 Aiwen Zhao
Aiwen Zhao Yuntao Lu
Yuntao Lu Qi Li
Qi Li Tao Li
Tao Li Jindong Zhao
Jindong Zhao- 1School of Science, Tibet University, Lhasa, China
- 2The State Key Laboratory of Freshwater Ecology, Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, China
- 3State Key Laboratory of Protein and Plant Genetic Engineering, College of Life Sciences, Peking University, Beijing, China
As the most important component of ecosystems, microbial communities play a significant role in global biogeochemical cycles. Geographical barriers created by topographic differences are proposed as one of the main factors to shape microbial diversity, functional composition and their evolution across aquatic ecosystem. There are few studies that compare the microbial community structure and functional potential of lakes with different terrain ladders (terrains of contrasting elevation levels), especially those involving Qinghai-Tibet Plateau. This study compared microbial 16S rRNA data from 51 lakes with different terrain ladders, showing that the composition of the dominant microbial community was similar, but the microbial abundance was quite different in different terrain ladders. Actinobacteria, Proteobacteria, Cyanobacteria, Planctomycete, Verrucomicrobia and Bacteroidetes were the dominant taxa. Through the correlation analysis between environmental factors and the microbial community structure, it was found that two environmental factors (elevation and salinity) had significant contributions to the microbial composition. Metagenomics of six representative lakes revealed the relationship between microbial composition and metabolic processes related to carbon, nitrogen and sulfur cycles. The comparative analysis of genes related to metabolism cycles showed that functional processes such as aerobic respiration, nitrogen assimilation, the mineralization of nitrogen and sulfur had highest metabolic potential in the ecosystems. The analysis of indicator species based on the metabolic process in the carbon, nitrogen and sulfur cycles showed that the microbial community structure is related to the dominant metabolic functions within biogeochemical cycles. Different dominant species play an important and distinct role in lakes with different gradients. Actinobacteria, Cyanobacteria and Proteobacteria were the most important indicator species on the Qinghai-Tibet Plateau, Yunnan-Guizhou Plateau and the middle and lower reaches of the Yangtze River Plain, respectively. In general, this study explored the functional distribution patterns of the dominant microbial communities in the lakes in biogeocycles.
Introduction
Carbon, nitrogen and sulfur in biosphere are the basic components of living organisms and important nutrient limiting factors (Tran et al., 2021), which are exchanged between water, atmosphere and soil in different forms. Hydrosphere, as the bond connecting diverse ecosystems, is habitat to more than half of the planet’s microbial populations that are important participants in the material cycle and energy flow in ecosystems (Williamson et al., 2009). For example, Water microbes help to slow down the greenhouse effect by fixing atmospheric carbon dioxide and releasing dimethyl sulfide (DMS) (Ding et al., 2021). The transformation of nitrogen by water microbes will alter carbon dynamics and biodiversity (Kuypers et al., 2018). The surface water body lies between the atmosphere and the sediment layer. It is an important scientific problem to explore how microbial community composition and functional diversity affect biogeochemical cycle.
With the iterative development of high-throughput sequencing technology, metagenomics technology can analyze lake microbial diversity and functional metabolism without isolating and culturing microbes. In recent years, many studies have focused on exploring the impact of environmental factors on the composition of microbial community (Yang et al., 2019; Ji et al., 2022; Shang et al., 2022; Zha et al., 2022). Salinity, pH, and geographic isolation are important factors of lake microbial distribution worldwide (Filker et al., 2016; Glasl et al., 2018). Lakes with different terrain ladders (terrains of contrasting elevation levels) have special living environments and abundant microbial taxa, which play diverse roles in energy metabolism and material circulation (Eiler et al., 2012; Xing et al., 2020; Tran et al., 2021). Many studies have shown that different microbial communities can complement each other in the biogeochemical cycle (Rathour et al., 2020; Liu et al., 2021). However, there are few studies on the lateral comparison of the microbial community structure among lakes in different ladders and their roles in biogeochemical cycles using functional metagenomics.
In order to figure out the microbial community of lakes with different topographical ladders and their role in biogeochemical cycles, this study took the three terrains of contrasting elevation levels formed on the eastern side of the Himalayas as the research object. The continuous collision of the Eurasian plate and the Indian plate formed the Qinghai-Tibet Plateau and the Himalayas, and this process of uplift has continued from 50 million years ago to the present (Tapponnier et al., 2001). From the east side of the Himalayas to the Pacific Ocean, there is an obvious three-level terrain ladder that located in different watersheds and climate zones. Therefore, it is of great significance to understand the relationship between microbial community diversity and metabolic potential in typical lakes according to terrain differences. Based on the metagenomics technology, this study conducted a horizontal comparative analysis on the surface water microbial community composition of 51 lakes in three terrain ladders, and focused on six typical lakes to deeply analyze the relationship between microbial composition and functional diversity. This study is the first to use metagenomic sequencing technology to reveal the microbial characteristics of the Ngari region on the Qinghai-Tibet Plateau. By comparing the microbes among lakes in different ladders, this study tries to answer two questions: 1) how environmental factors drive the difference of microbial composition in surface water among lakes; 2) how the microbial community structure is related to the dominant metabolic functions within biogeochemical cycles.
Materials and methods
Sample collection
According to the difference of the terrain ladder, the Qinghai-Tibet Plateau lakes of the first terrain ladder are labeled as TL1 (the elevation is over 4,000 m), the Yunnan-Guizhou Plateau lakes of the second terrain ladder are labeled as TL2 (the elevation is between 1,000 and 2000 m), and the lakes of the middle and lower reaches of the Yangtze River plain of the third terrain ladder are labeled as TL3 (the elevation is under 500 m).
The 16S rRNA gene of surface water of 51 lakes (16 in TL1, 11 in TL2, 24 in TL3, Figure 1) in three ladders was downloaded from National Center for Biotechnology Information (NCBI) according to relevant research (Hong et al., 2011; Liao et at., 2016; Liu et al., 2019; Bai et al., 2020). 51 lakes collected in the literature were sampled in July and September of 2017 and the surface water (top 50 cm) was collected at each lake. The filter membrane is made of polycarbonate with the pore size of 0.7 um (TL1) and 0.22 um (TL2 and TL3). This study collected and unified the geographical data (Supplementary Table S1) and the physicochemical variables of these lakes. The environmental factors used for subsequent analysis include temperature (Temp), salinity (Salinity), pH, dissolved oxygen (DO), total nitrogen (TN), total phosphorus (TP), and chlorophyll a content (chl a) (Supplementary Table S2).
