 Xiaofang Tian1
Xiaofang Tian1 Li Sun2Shengjie Guo1Liying Yuan1Tang Zhang1Chengqian Huang1Tingting He1
Li Sun2Shengjie Guo1Liying Yuan1Tang Zhang1Chengqian Huang1Tingting He1 Qianfeng Jiang3Yizhou Zeng4*
Qianfeng Jiang3Yizhou Zeng4*- 1Department of Nephrology, the First People's Hospital of Zunyi (The Third Affiliated Hospital of Zunyi Medical University), Zunyi, Guizhou, China
- 2Guizhou Aerospace Hospital, Zunyi, Guizhou, China
- 3Department of Cardiology, the First People's Hospital of Zunyi (The Third Affiliated Hospital of Zunyi Medical University), Zunyi, Guizhou, China
- 4Department of Urology, the First People's Hospital of Zunyi (The Third Affiliated Hospital of Zunyi Medical University), Zunyi, Guizhou, China
Metabolic syndrome (MetS) is a group of complex disorders characterized by abnormalities in the metabolism of proteins, fats, carbohydrates, and other substances in the human body. The kidney plays a vital role in these metabolic processes. Similarly, metabolic disorders can lead to renal damage, which can affect both its structure and function. The human intestinal tract possesses an abundant and diverse gut microbial community that significantly influences the physiology and pathology of the host. Growing evidence suggests that gut microbiota-derived metabolites exhibit multiple effects (anti-inflammatory, antioxidant, and improvement of lipid metabolism) in MetS. Particularly, considerable research has suggested that gut microbiota-derived short-chain fatty acids (SCFAs) have an intimate relationship with MetS-related nephropathy. The functions of SCFAs are involved in modulating energy metabolism, regulating immune and inflammatory responses, and inhibiting oxidative stress and mitochondrial damage, which are mainly through the activation of transmembrane G protein-coupled receptors (GPRs) and the inhibition of Histone deacetylase activity (HDAC). Regarding MetS-related nephropathy, therapeutic studies of SCFAs have been conducted in both clinical investigations and animal experiments. However, the role of SCFAs in kidney damage caused by various metabolic disorders has not been fully elucidated. The aim of this article is to review the role of SCFAs in MetS-related nephropathy, which will provide a prospective therapy strategy for MetS-related nephropathy.
1 Introduction
Metabolic syndrome (MetS) is a group of complex disorders involving abnormalities in the metabolism of the three major nutrients (proteins, fats, and carbohydrates) in the human body, including abdominal obesity or overweight, atherosclerotic dyslipidemia (hypertriglyceridemia and low HDL cholesterol), hypertension, and insulin resistance and/or glucose intolerance (1). Insulin resistance is the core of MetS and triggers a series of inflammatory responses, such as C-reactive protein and interleukin cytokines, causing damage to important organs (2). Similarly, the kidneys, as a crucial organ in metabolic processes, are sensitively impaired by metabolic disorders. Numerous previous studies have demonstrated that MetS augments an individual's susceptibility to developing chronic kidney disease (CKD) (3). Thus, MetS is considered a trigger for kidney injury in CKD, which magnifies the adverse impact of other insults.
Gut microbiota refers to the complex of various microorganisms in the human intestine. These microorganisms can produce a variety of metabolites, including short-chain fatty acids (SCFAs), ammonia, and sulfur compounds (4). The rich and diverse flora can use proteins, fats, carbohydrates, and other substances in the intestine to metabolize and release different metabolites. For example, protein metabolism can produce ammonia and sulfide, fat metabolism can produce fatty acids, and carbohydrate metabolism can produce sugars and SCFAs (5). Increasing data indicate that gut microbiota-derived metabolites exhibit multiple roles in the metabolic syndrome and may have significant deleterious and beneficial effects on host health. On the one hand, SCFAs are commonly acknowledged for their beneficial impacts on health. On the other hand, uremic toxins such as indoles, ammonia, and trimethylamine N-oxide, which are produced by the gut flora, are generally considered to be harmful substances (6). Specifically, extensive research has indicated that SCFAs generated from gut microbiota have a close association with nephropathy related to MetS.
The functions of SCFAs are involved in modulating energy metabolism, regulating immune and inflammatory responses, and inhibiting oxidative stress and mitochondrial damage, which are mainly achieved through the activation of transmembrane G protein-coupled receptors (GPRs) and the inhibition of histone acetylation (HDAC) (7). Currently, some therapeutic studies of SCFAs have been undertaken in both clinical investigation and animal experiments, such as those in obesity, diabetes, inflammatory bowel disease, hypertension, depression, and cancer (8–10). The growing number of therapeutic studies on SCFAs in clinical and fundamental research indicates a significant role for SCFAs in kidney diseases (11–13). Furthermore, emerging evidence indicates that SCFAs exerted some roles in MetS-related nephropathy (14–16). However, the underlying mechanism of SCFAs in MetS-related nephropathy has not been fully understood yet. This article aims to review the physiology and function of gut microbiota-derived SCFAs in MetS-related nephropathy and to propose prospective therapeutic strategies for MetS-related nephropathy.
2 MetS and kidney diseases
2.1 The definition of MetS
The concept of “MetS” was initially introduced by Grundy and colleagues in 2001 (17), which considerably arises from chronic inflammation caused by insulin resistance, along with disturbances in the metabolism of proteins, fats, carbohydrates, and other substances in the human body (18). Generally, the MetS is diagnosed based on five criteria proposed by the National Cholesterol Education Program-Adult Treatment Panel III (waist circumference, triglycerides, high-density lipoprotein, cholesterol, blood pressure, and glucose) (19). MetS is commonly influenced by internal genetic abnormalities or external surroundings. It is reported that a high-fat, high-carbohydrate diet structure, irregular lifestyle, and low physical activity are the main detrimental factors for MetS (20). There are various metabolic disorders in MetS, which mainly consist of insulin resistance, dyslipidemia, hypertension, obesity, high uric acid, and a high incidence of fatty liver (21). Following the increase in social burden and the spread of unhealthy living habits, the prevalence of MetS is gradually increasing, and MetS is regarded as an important contributor to a variety of diseases, including cardiovascular events, diabetes, cancer, and CKD (22–24).
2.2 The correlation between MetS and CKD
The kidney has a crucial role in metabolism, and an expanding body of evidence indicates that individuals with MetS are at a higher risk of developing CKD, which is characterized by impaired renal function and structural damage. Furthermore, the prevalence of MetS and its metabolic disorders is also much higher in patients with CKD than in the non-CKD population (25, 26). As shown in Table 1, there have been growing clinical investigations around the world evaluating the relationship between metabolic syndrome and CKD in the past decade. For example, a clinical study that enrolled 5800 individuals diagnosed with type-2 diabetes revealed that MetS served as an independent indicator for predicting the occurrence of CKD in new cases (27). A cross-sectional study conducted in 2012 found that the prevalence of CKD in China was 10.8% (10.2–11.3%), and the occurrence of renal damage was significantly associated with a variety of risk factors such as hypertension, diabetes mellitus, a history of cardiovascular disease, and hyperuricemia (28). Additionally, a survey of the 75,468 population in China also showed that the degree of MetS abnormality was positively correlated with the risk of CKD, and the prevalence of CKD in patients with MetS and those without MetS was 57% and 28%, respectively (29). Similarly, studies conducted in the United States have shown a correlation between MetS and a hastened advancement of CKD in individuals in stages 3 and 4 (30). In addition, a 10-year prospective cohort research was conducted on a sample of healthy individuals from the Republic of Korea to examine the connection between MetS and the occurrence of CKD (31). The findings revealed that MetS has emerged as an independent risk factor for the onset of CKD.
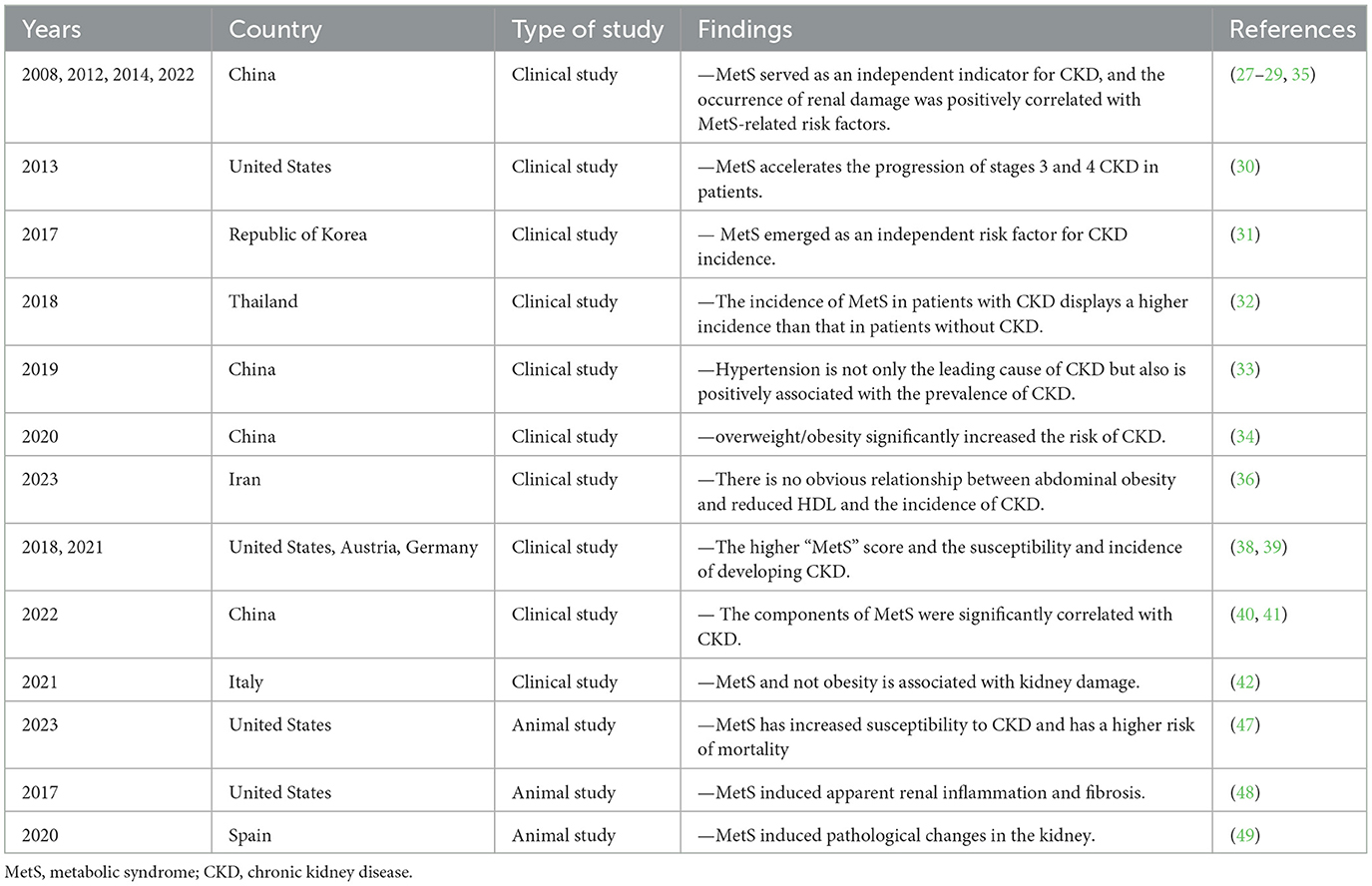
Table 1. The correlation between MetS and CKD.
Recently, there have been ongoing updates in domestic research on MetS and CKD aimed at providing more evidence in this field. Research in Thailand reported that the incidence of MetS in patients with CKD was 71.3%, displaying an evident higher incidence than that in patients without CKD (32). A multi-center and cross-sectional study involving 2,484 patients found that hypertension is not only the leading cause of CKD but also is positively associated with the prevalence of CKD (33). A cohort study of 15,229 participants from 2008 to 2013 demonstrated a significant increase in the risk of CKD due to overweight/obesity (34), and also found that MetS was independently associated with renal dysfunction (35). Interestingly, an investigation that recruited 8,987 participants showed that MetS promotes the development of CKD and is correlated with some strong indicators, including hypertension, diabetes, and age. There is no obvious relationship between abdominal obesity and reduced HDL and the incidence of CKD (36). In recent years, the “MetS score” and the “MetS factor” have been applied to identify MetS and its components (37). Previous studies confirmed that the higher the “MetS score” obtained, the higher the susceptibility and incidence of developing CKD (38, 39). When the components of MetS were analyzed separately with the incidence of CKD, several clinical investigations suggested that some metabolic components were still significantly correlated with CKD, including age, body mass index, waist circumference, systolic and diastolic blood pressure, serum triglyceride, serum glucose, serum uric acid, and C-reactive protein. Therefore, targeted screening and intervention tests among individuals were feasible and highly necessary (40–42). Considering the close correlation between MetS and CKD, gaining more understanding of their relationship can help us to further investigate the underlying mechanism.
2.3 The pathogenesis of MetS-related nephropathy
Insulin resistance is the critical feature of MetS, and insulin receptors are expressed in podocytes, tethered cells, renal microvascular endothelial cells, and tubular epithelial cells of the kidney (43). It is indicated that renal tissues in the MetS population predominantly exhibit chronic lesions, including varying degrees of glomerulosclerosis, tubular atrophy, interstitial fibrosis, and atherosclerosis (44, 45). It is suggested that glomerulosclerosis, cystic wall thickening, increased glomerular volume, and atherosclerotic vitriform degeneration of small renal arterioles were more pronounced in patients with MetS. In addition, the incidence of IgA nephropathy and focal segmental glomerulosclerosis was significantly higher in patients with MetS, and obesity may be an independent risk factor for IgA nephropathy (46). Additionally, multiple animal experiments have explored the changes and potential mechanisms of MetS-related nephropathy. For example, animal research confirmed that MetS increased susceptibility to CKD and risk of mortality (47). Moreover, significant renal inflammatory and fibrotic changes were observed in a porcine model of MetS (48). Similarly, it is reported that mesangial expansion, nodular glomerulosclerosis, and glomerulomegaly were validated in a model of metabolic syndrome that used Iberian pigs fed with fat-enriched food (49). The pathogenesis of MetS-related renal damage is very complex; obesity, insulin resistance, dyslipidemia, and hypertension usually serve as inducers to promote the progress of CKD. However, the underlying mechanism of MetS-related nephropathy has not been fully understood yet. It has been suggested that the activation of RAAS, production of inflammatory factors, and induction of reactive oxygen species (ROS) are crucial in the development of MetS (50, 51). Transforming growth factor-β1 (TGF-β1) is increased in renal inflammation and renal fibrosis, which contributes to the progression of MetS-related nephropathy (52, 53). TGF-β1 could activate the tumor suppressor p53 in diabetic insults; targeting p53 may have the efficacy for protecting MetS-related nephropathy (54). Similarly, several studies have suggested that the sterol regulatory element binding protein-1 (SREBP-1) is participating in promoting renal fibrosis of diabetic nephropathy (55). SREBP inhibition does not have a significant effect on ameliorating renal damage in the early stage of diabetic nephropathy (56).
