 Zejun Zhao1†
Zejun Zhao1† Bin Wang
Bin Wang- 1Department of Fetology, The First Affiliated Hospital of Soochow University, Suzhou, China
- 2McKusick-Zhang Center for Genetic Medicine, State Key Laboratory for Complex Severe and Rare Diseases, Institute of Basic Medical Sciences Chinese Academy of Medical Sciences, School of Basic Medicine Peking Union Medical College, Beijing, China
The fetal origins of adult disease hypothesis proposes that a variety of adverse stimuli during critical development stages can impair the structure and function of fetal organs, thereby increasing the risk of disease later in life. Iron affects fetal growth and development by facilitating oxygen and electron transport and by serving as a cofactor for enzymes that affect enzyme activity. Fetal iron deficiency (ID) can result from various factors during pregnancy, including inadequate maternal iron intake, maternal obesity, diabetes, smoking, prenatal stress, and prenatal alcohol exposure. These conditions disrupt fetal brain development and are associated with neurological disorders in offspring, such as cognitive impairment, anxiety, depression, schizophrenia, and autism. However, the mechanisms by which maternal iron deficiency leads to abnormal neurological development, as well as cognitive impairment and psychiatric disorders in the offspring, remain unknown. In this review, we summarize the causes of prenatal iron deficiency, the effects of iron deficiency on brain development and behavioral phenotypes, and the potential molecular mechanisms.
1 Introduction
Iron deficiency (ID) is a common nutritional deficiency worldwide, especially in women (1). Among pregnant women, the prevalence of ID is approximately 80% in developing countries and approximately 40% in developed countries (2). The global burden and inequality of ID continue to rise, which may be related to low utilization of public health intervention packages. Low socioeconomic status, low education levels, gender discrimination, religious beliefs, and frequency of antenatal care in countries with low Human Development Index (3–6). Iron–Folic Acid Supplementation (IFAS) is an effective strategy for preventing and managing prenatal iron deficiency anemia (IDA) during pregnancy. In Bangladesh, Ghana, the Philippines, and Northwest Ethiopia, the compliance with IFA intake among pregnant women is only 20%−50% (7–10). Even in developed countries such as Canada, there is a negative correlation between the socioeconomic status of pregnant women and the probability of a ferritin test (11). Currently, various healthcare systems lack effective policies for the detection and management of fetal iron deficiency (ID). The main indicators used to detect ID in pregnancy are ferritin, hemoglobin (Hb), and C-reactive protein (CRP) under inflammation (12). According to the US Preventive Services Task Force, there is insufficient evidence to support screening asymptomatic pregnant women for ID and IDA or treating them with iron supplements to prevent adverse maternal and infant health outcomes associated with IDA (13). The American College of Obstetricians and Gynecologists recommends screening hemoglobin levels for anemia rather than ID, universal supplementation with low-dose iron during pregnancy, and low-dose iron supplementation and prenatal vitamin therapy for pregnant women with IDA after determining the cause (13, 14). In Asia, most medical institutions use Hb concentration as a proxy for ID/IDA (14). They further diagnose ID using serum iron, total iron-binding capacity, and transferrin saturation (15). In line with WHO recommendation, pregnant women in Southeast Asia should take oral iron and folic acid supplements daily if the prevalence of anemia is exceeds 40%, or intermittently on a weekly basis if the prevalence is below 20% (14).
It is well-known that iron is crucial for maintaining the production of hemoglobin, which is the molecule that transports oxygen in the blood (16). In addition, iron is essential for maintaining cell development and metabolic function in the body, including DNA synthesis and repair, enzymatic activity, and mitochondrial function (17). The requirement for iron during pregnancy increases due to several factors: (1) the increased physiological plasma volume of pregnant women requires more iron to synthesize hemoglobin (18); (2) the fetus requires iron to synthesize endogenous reserves of iron as well as for its own oxygen transport and metabolism (19); and (3) the placenta, which is a metabolically active organ and a transporter between the maternal and fetal circuits, requires large amounts of iron (20).
It is a priority to meet fetal iron needs in the case of mild maternal ID. However, the women with severe ID and exposure to adverse factors in pregnancy could cause fetal ID (19). Fetal ID affects fetal brain development, including hippocampal neuronal differentiation and synaptic plasticity and monoamine neurotransmitter metabolism (21). Neurodevelopmental abnormalities and mental health disorders, such as impairments in learning, memory, and emotion, occur in maternal ID offspring (22–24). In this review, we summarize the factors leading to prenatal fetal ID, the effects of prenatal ID on brain development and behavior of the offspring, the animal models of prenatal ID, and the possible mechanisms (Figure 1 and Table 1).
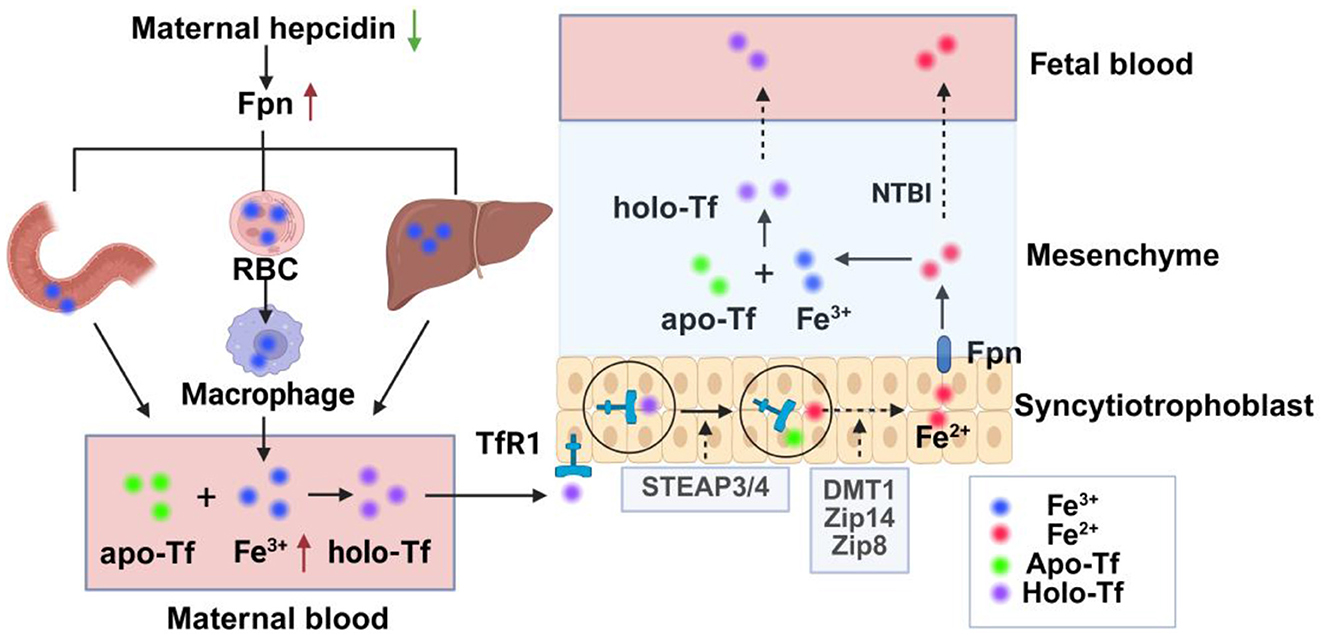
Figure 1. Schematic diagram of iron transport from mother to fetus. Maternal hepcidin is decreased during pregnancy, Fpn is increased, and iron flow into plasma is increased through increased intestinal iron absorption, macrophage iron recovery from aging red blood cells, and mobilization of ferritin in the liver. Fe3+ in plasma increases and binds to apo-Tf to form holo-Tf, which then binds to TfR1 on the apical membrane of placental trophoblastic cells to form the TFR1-transferrin complex, which is internalized by actin-coated vesicles. In vesicles, after the release of iron from transferrin, Fe3+ is reduced to Fe2+ by iron reductase and then transported from the vesicles to the interstitium by the iron transporters DMT1, Zip14, and Zip8. Fe2+ can be oxidized to Fe3+ after being transported by Fpn, which then combines with fetal Tf to form Holo-Tf and transported into the fetal blood, or directly into the fetal circulation in the form of NTBI. Fpn, ferroportin; RBC, red blood cell; STEAP3/4, 6-transmembrane epithelial antigen 3 and 4; Holo-Tf, holotransferrin, apo-Tf, iron-free transferrin; TfR1, transferrin receptor 1; DMT1, divalent metal transporter 1; Zip, Zrt/Irt-like protein; NTBI, non-transferrin-bound iron.
