CoV-2 diagnostics V.5. (2020). Available online at: https://www.protocols.io/view/salivadirect-rna-extraction-free-sars-cov-2-diagno-bkjgkujw?version_warning=no (Accessed September 1, 2020).
16. Vogels, CBF, Watkins, AE, Harden, CA, Brackney, DE, Shafer, J, Wang, J, et al. Yale IMPACT research team. SalivaDirect: a simplified and flexible platform to enhance SARS-CoV-2 testing capacity. Med. (2021) 2:e6:263–80. doi: 10.1016/j.medj.2020.12.010
17. Addetia, A, Lin, MJ, Peddu, V, Roychoudhury, P, Jerome, KR, and Greninger, AL. Sensitive recovery of complete SARS-CoV-2 genomes from clinical samples by use of swift biosciences’ SARS-CoV-2 multiplex amplicon sequencing panel. J Clin Microbiol. (2020) 59:e02226–20. doi: 10.1128/jcm.02226-20
18. Greninger Lab Covid Swift Pipepine. (2024). Available online at: https://github.com/greninger-lab/covid_swift_pipeline (Accessed January 15, 2023).
19. nCoV-2019 sequencing protocol v2 (GunIT) V.2. protocols.io. (2021). Available online at: https://www.protocols.io/view/ncov-2019-sequencing-protocol-v2-bp2l6n26rgqe/v2 (Accessed October 1, 2021).
20. nCoV-2019 Novel coronavirus bioinformatics protocol. ARTIC Network. (2020). Available online at: https://artic.network/ncov-2019/ncov2019-bioinformatics-sop.html (Accessed October 1, 2021).
21. Nextclade. Nextstrain. (2024). Available online at: https://clades.nextstrain.org (Accessed October 1, 2021).
22. COVID Data Tracker. Centers for Disease Control and Prevention. (2023). Available online at: https://covid.cdc.gov/covid-data-tracker (Accessed February 26, 2023).
23. Ling-Hu, T, Rios-Guzman, E, Lorenzo-Redondo, R, Ozer, EA, and Hultquist, JF. Challenges and opportunities for global genomic surveillance strategies in the COVID-19 era. Viruses. (2022) 14:2532. doi: 10.3390/v14112532
24. New WHO 10-year strategy aims to scale up genomic surveillance. Pan American health organization. (2022). Available online at: https://www.paho.org/en/news/31-3-2022-new-who-10-year-strategy-aims-scale-genomic-surveillance (Accessed April 3, 2023).
Keywords: SARS-CoV-2, COVID-19, saliva, genome sequencing, variant, surveillance
Citation: Schnaubelt AT, Brett-Major DM, Williamson J, Barcal B, Carstens J, Peer A, Wiley M and Broadhurst MJ (2025) SARS-CoV-2 genomic surveillance using self-collected saliva specimens during occupational testing programs. Front. Public Health. 13:1360862. doi: 10.3389/fpubh.2025.1360862
Edited by:
Deepak Y. Patil, National Institute of Virology (ICMR), IndiaReviewed by:
Sirajudheen Anwar, University of Hail, Saudi ArabiaEvenilton Pessoa Costa, Federal University of Rio de Janeiro, Brazil
Ritthideach Yorsaeng, Chulalongkorn University, Thailand
Copyright © 2025 Schnaubelt, Brett-Major, Williamson, Barcal, Carstens, Peer, Wiley and Broadhurst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Jana Broadhurst, amFuYS5icm9hZGh1cnN0QHVubWMuZWR1
†These authors have contributed equally to this work and share first authorship