Furthermore, lots of other potential molecular mechanisms were still explored in various studies recently, including insulin-like growth factor-1 (IGF-1), connective tissue growth factor (CTGF), HIF-1α, AMPK, and Nrf2 pathways, which were found to be correlated with MetS-related nephropathy (57–60). In vivo and in vitro experiments revealed that activation of the NLRP3 inflammasome exacerbated metabolic kidney injury by regulating ROS and mitochondrial damage (61, 62). Interestingly, emerging evidence suggested that gut microbiota and its metabolites played some roles in MetS-related kidney diseases (63–65). For example, an animal investigation on hyperuricemia-related nephropathy was performed and found that gut microbiota exhibited protective roles against renal inflammation in mice (66). Of course, there are numerous studies about the relevant mechanisms of metabolic kidney injury around the world, and we just depict the tip of the iceberg. A better understanding of the mechanisms of MetS-associated renal damage could guide potential therapeutic targets to prevent the development of MetS-related nephropathy.
3 Gut microbiota-derived SCFAs
3.1 The overview of intestinal flora
The intestinal microbiota has a large number of 10 trillion or even 100 trillion microorganisms, including bacteria, fungi, and viruses (67, 68), and the development of high-throughput sequencing technology has revealed that the gut flora contains far more genes than the human genome, leading to the term “second human genome” (69). In healthy adults, the phyla Firmicutes and Bacteroidetes are predominant. Factors such as age, medications, and allergens can affect the structure and number of intestinal flora, as well as cause changes in metabolites (70). Diseases such as obesity, type 2 diabetes, atherosclerosis, inflammatory bowel disease, and cancer have been linked to disruptions in the gut flora (71, 72). The term “gut-kidney axis” was originally used in 2011 to describe a theory that postulates the existence of a crosstalk-like relationship between the kidneys and the intestines that is controlled in both directions to create a gut–kidney axis balance and to maintain health (73). In recent years, studies have suggested a close relationship between the gut microbiota and kidney diseases. For example, during the course of CKD, uremic toxins gradually accumulate, which may be accompanied by chronic intestinal inflammation and epithelial dysfunction, promoting the translocation and structural changes of the intestinal flora. The change in the gut microbiota will, in turn, exacerbate the increase of uremic toxins, resulting in a vicious cycle within the gut–kidney axis (74). Some studies collected relevant literature from PubMed over the past 10 years for analysis and found that the impact of metabolomics in chronic kidney disease is higher than that of proteomics and transcriptomics. Moreover, the research results observed the enrichment of Eggerthella lenta, Enterobacteriaceae, and Clostridium spp., as well as the depletion of Bacteroides eggerthii, Roseburia faecis, and Prevotella spp. in the CKD model (75). Some studies analyzed the role of gut microbiota in acute kidney injury (AKI) (76) and also summarized the possible pathogenic pathways and mechanisms involved in the transition from AKI to CKD due to gut microbiota dysregulation (77). In addition, some literature reports that the gut microbiome may play a role in maintaining oxalate homeostasis and nephrolithiasis. Gut microbiota may also interact with symbiotic bacterial species in the urinary microbiome to jointly affect crystal formation and induce stone growth in the kidney (78). Furthermore, recent studies have particularly emphasized the close association between IgA nephropathy and gut microbiome dysregulation. Gut microbiome dysregulation is associated with an abnormal gut mucosal immune system, leading to the production of abnormal IgA1 antibodies by the body, which further form immune complexes and deposit in the kidneys, triggering kidney inflammation and damage (79). As a huge biological community, the gut microbiota plays a non-negligible role in kidney diseases, but the specific mechanism is not yet clear, which is worthy of our continuous in-depth exploration and discussion.
3.2 The definition of SCFAs
The intestinal flora produces a variety of metabolites through the breakdown of nutrients, including indolephenol sulfate, p-cresol sulfate, trimethylamine oxide, horse uric acid, and SCFAs. SCFAs are the products of fermentation of dietary fibers by the intestinal flora at the site of the cecal colon, which primarily consists of acetate, propanoate, butyrate, isobutyrate, valerate, isovalerate, hexanoate, and isohexanoate (7). SCFAs are usually present in the gut and can be absorbed into the bloodstream (80). Finally, the fraction of SCFAs was excreted via the breath, urine, and feces (81, 82). After entering the circulatory system, SCFAs could be distributed in many peripheral tissues, such as the heart, kidney, skin, and sympathetic ganglia (83–86), and modulate biological processes through binding to G-protein coupled receptors (GPR41, GPR43, and GPR109A) or inhibiting histone acetylation (87). GPR41, GPR43, and GPR109 are expressed in a variety of cells and tissues, serving as vital receptors activated by SCFAs for participating in some protective effects (88–90). Dietary fiber modulates microbial composition and influences the production of SCFAs (91), and alterations in gut flora directly affect acetate, propionate, and butyrate levels. The role of SCFAs in diseases remains controversial, and the underlying mechanisms involved are still unclear. For example, butyrate is thought to have antitumor, antifibrotic, and anti-inflammatory activities (92, 93). However, some studies do not support their protective role in disease (94).
3.3 The role of SCFAs in MetS
The core of MetS development is insulin resistance, which could be affected by gut microbiota-derived metabolites. To a certain extent, SCFAs play an important role in MetS. Similar to the diverse effects of SCFAs, considerable studies on SCFAs have been carried out in both clinical and animal studies. Although there may be a few discrepant opinions about the role of SCFAs in some diseases, the current mainstream view tends to consider SCFAs to play a beneficial role for individuals. Lucas S et al. reported that treatment of mice with SCFAs or feeding with a high-fiber diet increases bone mass and prevents postmenopausal and inflammation-induced bone loss (95). Moreover, accumulating evidence has suggested that SCFAs exhibit pivotal effects in MetS. Clinical research has found that the changes in SCFAs in the body after conservative weight loss and surgical intervention in obese patients are very interesting. It shows that the total and relative amounts of acetate, propanote, and butyrate decrease while the total and relative amounts of isobutyrate, isovalerate, and isohexanoate increase. This result suggests a shift in the proteolytic fermentation pattern, which has an adverse effect on health. SCFAs are related to diet, and the adverse effects can be offset through dietary intervention (96). SCFAs utilized in animal models of obesity have been shown to reduce lipogenesis and inhibit body weight gain, potentially by enhancing triglyceride hydrolysis and FFA oxidation in adipose tissue (97, 98). In addition, the levels of SCFAs have a close correlation with blood pressure (99). Recently, research showed that fecal levels of SCFAs were significantly decreased in patients with preeclampsia; pregnant rats of hypertension were utilized to explore the further mechanism, which demonstrated that SCFAs could directly regulate blood pressure and improve hypertension (100). The central features of diabetes are chronic inflammation and peripheral insulin resistance; accumulating evidence indicated that SCFAs could improve insulin sensitivity and prevent inflammation in in vivo and in vitro models of diabetes (101). However, the changes in SCFAs in preeclamptic patients are inconsistent with those in obese and other metabolic syndrome patients, which is a point worthy of great attention. We speculate that the shift in the metabolic pattern of nutrients may prompt changes in the gut microbiota structure, leading to a reduction in SCFA-producing bacteria. This may then give feedback and regulate the body to produce more SCFAs, resulting in an increase in the level of SCFAs in feces. At the same time, relatively high content of SCFAs in feces may also be secondary to a reduction in the intestinal absorption of SCFAs, leading to an obstacle in the process of SCFAs being absorbed into the bloodstream (102, 103). In preeclamptic patients, there is a relative disorder in the gut microbiota, such as a decrease in the abundance of Firmicutes and an increase in the abundance of Proteobacteria. Moreover, the fecal SCFA level in preeclamptic patients is positively correlated with the abundance of Firmicutes. The increase in the abundance of Proteobacteria may promote an increase in the production of LPS, thus triggering an excessive inflammatory response, resulting in a decrease in the relevant SCFAs in feces (104). However, the mechanisms involved in these differential manifestations are relatively complex and are not clear at present, awaiting further research.
3.4 The role of SCFAs in kidney diseases
Recently, accumulating evidence has suggested that SCFAs have an imitated relationship with CKD. Metagenomic analysis of the gut microbiome demonstrated that the evident changes of SCFAs can be observed in early CKD (105). Growing animal experiments illustrated that SCFAs exerted a substantial role in preventing the progress of CKD (106–111). However, the underlying mechanisms of SCFAs in renal disease remain unclear. It is currently proposed that SCFAs regulate the development of renal diseases primarily through the following aspects: (1) the effect of SCFAs on energy metabolism, where SCFAs play an important regulatory role in energy metabolism, including lipid, glucose, and insulin (112, 113). The metabolism of lipids, glucose, and insulin is regulated by SCFAs via activating GPRs and inhibiting HDACs (114, 115). These regulatory actions could have a significant impact on the onset and progression of CKD; (2) the effect of SCFAs on immunity and inflammation: SCFAs have anti-inflammatory effects and affect the immuno-inflammatory process (116). A clinical investigation in children with CKD found that gut barrier dysfunction and microbial metabolite imbalance apparently mediated the production of pro-inflammatory factors (IL-1β, IL-6, and TNF-α), as well as altered T-cell phenotype (117). Moreover, SCFAs significantly increased the level of M2 macrophage polarizing factor, limiting the progression of renal fibrosis (118). Thus, SCFAs may affect the development of renal diseases by modulating immuno-inflammatory processes; (3) the effect of SCFAs on oxidative stress and mitochondrial damage: superoxide dismutase, catalase, glutathione, and nitric oxide (NO) all contribute to ROS production in the process of oxidative stress. Treatment with SCFAs ameliorated the proximal tubule injury, which was associated with oxidative stress (84). In addition, autophagy and pyroptosis are critical for the accumulation of mitochondrial damage involved (119, 120). Previous studies revealed that SCFAs played a crucial role in the prevention of CKD, including the promotion of mitochondrial biogenesis and the reduction of mitochondrial damage (121, 122). Therefore, gut microbiota-derived SCFAs play some roles in kidney diseases, including energy metabolism, immune inflammation, oxidative stress, and mitochondrial damage, affecting the occurrence and development of kidney diseases (Figure 1). However, there are many issues that still require further investigation, such as the specific signaling pathways, as well as the type and distribution of receptors, and how SCFAs regulate immune-inflammatory responses and interact with other cytokines. Based on the above-mentioned roles of SCFAs in MetS and kidney diseases, we speculate that SCFAs play an important role in MetS-related nephropathy. Then, we will comprehensively analyze the progress of SCFAs in MetS-related nephropathy.
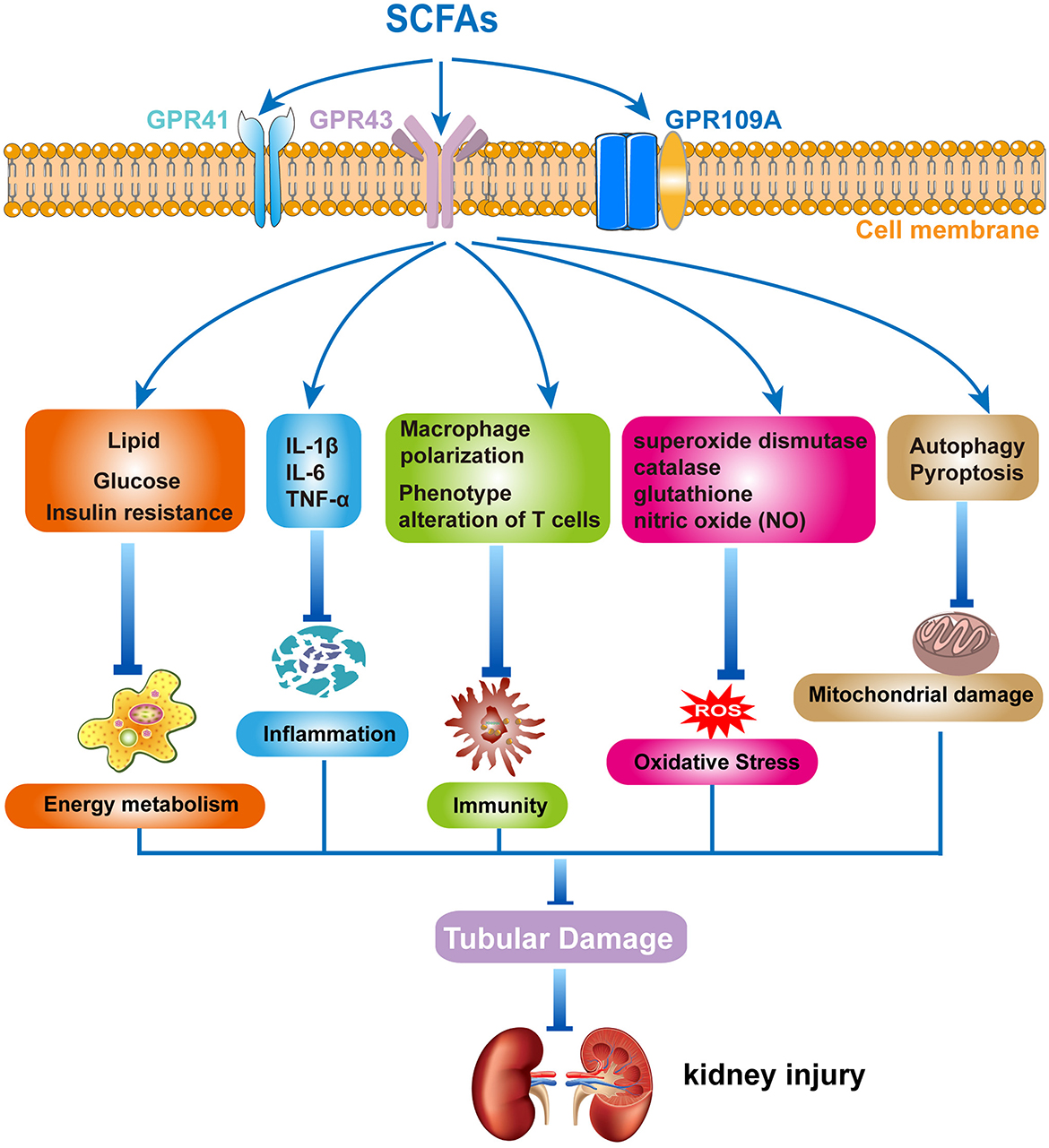
Figure 1. The mechanism of short-chain fatty acids (SCFAs) in kidney diseases. Gut microbiota-derived SCFAs exert protective effects in kidney diseases by modulating energy metabolism, regulating inflammation and immune response, and inhibiting oxidative stress and mitochondrial damage.