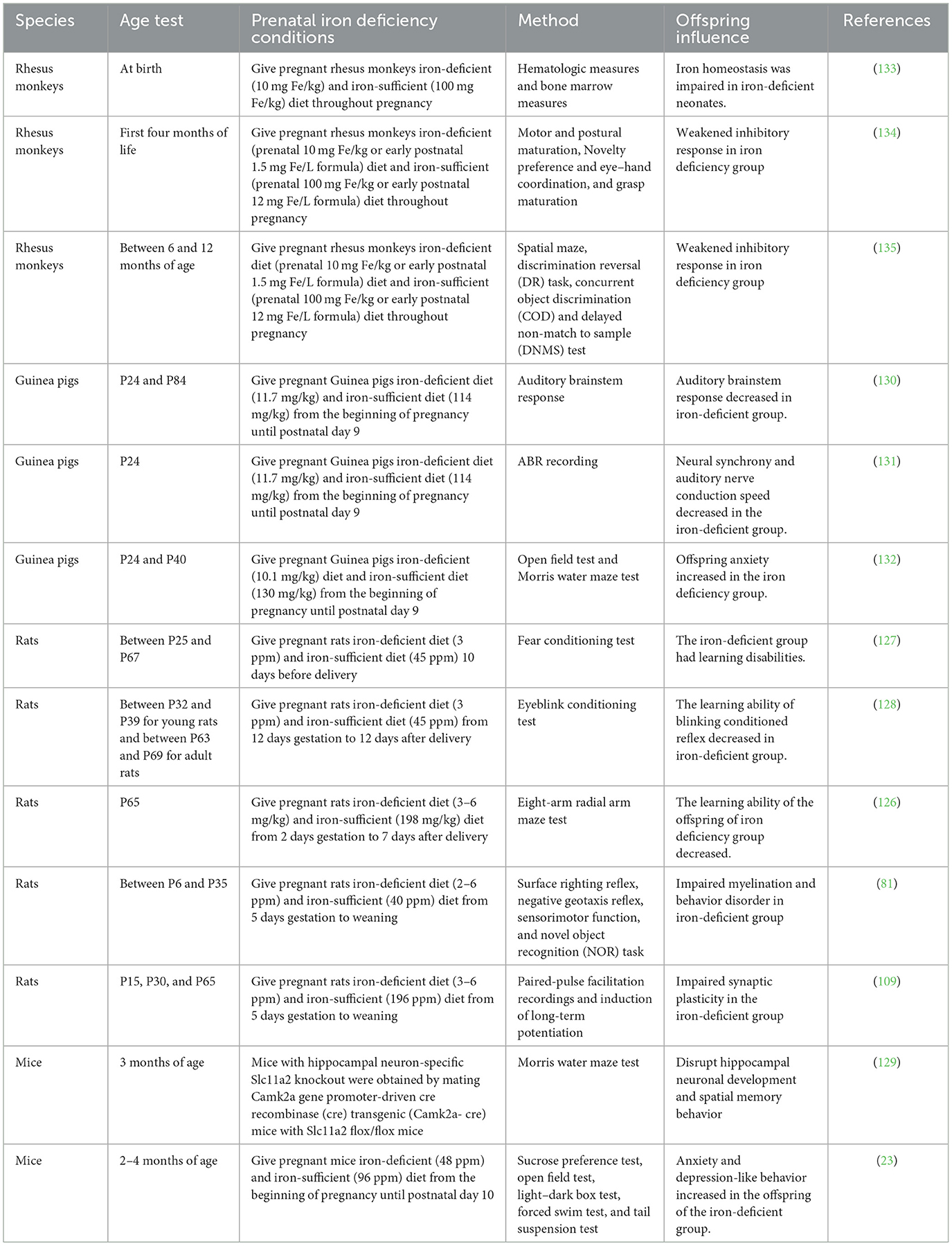
Table 1. Animal models used for maternal iron deficiency.
2 Iron homeostasis during pregnancy
Maternal physiological iron requirements rise significantly during pregnancy, with approximately 1 g of additional iron needed to maintain maternal iron homeostasis and to provide sufficient iron for fetal growth and development. Although maternal iron requirements are lower in the first trimester, they increase as the pregnancy progresses (25). Maternal hepcidin levels fall throughout the second and third trimesters of pregnancy, resulting in decreased binding and degradation of ferroportin (Fpn) (26, 27). Plasma iron is elevated through increased intestinal iron absorption, recycling of iron from senescent erythrocytes to macrophages, and mobilizing iron stores in the liver (26). In the interstitial fluid, iron ions bind to iron-free transferrin (apo-Tf) to form holo-transferrin (holo-Tf), which then binds to transferrin receptor 1 (TfR1) on the apical membrane of placental trophoblast cells to form the TfR1–transferrin complex. Upon binding, the TfR1–transferrin complex is internalized via clathrin-coated vesicles into an acidic environment (28). Then, the ferric iron (Fe3+) is separated from transferrin and reduced to ferrous iron (Fe2+) by iron reductases, such as the 6-transmembrane epithelial antigen of the prostate proteins 3 and 4 (STEAP3/4)(29). The specific pathway by which Fe2+ is transported from vesicles to the cytoplasm is unclear, and it may be related to iron transporters divalent metal transporter 1 (DMT1) and Zrt/Irt-like protein (ZIP) 8 (30, 31). Consequently, the TfR1–apolipoprotein complex returns to the membrane and is released (28). Fe2+ is exported from the syncytiotrophoblast by Fpn and oxidized to the Fe3+ by mammalian multicopper ferroxidases, such as ceruloplasmin, hephaestin, and zyklopen, which bind to fetal transferrin and are transported from endothelial cells to fetal circulation (Figure 2) (32–34).