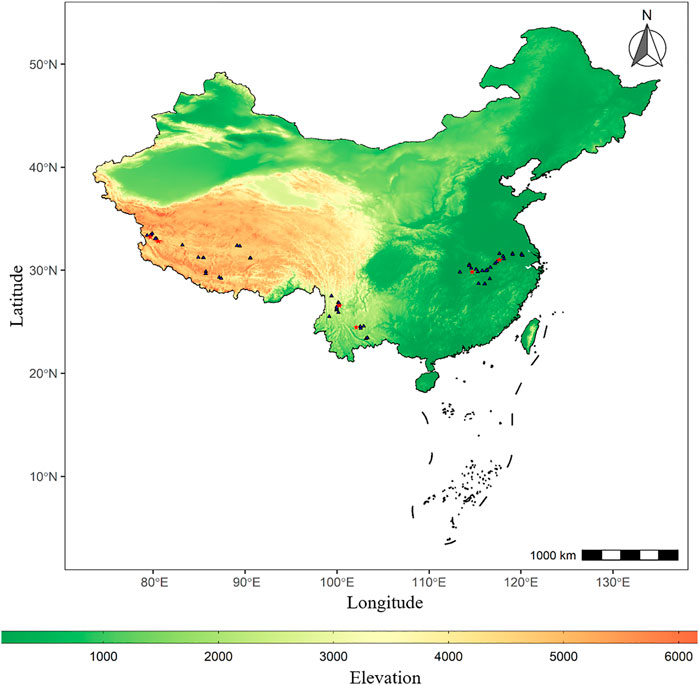
FIGURE 1. Distribution map of lake sites. The points (triangles and five-pointed stars) marked in the map represent the sites distribution of 51 lakes with 16S rRNA gene downloaded from National Center for Biotechnology Information (NCBI), among which the five-pointed stars (red) represent the site distribution of six typical lakes sampled.
In this study, 6 typical lakes from 51 lakes were selected to collect surface water samples. In September 2019, surface water samples from two lakes [Mandong Co (MD) and Kunzhong Co (KZ)] on Qinghai-Tibet Plateau and two lakes (Erhai (EH) and Fuxian Lake (FX)) on Yunnan-Guizhou Plateau were collected (Shen et al., 2019). In October 2019, surface water samples from two lakes (Bohu Lake (BH) and Daye Lake (DY)) in the middle and lower reaches of the Yangtze River were collected (Figure 1). For each lake, the seven sampling sites were in an S-shaped, equidistant distribution from the center of the lake. The surface water (top 50 cm) was collected at each sampling site. Water samples (2L) were 1) collected using disposable sterile bags; 2) mixed in a 20L sterile bucket; 3) vacuum filtered through polycarbonate membranes with pore sizes of 120 um and 0.22 um in turn; 4) put into sterile cryopreservation tubes and stored in liquid nitrogen; 5) transported back to the laboratory and stored in the laboratory at −80°C.
DNA extraction, sequencing and preprocessing
The DNA of total community was extracted from the sample according to the method of Xie (Xie et al., 2016). The three groups of lake samples were applied in the same method for DNA extraction. The metagenomic sequencing library was performed on the Illumina Genome IIx sequencing platform. More than 30G of raw data were generated from each library and preprocessed by FastQC software (v0.11.8, Brown et al., 2017) and Trimmomatic (v0.39, Bolger et al., 2014) for quality control.
Processing of sequence data
QIIME2 (v2022.8, Bolyen et al., 2019) was used to annotate the 16S amplicon data of 51 lakes: 1) Use the summarize plug-in to view the location distribution of low quality base and set parameters for filtering and reduced noise with Deblur algorithm; 2) Load and train the taxonomy classifier Greengene 13_8; 3) Perform species annotation and visualization.
The metagenomic data analysis was done as follows: 1) megahit (v1.2.9, Li et al., 2015) was used to splice reads into contigs of appropriate length; 2) Prodigal (v2.6.3, Hyatt et al., 2010) was used for gene prediction, and kaiju (v1.9.0, Menzel et al., 2016) was used for classification and statistics; 3) The functional analysis based on the prokaryotic microbial protein coding regions (CDSs) obtained by splicing; 4) Large (>2 kilobases) contig were clustered into metagenome-assembled genomes (MAGs) and read coverage using Metabat2 (v2.12.1, Kang et al., 2019;Kitzinger et al., 2019). Redundant bins were subsequently dereplicated and evaluated using CheckM(v1.1.3, Parks et al., 2014) with a completeness cutoff of 90%, contamination cutoff of 10% and minimum genome size of 900 kilobases; 5) CAT (v5.0.3, Pontius et al., 2007) was used for classification annotation; 6) emmapper (v2.0.0, Federhen, 2012) was used for functional annotation; 7) constructing the distance matrix based on assembly genome sequence information obtained from binning and CVTree (v3.9.6, Qi et al., 2004) to construct a genome-wide vector phylogenetic tree based on high-quality binning results.
Calculation of community metabolic potential
The analysis of community metabolic potential focused on the processes of the biogeochemical cycles of carbon, nitrogen and sulfur in lakes at three different ladders. In order to infer the metabolic potential of each lake, this study used to the metabolic marker genes (KO) and formulas summarized in relevant studies (Lanuro et al., 2011; Llorens-Mares et al., 2015; Kieft et al., 2018) for calculation. Transcripts per million (TPM) is usually used for the normalization of gene expression in transcriptomes, and it is also applicable to multidimensional data in metagenomics. This study used the TPM values of marker genes annotated in lake samples to calculate the metabolic potential of the microbial communities. The metabolic potential of the whole metabolic process is obtained by averaging the TPM values of genes with the same function and adding the TPM values of genes with other functions belonging to the same process. A total of 42 marker genes were used in the study, and the specific formulas and values are shown in Supplementary Tables S3, S4.
Statistical analysis
Based on Bray-Curtis similarity distance matrix, non-metric multidimensional scaling (NMDS) and analysis of similarity (ANOSIM) were used to compare and analyze the molecular data of microbial community structure of 51 lakes at different terrain ladders with R (v 4.2.2) vegan package. NMDS analysis was used to describe the difference of microbial community composition of 51 lake samples; ANOSIM analysis was used to test whether the difference between the three groups was significantly greater than the difference within the group. The distance of rank was used to determine whether differences exist between different groups, which was obtained with the equation:
Indicator species analysis was used for 51 lakes based on species abundance and functional abundance to calculate the indicator value based on the dplyr package. The indicator value quantifies the fidelity and specificity of species in relation to groups of sites in a user-specified classification of sites, and tests for the statistical significance of the associations by permutation. The indicator value of species i for group j was obtained with the equation:
Aij is specificity, i.e., the proportion of the individuals of species i that are in group j; Bij is fidelity, i.e., the proportion of sites in group j that contain species i.