4 SCFAs in MetS-related nephropathy
Abundant SCFA production can regulate the health status of the host and exert multiple protective effects against a variety of diseases, including diabetes, obesity, cardiovascular disease, and kidney diseases. This beneficial effect is due to the contribution of SCFAs in modulating energy metabolism, regulating inflammation and immune response, and inhibiting oxidative stress and mitochondrial damage. Studies on the role and related mechanisms of SCFAs in the prevention and treatment of MetS-related nephropathy are summarized in Figure 2.
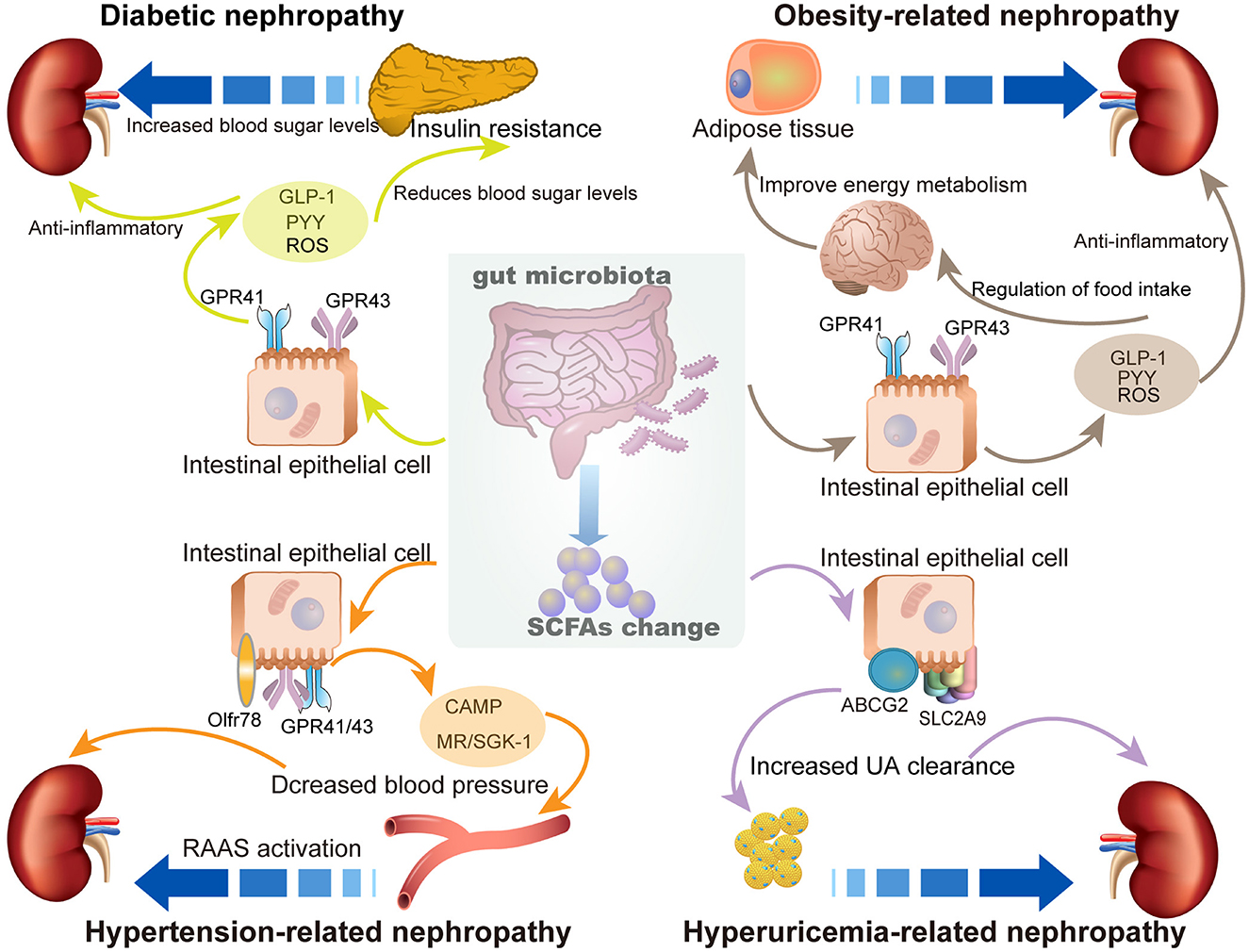
Figure 2. The role of SCFAs in metabolic syndrome (MetS)-related nephropathy. The enrichment of SCFAs exhibits multiple protective effects to prevent SCFAs MetS-related nephropathy, including diabetic nephropathy, obesity-related nephropathy, hypertension-related nephropathy, and hyperuricemia-related nephropathy.
4.1 Diabetic nephropathy
Diabetes mellitus (DM) is a group of metabolic diseases evaluated by hyperglycemia and insulin resistance, which often causes various chronic microvascular damage. Diabetic nephropathy (DN) is known as a serious microvascular complication of diabetes and is the leading cause of end-stage renal disease (ESRD) (123, 124), which is characterized by glomerulosclerosis, tubular atrophy, and fibrosis, concomitant with oxidative stress and NF-κB signaling activation (125). The pathological process of DN is extremely complex; particularly, the aberrant immune system and chronic inflammation have been regarded as pivotal regulators in the pathophysiological process and promote the onset and progression of DN (126, 127). Dysbiosis of gut microbiota is present in patients and rodents with DN, especially the decreased SCFAs-producing bacteria. The abundance of the gut microbiota in DN patients is reduced, and the relative abundance of Firmicutes is lower than that in the general healthy population (128). A 2022 study conducted a meta-analysis of the gut microbiota composition in healthy individuals and those with DN, DM, and non-diabetic nephropathy and found that the average abundance of Firmicutes in the gut microbiota of DN patients was reduced, while the average abundance of Actinobacteria increased. In addition, compared with patients without DN, the abundance of specific genera (such as Hungatella, Bilophila, and Escherichia) in the gut microbiota of DN patients changed significantly (129). Over the last few years, there has been an increasing amount of study focusing on the role of SCFAs in the DN (130). A clinical investigation provided evidence that the concentration of serum and fecal SCFA levels (fecal levels in particular) were lowered in individuals with DN, which are negatively correlated with renal function (131). Moreover, 308 subjects with type 1 diabetes maintained normal renal function, whose subsequent detection of metabolites displayed that the SCFAs are enriched in the majority of patients (at least 80%) (132). Similarly, a growing number of studies found that SCFAs directly exerted positive effects on DN, including attenuation of renal damage, prevention of insulin resistance, and renal function impairment (84, 90, 133).
The underlying molecular mechanism of the protective effect of SCFAs is worthy of further exploration and draws more attention. For instance, butyrate absorption by the intestinal epithelium is the major energy source for AMPK phosphorylation and promotion of glucagon-like peptide-1 (GLP1) release. Acetate and propionate exert essential functions through binding to G protein-coupled receptor 41 (GPR41) or G protein-coupled receptor 43 (GPR43) expressed on the intestinal epithelium. Activation of GPR41 promotes the secretion of peptide YY (PYY), which controls satiety and intestinal transport. In addition, GPR43 inhibits pro-inflammatory factor production and enhances the secretion of GLP1, which contributes to pancreatic β-cell proliferation, and thus, by lowering blood glucose levels, exerts a protective effect against DN (134). Decreased concentrations of SCFAs lead to decreased secretion of PYY and GLP1, thereby accelerating the development of DKD manifested by proteinuria, loss of renal structural integrity, and renal fibrosis (135). Furthermore, oxidative stress and NF-κB signaling were evidently activated in the pathophysiological process of DN, which could be significantly reversed by SCFAs. Moreover, the caspase1-GSDMD canonical pyroptosis pathway was also induced in the in vitro model treated with high glucose and presented a downregulated trend after SCFA intervention (136). In addition, several studies have shown that the activation of FFA2-mediated PI3K/Akt/mTOR signaling and the miR-7a-5p/P311/TGF-β1 pathway contributes to the positive regulation of SCFAs in DN (133, 137). In general, hyperglycemic stimulation triggers multiple factors to be disturbed, leading to intrarenal cellular abnormalities. Renal tubular injury is one of the important determinants of progressive renal failure in DN, and SCFAs significantly inhibited renal tubular cell apoptosis and oxidative stress induced by elevated glucose or H2O2 stimulation in in vitro experiments (138). Although considerable studies have demonstrated the protective effect of SCFAs on DN, there are still some controversies in this field, and their specific mechanisms in renal tubular cells, podocytes, and mesangial cells are not clear. For instance, it is suggested that increased levels of acetate related to disturbed gut microbiota enhance proteinuria, activate the renin-angiotensin-aldosterone system (RAAS), and aggravate renal intrinsic cell injury (139). The activation of RAAS is considered to be one of the important initial factors in the early development of DN. Acetate does not exist in germ-free mice. Under the stimulation of harmful factors, the intestinal flora will produce excessive short-chain fatty acids such as acetate, mediating the immune disorder and chronic inflammatory response of the host (140, 141). Acetate shows a dual role in DN. On the one hand, acetate can resist the damage of renal cells by external stimuli (such as anti-apoptosis and antioxidant stress) (138). On the other hand, SCFAs (such as acetate) can bind to receptors on renal arterioles and regulate renin secretion, participating in maintaining glomerular pressure. Due to the characteristics of early glomerular hypertension and glomerular ultrafiltration in DN, the disordered intestinal flora may produce excessive SCFAs, thus promoting the pathological changes of early DN (102). However, the role of SCFAs in DN still needs to be further investigated to provide more clinical and basic experimental evidence and to deeply explore its related mechanisms.
4.2 Obesity-related nephropathy
Obesity-related nephropathy (ORG) is a condition in which extreme obesity causes proteinuria, as first described by Weisinger et al. in 1974 (142). Obesity and CKD remain public health problems, and high BMI is an independent risk factor for the development of new-onset CKD. In obese patients, to meet the metabolic demands of the increased body weight, compensatory ultrafiltration occurs, increasing the intraglomerular pressure and damaging the kidneys through mechanisms such as lipotoxicity, chronic inflammation, and insulin resistance, increasing the risk of developing CKD (143). Recent evidence has highlighted other factors, including hemodynamic changes and gut microbiota dysbiosis, which exacerbate kidney dysfunction in obese patients, leading to histological changes known as ORG (144). Subsequently, an increasing number of clinical and experimental animal research studies focused on exploring the relationship between obesity and kidneys and showed that obesity substantially played a role in the structure and function of the kidney (145). Obese patients exhibited a faster reduction in glomerular filtration rate, making them more likely to develop ESRD or even death. Treating obesity proceeds to improve the aforementioned renal outcomes and significantly enhance the quality of life for patients (146). However, the underlying molecular mechanism of the ORG still remains unclear. It is commonly assumed to be linked to hyperglycemia and insulin resistance, the role of adipocytokines, irregular activation of RAAS, release of inflammatory factors, aberrant lipid metabolism, and renal structural damage caused by obesity itself (147). It was suggested that imbalanced gut microbiota and aberrant metabolites contribute to the progression of obesity, which is linked to the modulation of energy homeostasis, fat accumulation, and decreased lipoprotein lipase activity (148). High-fat diet (HFD) was utilized in feeding germ-free rodents to establish obesity-related changes in gut microbiota (149). Compared to the healthy group, microbial diversity was reduced in obese individuals (150, 151). A systematic review and meta-analysis showed that obese individuals had significantly higher SCFA levels and reduced richness of gut microbiota at the phylum level (152). This suggests that we cannot single-handedly use high or low levels of SCFAs to judge the status of obese patients, and to some extent, SCFAs may play an unfavorable role in obesity. In addition, the type of diet and the possibility of the genetic background of the experimental animals may also lead to different results in the data related to obesity-induced changes in the gut microbiota profile (153). For example, the investigation of children suggested that excessive SCFAs produced by a particular gut microbiota represent an additional energy source and may cause a disturbance of energy balance, contributing to obesity (154). Besides, a study of 441 community-dwelling adults reported that higher SCFA concentrations were associated with gut abnormal permeability, metabolic disturbance, and obesity (155).
SCFAs contributed to the inhibition of diet appetite, metabolic rate improvement, and loss of weight in mice and humans because of increasing release of leptin from adipose tissue (156). Endocrine hormones, such as GLP-1, PYY, and leptin, produced by the combination of SCFAs and GPRs, can increase satiety and improve obesity. Animal experiments found that inulin could stimulate the production of SCFAs in wild-type or FFAR2-/- mice, drive an increase in the number of cells that secrete PYY, increase the release of PYY, and then suppress appetite to control obesity (157). However, PYY can also slow down intestinal peristalsis, causing food to stay in the intestine for a longer time and increase energy absorption, thus leading to obesity. Increased GLP-1 hormone increases insulin sensitivity and inhibits fat accumulation in adipose tissue, thereby maintaining energy homeostasis in the body (158). SCFA production can promote leptin secretion from adipocytes by activating GPR43 receptors. In a mouse model of diet-induced obesity, the expression of GPR43 in adipose tissue is downregulated by SCFAs (159). In addition, GPR43-deficient mice prefer to gain weight regardless of diet, while overexpressing GPR43 in adipose tissue could reverse the phenomenon independent of HFD (160). However, in the fasting state, gastric tissue-derived leptin can promote appetite by attenuating the sensitivity of afferent nerves. It can be seen that leptin promotes satiety or appetite, depending on the state of food intake. Thus, it is obvious that obese patients need to increase the expression of GLP-1, PYY, leptin, and GPR43 for adaptive purposes, which may be responsible for the feedback elevation of SCFAs. Moreover, alterations in some of the key downstream components may also directly affect organismal performance through other pathways. For example, dietary SCFA supplementation prevented and reversed HFD-induced obesity in mice by downregulating PPARγ. Notably, the SCFA effect on lost weight was abrogated in mice with adipose-specific disruption of PPARγ, indicating that PPARγ acted as a critical mediator of the beneficial effects of SCFAs in MetS (160). Collectively, the factors contributing to obesity in the body are complex, involving genes, environment, and comorbidities. Generally, differences in diet, host genes, or the composition of the microbiota can also lead to certain differential manifestations of SCFAs in obese patients. There is still some controversy about whether SCFAs are beneficial or detrimental to obesity between humans and rodents; further studies should be conducted to validate the effect of SCFAs and the underlying mechanism.