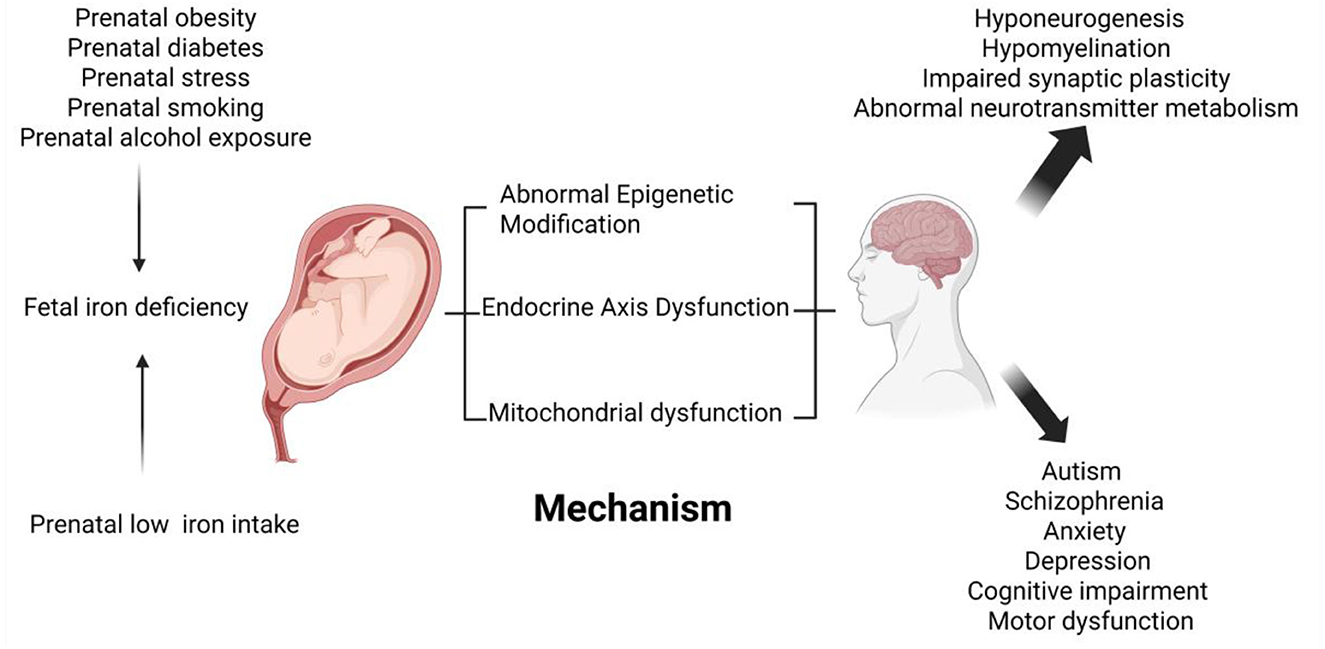
Figure 2. Schematic diagram of the effects of fetal iron deficiency on neurological development and related diseases. Low maternal iron intake during pregnancy, maternal obesity, maternal diabetes, prenatal stress, maternal smoking, and prenatal alcohol exposure have all been linked to fetal iron deficiency. Fetal iron deficiency affects fetal brain development, such as neurogenesis, emerging myelination, synaptic plasticity, and neurotransmitter metabolism, through epigenetics, endocrine axis dysfunction, and mitochondrial oxidative damage. Fetal iron deficiency is also associated with neurocognitive and mental health disorders in the offspring, such as depression and anxiety behaviors increased, impaired psychomotor development, learning and memory decline, autism, and schizophrenia.
When ID occurs during pregnancy, the expression levels of the molecules mediating placental iron uptake (TfR1) and output (Fpn) are altered. In mice, TfR1 increases, while Fpn decreases to maintain placental iron content in ID fetuses. In the placentas of pregnant women with mild ID, TfR1 expression is elevated, whereas Fpn remains unchanged. When severe ID is induced in human trophoblasts in vitro, TfR1 increases while Fpn decreases. The placenta cannot compensate for maternal ID to maintain fetal iron levels (35). In rats, fetuses exposed to prenatal ID can adaptively increase the expression of iron regulatory proteins (IRP-1 and IRP-2) and iron transport proteins (TfR and DMT1) in the hippocampus and the cerebral cortex to maintain brain iron requirements (36).
3 Causes of prenatal iron deficiency
3.1 Low maternal iron intake
Women with low iron intake, such as vegetarians or patients with gastrointestinal disorders, are at an increased risk of developing ID due to increased physiological iron requirements during pregnancy. Daily oral iron supplementation can decrease maternal anemia and full-term iron deficiency (37). When maternal ID is mild, iron is supplied preferentially to the fetus to ensure adequate fetal iron stores. However, when maternal ID is moderate or severe, fetal iron homeostasis is disrupted (2). Currently, it is believed that ID in pregnancy is defined as serum ferritin (SF) <30 μg/L (38). Fetal ID will occur when maternal ferritin concentration is less than 12 μg/L (39). Severe maternal ID is clinically manifested as iron deficiency anemia (IDA). When the maternal hemoglobin (Hb) concentration is <85 g/L, umbilical cord serum ferritin is <60 μg/L, indicating impaired fetal iron stores. When maternal Hb is <60 g/L, umbilical cord serum ferritin concentration is <30 μg/L, and umbilical cord Hb concentration is also decreased, indicating a progressive decline in umbilical cord ferritin levels as maternal anemia severity increases (40).