The correlation between environmental factors was tested by spearman correlation analysis, and the correlation between environmental factors and microbial community was tested by Mantel test. The significance level was taken as p < 0.05.
Results
Comparison of microbial diversity in surface water with different ladder lakes
Based on the 16S rRNA gene information of 51 lakes, NMDS analysis and ANOSIM analysis were performed on the microbial composition of the lakes. The results showed that there were significant differences in the microbial composition among the terrain ladders (R = 0.927, p = 0.001, Figure 2B), while the microbial composition of lakes with the same ladder was similar (stress = 0.092, Figure 2A). The analysis results based on the indicator species (Figure 2C) showed that Verrucomicrobia was the indicator species that caused the difference between TL1 and the other two groups; Planctomycota was the indicator species that caused the difference between TL2 and the other two groups. Proteobacteria and Bacteroidetes were the indicator species that caused the difference between plateau lakes (TL1 and TL2) and plain lakes (TL3). In general, there were different indicator species in the lakes with different ladders (Supplementary Table S5).

FIGURE 2. Microbial community diversity and its correlation with environmental drivers in surface water samples from 51 lakes. (A) Results of NMDS analysis of microbial composition based on Bray-Curtis similarities. The five-pointed star site represents six typical lakes selected for sampling (Mandong Co (MD), Kunzhong Co (KZ), Erhai (EH), Fuxian Lake (FX), Bohu Lake (BH) and Daye Lake (DY)). (B) Box plots of the ANOSIM results on lake microbial community composition in individual terrain groups and between the groups; Borders represent interquartile range (IQR), whiskers represent the 1.5 times IQR range within the upper and lower quartiles respectively, points represent outliers that exceed 1.5 times IQR. (C) Indicator species analysis was used to reveal the species that cause significant differences between groups. The bars show the indicator values of the phyla in the groups. (D) Correlation analysis of environmental factors and microbial composition. Spearman correlation coefficient was used to measure the correlation between environmental factors, the color gradient and box size represent the degree of the correlation, red indicates positive correlation, and blue indicates negative correlation. Mantel test was used to measure the correlation between community composition and various environmental factors. Line width corresponds to Mantel’s r-statistic for distance correlations, line color denotes statistical significance based on 9,999 permutations.
The physical and chemical data of the three groups of lakes were different. The salinity increased with the altitude gradient (TL1>TL2>TL3), the temperature and chlorophyll decreased with the altitude gradient (TL3>TL2>TL1), and the total nitrogen of plain lakes was higher than that of plateau lakes (TL1>TL3>TL2) (Supplementary Table S2). Based on Spearman correlation analysis (Figure 2D), elevation was significantly positively correlated with total phosphorus (TP) and salinity (p < 0.05); chlorophyll (chl a) and temperature (Temp) was significantly negatively correlated (p < 0.05). The Mantel test results showed that elevation and salinity were important drivers of microbial community structure composition (p < 0.01).
Microbial species analysis based on assembly genomes
Based on the significant differences in the microbial community composition of lakes among terrain ladders, two lakes in each group were selected for sampling. 146 metagenome-assembled genomes (MAGs) from sampling lakes were obtained based on metagenome data. Six dominant phyla were determined by calculating the abundance of each MAGs and constructing a phylogenetic tree [Proteobacteria (47), Actinobacteria (33), Plantomycetes (21), Bacteroidetes (15), Cyanobacteria (12) and Verrucomicrobia (11)], which accounted for more than 80% of MAGs (Supplementary Table S6; Figure 3A). Species indicative of differences in microbial composition in the 51 lakes (Figure 2C) were dominant in the six selected study lakes. The microbial composition of the six typical lakes selected to sample was representative among the 51 lakes.
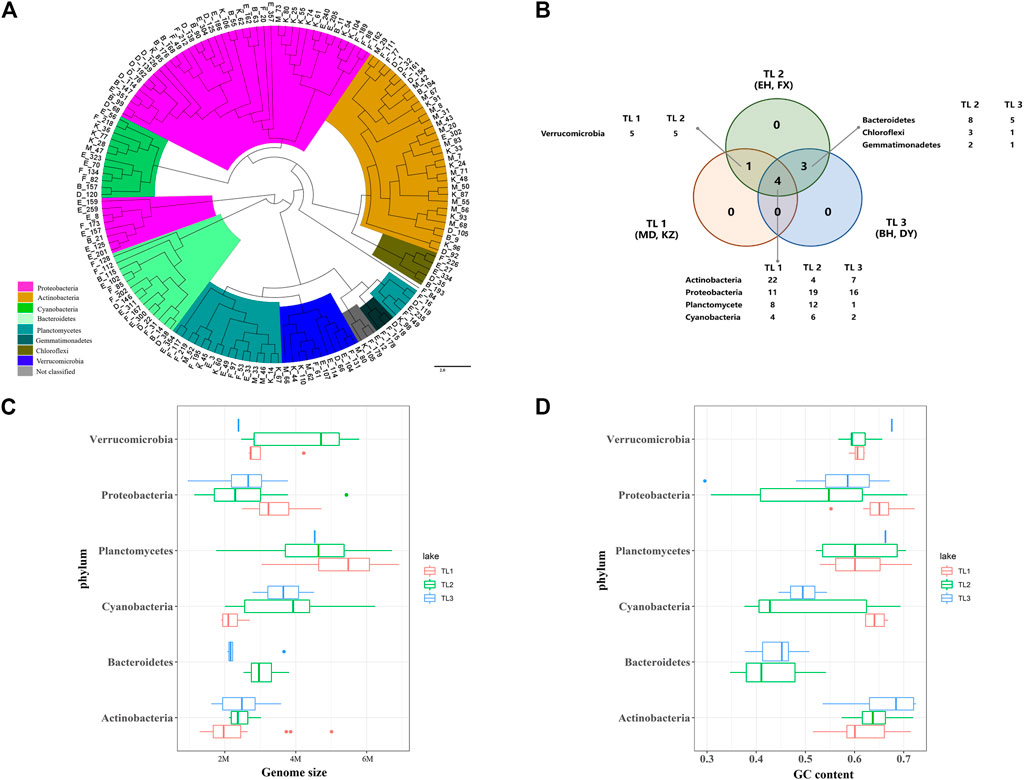
FIGURE 3. High-quality genomic characterization. (A) Whole-genome phylogenetic tree. Different colors represent different phyla. (B) Venn diagram of the distribution of species based on MAGs, the number after species is the count of MAG. (C) Grouping box diagram of the relationship between genome size and phylum. (D) Grouping box diagram of the relationship between GC content and phylum.