4.3 Hypertension-related nephropathy
Hypertension-related nephropathy (HN) is a condition in which sustained hypertension causes damage to renal structure and function. High blood pressure increases the blood pressure in the blood vessels, resulting in the leakage of protein into the urine and causing damage to the renal filter system. Prolonged, poorly controlled hypertension further causes irreversible kidney damage. The initiation and progression of hypertension are attributed to dysregulation of the RAAS, ANS, and immune system (161, 162). Emerging evidence indicates that the gut microbiota and its metabolism play a role in hypertension development (163–165). The reduction of SCFAs-producing bacteria changed the gut environment, involving a decrease of the hypoxic gut profile and deterioration of the microbial balance, resulting in damage to epithelial barrier integrity, gut inflammation, dysregulation of blood pressure, and impairment of renal function. Consequently, impaired renal function leads to the accumulation of uremic toxins that reach the intestine and cause alterations in bacteria composition, which induces positive feedback that triggers the endotoxins to translocate into the bloodstream, enhances local kidney inflammation, and exacerbates kidney injury (166). In hemodialysis patients with renal disorders, sodium propionate supplementation resulted in a 10% drop in systolic blood pressure, while diastolic blood pressure remained constant (167). Dysbiosis of the intestinal flora has been reported in corresponding animal models of hypertension (168, 169) and in hypertensive patients (170, 171). For instance, spontaneously hypertensive rats (SHRs) displayed pathophysiological changes in the gut, including decreased numbers of goblet cells and villi length and increased fibrosis, compared to age-matched normotensive Wistar Kyoto (WKY) controls (172). Increasingly studies demonstrated the direct effect of gut dysbiosis on the initiation and progression of hypertension; specifically, fecal microbiota transplantation (FMT) experiments were conducted, including transferring dysbiotic fecal samples from patients with hypertension to germ-free mice (171) or feces from hypertensive stroke-prone SHRs to normotensive WKY rats, resulting in increased blood pressure in the recipients (173). The male SHR model showed that intestinal flora and its metabolites are closely associated with hypertension-related nephropathy, and the results indicate that reduced levels of SCFAs, inflammatory factor release, and blood pressure disturbances occur in the process (174). Mechanistically, a variety of SCFA receptors are seen to be expressed in human kidneys, and it was found that olfactory receptor 78 (Olfr78) was expressed mainly on afferent small arterioles (paraglomerular apparatus), which can elevate blood pressure by mediating renin secretion and subsequent vasoconstriction (165). SCFAs such as butyrate can target the GPR41 and olfactory receptor 78 (Olfr78) to prevent programmed hypertension by regulating cAMP (102, 175). Moreover, butyrate could attenuate DOCA/salt-induced hypertension and renal damage by inhibiting the MR/SGK1 pathway (176). Thus, it is clear that in HN, SCFAs can function through Olfr78, MR, etc., in addition to their usual function of binding to GPRs. However, it is obvious that other SCFA receptors, such as GPR41, are expressed in the renal microvascular system (especially the smooth muscle cells of small resistance vessels), but there is still some controversy about their role in regulating blood pressure (177–179). Currently, non-coding RNAs have become a hotspot in medical research; for example, microRNA (miRNA) is a kind of endogenous non-coding RNA with regulatory functions in eukaryotic organisms. miRNAs have been found to play a role in the regulation of blood pressure by binding to SCFAs receptors. For example, in hypertension-related nephropathy, miR-329 and miR-132 are upregulated, whereas miR-129 is downregulated. Furthermore, miR-329 and miR-132 can target GPR41 and GPR43, respectively, while miR-129 is expected to potentially target Olfr78 (180, 181). Despite the emerging evidence that SCFAs may play a role in the development of hypertension-related nephropathy, the underlying mechanisms of regulation of these receptors remain unclear and will require further studies to elucidate.
4.4 Hyperuricemia-related nephropathy
Uric acid (UA) is the end product of purine metabolism by xanthine oxidase, which belongs to anionic organic acid and is slightly soluble in water. Approximately 70% of UA in the normal human body is excreted through the kidney, and the remaining 30% is excreted through the bile duct and intestine (182). The increased generation or decreased excretion of UA results in the accumulation of UA in the body, and disturbances in UA in individuals will lead to hyperuricemia (HUA). HUA often contributes to renal dysfunction, for example, renal tubulointerstitial inflammation, kidney stones, renal fibrosis, and polycystic kidney disease (183). A study reported that the prevalence of nephropathy was 15.11% among 266 patients with HUA, while that was only 2.19% in the population with normouricemia (184). An investigation was conducted to explore the relationship between UA and subsequent decline in renal function. 13,338 participants with intact renal function in a community cohort were followed up and analyzed, and it was found that elevated serum UA level is an independent risk factor for kidney disease in the general population (185). Interestingly, it is reported that asymptomatic HUA could not affect CKD progression unless UA crystallizes and is deposited in kidney tissues. Following the development of UA crystal granulomas, renal interstitial inflammation, and fibrosis contribute to CKD progression, involving M1-like macrophage polarization (186). Currently, relevant studies have suggested a close relationship between the gut microbiota and HUA. A variety of obligate anaerobic and facultative anaerobic human and murine gut microbiota (mainly Firmicutes) have a higher ability to lower UA. This microbiota can drive the conversion of UA into lactic acid or anti-inflammatory SCFAs. Animal studies have shown that when there is a lack of hepatic uricase in the body, the gut microbiota structure changes significantly and the cecal and serum UA levels increase significantly (187). Moreover, HUA-related nephropathy is found to be usually accompanied by disordered intestinal flora, such as the overgrowth of opportunistic pathogens in HUA-related nephropathy, including Escherichia-Shigella and Bacteroides, and reduction of bacteria-producing SCFAs, such as Lactobacillus and Ruminococcaceae (188). The gut bacterial diversity and SCFAs in the HUA group reduced significantly, and the structure and function of the gut microbiota and the levels of SCFAs between patients with HUA and healthy controls have altered apparently (189). There are a large number of intestinal bacteria in the intestine that can consume UA under anaerobic conditions and convert it into xanthine or lactic acid and SCFAs, thereby changing the production and excretion of UA. The intestine has a variety of UA transporters, such as adenosine triphosphate-binding cassette Transporter G2 (ABCG2) and urate transporter soluble carrier protein 2 family member 9 (SLC2A9) (190). Then, what role do SCFAs play in HUA-associated nephropathy? Similarly, an animal research showed that the gut microbiota dysbiosis and decreased production of SCFAs in HUA-related nephropathy, and further experiments with FMT demonstrated that an increase in SCFAs could alleviate HUA-associated renal injury (66). Furthermore, an extra supplement of SCFAs could prevent cardiorenal lipotoxicity and glucometabolic dysregulation by suppressing UA accumulation (191). Mechanistically, several studies indicated that renal osteopontin, CD44, and TLR4/MyD88/NF-κB signaling were involved in the renal damage of HUA (192, 193). Overall, for the treatment and prevention of HUA-related nephropathy, previous studies inspire us not only to focus on the basic treatment of UA but also to pay attention to the reduction of SCFAs caused by diet and intestinal flora disorders of the patients. Moreover, the effort to find the therapeutic targets from multiple dimensions may provide new ideas for the prevention and treatment of HUA-related nephropathy.
5 Potential interventions
5.1 Diet
In recent years, studies have suggested that diet is closely related to the gut microbiota (194). Previous research analysis found that there are differences in the gut microbiota structure between healthy groups and CKD patients. CKD patients showed a decrease in the abundance of Firmicutes (128). Some studies have shown that supplementing dietary fiber can optimize the gut microbiota structure, promote the growth of glycolytic bacteria, increase the production of SCFAs, reduce gut microbiota-derived uremic toxins (such as p-cresol sulfate (p-CS), indoxyl sulfate (IS), and trimethylamine N-oxide), and have beneficial effects such as maintaining intestinal barrier integrity and mucus production to resist inflammation (195). Further animal studies have shown that a high-fiber diet can reduce kidney injury in animal models of MetS-related kidney disease (90, 196). However, there is currently a lack of relevant clinical studies to confirm the direct relationship between diet and the gut microbiota, and it is not clear how these diets affect the gut microbiota.
5.2 Probiotics, prebiotics, synbiotics, or postbiotics
Some research scholars have gradually paid attention to the prevention and treatment applications of gut microbiota and their metabolites in CKD (197). For example, well-known ones include probiotics (gut microbiota beneficial to the host), prebiotics (metabolites of gut microbiota beneficial to the host), synbiotics (gut microbiota and metabolites beneficial to the host), etc. Some studies have demonstrated through animal experiments that after the probiotic intervention, kidney inflammation and tubular cell apoptosis in mice with acute kidney injury were alleviated. Subsequently, a 1-year clinical trial was also conducted, and the results showed that probiotics slowed down the decline of kidney function in individuals with stages 3–5 CKD, both acute and chronic kidney injuries (106). A meta-analysis on the impact of probiotics on chronic kidney disease indicated that probiotics might also reduce the level of p-CS and increase the level of interleukin-6 (IL-6), thereby improving gastrointestinal symptoms (198). However, the impact of probiotics on the long-term prognosis of CKD patients remains to be studied. Additionally, a network meta-analysis of probiotics, prebiotics, and synbiotics in dialysis patients was conducted, and the results showed that prebiotics were the most effective in reducing tumor necrosis factor-α, urea, IS, and IL-6 (199). There are also newly emerging postbiotics (certain specific gut microbiota and their metabolites beneficial to the host), but the evidence regarding the application of postbiotics in kidney diseases is relatively lacking, and further research is needed (200).
5.3 FMT or smart bacteria
FMT refers to the transplantation of gut microbiota samples from healthy individuals into individuals with dysbacteriosis. In recent years, studies have found that FMT can alleviate the progression of kidney diseases (201). However, the vast majority of these studies were conducted in animal experiments, and the therapeutic efficacy has not been evaluated in clinical patients. There are relatively many applications of FMT in clinical MetS, such as diabetes, obesity, and gout (202). However, the research on FMT in MetS-related kidney diseases is still insufficient, and further studies are needed to confirm it. Additionally, some studies have found that intelligent bacteria can improve the gut microbiota structure of the body, reduce the level of uremic toxins in the body, and thus delay the progression of kidney diseases. For example, after oral administration of microcapsules containing Escherichia coli DH5 with urease-producing ability in uremic rats, the serum urea level decreased significantly (203). However, so far, no human studies have been conducted, and the issues of safety and biosafety have not been resolved (204).
5.4 SCFAs
SCFAs play important roles in influencing the body's energy homeostasis, immune function, and microbial signal transduction and have also become key risk factors affecting the development and prognosis of CKD (13). In recent years, studies have confirmed the potential therapeutic effects of SCFAs in patients with CKD. It has been found that during the process of CKD, the levels of fecal propionate and butyrate in patients gradually decrease. Through genome-wide expression assay analysis, it was found that propionate and butyrate jointly downregulated the expression of 103 genes related to the inflammatory process of tubular cells triggered by TNF-α and the activation of the immune system. Administration of propionate and butyrate either before or shortly after kidney injury in animal models prevented the occurrence and progression of kidney injury (110). Administration of butyrate intervention in a CKD mouse model has been shown to reduce kidney fibrosis (109). Interestingly, some researchers have proposed combining SCFAs with monotherapy, with SCFAs acting as drug carriers. SCFAs act on host gene expression by inhibiting HDAC inhibition (113). However, many studies have not focused on this area, and further research is still needed on the molecular mechanisms involved in the treatment of kidney diseases by SCFAs.
6 Conclusion
MetS is a group of clinical syndromes with chronic inflammation and metabolic disorders, which is an independent risk factor for CKD. Recently, the function of gut microbiota-derived SCFAs in kidney diseases has become an exciting area. Emerging evidence suggests that SCFAs have a wide range of roles in MetS-related nephropathy, including reprogramming of lipid, glucose, and UA metabolism, modulation of immune inflammation, and regulation of blood pressure. Existing clinical studies and animal experiments have indicated that SCFA treatment has potential therapeutic effects in MetS-related nephropathy, which provides prospective treatment of MetS-related nephropathy (Figure 3).