3.2 Maternal obesity
Maternal obesity and rapid weight gain are independent risk factors for fetal ID and are associated with elevated hepcidin levels (41–43). During pregnancy, high maternal body mass index (BMI) could induce maternal inflammatory responses, such as increased concentrations of interleukin-6 (IL-6) and C-reactive protein (CRP), which in turn lead to overexpression of hepcidin (44, 45). The increased number of macrophages in the placenta suggests that the inflammatory response in obese mothers extends to the uterus (46). Ultimately, fetal iron status is impaired. The released IL-6 forms a complex with the IL-6 receptor and glycoprotein 130 (gp130) to activate Janus kinase (JAK) (47). JAK phosphorylates tyrosine residues, which activates signal transducer and activator of transcription 3 (STAT3), and then enters the nucleus, binds to the hepcidin promoter, and induces hepcidin expression (48). In addition, maternal obesity increases the size of fat cells to produce more leptin, which induces hepcidin overexpression (49). In conclusion, obese women have a smaller decrease in hepcidin levels during pregnancy than non-obese women. Fpn located in intestinal cells, reticulocytes, and hepatocytes can bind more to hepcidin and be internalized and hydrolyzed by lysosomes, which can increase iron concentration in cells and reduce iron transport to plasma. However, some studies have shown that maternal obesity does not affect maternal and fetal iron status (50, 51), which may be related to the different degrees of maternal obesity and race.
3.3 Maternal diabetes
The offspring of diabetic mothers have abnormal iron distribution and decreased brain iron concentration, which may be associated with impaired iron transport (52, 53). In pregnant women with insulin-dependent diabetes, increased N-glycosylation of transferrin receptor (TfR) released from the placenta can reduce its binding capacity to transferrin (Tf), thereby reducing iron transport in the placenta (54). Then, decreased fetal iron reserve leads to increased expression of placental iron regulatory protein-1 (IRP-1), which binds to iron-responsive elements on the TfR mRNA and plays a stabilizing role in the upregulation of its expression (55, 56). However, Yang et al. (57) found that maternal iron transport to the fetus was reduced in gestational diabetes but was not associated with TfR expression.
3.4 Prenatal stress
Prenatal stress can lead to sex-specific fetal ID, which occurs predominantly in male fetuses and is associated with the fetal stress response system (58–63). Chronic stress can alter maternal expression of the acetylcholinesterase (AChE) gene, thereby converting the normal AChE-S splicing variant into an unstable AChE-R variant. On the one hand, the ratio of AChE-S to AChE-R can downregulate the expression of Fpn and metal ion transporters by modulating cholinergic pathway signaling via microglial α7 nicotinic acetylcholine receptors (α7nAChR) (64). On the other hand, elevated AChE-R is associated with chronic inflammation, which may lead to an increase in hepcidin (65). The above changes can reduce extracellular iron, thus affecting maternal and fetal iron homeostasis.
3.5 Maternal smoking
Fetal ID is associated with maternal smoking and is influenced by the frequency and number of days smoked. Iron stores might not be significantly impaired in pregnant women who smoke, but they are reduced in newborns. First, maternal smoking increases catecholamines in the maternal blood, which affects blood flow and vascular resistance in the placenta, as well as reduces blood nutrients and oxygen delivered to the fetus (66). Second, carbon monoxide in tobacco causes carboxyhemoglobinemia, which reduces the supply of hemoglobin to the fetus (67). Third, cyanide compounds contained in tobacco can exacerbate fetal hypoxia by impairing fetal oxidative mechanisms (67). Fourth, maternal smoking is positively associated with increased fetal lead concentration, which may contribute to hypoxia by interfering with hemoglobin synthesis and reducing the number of red blood cells (68). Conversely, a study showed that fetal iron homeostasis was not affected when women were exposed to smokeless tobacco during pregnancy (69).
3.6 Prenatal alcohol exposure
In pregnant women, prenatal alcohol exposure (PAE) increases maternal ferritin levels and decreases maternal hemoglobin-to-log (ferritin) ratio (70). In the fetus, PAE can decrease iron concentration and iron utilization in the brain (71). This may be related to elevated maternal and fetal hepcidin and the transfer of iron from the fetal brain and erythrocytes to the liver for storage. PAE can increase maternal and fetal inflammatory cytokines, such as IL-6, and activate hepcidin transcription, which impairs fetal iron homeostasis through the JAK/STAT signaling pathway (72). Due to PAE, serum ferritin (SF), Tf, and TfR cannot be upregulated in time, while fetal brain iron level drops (73). In addition, PAE upregulates the expression of the IL-1β gene in the placenta, which increases iron storage via promoting iron uptake into macrophages, the destruction of erythrocytes, and ferritin biosynthesis, as well as blocks the transport of iron in cells by inhibiting FPN-1 (74).
3.7 Gene–environment interactions
Gene–diet interactions and diseases indicate that genetic variations can also influence iron absorption and utilization. Haptoglobin (Hp) prevents oxidative damage mediated by free heme iron by removing it from cells (75). Hp gene polymorphisms constitute three main phenotypes: Hp 1-1, Hp 2-1, and Hp 2-2 (76). During mid-to-late stages of pregnancy, Hp phenotypes may increase susceptibility to ID in pregnant women. Pregnant women carrying the Hp 1 alleles may have increased susceptibility to ID if they do not have sufficient dietary iron intake or use prenatal supplements related to erythropoiesis. Additionally, obese women carrying the Hp 2-2 phenotype may have an increased risk of developing functional ID (77).
4 Consequences of iron deficiency on the nervous system
In fetal iron homeostasis, iron allocation is prioritized for red blood cells, rendering the brain susceptible to ID-mediated impairment even when hemoglobin levels remain within the normal ranges (21). Adequate iron during fetal brain development is necessary for neurogenesis, myelination, synaptic plasticity, and energy metabolism in neuronal and glial cells. Different durations and degrees of ID and the developmental stage at which ID occurs have various effects on brain development and function (21).
4.1 Reduced neurogenesis
Prenatal ID is associated with the inhibition of neurogenesis in the hippocampus of offspring mice and a reduction in the number of pyramidal cells and granule cells. The occurrence of ID at different stages of pregnancy may contribute to selectively change the volume of different parts of the fetal hippocampus and affect corresponding memory function. The critical time for susceptibility of the CA1 region of the hippocampus to ID is prenatal, and the dentate gyrus region of the hippocampus is susceptible to ID both prenatally and postnatally. Prenatal ID induces reduced neurogenesis and altered hippocampal volumes in the offspring, which may be associated with reduced brain-derived neurotrophic factor (BDNF) signaling (78).