The distribution of dominant species showed that the four dominant species phyla were distributed in the three groups of lakes (Figure 3B), the species richness of TL2 was highest, and the species richness of TL1 was lowest. The genome size and GC content of dominant microbial groups in the six sample lakes were different (Figures 3C, D). Actinobacteria and Proteobacteria had relatively small genomes in the three groups. Plantomycetes had larger genome, which in TL1 (5.5 M) was larger than other groups (5 M). The genomic GC content was mostly distributed between 0.55 and 0.70. The evolutionary trend that microbes tend to have larger genomes and a higher GC content may be driven by energy and nutrients (Mende et al., 2017). But the Actinobacteria in this study had smaller genomes and a higher GC content. Microbes are often adapted to oligotrophic environments through metabolic lean strategies that reduce the GC content and maintain a smaller cell morphology (Haro-Moreno et al., 2018). The lake microbes in this study did not show an obvious metabolic reduction strategy, and the GC content of the dominant species was approximately 0.6.
Biogeochemical cycles driven by lake surface microbial communities
Based on the functional gene clustering of carbon, nitrogen and sulfur obtained from dominant microbes (Supplementary Figure S1), it was found that lakes of different ladders were clustered from a functional perspective. In this study, the TPM values of 42 marker genes (Lanuro et al., 2011; Llorens-Mares et al., 2015; Kieft et al., 2018) present in the lake samples were used to calculate the metabolic potential in the carbon, nitrogen and sulfur cycles, and converted to the relative metabolic potential of 100% in each cycle (Figure 4; Supplementary Tables S3, S4). Based on the analysis of indicator species, it was found that the functional distribution of microbes was significantly different in 12 metabolic processes (Figure 5; Table 1).
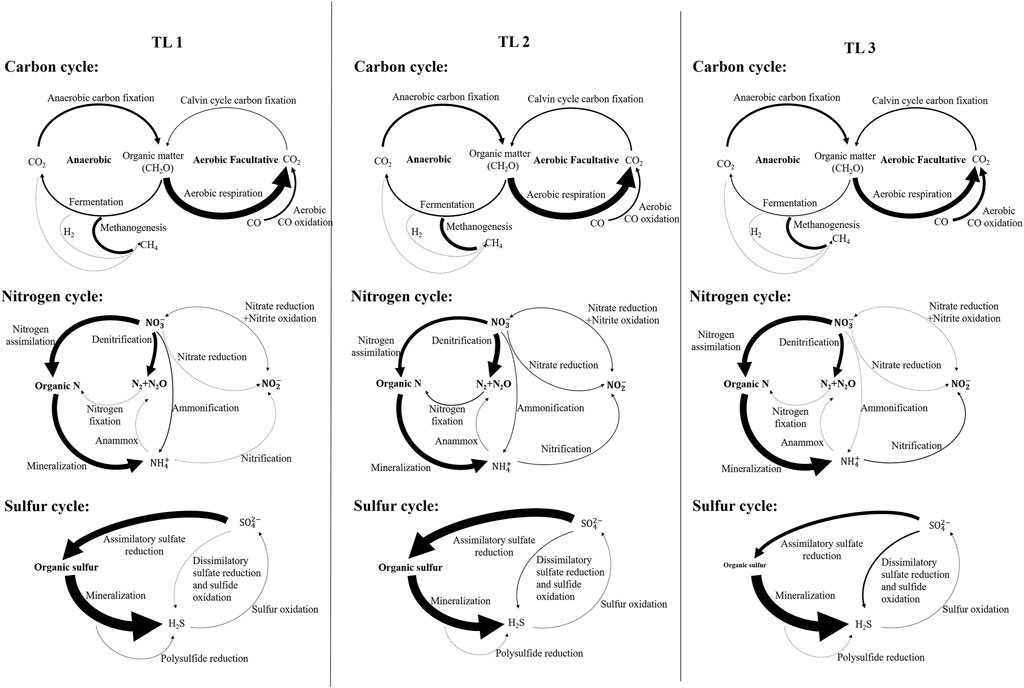
FIGURE 4. Schematic diagram of metabolic potential of lakes in each terrain ladder participating in carbon, nitrogen and sulfur cycles. Width of arrow is proportional to the relative metabolic potential of the pathways (100% values of each cycle, see Supplementary Table S4). Dotted lines indicate that marked genes were not detected.
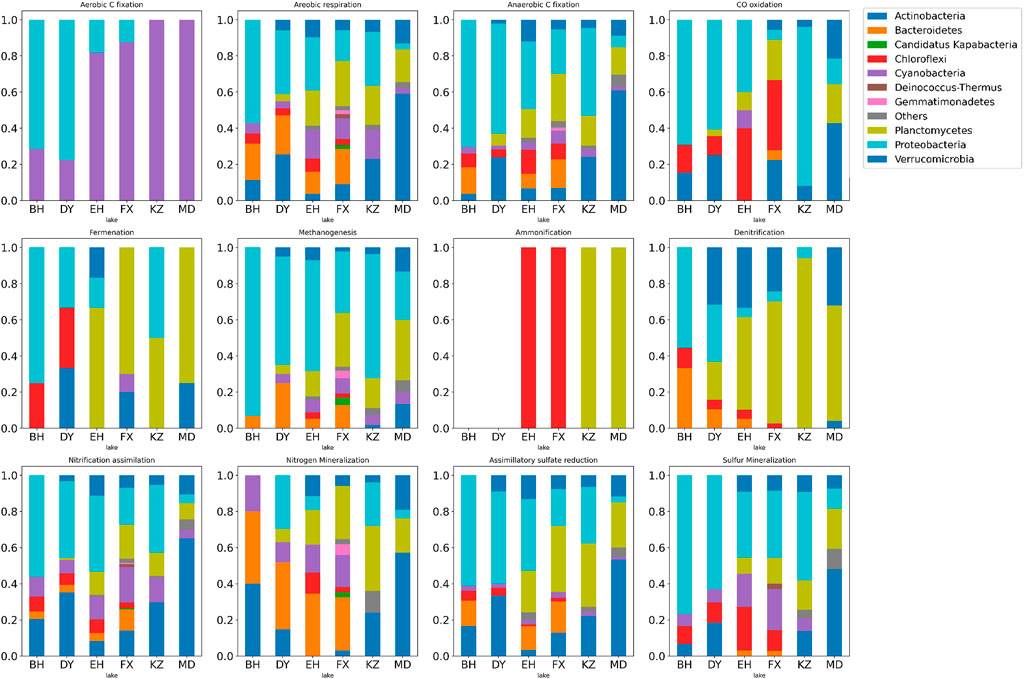
FIGURE 5. Stacked histogram of relative abundance of microbial functions in six typical lakes during 12 metabolic processes of nitrogen, and sulfur cycles. The horizontal axis represents each lake, the vertical axis represents the percentage of relative abundance of functions and the color represents phylum.