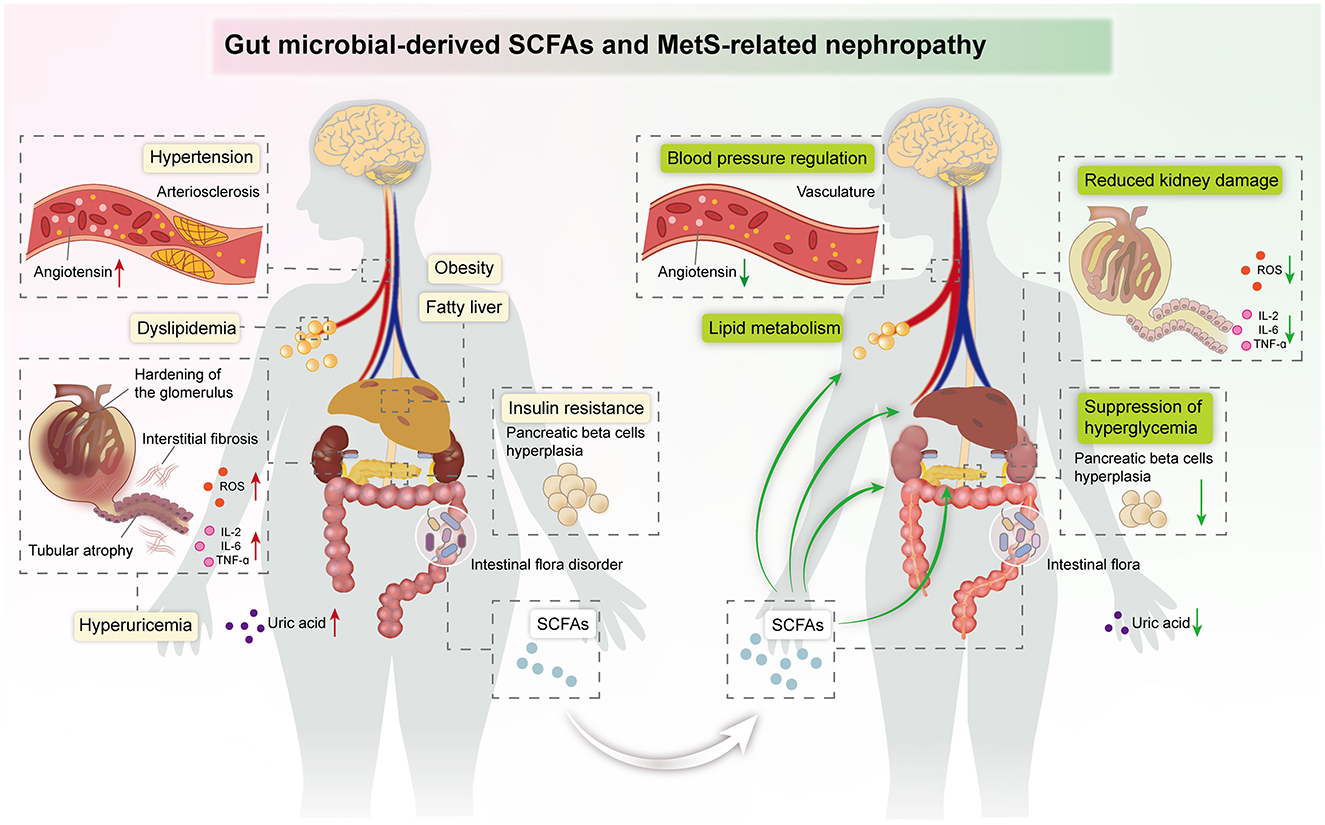
Figure 3. Summary image: SCFAs exert beneficial effects against kidney damage in the host, including regulation of blood pressure and lipid metabolism, suppression of hyperglycemia, and improvement of insulin resistance.
However, the specific role of SCFAs in MetS-related nephropathy has not yet been fully elucidated. The limitations of existing research are mainly reflected in the following aspects: First, most studies are based on animal models or clinical studies of small samples, lacking large samples and multi-center randomized controlled trials. Second, there are still some unknown issues regarding the specific mechanism of action of SCFAs, such as whether different types of SCFAs have different effects on the kidneys and how host genetics influence SCFA-mediated protection. In addition, the impact of diet-microbiota interactions (e.g., high-fat vs. high-fiber diets) on SCFA production, the interaction between SCFAs and other factors (such as diet, genetics, etc.), and its impact on the development of kidney diseases also need to be further explored. Therefore, future research directions should include more large-sample, multi-center, randomized controlled clinical trials, as well as in-depth research on the mechanism of action of SCFAs and their interaction with other factors. Furthermore, with the help of high-throughput sequencing technology and systems biology methods, the mechanism of action of different types of SCFAs in MetS-related nephropathy, as well as the impact of the overall structure and function of the gut microbiota on kidney disease, can be further investigated. The extensive efforts will allow us to gain a better understanding of the role of SCFAs in MetS-related nephropathy and give a more scientific basis for their clinical application.
Author contributions
XT: Writing – original draft, Writing – review & editing, Funding acquisition. LS: Writing – original draft. SG: Writing – original draft. LY: Writing – review & editing, Supervision. TZ: Writing – review & editing. CH: Writing – original draft. TH: Writing – original draft. QJ: Writing – review & editing, Supervision. YZ: Supervision, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from Science and Technology of Guizhou Province (Guizhou Science and Technology Foundation-ZK [2024] General 679) and Zunyi City Science and Technology Planning Project (Zunyi Kehe HZ Zi [2023] No. 11).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Endocr Rev. (2008) 29:777–822. doi: 10.1210/er.2008-0024
2. Gluvic Z, Zaric B, Resanovic I, Obradovic M, Mitrovic A, Radak D, et al. Link between metabolic syndrome and insulin resistance. Curr Vasc Pharmacol. (2017) 15:30–9. doi: 10.2174/1570161114666161007164510
3. Zhang X, Lerman LO. The metabolic syndrome and chronic kidney disease. Transl Res. (2017). 183:14–25. doi: 10.1016/j.trsl.2016.12.004
4. Arenas-Gómez CM, Garcia-Gutierrez E, Escobar JS, Cotter PD. Human gut homeostasis and regeneration: the role of the gut microbiota and its metabolites. Crit Rev Microbiol. (2023) 49:764–85. doi: 10.1080/1040841X.2022.2142088
5. Perler BK, Friedman ES, Wu GD. The role of the gut microbiota in the relationship between diet and human health. Annu Rev Physiol. (2023) 85:449–68. doi: 10.1146/annurev-physiol-031522-092054
6. Debnath N, Kumar R, Kumar A, Mehta PK, Yadav AK. Gut-microbiota derived bioactive metabolites and their functions in host physiology. Biotechnol Genet Eng Rev. (2021) 37:105–53. doi: 10.1080/02648725.2021.1989847
7. Morrison D J, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. (2016) 7:189–200. doi: 10.1080/19490976.2015.1134082
8. Patil RS, Tupe RS. Communal interaction of glycation and gut microbes in diabetes mellitus, Alzheimer's disease, and Parkinson's disease pathogenesis. Med Res Rev. (2023) 44:365–405. doi: 10.1002/med.21987
9. Yang F, Chen H, Gao Y, An N, Li X, Pan X, et al. Gut microbiota-derived short-chain fatty acids and hypertension: Mechanism and treatment. Biomed Pharmacother. (2020) 130:110503. doi: 10.1016/j.biopha.2020.110503
10. Hou H, Chen D, Zhang K, Zhang W, Liu T, Wang S, et al. Gut microbiota-derived short-chain fatty acids and colorectal cancer: ready for clinical translation? Cancer Lett. (2022) 526:225–35. doi: 10.1016/j.canlet.2021.11.027
11. Esgalhado M, Kemp JA, Damasceno NR, Fouque D, Mafra D. Short-chain fatty acids: a link between prebiotics and microbiota in chronic kidney disease. Future Microbiol. (2017). 12(1413-1425). doi: 10.2217/fmb-2017-0059
12. Li LZ, Tao SB, Ma L, Fu P. Roles of short-chain fatty acids in kidney diseases. Chinese Med J. (2019) 132:1228–32. doi: 10.1097/CM9.0000000000000228
13. He M, Wei W, Zhang Y, Xiang Z, Peng D, Kasimumali A, et al. Gut microbial metabolites SCFAs and chronic kidney disease. J Transl Med. (2024) 22:172. doi: 10.1186/s12967-024-04974-6
14. Li Y, Qin GQ, Wang WY, Liu X, Gao XQ, Liu JH, et al. Short chain fatty acids for the risk of diabetic nephropathy in type 2 diabetes patients. Acta Diabetol. (2022) 59:901–9. doi: 10.1007/s00592-022-01870-7
15. Li H, Zhang L, Huang R, Ren Q, Guo F, Shi M, et al. Sichuan dark tea-based medicated dietary formula improves obesity-induced renal lipid metabolism disorder in mice by remodeling gut microbiota and short-chain fatty acid metabolism. Sichuan Da Xue Xue Bao Yi Xue Ban. (2023) 54:1112−20. doi: 10.12182/20231160208
16. Chen YJ, Guo ZT, Chen HQ, Zhang SF, Bao YX, Xie Z, et al. Salinomycin, a potent inhibitor of XOD and URAT1, ameliorates hyperuricemic nephropathy by activating NRF2, modulating the gut microbiota, and promoting SCFA production. Chem Biol Interact. (2024) 403:111220. doi: 10.1016/j.cbi.2024.111220
17. Expert Panel on Detection, Evaluation, and and Treatment of High Blood Cholesterol in Adults. Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA. (2001) 285:2486–97. doi: 10.1001/jama.285.19.2486
18. Schlaich M, Straznicky N, Lambert E. Lambert G. Metabolic syndrome: a sympathetic disease? Lancet Diabetes Endocrinol. (2015) 3:148–57. doi: 10.1016/S2213-8587(14)70033-6
19. Despres JP, Lemieux I. Abdominal obesity and metabolic syndrome. Nature. (2006) 444:881–7. doi: 10.1038/nature05488
20. Grundy SM. Metabolic syndrome: connecting and reconciling cardiovascular and diabetes worlds. J Am Coll Cardiol. (2006) 47:1093–100. doi: 10.1016/j.jacc.2005.11.046
21. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. (2005) 365:1415–28. doi: 10.1016/S0140-6736(05)66378-7
22. Grundy SM. Pre-diabetes, metabolic syndrome, and cardiovascular risk. J Am Coll Cardiol. (2012) 59:635–43. doi: 10.1016/j.jacc.2011.08.080
23. Hudish LI, Reusch JE, Sussel L. beta Cell dysfunction during progression of metabolic syndrome to type 2 diabetes. J Clin Invest. (2019) 129:4001–8. doi: 10.1172/JCI129188
24. Bishehsari F, Voigt RM, Keshavarzian A. Circadian rhythms and the gut microbiota: from the metabolic syndrome to cancer. Nat Rev Endocrinol. (2020) 16:731–9. doi: 10.1038/s41574-020-00427-4
25. Locatelli F, Pozzoni P, Del Vecchio L. Renal manifestations in the metabolic syndrome. J Am Soc Nephrol. (2006) 17: S81-85. doi: 10.1681/ASN.2005121332
26. Scurt FG, Ganz MJ, Herzog C, Bose K, Mertens PR, Chatzikyrkou C, et al. Association of metabolic syndrome and chronic kidney disease. Obes Rev. (2024) 25:e13649. doi: 10.1111/obr.13649
27. Luk AO, So WY, Ma RC, Kong AP, Ozaki R, Ng VS, et al. Metabolic syndrome predicts new onset of chronic kidney disease in 5,829 patients with type 2 diabetes: a 5-year prospective analysis of the Hong Kong Diabetes Registry. Diabetes Care. (2008) 31:2357–61. doi: 10.2337/dc08-0971
28. Zhang L, Wang F, Wang L, Wang W, Liu B, Liu J, et al. Prevalence of chronic kidney disease in China: a cross-sectional survey. Lancet. (2012) 379:815–22. doi: 10.1016/S0140-6736(12)60033-6
29. Song H, Wang X, Cai Q, Ding W, Huang S, Zhuo L, et al. Association of metabolic syndrome with decreased glomerular filtration rate among 75,468 Chinese adults: a cross-sectional study. PLoS ONE. (2014) 9:e113450. doi: 10.1371/journal.pone.0113450
30. Navaneethan SD, Schold JD, Kirwan JP, Arrigain S, Jolly SE, Poggio ED, et al. Metabolic syndrome, ESRD, and death in CKD. Clin J Am Soc Nephrol. (2013) 8:945–52. doi: 10.2215/CJN.09870912
31. Huh JH, Yadav D, Kim JS, Son JW, Choi E, Kim SH, et al. An association of metabolic syndrome and chronic kidney disease from a 10-year prospective cohort study. Metabolism. (2017) 67:54–61. doi: 10.1016/j.metabol.2016.11.003
32. Kittiskulnam P, Thokanit NS, Katavetin P, Susanthitaphong P, Srisawat N, Praditpornsilpa K, et al. The magnitude of obesity and metabolic syndrome among diabetic chronic kidney disease population: a nationwide study. PLoS ONE. (2018) 13:e0196332. doi: 10.1371/journal.pone.0196332
33. Xie K, Bao L, Jiang X, Ye Z, Bing J, Dong Y, et al. The association of metabolic syndrome components and chronic kidney disease in patients with hypertension. Lipids Health Dis. (2019) 18:229. doi: 10.1186/s12944-019-1121-5
34. Wang Y, Sun B, Sheng LT, Pan XF, Zhou Y, Zhu J, et al. Association between weight status, metabolic syndrome, and chronic kidney disease among middle-aged and elderly Chinese. Nutr Metab Cardiovasc Dis. (2020) 30:2017–26. doi: 10.1016/j.numecd.2020.06.025
35. Chu SC, Wang PH, Lu KY, Ko CC, She YH, Lee CC, et al. Relationships between metabolic body composition status and rapid kidney function decline in a community-based population: a prospective observational study. Front Public Health. (2022) 10:895787. doi: 10.3389/fpubh.2022.895787
36. Fanaei SM, Mehran L, Amouzegar A, Masoumi S, Amouzegar A, Azizi F. The impact of metabolic syndrome on chronic kidney disease development. Insights from a big prospective study. Eur J Clin Invest. (2023) 53:e13945. doi: 10.1111/eci.13945
37. Wu M, Shu Y, Wang L, Song L, Chen S, Liu Y, et al. Metabolic syndrome severity score and the progression of CKD. Eur J Clin Invest. (2022) 52:e13646. doi: 10.1111/eci.13646
38. DeBoer MD, Filipp SL, Musani SK, Sims M, Okusa MD, Gurka M. Metabolic syndrome severity and risk of CKD and Worsened GFR: the jackson heart study. Kidney Blood Press Res. (2018) 43:555–67. doi: 10.1159/000488829
39. Pammer LM, Lamina C, Schultheiss UT, Kotsis F, Kollerits B, Stockmann H, et al. Association of the metabolic syndrome with mortality and major adverse cardiac events: a large chronic kidney disease cohort. J Intern Med. (2021) 290:1219–32. doi: 10.1111/joim.13355
40. Xiao H, Shao X, Gao P, Zou H, Zhang X. Metabolic syndrome components and chronic kidney disease in a community population aged 40 years and older in southern china: a cross-sectional study. Diabetes Metab Syndr Obes. (2022) 15:839–48. doi: 10.2147/DMSO.S353305
41. Xu L, Liu J, Li D, Yang H, Zhou Y, Yang J, et al. Association between metabolic syndrome components and chronic kidney disease among 37,533 old Chinese individuals. Int Urol Nephrol. (2022) 54:1445–54. doi: 10.1007/s11255-021-03013-3
42. Ciardullo S, Ballabeni C, Trevisan R, Perseghin G. Perseghin G. Metabolic syndrome, and not obesity, is associated with chronic kidney disease. Am J Nephrol. (2021) 52:666–72. doi: 10.1159/000518111
43. Artunc F, Schleicher E, Weigert C, Fritsche A, Stefan N, Häring HU, et al. The impact of insulin resistance on the kidney and vasculature. Nat Rev Nephrol. (2016) 12:721–37. doi: 10.1038/nrneph.2016.145
44. Ohashi Y, Thomas G, Nurko S, Stephany B, Fatica R, Chiesa A, et al. Association of metabolic syndrome with kidney function and histology in living kidney donors. Am J Transplant. (2013) 13:2342–51. doi: 10.1111/ajt.12369
45. Joyce T, Chirino YI, Natalia MT, Jose PC. Renal damage in the metabolic syndrome (MetSx): disorders implicated. Eur J Pharmacol. (2018) 818:554–68. doi: 10.1016/j.ejphar.2017.11.032
46. Chen J, Kong X, Jia X, Li W, Wang Z, Cui M, et al. Association between metabolic syndrome and chronic kidney disease in a Chinese urban population. Clinica Chimica Acta. (2017) 470:103–8. doi: 10.1016/j.cca.2017.05.012