4.2 Inhibition of myelin regeneration
ID is associated with myelin degeneration in both human studies and animal models. In human studies, the latency of auditory brainstem potentials as indirect markers of myelination is prolonged in infants with ID (79). In a rat model, severe ID may lead to persistent hypomyelination, the production of immature astrocytes, and increased pericyte permeability in offspring exposed to a maternal iron-deficient diet (80). Delayed myelination in specific parts of the brain is associated with behavioral disorders in rats (81). Oligodendrocyte progenitor cells (OPCs) and mature myelin oligodendrocytes are rich in iron and play an important role in myelination. In the brain, insufficient iron supply affects enzyme synthesis, which further affects the proliferation and differentiation of OPCs and myelin synthesis (82, 83). On the one hand, TfR expression on OPCs peaks during oligodendrocyte maturation and declines in mature myelinating cells to maintain iron homeostasis and development (84–86). Due to ID in the brain, the binding of apo-Tf produced by oligodendrocytes and epithelial choroid plexus cells to iron is decreased. This impairs holo-Tf formation, which is required for high-affinity binding to TfR, ultimately leading to a decreased iron uptake by OPCs (87). The effects of apo-Tf on oligodendrocyte maturation and myelination may be mediated by the following signaling pathways: (1) Apo-Tf injection improves oligodendrocyte maturation and myelination by the Notch signaling pathway, which participates in focal demyelination and regeneration by increasing the F3/contact protein levels and Hes5 expression (88, 89). (2) The Fyn/MEK/ERK and PI3K/Akt pathways are also active post apo-Tf treatment (90, 91). Iron-related pathways (e.g., Fyn/MEK/ERK, PI3K/Akt, Notch) are closely related to neurological diseases, such as cognitive impairment and schizophrenia (92–94). On the other hand, ferritin in oligodendrocytes consists of an equal combination of heavy chain (Fth) and light chain (Ftl) (87). As an antioxidant protein, Fth may prevent the formation of reactive oxygen species, but it also increases cytoplasmic iron levels and oxidative stress (87). In oligodendrocyte-specific Fth1 KO mice, knocking out Fth in oligodendrocytes leads to neuronal loss and oxidative damage, thus affecting myelination (95).
Under ID, the increased proliferation of astrocytes and the decreased expression of glial fibrillary acidic protein (GFAP) and connexin 43 (CX43) suggest that astrocyte maturation is impeded (96). Astrocytes can inhibit remyelination by secreting cytotoxic factors and conversely promote myelin repair by secreting trophic factors, such as tumor necrosis factor-α (TNF-α) and cytokines interleukin-1β (IL-1β) (97–99). Insulin-like growth factor 1 (IGF-1) in response to TNF-α and fibroblast growth factor 2 (FGF-2) in response to IL-1β are important for myelination (97, 100). Astrocytes have high expression of iron influx proteins and iron efflux proteins and, thus can safely uptake and recycle iron in the brain during demyelination (101). Iron distribution in astrocytes is critical for the remyelination process. When the iron efflux transporter Fpn is knocked out in astrocytes, there is a decrease in the proliferation of OPCs and a decrease in the expression of IL-1β and IGF-1, which are associated with decreased remyelination (102). When multi-copper ferroxidases are knocked out in astrocytes, iron efflux is impaired and free radical production is increased, which ultimately drives myelin damage (103).
4.3 Impaired synaptic plasticity
Synaptic plasticity is defined as the ability of synapses to change their structure, connectivity, and function in response to internal or external stimuli (104). In newborns, a previous study has shown that low maternal iron intake accelerates the decline of fractional anisotropy (FA) values in gray matter, indicating reduced synaptic formation and dendritic arborization in offspring (105). In rodents, fetal ID regionally affects dendrite morphology and branching before adulthood, despite subsequent iron supplementation in the brain (106–108). ID reduces the basal dendrite length of pyramidal neurons in the hippocampus without affecting branch complexity and increases the proximal branches of apical dendrites without affecting total length. In contrast, both apical and basal dendrite branch complexity are reduced in cortical neurons, but total length remained unchanged (107). The decreased long-term potentiation (LTP) indicates abnormal synaptic plasticity in fetal iron-deficient mice (109). ID leads to the reduction of four synaptic proteins: CaMKIIα, PSD-95, Fkbp1a, and Vamp1 (110). During repeated stimulation, iron deficiency can change synaptic plasticity by keeping the content of synaptic vesicles constant but reducing their release, and iron supplementation partially reverses this (111).
4.4 Abnormal neurotransmitter metabolism
Iron is involved in the synthesis of neurotransmitters as a cofactor of enzymes such as tyrosine hydroxylase and tryptophan hydroxylase (112). The striatum, one of the basal ganglia of the brain, delivers dopamine-rich substances to the prefrontal cortex and is involved in cognitive and motor functions (8). Altered dopamine function has been associated with an increased risk of schizophrenia in adult offspring of maternal ID (113). In the striatum, dopamine concentration is high due to high iron concentration (114). When ID occurs, cellular uptake of dopamine is reduced because the density and function of dopamine transporters as well as dopamine receptors are reduced in the caudate-putamen (115, 116). Injection of physiological iron concentrations into the ventral midbrain (VMB) alleviates ID-induced decrease in dopamine concentration in the striatum (117). Thy-1 is a cell adhesion molecule that regulates the release of neurotransmitter vesicles, and the fact that ID leads to a decrease in Thy-1 provides a new explanation for impaired dopaminergic transmission in the brain (118). Changes in local monoamine metabolism across various brain regions are sensitive, proportional to the degree of ID, and occur prior to the severe decrease in brain iron concentration (114, 119). Prenatal ID is associated with impaired monoamine metabolism in the offspring's brain, leading to abnormalities in learning and memory functions. These changes cannot be treated by postpartum iron supplementation (120). In addition, ID can also alter the density of serotonin transporters and norepinephrine transporters, which is more pronounced in male offspring (121).
5 Animal models used for iron deficiency
To better understand the effects of iron deficiency during pregnancy on brain development and behavioral phenotypes in animal offspring and the possible mechanisms involved, multiple animal models of ID in pregnancy have been established, as shown in Table 1.
In terms of pregnancy physiology, mice and rats have shorter gestation periods and multiple pregnancies, with fetuses being born with underdeveloped organs (122, 123). Regarding endocrinology, the entire pregnancy period is highly dependent on ovarian progesterone production to maintain pregnancy (123). With regard to the structure and efficiency of the placenta, there are uterine endothelial cells, maternal capillary endothelial cells, trophoblast cells, and fetal capillary endothelial cells between maternal blood and fetal tissue, resulting in low efficiency of material exchange (122). Guinea pigs have a longer gestation period than rats and mice, produce fewer offspring, and experience a rapid phase of brain development at birth (123, 124). Guinea pig placenta is discoid, labyrinthine, and haemomonochorial, resulting in higher efficiency of material exchange across the placenta than mice and rats (122). Rhesus monkeys are also similar to humans because they experience single-offspring pregnancies, have similar hematological changes during pregnancy (125), and share characteristics with humans in terms of placental transport, relative fetal growth, and regional brain development (21).