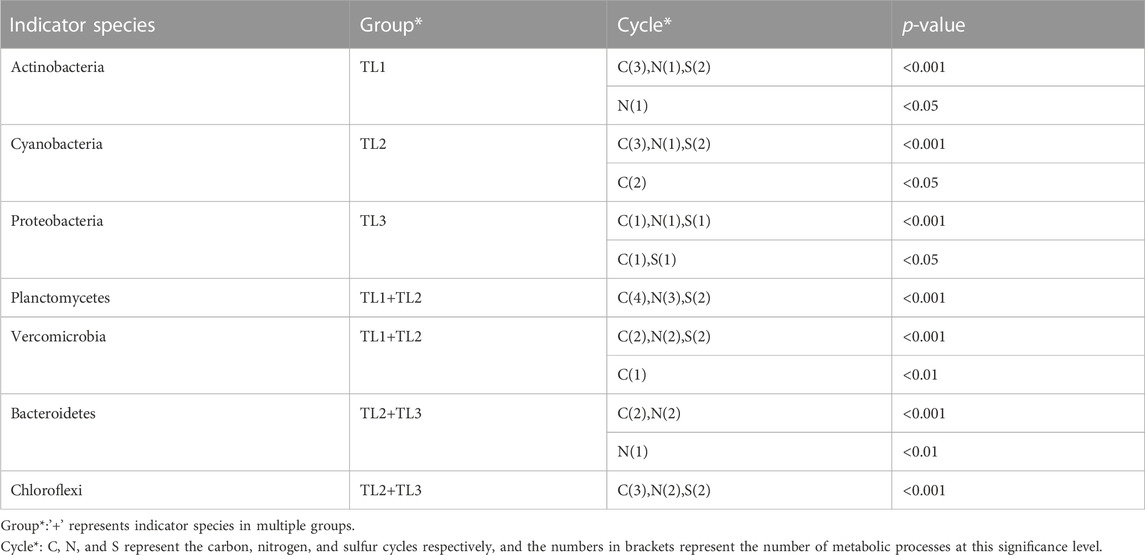
TABLE 1. Statistics of indicator species in metabolic processes.
The six dominant species in this study played important and distinct roles in multiple metabolic processes of the nitrogen and sulfur cycles in lakes with different ladders. Actinobacteria was the important indicator species that distinguish Qinghai-Tibet Plateau lakes from the other two groups. Cyanobacteria was the important indicator species that distinguish Yunnan-Guizhou Plateau lakes from the other two groups. Planctomycetes and Verrucomicrobia were important indicator species that distinguish plateau lakes from plain lakes. Proteobacteria was the important indicator species that distinguish plain lakes from plateau (especially Qinghai-Tibet Plateau) lakes. Functional genes of Chloroflexi and Bacteroidetes were rarely detected in lakes of the Qinghai-Tibet Plateau, but they were indicator species of multiple metabolic processes in lakes of Yunnan-Guizhou Plateau and plains.
For the carbon cycle, the aerobic respiration and fermentation metabolic potential of plateau lakes (TL1 and TL2) were higher than plain lakes (TL3). The fermentation process was mainly driven by Planctomycetes (p < 0.001) in plateau lakes and was jointly driven by Proteobacteria (p < 0.001) and Chloroflexi (p < 0.001) in plain lakes. The metabolic potential of CO oxidation and aerobic C fixation showed a gradient change (TL3>TL2>TL1). Chloroflexi (p < 0.001) in TL2 and TL3 played an important role in the CO oxidation process. The aerobic C fixation process was driven by Cyanobacteria (p < 0.001) in plateau lakes and by Proteobacteria (p < 0.001) in plain lakes. Overall, Proteobacteria played a significant role in carbon cycle in plain lakes; the carbon cycle process was driven by a variety of dominant microbes in plateau lakes.
For the nitrogen cycle, the processes of nitrogen assimilation and mineralization were driven by Actinobacteria (p < 0.001) in TL1 and Bacteroidetes (p < 0.001) in TL2 and TL3. In the process of denitrification, the plateau lakes were mainly driven by the Planctomycetes (p < 0.001), while the plain lakes were coordinated by Proteobacteria (p < 0.001) and Bacteroidetes (p < 0.001). Ammonification was absent in TL3 and was driven by Planctomycetes (p < 0.001) in TL1 and Chloroflexi (p < 0.001) in TL2. The relatively complete nitrogen cycle process was annotated in TL2, and the processes of nitrogen fixation, anammox and nitrate reduction were only found in TL2. There were relatively few nitrogen cycle processes annotated in TL3, but the metabolic potentials of nitrogen assimilation, mineralization and nitrification were higher than plateau lakes.
For the sulfur cycle, polysulfide reduction was a unique metabolic process in TL1 driven by Actinobacteria. The metabolic potential of the assimilatory sulfate reduction process in plateau lakes was significantly higher than that in plain lakes; the metabolic potential of dissimilatory sulfate reduction and sulfide oxidation in plain lakes is higher than plateau lakes. These two processes were mainly driven by Proteobacteria (p < 0.05) in the plain lakes, and Planctomycota and Verrucomicrobia (p < 0.001) in the plateau lakes.
Discusssion
Lake microbial community diversity response to environmental factors
Based on metagenomic sequencing technology, this study compared the differences in microbial community composition of 51 representative lakes at different terrain ladders. The composition of dominant microbial species in the 51 lakes of the three terrain ladders was similar. At the phylum level, the top six dominant phyla accounted for more than 60% of the lake microbial abundance. Researches have shown that the microbes in oligotrophic lakes on the northern Qinghai-Tibet Plateau are dominated by Actinobacteria and Verrucomicrobia (Liu et al., 2019; Gu et al., 2021), which was consistent with the results of this study using selected lakes on the Qinghai Tibet Plateau. The microbial composition of Erhai, Fuxian Lake, Bohu Lake, and Daye Lake also corroborates previous literature records (Hong et al., 2011; Bai et al., 2020). Global-scale studies of lake microbes have shown that there are biogeographical patterns of microbial distribution existed in lakes and microbial dispersal limitations can be used to explain this biogeographical pattern (Martitny et al., 2011). This point of view was mutually supported by the result that the microbial abundance in the surface water of lakes with different ladders is significantly different (Supplementary Figure S2). Furthermore, the three terrain ladders are located in different watersheds and climate zones, and the differences in water and air conditions caused by different watersheds and monsoons may be an important reason for the formation of microbial community diversity.