47. Andres-Hernando A, Orlicky DJ, Cicerchi C, Kuwabara M, Garcia GE, Nakagawa T, et al. High fructose corn syrup accelerates kidney disease and mortality in obese mice with metabolic syndrome. Biomolecules. (2023) 13:780. doi: 10.3390/biom13050780
48. Eirin A, Zhu XY, Puranik AS, Tang H, McGurren KA, van Wijnen AJ, et al. Mesenchymal stem cell-derived extracellular vesicles attenuate kidney inflammation. Kidney Int. (2017) 92:114–24. doi: 10.1016/j.kint.2016.12.023
49. Rodríguez RR, González-Bulnes A, Garcia-Contreras C, Elena Rodriguez-Rodriguez A, Astiz S, Vazquez-Gomez M, et al. The Iberian pig fed with high-fat diet: a model of renal disease in obesity and metabolic syndrome. Int J Obes. (2020) 44:457–65. doi: 10.1038/s41366-019-0434-9
50. Eley VA, Thuzar M, Navarro S, Dodd BR, van Zundert AA. Obesity, metabolic syndrome, and inflammation: An update for anaesthetists caring for patients with obesity. Anaesth Crit Care Pain Med. (2021) 40:100947. doi: 10.1016/j.accpm.2021.100947
51. Gherghina ME, Peride I, Tiglis M, Neagu TP, Niculae A, Checherita IA. Uric Acid and Oxidative Stress-Relationship with Cardiovascular, Metabolic, and Renal Impairment. Int J Mol Sci. (2022) 23:3188. doi: 10.3390/ijms23063188
52. Eid BG, Neamatallah T, Hanafy A, El-Bassossy HM, Binmahfouz L, Aldawsari HM, et al. Interference with TGFbeta1-mediated inflammation and fibrosis underlies reno-protective effects of the CB1 receptor neutral antagonists AM6545 and AM4113 in a rat model of metabolic syndrome. Molecules. (2021) 26:866. doi: 10.3390/molecules26040866
53. Qiu D, Song S, Chen N, Bian Y, Yuan C, Zhang W, et al. NQO1 alleviates renal fibrosis by inhibiting the TLR4/NF-kappaB and TGF-beta/Smad signaling pathways in diabetic nephropathy. Cell Signal. (2023) 108:110712. doi: 10.1016/j.cellsig.2023.110712
54. Overstreet JM, Gifford CC, Tang J, Higgins PJ, Samarakoon R. Emerging role of tumor suppressor p53 in acute and chronic kidney diseases. Cell Mol Life Sci. (2022) 79:474. doi: 10.1007/s00018-022-04505-w
55. Li L, Yang J, Li F, Gao F, Zhu L, Hao J, et al. FBXW7 mediates high glucose-induced SREBP-1 expression in renal tubular cells of diabetic nephropathy under PI3K/Akt pathway regulation. Mol Med Rep. (2021) 23:233. doi: 10.3892/mmr.2021.11872
56. Van Krieken R, Marway M, Parthasarathy P, Mehta N, Ingram AJ, Gao B, et al. Inhibition of SREBP with fatostatin does not attenuate early diabetic nephropathy in male mice. Endocrinology. (2018) 159:1479–95. doi: 10.1210/en.2018-00093
57. Liu GW, Zeng JE, Li LF. Correlation analysis of serum IGF-1 and IL-6 and urinary albumin/creatinine ratio in patients with type 2 diabetic kidney disease. Front Endocrinol. (2022) 13:1082492. doi: 10.3389/fendo.2022.1082492
58. Toda N, Mukoyama M, Yanagita M, Yokoi H. CTGF in kidney fibrosis and glomerulonephritis. Inflammat Regenerat. (2018) 38:14. doi: 10.1186/s41232-018-0070-0
59. Cai T, Ke Q, Fang Y, Wen P, Chen H, Yuan Q, et al. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1alpha-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis. (2020) 11:390. doi: 10.1038/s41419-020-2544-7
60. Li Z, Guo H, Li J, Ma T, Zhou S, Zhang Z, et al. Sulforaphane prevents type 2 diabetes-induced nephropathy via AMPK-mediated activation of lipid metabolic pathways and Nrf2 antioxidative function. Clin Sci. (2020) 134:2469–87. doi: 10.1042/CS20191088
61. Rampanelli E, Orsó E, Ochodnicky P, Liebisch G, Bakker PJ, Claessen N, et al. Metabolic injury-induced NLRP3 inflammasome activation dampens phospholipid degradation. Sci Rep. (2017) 7:2861. doi: 10.1038/s41598-017-01994-9
62. Ren C, Bao X, Lu X, Du W, Wang X, Wei J, et al. Complanatoside A targeting NOX4 blocks renal fibrosis in diabetic mice by suppressing NLRP3 inflammasome activation and autophagy. Phytomedicine. (2022) 104:154310. doi: 10.1016/j.phymed.2022.154310
63. Plata C, Cruz C, Cervantes LG, Ramírez V. The gut microbiota and its relationship with chronic kidney disease. Int Urol Nephrol. (2019) 51:2209–26. doi: 10.1007/s11255-019-02291-2
64. Yu W, Shang J, Guo R, Zhang F, Zhang W, Zhang Y, et al. The gut microbiome in differential diagnosis of diabetic kidney disease and membranous nephropathy. Ren Fail. (2020) 42:1100–10. doi: 10.1080/0886022X.2020.1837869
65. Lin L, Tan W, Pan X, Tian E, Wu Z, Yang J. Metabolic syndrome-related kidney injury: a review and update. Front Endocrinol. (2022) 13:904001. doi: 10.3389/fendo.2022.904001
66. Han J, Wang X, Tang S, Lu C, Wan H, Zhou J, et al. Protective effects of tuna meat oligopeptides (TMOP) supplementation on hyperuricemia and associated renal inflammation mediated by gut microbiota. FASEB J. (2020) 34:5061–76. doi: 10.1096/fj.201902597RR
67. Kamada N, Seo SU, Chen GY, Núñez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. (2013) 13:321–35. doi: 10.1038/nri3430
68. Giambra V, Pagliari D, Rio P, Totti B, Di Nunzio C, Bosi A, et al. Gut microbiota, inflammatory bowel disease, and cancer: the role of guardians of innate immunity. Cells. (2023) 12:2654. doi: 10.3390/cells12222654
69. Almeida A, Nayfach S, Boland M, Strozzi F, Beracochea M, Shi ZJ, et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat Biotechnol. (2021) 39:105–14. doi: 10.1038/s41587-020-0603-3
70. Huang Y, Xin W, Xiong J, Yao M, Zhang B, Zhao J. The intestinal microbiota and metabolites in the gut-kidney-heart axis of chronic kidney disease. Front Pharmacol. (2022) 13:837500. doi: 10.3389/fphar.2022.837500
71. Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. (2010) 90:859–904. doi: 10.1152/physrev.00045.2009
72. Gomaa EZ. Human gut microbiota/microbiome in health and diseases: a review. Antonie Van Leeuwenhoek. (2020) 113:2019–40. doi: 10.1007/s10482-020-01474-7
73. Feng Z, Zhang Y, Lai Y, Jia C, Wu F, Chen D. Causal relationship between gut microbiota and kidney diseases: a two-sample Mendelian randomization study. Front Immunol. (2023) 14:1277554. doi: 10.3389/fimmu.2023.1277554
74. Rysz J, Franczyk B, Ławiński J, Olszewski R, Ciałkowska-Rysz A, Gluba-Brzózka A. The impact of CKD on uremic toxins and gut microbiota. Toxins. (2021) 13:252. doi: 10.3390/toxins13040252
75. Lohia S, Vlahou A, Zoidakis J. Microbiome in Chronic Kidney Disease (CKD): an omics perspective. Toxins. (2022) 14:176. doi: 10.3390/toxins14030176
76. Yang K, Du G, Liu J, Zhao S, Dong W, Zhao S, et al. Gut microbiota and neonatal acute kidney injury biomarkers. Pediatr Nephrol. (2023) 38:3529–47. doi: 10.1007/s00467-023-05931-z
77. Saranya GR, Viswanathan P. Gut microbiota dysbiosis in AKI to CKD transition. Biomed Pharmacother. (2023) 161:114447. doi: 10.1016/j.biopha.2023.114447
78. Miller AW, Penniston KL, Fitzpatrick K, Agudelo J, Tasian G, Lange D. Mechanisms of the intestinal and urinary microbiome in kidney stone disease. Nat Rev Urol. (2022) 19:695–707. doi: 10.1038/s41585-022-00647-5
79. Fan Y, Wang Y, Xiao H, Sun H. Sun H. Advancements in understanding the role of intestinal dysbacteriosis mediated mucosal immunity in IgA nephropathy. BMC Nephrol. (2024) 25:203. doi: 10.1186/s12882-024-03646-3
80. Pomare EW, Branch WJ, Cummings JH. Carbohydrate fermentation in the human colon and its relation to acetate concentrations in venous blood. J Clin Invest. (1985) 75:1448–54. doi: 10.1172/JCI111847
81. Boets E, Gomand SV, Deroover L, Preston T, Vermeulen K, De Preter V, et al. Systemic availability and metabolism of colonic-derived short-chain fatty acids in healthy subjects: a stable isotope study. J Physiol. (2017) 595:541–55. doi: 10.1113/JP272613
82. Zhang S, Wang H, Zhu MJ. A sensitive GC/MS detection method for analyzing microbial metabolites short chain fatty acids in fecal and serum samples. Talanta. (2019) 196:249–254. doi: 10.1016/j.talanta.2018.12.049
83. Carley AN, Maurya SK, Fasano M, Wang Y, Selzman CH, Drakos SG, et al. Short-chain fatty acids outpace ketone oxidation in the failing heart. Circulation. (2021) 143:1797–808. doi: 10.1161/CIRCULATIONAHA.120.052671
84. Huang W, Man Y, Gao C, Zhou L, Gu J, Xu H, et al. Short-chain fatty acids ameliorate diabetic nephropathy via GPR43-mediated inhibition of oxidative stress and NF-kappaB signaling. Oxid Med Cell Longev. (2020) 2020:4074832. doi: 10.1155/2020/4074832
85. Schwarz A, Bruhs A, Schwarz T. The short-chain fatty acid sodium butyrate functions as a regulator of the skin immune system. J Invest Dermatol. (2017) 137:855–64. doi: 10.1016/j.jid.2016.11.014
86. Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, et al. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc Natl Acad Sci U S A. (2011) 108:8030–5. doi: 10.1073/pnas.1016088108
87. Andrade-Oliveira V, Amano MT, Correa-Costa M, Castoldi A, Felizardo RJ, de Almeida DC, et al. Gut bacteria products prevent AKI induced by ischemia-reperfusion. J Am Soc Nephrol. (2015) 26:1877–88. doi: 10.1681/ASN.2014030288
88. Ulven T. Short-chain free fatty acid receptors FFA2/GPR43 and FFA3/GPR41 as new potential therapeutic targets. Front Endocrinol. (2012) 3:111. doi: 10.3389/fendo.2012.00111
89. Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology. (2013) 145:396–406. doi: 10.1053/j.gastro.2013.04.056
90. Li YJ, Chen X, Kwan TK, Loh YW, Singer J, Liu Y, et al. Dietary fiber protects against diabetic nephropathy through short-chain fatty acid-mediated activation of g protein-coupled receptors GPR43 and GPR109A. J Am Soc Nephrol. (2020) 31:1267–81. doi: 10.1681/ASN.2019101029
91. Deehan EC, Yang C, Perez-Muñoz ME, Nguyen NK, Cheng CC, Triador L, et al. Precision microbiome modulation with discrete dietary fiber structures directs short-chain fatty acid production. Cell Host Microbe. (2020) 27:389–404. doi: 10.1016/j.chom.2020.01.006
92. Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F. Bäckhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell. (2016) 165:1332–45. doi: 10.1016/j.cell.2016.05.041
93. Huang W, Zhou L, Guo H, Xu Y, Xu Y. The role of short-chain fatty acids in kidney injury induced by gut-derived inflammatory response. Metabolism. (2017) 68:20–30. doi: 10.1016/j.metabol.2016.11.006
94. Gryp T, De Paepe K, Vanholder R, Kerckhof FM, Van Biesen W, Van de Wiele T, et al. Gut microbiota generation of protein-bound uremic toxins and related metabolites is not altered at different stages of chronic kidney disease. Kidney Int. (2020) 97:1230–42. doi: 10.1016/j.kint.2020.01.028
95. Lucas S, Omata Y, Hofmann J, Böttcher M, Iljazovic A, Sarter K, et al. Short-chain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat Commun. (2018) 9:55. doi: 10.1038/s41467-017-02490-4
96. Farup P G, Valeur J. Changes in faecal short-chain fatty acids after weight-loss interventions in subjects with morbid obesity. Nutrients. (2020) 12:802. doi: 10.3390/nu12030802
97. Jiao A, Yu B, He J, Yu J, Zheng P, Luo Y, et al. Short chain fatty acids could prevent fat deposition in pigs via regulating related hormones and genes. Food Funct. (2020) 11:1845–55. doi: 10.1039/C9FO02585E
98. Lu Y, Fan C, Li P, Lu Y, Chang X, Qi K. Short chain fatty acids prevent high-fat-diet-induced obesity in mice by regulating G protein-coupled receptors and gut microbiota. Sci Rep. (2016) 6:37589. doi: 10.1038/srep37589
99. Huart J, Leenders J, Taminiau B, Descy J, Saint-Remy A, Daube G, et al. Gut microbiota and fecal levels of short-chain fatty acids differ upon 24-hour blood pressure levels in men. Hypertension. (2019) 74:1005–13. doi: 10.1161/HYPERTENSIONAHA.118.12588
100. Chang Y, Chen Y, Zhou Q, Wang C, Chen L, Di W, et al. Short-chain fatty acids accompanying changes in the gut microbiome contribute to the development of hypertension in patients with preeclampsia. Clin Sci. (2020) 134:289–302. doi: 10.1042/CS20191253
101. Roy R, Nguyen-Ngo C, Lappas M. Short-chain fatty acids as novel therapeutics for gestational diabetes. J Mol Endocrinol. (2020) 65:21–34. doi: 10.1530/JME-20-0094
102. Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc Natl Acad Sci U S A. (2013) 110:4410–5. doi: 10.1073/pnas.1215927110
103. Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci U S A. (2008) 105:16767–72. doi: 10.1073/pnas.0808567105
104. Wu D, Cao M, Li N, Zhang A, Yu Z, Cheng J, et al. Effect of trimethylamine N-oxide on inflammation and the gut microbiota in Helicobacter pylori-infected mice. Int Immunopharmacol. (2020) 81:106026. doi: 10.1016/j.intimp.2019.106026