The animal model of ID during pregnancy has been established by restricting dietary iron intake of pregnant mothers to study its effect on adverse outcomes in offspring. Rats and mice are the most common animal models. The rodent brain at 10 days of gestation is considered equivalent to the human brain at full-term birth. Therefore, most rodent models of maternal ID are given an ID diet from pregnancy to about postnatal day 7, followed by which they are given an iron-sufficient diet (21). The pregnant rats are given an iron-deficient diet until after delivery, which decreases offspring's brain iron concentration, delays myelination, and impairs synaptic plasticity (81, 109, 126). Moreover, the offspring of rats exposed to maternal ID have impaired cognitive development, such as poor hippocampus-mediated spatial recognition learning and hippocampus-dependent trace fear conditioning and eyeblink conditioning (126–128). In addition, there are behavioral impairments in tests such as surface correction reflex and novel object recognition task (81). In the maternal iron-deficient mouse model, decreased iron level in the brain of the offspring is associated with anxiety and depression in adulthood (23). The mouse model with hippocampal neuron-specific knockout of Slc11a2, a gene responsible for iron uptake, showed that reduced iron content impaired the memory function by affecting the hippocampal neurodevelopment, including energy metabolism and dendrite morphology (129). When ID occurs in pregnant guinea pigs, the hearing function of offspring is impaired by affecting neural synchronization and auditory nerve conduction velocity (130, 131). Offspring of prenatal iron-deficient guinea pigs have increased locomotor activity, suggesting increased nervousness due to anxiety (132). Rhesus monkeys in the experimental group were fed a low iron diet (10 mg Fe/g) from 28 to 30 days of gestation until delivery, while controls were fed an iron-rich diet (100 mg Fe/g) (133). Newborns of pregnant Rhesus monkeys with ID were born with reduced hemoglobin, volume, and number of red blood cells but without neurobehavioral abnormalities (133). Other studies have shown that the offspring of prenatal ID in rhesus monkeys have reduced spontaneous activity in a new environment and behavioral disorders such as reduced inhibitory responses (134, 135). Moreover, the offspring of prenatal ID in rhesus monkey showed more active exploration in a new environment and in the manipulation of new objects than the control group, suggesting the presence of impulsive behavior syndrome (134).
6 Abnormal neurological behaviors
6.1 Motor function
Psychomotor development mainly encompasses gross and fine motor skills, and if impaired, it can affect an individual's cognitive and emotional development (136). The gross motor scores consist of three parts: reflexes, locomotion, and stationary subscales. Reflexes are automatic responses to environmental changes, such as the righting reflex. The assessment of locomotion is based on the ability to move from one place to another. In addition, the assessment of stationary is based on the ability to control the center of gravity and maintain balance (137). Fine movement mainly focuses on the use of body muscles to complete specific actions, such as finger movements and hand–eye coordination (138). During pregnancy, the offspring of anemic mothers with low hemoglobin concentration have slightly low gross and fine motor scores, which are positively correlated when maternal hemoglobin is below 110 g/L (139). Children whose mothers had a low dietary iron intake or low umbilical cord ferritin concentration during pregnancy have lower gross motor and fine motor scores than children whose mothers had a diet rich in protein and micronutrients (22, 140). The severity of impaired motor development is related to the timing and duration of ID. When ID occurs in the third trimester of pregnancy, the Peabody Developmental Motor Scales, Second Edition (PDMS-2) gross motor scores are lower (141). However, a study showed that prenatal ID or IDA were not associated with motor development in the offspring despite low umbilical serum ferritin concentration, possibly because iron supplementation was not considered during pregnancy (142).
6.2 Learning and memory
Prenatal IDA or low cord ferritin concentration is associated with impaired cognitive and intellectual development in offspring (22, 143–146). Memory is categorized into two distinct types: explicit memory, employed to recall past events, and implicit memory, related to motor and skill tasks and cognition (147). The explicit memory is assessed by electrophysiological measurements and behavioral memory performance, which include evoked imitation of immediate recall and delayed imitation of 1-week delayed recall. The study shows that prenatal ID can have a lasting effect on memory function in offspring, as evidenced by impaired recall and compromised encoding and retrieval processes (148). Fetuses with low cord ferritin or high ratio of porphyrin/heme zinc in the umbilical cord blood allocated more attentional resources to mother's voice and face recognition memory (24, 149, 150). Both the timing of the onset of ID and the age at which the infant's recognition memory is assessed influence the results (150). In the animal model, prenatal ID in rats impairs hippocampus-dependent trace fear conditioning and eye-blinking conditioning in offspring, indicating implicit memory is impaired (127, 128). The offspring of prenatal iron-deficient rats are more likely to rely on the striatum to navigate spatial memory tasks (126).
6.3 Affective and neurodevelopmental disorders
Accumulating evidence suggests that prenatal ID is associated with affective disorders in the offspring. In mice, prenatal ID reduces brain iron levels and increases susceptibility to anxiety- and depression-like behaviors in offspring (23). In rats, prenatal ID is associated with autism-like and schizophrenia-like behaviors in offspring, exhibiting abnormal pre-pulse inhibition of offspring's acoustic shock and sensitivity in novel environments (151). In humans, low maternal iron intake or ferritin level during pregnancy are related to an increased risk of autism in offspring (152, 153). In addition, offspring with low maternal hemoglobin concentration or anemia during pregnancy have an increased risk of schizophrenia, suggesting that maternal ID is a risk factor for schizophrenia in offspring (154).
7 Possible mechanisms underlying the neurological disorders caused by prenatal iron deficiency
7.1 Abnormal epigenetic modification
Epigenetic regulation refers to chemical modifications of DNA and histones that affect gene expression without altering the genetic code, such as DNA methylation, histone modification, regulation of non-coding RNA, and chromatin remodeling (155, 156). In animal models, maternal ID-induced dysregulation of gene expression in hippocampal neuronal development and functional pathways is related to aberrant DNA methylation (157). These pathways are the β-adrenergic signaling pathway, the CAMP-PKA signaling pathway, Rho GTPase signaling, and reelin signaling, all of which are involved in synaptogenesis and synaptic plasticity (157). In humans, lower levels of DNA methylation in the umbilical cord are related to lower maternal serum ferritin concentrations during the first trimester, and these relationships partially persist in children (158). The concentration of transferrin in pregnant women is associated with increased DNA methylation at cg09996156 (KIAA1324L), a regulator of the bone morphogenetic protein (BMP) pathway, which participates in apoptosis and autophagy and affects the development of the embryonic nervous system (158, 159). Iron is involved in two families of epigenetic modifications—Ten-Eleven Translocation (TET) proteins and Jumonji and AT-rich interaction domain-containing (JARID) proteins—both of which regulate gene expression during critical periods of brain development (160). The TET enzyme demethylates DNA by catalyzing the oxidation of 5-methylcytosine to form 5-hydroxymethylcytosine (5hmC), which serves as a stable epigenetic marker for neurons (161, 162). Syt1 and Nav2 are genes with high levels of 5hmC that play a role in neurogenesis and synaptic transmission (162, 163).