Based on the correlation analysis, the three environmental factors (elevation, salinity and chl a) had significant correlations to composition of microbial community (p < 0.01). For the lakes, the most direct changes brought about by elevation is temperature. Temperature changes have been shown to be an important factor in microbial changes (Glasl et al., 2018). Global-scale lake researches have shown that the microbial community composition in lake is significantly correlated with geographic distance, salinity and pH (Glasl et al., 2018). Microbes have specific adaptation strategies to the environmental stress of salinity (Oren, 2011), which is consistent with our finding that salinity is a significant factor affecting the composition of microbial communities and functional composition in this study.
In the oligotrophic environment, there are more species of microbes in lakes (Yao et al., 2017; Yanez-Montalvo et al., 2022). In this study, the lakes in Yunnan Guizhou Plateau are obviously the most abundant in microbial composition, which may be due to the oligotrophic condition and large water depth span providing a suitable living environment for microbes. The distribution of microbe in the middle and lower reaches of the Yangtze River may be concentrated due to human activities. Although the Qinghai Tibet Plateau has an oligotrophic environment, the harsh natural conditions in Ngari region make many microorganisms lose their suitable niches (Yao et al., 2017; Liu L. et al., 2021). The genome size and GC content of microbes should be affected by a variety of factors (Mende et al., 2017). It is speculated that under the stress of special environments, microbes in plateau lakes can adapt to the plateau environment by increasing the GC content to maintain a higher bond energy; plain lakes require more complex metabolic strategies due to long-term human disturbance (Mende et al., 2017; Haro-oreno et al., 2018).
Relationships between metabolic potential and dominant microbial species
Based on different dominant microbes acting as indicator species in multiple metabolic processes in the lakes of the three groups, it was speculated that microbial mediated biogeochemical cycles are unique in aquatic ecosystems with different topographical environments.
The CO oxidation process played an important role in the carbon cycle of different ladder lakes. CO in water mainly comes from the photolysis or oxidation of organic matter, and the intensification of water eutrophication will lead to the accelerated production of CO (Kieft et al., 2018), which verifies the view that plain lakes with higher trophic status have a relatively higher potential for CO oxidation metabolism. A reaearch (Llorens-Mares et al., 2015) has shown that Proteobacteria is the main driver of CO oxidation in aquatic ecosystems, which is consistent with the finding that Chloroflexi is the main driver of the CO oxidation process in the lakes of Yunnan-Guizhou Plateau in this study. As a special phylum of photosynthetic bacteria, Chloroflexi achieves CO2 fixation through the dual-cycle pathway of 3-hydroxypropionate. This study supports a view (Rao et al., 2022) that Chloroflexi can compensate for the lack of CO2 through the CO oxidation pathway.
Shen (Shen et al., 2019) had shown that Cyanobacteria use fermentation as an alternative means of producing energy under anoxic conditions in eutrophic lakes. The fermentation was mainly driven by the Planctomycetes in the plateau lakes and were jointly driven by the Proteobacteria and Chloroflexi in the plain lakes, which may because the microbes are relatively structurally stable and not affected by the anoxic conditions caused by algal blooms in relatively oligotrophication lakes. There is functional redundancy in the Calvin cycle that the microbes that drive the process gradually change from Cyanobacteria to Proteobacteria as the relative abundance of cyanobacteria decreased.
The metabolic potential of ammoniation in the lakes of Qinghai-Tibet Plateau is significantly higher than the lakes in other terrain groups, presumably due to the high salinity in the lakes of Qinghai-Tibet Plateau that promotes the transformation of nitrate into ammonium by Planctomycetes (Peeters et al., 2020). There is a complete nitrogen metabolism process in the lakes of Yunnan-Guizhou Plateau. This may be because the vast water area and deeper depth span provide microbes a larger and more complex living space (Shen et al., 2019; Xing et al., 2020), which makes microbes need a more complete functional system to adapt to the environment. In plain lakes with more frequent anthropogenic disturbance, there are few nitrogen metabolism pathways, but the metabolic potential of the existing pathways is high, which may reflect the different functions of the microbes. The two metabolic processes of assimilating sulfate reduction and sulfur mineralization in Mandong Co on the Qinghai-Tibet Plateau were driven by Actinobacteria, while those in other lakes were mostly driven by Proteobacteria. This may be because Mandong Co provides a unique living environment for Actinobacteria, which consolidates the dominant position of Actinobacteria and drives it to complete multiple metabolic processes. The potential of dissimilatory sulfate reduction and sulfide oxidation metabolism that mediated by Proteobacteria in plain lakes was significantly higher than that in plateau lakes, which may lead to the release of H2S from plain lakes (Arora-Williams et al., 2018).
The abundances of functional gene for CO oxidation, fermentation, and ammonification in Chloroflexi were significantly inconsistent with its relative community abundances in lakes (Rao et al., 2022). This finding demonstrates the important contribution of low-abundance bacteria to geochemical cycles, which also confirmed by the performance of Planctomycetes in the processes of fermentation, ammonification and denitrification (Peeters et al., 2020). The compositional abundance of microbial communities in different ladder lakes was significantly different but the differences of relative metabolic potential within lakes were similar, indicating that each lake has a unique structure of community taxonomic and functional redundancy formed internally.
Conclusion
In summary, the microbial communities within the same terrain ladder lakes have high homogeneity. The dispersion of mircobes on the large spatial scale has been limited by the geographical barrier caused by elevation and the physicochemical differences of water. This study complements other studies of microbial diversity and reveals that the microbial community structure is related to the dominant metabolic functions within biogeochemical cycles by metagenomic methods. In the future, more research will focus on the mechanism of microbes in the geochemical cycle and the interaction between microbes, habitats and human activities from a larger spatial scale.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: www.ncbi.nlm.nih.gov/sra/?term=PRJNA901345.
Author contributions
AZ and YL carried out data analysis and wrote the manuscript. JZ, TL, and QL supervised the project. All authors read and approved the final manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (Grant No. 92251304 and No. 31900096), and the National Key R&D Program of China (Grant No. 2018YFA0903100 and No. 2020YFA0907402).