105. Sato N, Kakuta M, Hasegawa T, Yamaguchi R, Uchino E, Murashita K, et al. Metagenomic profiling of gut microbiome in early chronic kidney disease. Nephrol Dial Transplant. (2021) 36:1675–84. doi: 10.1093/ndt/gfaa122
106. Zhu H, Cao C, Wu Z, Zhang H, Sun Z, Wang M, et al. The probiotic L. casei Zhang slows the progression of acute and chronic kidney disease. Cell Metab. (2021) 33:1926–42. doi: 10.1016/j.cmet.2021.06.014
107. Mikami D, Kobayashi M, Uwada J, Yazawa T, Kamiyama K, Nishimori K, et al. Short-chain fatty acid mitigates adenine-induced chronic kidney disease via FFA2 and FFA3 pathways. Biochim Biophys Acta Mol Cell Biol Lipids. (2020) 1865:158666. doi: 10.1016/j.bbalip.2020.158666
108. Wang S, Lv D, Jiang S, Jiang J, Liang M, Hou F, et al. Quantitative reduction in short-chain fatty acids, especially butyrate, contributes to the progression of chronic kidney disease. Clin Sci). (2019) 133:1857–70. doi: 10.1042/CS20190171
109. Tian X, Zeng Y, Tu Q, Jiao Y, Yao S, Chen Y, et al. Butyrate alleviates renal fibrosis in CKD by regulating NLRP3-mediated pyroptosis via the STING/NF-kappaB/p65 pathway. Int Immunopharmacol. (2023) 124:111010. doi: 10.1016/j.intimp.2023.111010
110. Corte-Iglesias V, Saiz ML, Andrade-Lopez AC, Salazar N, Bernet CR, Martin-Martin C, et al. Propionate and butyrate counteract renal damage and progression to chronic kidney disease. Nephrol Dial Transplant. (2024) 40:133–50. doi: 10.1093/ndt/gfae118
111. Liu J, Gu QH, Cui Z, Zhao MH, Jia XY. Short-chain fatty acids ameliorate experimental anti-glomerular basement membrane disease. Clini immunol. (2024) 259:109903. doi: 10.1016/j.clim.2024.109903
112. Portincasa P, Bonfrate L, Vacca M, De Angelis M, Farella I, Lanza E, et al. Gut microbiota and short chain fatty acids: implications in glucose homeostasis. Int J Mol Sci. (2022) 23:1105. doi: 10.3390/ijms23031105
113. Rekha K, Venkidasamy B, Samynathan R, Nagella P, Rebezov M, Khayrullin M, et al. Short-chain fatty acid: an updated review on signaling, metabolism, and therapeutic effects. Crit Rev Food Sci Nutr. (2022) 64:2461–89. doi: 10.1080/10408398.2022.2124231
114. Marchix J, Goddard G, Helmrath MA. Host-gut microbiota crosstalk in intestinal adaptation. Cell Mol Gastroenterol Hepatol. (2018) 6:149–62. doi: 10.1016/j.jcmgh.2018.01.024
115. Di Ciaula A, Baj J, Garruti G, Celano G, De Angelis M, Wang HH, et al. Liver steatosis, gut-liver axis, microbiome and environmental factors. A never-ending bidirectional cross-talk. J Clini Med. (2020) 9:2648. doi: 10.3390/jcm9082648
116. Magliocca G, Mone P, Di Iorio BR, Heidland A, Marzocco S. Short-chain fatty acids in chronic kidney disease: focus on inflammation and oxidative stress regulation. Int J Mol Sci. (2022) 23:5354. doi: 10.3390/ijms23105354
117. Holle J, Bartolomaeus H, Löber U, Behrens F, Bartolomaeus TUP, Anandakumar H, et al. Inflammation in children with CKD linked to gut dysbiosis and metabolite imbalance. J Am Soc Nephrol. (2022) 33:2259–75. doi: 10.1681/ASN.2022030378
118. Kim H, Nam BY, Park J, Song S, Kim WK, Lee K, et al. Lactobacillus acidophilus KBL409 reduces kidney fibrosis via immune modulatory effects in mice with chronic kidney disease. Mol Nutr Food Res. (2022) 66:e2101105. doi: 10.1002/mnfr.202101105
119. Harrington JS, Ryter SW, Plataki M, Price DR, Choi AMK. Mitochondria in health, disease, and aging. Physiol Rev. (2023) 103:2349–422. doi: 10.1152/physrev.00058.2021
120. Neel DV, Basu H, Gunner G, Bergstresser MD, Giadone RM, Chung H, et al. Gasdermin-E mediates mitochondrial damage in axons and neurodegeneration. Neuron. (2023) 111:1222–40. doi: 10.1016/j.neuron.2023.02.019
121. Nicese MN, Bijkerk R, Van Zonneveld AJ, Van den Berg BM, Rotmans JI. Sodium butyrate as key regulator of mitochondrial function and barrier integrity of human glomerular endothelial cells. Int J Mol Sci. (2023) 24:13090. doi: 10.3390/ijms241713090
122. Kawabata C, Hirakawa Y, Inagi R, Nangaku M. Nangaku M. Acetate attenuates kidney fibrosis in an oxidative stress-dependent manner. Physiol Rep. (2023) 11:e15774. doi: 10.14814/phy2.15774
123. Marshall CB. Rethinking glomerular basement membrane thickening in diabetic nephropathy: adaptive or pathogenic? Am J Physiol Renal Physiol. (2016) 311:F831–43. doi: 10.1152/ajprenal.00313.2016
124. Gaddy A, Elrggal M, Madariaga H, Kelly A, Lerma E, Colbert GB. Diabetic kidney disease. Dis Mon. (2025) 71:101848. doi: 10.1016/j.disamonth.2024.101848
125. Kawanami D, Matoba K, Utsunomiya K. Signaling pathways in diabetic nephropathy. Histol Histopathol. (2016) 31:1059–67. doi: 10.1186/s41100-016-0028-0
126. Tuttle K R. Linking metabolism and immunology: diabetic nephropathy is an inflammatory disease. J Am Soc Nephrol. (2005) 16:1537–8. doi: 10.1681/ASN.2005040393
127. Dwivedi S, Sikarwar MS. Diabetic nephropathy: pathogenesis, mechanisms, and therapeutic strategies. Horm Metab Res. (2025) 57:7–17. doi: 10.1055/a-2435-8264
128. Wang Y, Zhao J, Qin Y, Yu Z, Zhang Y, Ning X, et al. The specific alteration of gut microbiota in diabetic kidney diseases-a systematic review and meta-analysis. Front Immunol. (2022) 13:908219. doi: 10.3389/fimmu.2022.908219
129. Han S, Chen M, Cheng P, Zhang Z, Lu Y, Xu Y, et al. A systematic review and meta-analysis of gut microbiota in diabetic kidney disease: comparisons with diabetes mellitus, non-diabetic kidney disease, and healthy individuals. Front Endocrinol. (2022) 13:1018093. doi: 10.3389/fendo.2022.1018093
130. Tian E, Wang F, Zhao L, Sun Y, Yang J. The pathogenic role of intestinal flora metabolites in diabetic nephropathy. Front Physiol. (2023) 14:1231621. doi: 10.3389/fphys.2023.1231621
131. Zhong C, Dai Z, Chai L, Wu L, Li J, Guo W, et al. The change of gut microbiota-derived short-chain fatty acids in diabetic kidney disease. J Clin Lab Anal. (2021) 2021:e24062. doi: 10.21203/rs.3.rs-378090/v1
132. Moon S, Tsay JJ, Lampert H, Md Dom ZI, Kostic AD, Smiles A, et al. Circulating short and medium chain fatty acids are associated with normoalbuminuria in type 1 diabetes of long duration. Sci Rep. (2021) 11:8592. doi: 10.1038/s41598-021-87585-1
133. Du Y, Yang YT, Tang G, Jia JS, Zhu N, Yuan WJ. Butyrate alleviates diabetic kidney disease by mediating the miR-7a-5p/P311/TGF-beta1 pathway. FASEB J. (2020) 34:10462–75. doi: 10.1096/fj.202000431R
134. Everard A, Cani P D. Gut microbiota and GLP-1. Rev Endocr Metab Disord. (2014) 15:189–96. doi: 10.1007/s11154-014-9288-6
135. Mandaliya D K, Seshadri S. Short chain fatty acids, pancreatic dysfunction and type 2 diabetes. Pancreatology. (2019) 19:280–4. doi: 10.1016/j.pan.2019.01.021
136. Gu J, Huang W, Zhang W, Zhao T, Gao C, Gan W, et al. Sodium butyrate alleviates high-glucose-induced renal glomerular endothelial cells damage via inhibiting pyroptosis. Int Immunopharmacol. (2019) 75:105832. doi: 10.1016/j.intimp.2019.105832
137. Tang G, Du Y, Guan H, Jia J, Zhu N, Shi Y, et al. Butyrate ameliorates skeletal muscle atrophy in diabetic nephropathy by enhancing gut barrier function and FFA2-mediated PI3K/Akt/mTOR signals. Br J Pharmacol. (2021) 179:159–78. doi: 10.1111/bph.15693
138. Du Y, Tang G, Yuan W. Suppression of HDAC2 by sodium butyrate alleviates apoptosis of kidney cells in db/db mice and HGinduced NRK52E cells. Int J Mol Med. (2020) 45:210–22. doi: 10.3892/ijmm.2019.4397
139. Lu CC, Hu ZB, Wang R, Hong ZH, Lu J, Chen PP, et al. Gut microbiota dysbiosis-induced activation of the intrarenal renin-angiotensin system is involved in kidney injuries in rat diabetic nephropathy. Acta Pharmacol Sin. (2020) 41:1111–8. doi: 10.1038/s41401-019-0326-5
140. Vinolo MA, Rodrigues HG, Nachbar RT, Curi R. Regulation of inflammation by short chain fatty acids. Nutrients. (2011) 3:858–76. doi: 10.3390/nu3100858
141. Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, et al. Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature. (2016) 534:213–7. doi: 10.1038/nature18309
142. Weisinger JR, Kempson RL, Eldridge FL, Swenson RS. The nephrotic syndrome: a complication of massive obesity. Ann Intern Med. (1974) 81:440–7. doi: 10.7326/0003-4819-81-4-440
143. Kovesdy CP, Furth SL, Zoccali C, World Kidney Day Steering Committee. Obesity and kidney disease: Hidden consequences of the epidemic. Saudi J Kidney Dis Transpl. (2017) 28:241–52. doi: 10.4103/1319-2442.202776
144. Kounatidis D, Vallianou NG, Stratigou T, Voukali M, Karampela I, Dalamaga M. The kidney in obesity: current evidence, perspectives and controversies. Curr Obes Rep. (2024) 13:680–702. doi: 10.1007/s13679-024-00583-y
145. Câmara NO, Iseki K, Kramer H, Liu ZH, Sharma K. Kidney disease and obesity: epidemiology, mechanisms and treatment. Nat Rev Nephrol. (2017) 13:181–90. doi: 10.1038/nrneph.2016.191
146. Jacob P, Mccafferty K. Assessment and management of chronic kidney disease in people living with obesity. Clin Med. (2023) 23:353–6. doi: 10.7861/clinmed.2023-0195
147. Wahba I M, Mak RH. Obesity and obesity-initiated metabolic syndrome: mechanistic links to chronic kidney disease. Clin J Am Soc Nephrol. (2007) 2:550–62. doi: 10.2215/CJN.04071206
148. Munoz-Garach A, Diaz-Perdigones C, Tinahones FJ. Gut microbiota and type 2 diabetes mellitus. Endocrinol Nutr. (2016) 63:560–8. doi: 10.1016/j.endoen.2016.07.004
149. Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen YY, et al. High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology. (2009) 137:1716–1724. doi: 10.1053/j.gastro.2009.08.042
150. Da Silva CC, Monteil MA, Davis EM. Overweight and obesity in children are associated with an abundance of firmicutes and reduction of bifidobacterium in their gastrointestinal microbiota. Child Obes. (2020) 16:204–10. doi: 10.1089/chi.2019.0280
151. Gao R, Zhu C, Li H, Yin M, Pan C, Huang L, et al. Dysbiosis signatures of gut microbiota along the sequence from healthy, young patients to those with overweight and obesity. Obesity. (2018) 26:351–61. doi: 10.1002/oby.22088
152. Kim KN, Yao Y, Ju SY. Short chain fatty acids and fecal microbiota abundance in humans with obesity: a systematic review and meta-analysis. Nutrients. (2019) 11:2512. doi: 10.3390/nu11102512
153. Bagarolli RA, Tobar N, Oliveira AG, Araújo TG, Carvalho BM, Rocha GZ, et al. Probiotics modulate gut microbiota and improve insulin sensitivity in DIO mice. J Nutr Biochem. (2017) 50:16–25. doi: 10.1016/j.jnutbio.2017.08.006
154. Murugesan S, Nirmalkar K, Hoyo-Vadillo C, García-Espitia M, Ramírez-Sánchez D, García-Mena J, et al. Gut microbiome production of short-chain fatty acids and obesity in children. Eur J Clin Microbiol Infect Dis. (2018) 37:621–5. doi: 10.1007/s10096-017-3143-0
155. de la Cuesta-Zuluaga J, Mueller NT, Álvarez-Quintero R, Velásquez-Mejía EP, Sierra JA, Corrales-Agudelo V, et al. Higher fecal short-chain fatty acid levels are associated with gut microbiome dysbiosis, obesity, hypertension and cardiometabolic disease risk factors. Nutrients. (2018) 11:51. doi: 10.3390/nu11010051
156. Arora T, Sharma R, Frost G. Propionate. Anti-obesity and satiety enhancing factor? Appetite. (2011) 56:511–5. doi: 10.1016/j.appet.2011.01.016
157. Brooks L, Viardot A, Tsakmaki A, Stolarczyk E, Howard JK, Cani PD, et al. Fermentable carbohydrate stimulates FFAR2-dependent colonic PYY cell expansion to increase satiety. Mol Metabol. (2017) 6:48–60. doi: 10.1016/j.molmet.2016.10.011
158. Steinert RE, Feinle-Bisset C, Asarian L, Horowitz M, Beglinger C, Geary N. Ghrelin, CCK, GLP-1, and PYY(3-36): secretory controls and physiological roles in eating and glycemia in health, obesity, and after RYGB. Physiol Rev. (2017) 97:411–63. doi: 10.1152/physrev.00031.2014
159. Guo Y, Liu C, Zhao X, Zhang X, Wu Q, Wang Z, et al. Changes in gut microbiota, metabolite SCFAs, and GPR43 expression in obese diabetic mice after sleeve gastrectomy. J Appl Microbiol. (2022) 133:555–68. doi: 10.1111/jam.15583
160. Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun. (2013) 4:1829. doi: 10.1038/ncomms2852
161. Young CN, Davisson RL. Angiotensin-II, the brain, and hypertension: an update. Hypertension. (2015) 66:920–6. doi: 10.1161/HYPERTENSIONAHA.115.03624