Histone modifications include methylation, acetylation, phosphorylation, and ubiquitination, with methylation and acetylation being the most common (164). The expression of JARID1B gene is downregulated in the hippocampus of the offspring of maternal iron-deficient rats (165) due to enrichment of histone deacetylase 1 (HDAC1) at the JARID1B promoter and the low acetylation level of H3K9 (166). Proteins containing JmjC domain are known as demethylases and can regulate transcription by removing methyl groups from lysine residues in the tail of histones (167, 168). Low JARID1B (Kdm5b) demethylation from trimethylated and dimethylated histone H3 lysine 4 (H3K4me1/2) leads to an increase in chromatin compaction and a decrease in transcriptional activity, whereas low JMJD3 (Kdm6b) and JHDM1d demethylates from H3K9me3 and H3K27me3 leads to a decrease in transcriptional repression of chromatin conformation (169–171). Therefore, alteration of JARID1B expression under ID can regulate transcription levels by altering chromatin structure, such as BDNF-related genes (165). Increased levels of H3K27me3 labeling are associated with promoter inhibition, and increased levels of H3K4me3 labeling are associated with promoter activity (172). Iron-deficient fetuses have an increased concentration of H3K27me3 and a decreased concentration of H3K4me3 in the hippocampus, which may be one of the mechanisms for decreased transcriptional activity of Bdnf-P4 (165, 173). Iron supplementation during the critical period of hippocampal development could partially increase JARID, but the recovery ability is limited (166). Choline supplementation during pregnancy reduces the expression of histone methyltransferase G9a and Suv39h1 in the hippocampus, which may be a potential mechanism for reversing maternal ID-induced HDAC1 enrichment and reduced H3K4me3 levels (173, 174).
MicroRNAs are non-coding single-stranded RNAs of approximately 22 nucleotides in length that are widely involved in the regulation of neurogenesis, development, apoptosis, cell differentiation, proliferation, and other biological processes by inhibiting the translation of messenger RNAs or promoting mRNA degradation (175). Prenatal ID alters miRNA expression in the brain, such as miR-200a and miR-200b, which may increase the risk of depression and anxiety-like behaviors in the offspring (23).
7.2 Mitochondrial dysfunction
Iron is involved in enzymes that make up the electron transport chain and the tricarboxylic acid cycle; therefore, it can influence brain development through energy metabolism (176). ID in hippocampal neurons alters the mRNA levels of genes related to mitochondrial function and energy metabolism, causing impaired mitochondrial respiration and glycolysis, which in turn affects the dendritic growth and branching (177). In the early stage of ID, only the oxidative capacity of mitochondria is affected, but in the later stage, the density of mitochondria is reduced, suggesting that there may be long-term effects on neurons (177). There are three main approaches in which chronic ID alters dendritic mitochondrial movement: first, an increase in the frequency of dendritic mitochondrial pauses decreases the speed of mitochondrial motion (178). On the one hand, a reduction in localized transient ATP may influence the ATPase activity utilized by motor proteins, such as dynein motor proteins and dynamins in transporting mitochondria (179, 180). On the other hand, ID may enhance the mRNA expression levels of blood–brain barrier and neuronal glucose transporters in the hippocampus (181). Extracellular glucose alters Milton GlcNAcylation, which regulates the mitochondrial motility by O-GlcNAc transferase (OGT) (182). Second, changes in mitochondrial fusion and fission gene expression in response to ID can reduce mitochondrial size by inhibiting OPA1-mediated fusion and stimulating DRP1-mediated fission (183, 184). Third, reduced anterograde mitochondrial movement and increased retrograde segmental velocity are observed in ID, while overall retrograde motion remains unchanged (178). Therefore, mitochondrial malfunction due to ID may contribute to long-term neurological damage and psychiatric disorders in offspring.
7.3 HPA axis dysfunction
Stress leads to activation of the hypothalamic–pituitary–adrenal (HPA) axis, which elevates glucocorticoid (GC) concentrations (185). Elevated GC leads to the apoptosis and atrophy of hippocampal neurons, which impairs neuroplasticity and leads to abnormal behavior (186). Glucocorticoid receptor (GR) in the hippocampus can regulate glucocorticoid levels through a negative feedback loop (187). GR binds to the cytoplasmic heat shock protein (Hsp) 40 and Hsp70 to form a GR–Hsp40/Hsp70 complex, which promotes GR folding and localization to the intermediate domain of Hsp90. The GR–Hsp90 complex alters the structure of the protein to allow it to bind GC (188–190). The binding of FK506-binding protein (FKBP51) and p23 to the GR–Hsp90 complex increases the binding affinity of GR to GC. After GC binds to the GR–Hsp90 complex, FKBP51 is replaced by FKBP52, which assists in nuclear translocation of the GC–GR heterocomplex and inhibits gene transcription of corticotropin-releasing hormone (CRH) in the nucleus (191, 192). On the one hand, when GC concentration is elevated, the increased expression of FKBP5 gene inhibits GR activity by limiting the translocation of the receptor complex to the nucleus (193). On the other hand, alterations in the self-phosphorylation state of GR regulate its transport from the cytoplasm to the nucleus (193, 194). The cognitive impairment of iron-deficient offspring may be related to the elevated serum glucocorticoid level and the reduced GR activity. In a mouse model of the offspring of iron-deficient mothers, serum glucocorticoid level is increased and GR activity is significantly reduced (195). In the case of ID in the brain, the hippocampus GC–GR signaling pathway can be inhibited by impaired GR–HSP90 complex formation and nuclear translocation, which affects the negative feedback regulation function of GR and hyperactivates the HPA axis (195). Therefore, HPA axis dysfunction due to prenatal ID may also be one of the important pathways for neurodevelopmental and behavioral abnormalities in offspring.
8 Future prospects
Although previous studies have shown associations between maternal obesity, diabetes, smoking, alcohol exposure, stress and fetal ID, the underlying mechanisms remain unclear. Identifying these factors associated with fetal ID can help prevent neuronal dysfunction-related diseases. Currently, because the gestation process in rodents is different from that of humans, they cannot provide adequate models for neurodevelopment, disorders, and mechanisms of ID in pregnant offspring. Rhesus monkeys with similar pregnancy cycles to humans are used as animal models for ID in pregnancy. In human cohort studies, most researchers have linked fetal ID to disorders such as cognitive impairment, autism, and schizophrenia. However, perhaps due to the different diagnostic criteria for fetal ID and cognitive function, studies have shown no association with cognitive function changes in adulthood (196–198). In addition, further research is needed to determine whether there is a clear link between fetal ID and ADHD.