Acknowledgments
The authors would like to thank the support from Yan Lin.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvs.2023.1121775/full#supplementary-material
References
Arora-Williams, K., Olesen, S. W., Scandella, B. P., Delwiche, K., Spencer, S. J., Myers, E. M., et al. (2018). Dynamics of microbial populations mediating biogeochemical cycling in a freshwater lake. Microbiome 6 (1), 165–216. doi:10.1186/s40168-018-0556-7
Bai, C., Cai, J., Zhou, L., Jiang, X., Hu, Y., Dai, J., et al. (2020). Geographic patterns of bacterioplankton among lakes of the middle and lower reaches of the Yangtze River Basin, China. Appl. Environ. Microbiol. 86 (6), 024233–e2519. doi:10.1128/aem.02423-19
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30 (15), 2114–2120. doi:10.1093/bioinformatics/btu170
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37 (8), 852–857. doi:10.1038/s41587-019-0209-9
Brown, J., Pirrung, M., and McCue, L. A. (2017). FQC dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33 (19), 3137–3139. doi:10.1093/bioinformatics/btx373
Ding, C., Wu, C., Guo, C., Gui, J., Wei, Y., and Sun, J. (2021). The composition and primary metabolic potential of microbial communities inhabiting the surface water in the equatorial Eastern Indian Ocean. Biology 10 (3), 248. doi:10.3390/biology10030248
Eiler, A., Heinrich, F., and Bertilsson, S. (2012). Coherent dynamics and association networks among lake bacterioplankton taxa. ISME J. 6 (2), 330–342. doi:10.1038/ismej.2011.113
Federhen, S. (2012). The NCBI taxonomy database. Nucleic Acids Res. 40 (D1), D136–D143. doi:10.1093/nar/gkr1178
Filker, S., Sommaruga, R., Vila, I., and Stoeck, T. (2016). Microbial eukaryote plankton communities of high-mountain lakes from three continents exhibit strong biogeographic patterns. Mol. Ecol. 25 (10), 2286–2301. doi:10.1111/mec.13633
Glasl, B., Smith, C. E., Bourne, D. G., and Webster, N. S. (2018). Exploring the diversity-stability paradigm using sponge microbial communities. Sci. Rep. 8 (1), 8425–8429. doi:10.1038/s41598-018-26641-9
Gu, Z., Liu, K., Pedersen, M. W., Wang, F., Chen, Y., Zeng, C., et al. (2021). Community assembly processes underlying the temporal dynamics of glacial stream and lake bacterial communities. Sci. Total Environ. 761, 143178. doi:10.1016/j.scitotenv.2020.143178
Haro-Moreno, J. M., López-Pérez, M., de la Torre, J. R., Picazo, A., Camacho, A., and Rodriguez-Valera, F. (2018). Fine metagenomic profile of the Mediterranean stratified and mixed water columns revealed by assembly and recruitment. Microbiome 6 (1), 128–219. doi:10.1186/s40168-018-0513-5
Hong, Z., Hailin, L., and Zhen, C. (2011). Analysis of land use dynamic change and its impact on the water environment in yunnan plateau lake area––A case study of the dianchi lake drainage area. Procedia Environ. Sci. 10, 2709–2717. doi:10.1016/j.proenv.2011.09.421
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinforma. 11 (1), 119–211. doi:10.1186/1471-2105-11-119
Ji, L., Zhang, L., Wang, Z., Zhu, X., and Ning, K. (2022). High biodiversity and distinct assembly patterns of microbial communities in groundwater compared with surface water. Sci. Total Environ. 834, 155345. doi:10.1016/j.scitotenv.2022.155345
Kang, D. D., Li, F., Kirton, E., Thomas, A., Egan, R., An, H., et al. (2019). MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359. doi:10.7717/peerj.7359
Kieft, B., Li, Z., Bryson, S., Crump, B. C., Hettich, R., Pan, C., et al. (2018). Microbial community structure–function relationships in yaquina bay estuary reveal spatially distinct carbon and nitrogen cycling capacities. Front. Microbiol. 9, 1282. doi:10.3389/fmicb.2018.01282
Kitzinger, K., Padilla, C. C., Marchant, H. K., Hach, P. F., Herbold, C. W., Kidane, A. T., et al. (2019). Cyanate and urea are substrates for nitrification by Thaumarchaeota in the marine environment. Nat. Microbiol. 4 (2), 234–243. doi:10.1038/s41564-018-0316-2
Kuypers, M. M., Marchant, H. K., and Kartal, B. (2018). The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 16 (5), 263–276. doi:10.1038/nrmicro.2018.9
Lauro, F. M., DeMaere, M. Z., Yau, S., Brown, M. V., Ng, C., Wilkins, D., et al. (2011). An integrative study of a meromictic lake ecosystem in Antarctica. ISME J. 5 (5), 879–895. doi:10.1038/ismej.2010.185
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). Megahit: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31 (10), 1674–1676. doi:10.1093/bioinformatics/btv033
Liao, J., Zhao, L., Cao, X., Sun, J., Gao, Z., Wang, J., et al. (2016). Cyanobacteria in lakes on Yungui Plateau, China are assembled via niche processes driven by water physicochemical property, lake morphology and watershed land-use. Sci. Rep. 6 (1), 36357–36410. doi:10.1038/srep36357
Liu, K., Hou, J., Liu, Y., Hu, A., Wang, M., Wang, F., et al. (2019). Biogeography of the free-living and particle-attached bacteria in Tibetan lakes. FEMS Microbiol. Ecol. 95 (7), fiz088. doi:10.1093/femsec/fiz088
Liu, K., Yao, T., Pearce, D. A., Jiao, N., Zeng, Y., Guo, B., et al. (2021a). Bacteria in the lakes of the Tibetan Plateau and polar regions. Sci. Total Environ. 754, 142248. doi:10.1016/j.scitotenv.2020.142248
Liu, L., Sun, F., Zhao, H., Mi, H., He, S., Chen, Y., et al. (2021b). Compositional changes of sedimentary microbes in the Yangtze River Estuary and their roles in the biochemical cycle. Sci. Total Environ. 760, 143383. doi:10.1016/j.scitotenv.2020.143383