162. Harrison DG. The immune system in hypertension. Trans Am Clin Climatol Assoc. (2014) 125:130–138.
163. Yang T, Richards EM, Pepine CJ, Raizada MK. The gut microbiota and the brain-gut-kidney axis in hypertension and chronic kidney disease. Nat Rev Nephrol. (2018) 14:442–56. doi: 10.1038/s41581-018-0018-2
164. Karbach SH, Schönfelder T, Brandão I, Wilms E, Hörmann N, Jäckel S, et al. Gut microbiota promote angiotensin II-induced arterial hypertension and vascular dysfunction. J Am Heart Assoc. (2016) 5:e003698. doi: 10.1161/JAHA.116.003698
165. Natarajan N, Hori D, Flavahan S, Steppan J, Flavahan NA, Berkowitz DE, et al. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol Genomics. (2016) 48:826–34. doi: 10.1152/physiolgenomics.00089.2016
166. Felizardo RJF, Watanabe IKM, Dardi P, Rossoni LV, Câmara NOS. The interplay among gut microbiota, hypertension and kidney diseases: the role of short-chain fatty acids. Pharmacol Res. (2019) 141:366–77. doi: 10.1016/j.phrs.2019.01.019
167. Marzocco S, Fazeli G, Di Micco L, Autore G, Adesso S, Dal Piaz F, et al. Supplementation of short-chain fatty acid, sodium propionate, in patients on maintenance hemodialysis: beneficial effects on inflammatory parameters and gut-derived uremic toxins, a pilot study (PLAN Study). J Clin Med. (2018) 7:315. doi: 10.3390/jcm7100315
168. Mell B, Jala VR, Mathew AV, Byun J, Waghulde H, Zhang Y, et al. Evidence for a link between gut microbiota and hypertension in the Dahl rat. Physiol Genomics. (2015) 47:187–97. doi: 10.1152/physiolgenomics.00136.2014
169. Durgan DJ, Ganesh BP, Cope JL, Ajami NJ, Phillips SC, Petrosino JF, et al. Role of the gut microbiome in obstructive sleep apnea-induced hypertension. Hypertension. (2016) 67:469–74. doi: 10.1161/HYPERTENSIONAHA.115.06672
170. Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, et al. Gut dysbiosis is linked to hypertension. Hypertension. (2015) 65:1331–40. doi: 10.1161/HYPERTENSIONAHA.115.05315
171. Li J, Zhao F, Wang Y, Chen J, Tao J, Tian G, et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome. (2017) 5:14. doi: 10.1186/s40168-016-0222-x
172. Santisteban MM, Qi Y, Zubcevic J, Kim S, Yang T, Shenoy V, et al. Hypertension-linked pathophysiological alterations in the gut. Circ Res. (2017) 120:312–23. doi: 10.1161/CIRCRESAHA.116.309006
173. Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. (2017) 551:585–9. doi: 10.1038/nature24628
174. Guan Y, Chen K, Quan D, Kang L, Yang D, Wu H, et al. The combination of scutellaria baicalensis georgi and sophora japonica l. ameliorate renal function by regulating gut microbiota in spontaneously hypertensive rats. Front Pharmacol. (2020) 11:575294. doi: 10.3389/fphar.2020.575294
175. Chao YM, Tain YL, Lee WC, Wu KLH, Yu HR, Chan JYH. Protection by -biotics against hypertension programmed by maternal high fructose diet: rectification of dysregulated expression of short-chain fatty acid receptors in the hypothalamic paraventricular nucleus of adult offspring. Nutrients. (2022) 14:4306. doi: 10.3390/nu14204306
176. Wu C, Chen Z, Zhang L, Zhu Y, Deng M, Huang C, et al. Sodium butyrate ameliorates deoxycorticosterone acetate/salt-induced hypertension and renal damage by inhibiting the MR/SGK1 pathway. Hypertens Res. (2021) 44:168–78. doi: 10.1038/s41440-020-00548-3
177. Pluznick J. A novel SCFA receptor, the microbiota, and blood pressure regulation. Gut Microbes. (2014) 5:202–7. doi: 10.4161/gmic.27492
178. Pluznick JL. Gut microbiota in renal physiology: focus on short-chain fatty acids and their receptors. Kidney Int. (2016) 90:1191–8. doi: 10.1016/j.kint.2016.06.033
179. Pluznick JL. Microbial short-chain fatty acids and blood pressure regulation. Curr Hypertens Rep. (2017) 19:25. doi: 10.1007/s11906-017-0722-5
180. Weber GJ, Foster J, Pushpakumar SB, Sen U. Altered microRNA regulation of short chain fatty acid receptors in the hypertensive kidney is normalized with hydrogen sulfide supplementation. Pharmacol Res. (2018) 134:157–65. doi: 10.1016/j.phrs.2018.06.012
181. Weber G J, Pushpakumar S B, Sen U. Hydrogen sulfide alleviates hypertensive kidney dysfunction through an epigenetic mechanism. Am J Physiol Heart Circ Physiol. (2017) 312:H874–85. doi: 10.1152/ajpheart.00637.2016
182. Mandal AK, Mount DB. The molecular physiology of uric acid homeostasis. Annu Rev Physiol. (2015) 77:323–345. doi: 10.1146/annurev-physiol-021113-170343
183. Braga TT, Foresto-Neto O, Camara NOS. The role of uric acid in inflammasome-mediated kidney injury. Curr Opin Nephrol Hypertens. (2020) 29:423–31. doi: 10.1097/MNH.0000000000000619
184. Toprak O, Cirit M, Esi E, Postaci N, Yesil M, Bayata S, et al. Hyperuricemia as a risk factor for contrast-induced nephropathy in patients with chronic kidney disease. Catheter Cardiovasc Interv. (2006) 67:227–35. doi: 10.1002/ccd.20598
185. Weiner DE, Tighiouart H, Elsayed EF, Griffith JL, Salem DN, Levey AS. Uric acid and incident kidney disease in the community. J Am Soc Nephrol. (2008) 19:1204–11. doi: 10.1681/ASN.2007101075
186. Sellmayr M, Hernandez Petzsche MR, Ma Q, Krüger N, Liapis H, Brink A, et al. Only hyperuricemia with crystalluria, but not asymptomatic hyperuricemia, drives progression of chronic kidney disease. J Am Soc Nephrol. (2020) 31:2773–92. doi: 10.1681/ASN.2020040523
187. Terkeltaub R, Dodd D. The gut microbiome in hyperuricemia and gout. Arthritis Rheumatol. (2025). doi: 10.1002/art.43118. [Epub ahead of print].
188. Xu X, Wang H, Guo D, Man X, Liu J, Li J, et al. Curcumin modulates gut microbiota and improves renal function in rats with uric acid nephropathy. Ren Fail. (2021) 43:1063–75. doi: 10.1080/0886022X.2021.1944875
189. Sheng S, Chen J, Zhang Y, Qin Q, Li W, Yan S, et al. Structural and functional alterations of gut microbiota in males with hyperuricemia and high levels of liver enzymes. Front Med. (2021). 8:779994. doi: 10.3389/fmed.2021.779994
190. Park JW, Noh JH, Kim JM, Lee HY, Kim KA, Park JY. Gene dose-dependent and additive effects of ABCG2 rs2231142 and SLC2A9 rs3733591 genetic polymorphisms on serum uric acid levels. Metabolites. (2022) 12:1192. doi: 10.3390/metabo12121192
191. Michael OS, Dibia CL, Soetan OA, Adeyanju OA, Oyewole AL, Badmus OO, et al. Sodium acetate prevents nicotine-induced cardiorenal dysmetabolism through uric acid/creatine kinase-dependent pathway. Life Sci. (2020) 257:118127. doi: 10.1016/j.lfs.2020.118127
192. Zhou X, Zhang B, Zhao X, Lin Y, Wang J, Wang X, et al. Chlorogenic acid supplementation ameliorates hyperuricemia, relieves renal inflammation, and modulates intestinal homeostasis. Food Funct. (2021) 12:5637–49. doi: 10.1039/D0FO03199B
193. Wei Z, Cui Y, Tian L, Liu Y, Yu Y, Jin X, et al. Probiotic Lactiplantibacillus plantarum N-1 could prevent ethylene glycol-induced kidney stones by regulating gut microbiota and enhancing intestinal barrier function. FASEB J. (2021) 35:e21937. doi: 10.1096/fj.202100887RR
194. Ikee R, Sasaki N, Yasuda T, Fukazawa S. Chronic kidney disease, gut dysbiosis, and constipation: a burdensome triplet. Microorganisms. (2020) 8:1862. doi: 10.3390/microorganisms8121862
195. Tomova A, Bukovsky I, Rembert E, Yonas W, Alwarith J, Barnard ND, et al. The effects of vegetarian and vegan diets on gut microbiota. Front Nutr. (2019) 6:47. doi: 10.3389/fnut.2019.00047
196. Melekoglu E, Samur F G. Dietary strategies for gut-derived protein-bound uremic toxins and cardio-metabolic risk factors in chronic kidney disease: a focus on dietary fibers. Crit Rev Food Sci Nutr. (2023) 63:3994–4008. doi: 10.1080/10408398.2021.1996331
197. Lambert K, Rinninella E, Biruete A, Sumida K, Stanford J, Raoul P, et al. Targeting the gut microbiota in kidney disease: the future in renal nutrition and metabolism. J Renal Nutr. (2023) 33:S30–39. doi: 10.1053/j.jrn.2022.12.004
198. Koppe L, Mafra D, Fouque D. Probiotics and chronic kidney disease. Kidney Int. (2015) 88:958–66. doi: 10.1038/ki.2015.255
199. Yu Z, Zhao J, Qin Y, Wang Y, Zhang Y, Sun S. Probiotics, prebiotics, and synbiotics improve uremic, inflammatory, and gastrointestinal symptoms in end-stage renal disease with dialysis: a network meta-analysis of randomized controlled trials. Front Nutr. (2022) 9:850425. doi: 10.3389/fnut.2022.850425
200. Cooper TE, Khalid R, Chan S, Craig JC, Hawley CM, Howell M, et al. Synbiotics, prebiotics and probiotics for people with chronic kidney disease. Cochrane Database Syst Rev. (2023) 10:CD013631. doi: 10.1002/14651858.CD013631.pub2
201. Barba C, Soulage CO, Caggiano G, Glorieux G, Fouque D, Koppe L. Effects of fecal microbiota transplantation on composition in mice with CKD. Toxins. (2020) 12:12. doi: 10.3390/toxins1212074
202. Aslam MR, Perala A, Wishart AV, Hamouda RK, Elsaady E, Khan S. Therapeutic potential of fecal microbiota transplantation in type 2 diabetes mellitus: a systematic review. Cureus. (2024) 16:e70642. doi: 10.7759/cureus.70642
203. Prakash S, Chang T M. Microencapsulated genetically engineered live E. coli DH5 cells administered orally to maintain normal plasma urea level in uremic rats. Nat Med. (1996) 2:883–7. doi: 10.1038/nm0896-883
Keywords: gut microbiota, short-chain fatty acids, metabolic syndrome, chronic kidney disease, energy metabolism
Citation: Tian X, Sun L, Guo S, Yuan L, Zhang T, Huang C, He T, Jiang Q and Zeng Y (2025) Gut microbiota-derived SCFAs and MetS-related nephropathy. Front. Nutr. 12:1561271. doi: 10.3389/fnut.2025.1561271
Received: 19 February 2025; Accepted: 10 June 2025;
Published: 08 July 2025.
Edited by:
Ruixia Dong, Jinling Institute of Technology, ChinaReviewed by:
Cristina Torres Fuentes, University of Rovira i Virgili, SpainMithun Rudrapal, Vignan's Foundation for Science, Technology and Research, India
Shivani Sharma, Mercer University, United States
Copyright © 2025 Tian, Sun, Guo, Yuan, Zhang, Huang, He, Jiang and Zeng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yizhou Zeng, b21lZ2FtYW5tYW5AMTYzLmNvbQ==