Llorens-Marès, T., Yooseph, S., Goll, J., Hoffman, J., Vila-Costa, M., Borrego, C. M., et al. (2015). Connecting biodiversity and potential functional role in modern euxinic environments by microbial metagenomics. ISME J. 9 (7), 1648–1661. doi:10.1038/ismej.2014.254
Martiny, J. B., Eisen, J. A., Penn, K., Allison, S. D., and Horner-Devine, M. C. (2011). Drivers of bacterial β-diversity depend on spatial scale. Proc. Natl. Acad. Sci. 108 (19), 7850–7854. doi:10.1073/pnas.1016308108
Mende, D. R., Bryant, J. A., Aylward, F. O., Eppley, J. M., Nielsen, T., Karl, D. M., et al. (2017). Environmental drivers of a microbial genomic transition zone in the ocean’s interior. Nat. Microbiol. 2 (10), 1367–1373. doi:10.1038/s41564-017-0008-3
Menzel, P., Ng, K. L., and Krogh, A. (2016). Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7 (1), 11257–11259. doi:10.1038/ncomms11257
Oren, A. (2011). Thermodynamic limits to microbial life at high salt concentrations. Environ. Microbiol. 13 (8), 1908–1923. doi:10.1111/j.1462-2920.2010.02365.x
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2014). CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi:10.1101/gr.186072.114
Peeters, S. H., Wiegand, S., Kallscheuer, N., Jogler, M., Heuer, A., Jetten, M. S., et al. (2020). Three marine strains constitute the novel genus and species Crateriforma conspicua in the phylum Planctomycetes. Ant. Van Leeuwenhoek 113 (12), 1797–1809. doi:10.1007/s10482-019-01375-4
Pontius, J. U., Mullikin, J. C., Smith, D. R., Team, A. S., Lindblad-Toh, K., Gnerre, S., et al. NISC Comparative Sequencing Program (2007). Initial sequence and comparative analysis of the cat genome. Genome Res. 17 (11), 1675–1689. doi:10.1101/gr.6380007
Qi, J., Luo, H., and Hao, B. (2004). CVTree: A phylogenetic tree reconstruction tool based on whole genomes. Nucleic acids Res. 32 (2), W45–W47. doi:10.1093/nar/gkh362
Rao, M. P. N., Luo, Z. H., Dong, Z. Y., Li, Q., Liu, B. B., Guo, S. X., et al. (2022). Metagenomic analysis further extends the role of Chloroflexi in fundamental biogeochemical cycles. Environ. Res. 209, 112888. doi:10.1016/j.envres.2022.112888
Rathour, R., Gupta, J., Mishra, A., Rajeev, A. C., Dupont, C. L., and Thakur, I. S. (2020). A comparative metagenomic study reveals microbial diversity and their role in the biogeochemical cycling of Pangong lake. Sci. Total Environ. 731, 139074. doi:10.1016/j.scitotenv.2020.139074
Shang, Y., Wu, X., Wang, X., Wei, Q., Ma, S., Sun, G., et al. (2022). Factors affecting seasonal variation of microbial community structure in Hulun Lake, China. Sci. Total Environ. 805, 150294. doi:10.1016/j.scitotenv.2021.150294
Shen, M., Li, Q., Ren, M., Lin, Y., Wang, J., Chen, L., et al. (2019). Trophic status is associated with community structure and metabolic potential of planktonic microbiota in plateau lakes. Front. Microbiol. 10, 2560. doi:10.3389/fmicb.2019.02560
Tapponnier, P., Zhiqin, X., Roger, F., Meyer, B., Arnaud, N., Wittlinger, G., et al. (2001). Oblique stepwise rise and growth of the Tibet Plateau. science 294 (5547), 1671–1677. doi:10.1126/science.105978
Tran, P. Q., Bachand, S. C., McIntyre, P. B., Kraemer, B. M., Vadeboncoeur, Y., Kimirei, I. A., et al. (2021). Depth-discrete metagenomics reveals the roles of microbes in biogeochemical cycling in the tropical freshwater Lake Tanganyika. ISME J. 15 (7), 1971–1986. doi:10.1038/s41396-021-00898-x
Williamson, C. E., Saros, J. E., Vincent, W. F., and Smol, J. P. (2009). Lakes and reservoirs as sentinels, integrators, and regulators of climate change. Limnol. Oceanogr. 54 (62), 2273–2282. doi:10.4319/lo.2009.54.6_part_2.2273
Xie, M., Ren, M., Yang, C., Yi, H., Li, Z., Li, T., et al. (2016). Metagenomic analysis reveals symbiotic relationship among bacteria in Microcystis-dominated community. Front. Microbiol. 7, 56. doi:10.3389/fmicb.2016.00056
Xing, P., Tao, Y., Luo, J., Wang, L., Li, B., Li, H., et al. (2020). Stratification of microbiomes during the holomictic period of Lake Fuxian, an alpine monomictic lake. Limnol. Oceanogr. 65, S134–S148. doi:10.1002/lno.11346
Yanez-Montalvo, A., Aguila, B., Gómez-Acata, E. S., Guerrero-Jacinto, M., Oseguera, L. A., Falcón, L. I., et al. (2022). Shifts in water column microbial composition associated to lakes with different trophic conditions:“Lagunas de Montebello” National Park, Chiapas, México. PeerJ 10, e13999. doi:10.7717/peerj.13999
Yang, J., Jiang, H., Dong, H., and Liu, Y. (2019). A comprehensive census of lake microbial diversity on a global scale. Sci. China Life Sci. 62 (10), 1320–1331. doi:10.1007/s11427-018-9525-9
Yao, F., Yang, S., Wang, Z., Wang, X., Ye, J., Wang, X., et al. (2017). Microbial taxa distribution is associated with ecological trophic cascades along an elevation gradient. Front. Microbiol. 8, 2071. doi:10.3389/fmicb.2017.02071
Keywords: microbial community, plateau lake, plain lake, metagenomics, metabolic potential, biogeochemical cycles
Citation: Zhao A, Lu Y, Li Q, Li T and Zhao J (2023) Metagenomics reveals the diversity and role of surface-water microbes in biogeochemical cycles in lakes at different terrain ladders. Front. Environ. Sci. 11:1121775. doi: 10.3389/fenvs.2023.1121775
Received: 12 December 2022; Accepted: 15 March 2023;
Published: 23 March 2023.
Edited by:
Danny Chun Pong Lau, Swedish University of Agricultural Sciences, SwedenReviewed by:
Peng Xing, Nanjing Institute of Geography and Limnology (CAS), ChinaJianjun Wang, Nanjing Institute of Geography and Limnology (CAS), China
Copyright © 2023 Zhao, Lu, Li, Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qi Li, bGlxaUBpaGIuYWMuY24=; Tao Li, bGl0YW9AaWhiLmFjLmNu