Although it has been demonstrated that fetal ID affects the brain development of offspring, the relationship between neurological disorders and the specific molecular mechanism is still unclear. The study of maternal iron deficiency models will help us to further understand and lay a good foundation for treatment. Dietary therapy can enhance iron status in pregnant women at risk of or with mild ID during pregnancy. Pregnant women should eat more foods rich in ascorbic acid and carotenoids, such as kiwi fruit, and reduce their intake of foods that inhibit iron absorption, such as coffee, tea, and phytic acid in grains (199). In animal models, prenatal choline supplementation mitigated the expression of genes associated with ID-induced psychological disorders, such as schizophrenia, autism, and anxiety (200). Iron-deficient neurons treated with choline can stimulate dendritic growth, restore dendritic complexity, and improve ATP production rate and glycolysis but not be fully restored to normal (201). These changes are more pronounced in female rats (202). However, prenatal choline supplementation in iron-sufficient rats can dysregulate the expression of genes associated with cognitive and psychological disorders and promote epithelial to mesenchymal transformation by inhibiting fatty acid metabolism and oxidative phosphorylation activity, leading to cell adhesion and migration, which is similar to the adverse effects of ID (202). However, these changes do not affect the complexity of dendrites and the structural development of neurons (201). Prenatal choline supplementation attenuated ID-induced ADORA2 gene network in women and FEV gene network in men, which are associated with depression and attention disorders, respectively (202). In humans, existing studies indicate that choline supplementation during pregnancy may improve cognitive function in offspring (203, 204). In terms of the choline supplementation window during pregnancy, the cognitive ability improvement is lower in early pregnancy than in mid-pregnancy when choline intake is the same (204). Choline supplementation in late pregnancy enhances the attention maintenance ability of offspring (203). The recommended daily intake of choline for pregnant women is 450 mg/day (205). In the late stages of pregnancy, offspring with daily intakes of 930 mg/day demonstrated higher cognitive abilities than those with daily intakes of 480 mg/day (203). Additionally, maternal plasma choline levels were positively correlated with cognitive development in full-term infants (206). However, the dosage and duration of choline supplementation to alleviate the adverse reactions caused by ID during the prenatal period have not yet been conclusive. Moreover, in terms of human health, nearly 40%−50% of children with prenatal ID continue to experience intellectual disability and long-term neurological impairment despite iron supplementation (207). Early-life mitochondrial dysfunction is recognized as one of the potential factors for these psychiatric disorders (178). Supplementing dietary selenium can improve mitochondrial function by increasing the expression of selenoprotein K in the endoplasmic reticulum of neurons, promoting TfR-1 palmitoylation, and increasing intracellular iron levels (208). Idebenone is a Coenzyme Q10 analog that protects the mitochondria by acting as an antioxidant and increasing ATP production, thereby alleviating cognitive impairment. However, whether they can improve cognitive impairment in offspring of prenatal ID remains to be studied (209). Therefore, mitochondria are an attractive target for the design of alternative therapeutic interventions to prevent long-term neuropathology in many children, in addition to timely iron supplementation.
Various models (cells, organ tissues, and animals) will be utilized to rigorously validate whether target regulation can rescue or improve neurodevelopmental phenotypes through genetic manipulation (overexpression, knockout, and knockdown) and pharmacological intervention. Conducting standard reproductive toxicity and developmental toxicity studies in at least two animal species (typically one rodent and one non-rodent species such as non-human primates) to evaluate how different doses and administration timings (corresponding to various developmental stages) affect maternal and fetal health (including all organ systems, particularly the nervous and reproductive systems), as well as the long-term developmental outcomes of offspring. With strict regulatory and ethical oversight, the safety, tolerability, pharmacokinetic characteristics, and preliminary efficacy of fetal interventions in humans should be assessed, along with long-term postnatal follow-up. Professional societies should develop evidence-based clinical practice guidelines for fetal intervention procedures, clearly defining the indications, contraindications, operational standards, monitoring requirements, and long-term follow-up protocols. The following five aspects encompass ethical considerations regarding interventions during fetal development: the moral status of the fetus as a patient; the extreme uncertainty in risk-benefit assessments; the complexity of informed consent; equity, accessibility, and resource allocation; and the establishment of regulatory and oversight frameworks.
Author contributions
ZZ: Writing – original draft, Data curation. YS: Data curation, Writing – original draft. MS: Writing – review & editing, Supervision. BW: Writing – review & editing, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the National Key R&D Program of China (2022YFC2703700 and 2019YFA0802600) and the National Natural Science Foundation of China (81974244). Suzhou Basic Research Pilot Program (SSD2024069) and Suzhou Gusu Health Talents Project (GSWS2022010). We appreciate the support of Suzhou city “Gusu Talent Program” (2021057).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ID, iron deficiency; Fpn, ferroportin; Apo-Tf, iron-free transferrin; holo-Tf, holotransferrin; TfR1, transferrin receptor 1; Fe3+, ferric iron; Fe2+, ferrous iron; STEAP3/4, six-transmembrane epithelial antigen of the prostrate protiens 3 and 4; DMT1, divalent metal transporter; ZIP, Zrt/IRt-like protein; IDA, iron deficiency anemia; Hb, hemoglobin; BMI, body mass index; IL-6, interleukin-6; CRP, C-reactive protein; Gp130, glycoprotein 130; JAK, Janus kinase; STAT, signal transducer and activator of transcription; Tf, transferrin; IRP-1, iron regulatory protein-1; AChE, acetylcholinesterase; α7nAChR, α7 nicotinic acetylcholine receptors; PAE, prenatal alcohol exposure; SF, serum ferritin; BDNF, brain-derived neurotrophic factor; OPC, oligodendrocyte progenitor cell; Fth, heavy chain; Ftl, light chain; GFAP, glial fibrillary acidic protein; CX43, connexin43; TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; IGF-1, insulin-like growth factor 1; FA, fractional anisotropy; LTP, long-term potentiation; VMB, ventral midbrain; Hct, hematocrit; PDMS-2, Peabody Developmental Motor Scale; BMP, bone morphogenetic protein; TET, Ten-Eleven translocation; JARID, jumonji and AT-rich interaction domain; 5mC, 5-methylcytosine; 5hmC, 5-hydroxymethylcytosine; JmjC, Jumonji C; HDAC1, histone deacetylase 1; H3K4 me1/2, histone H3 lysine 4; miRNA, microRNA; OGT, O-GlcNAc Transferase; HPA, hypothalamic–pituitary–adrenal; GR, glucocorticoid receptor; GC, glucocorticoid; Hsp, heat shock protein; CRH, corticotropin-releasing hormone; ADHD, attention deficit/hyperactivity disorder; ERK, extracellular signal-regulated kinase; PI3K, phosphoinositide 3-kinase; UMP, uridine monophosphate; IFAS, iron–folate acid supplementation; Hp, Haptoglobin.
References
1. Johnson-Wimbley TD, Graham DY. Diagnosis and management of iron deficiency anemia in the 21st century. Therap Adv Gastroenterol. (2011) 4:177–84. doi: 10.1177/1756283X11398736
2. Georgieff MK. Iron deficiency in pregnancy. Am J Obstet Gynecol. (2020) 223:516–24. doi: 10.1016/j.ajog.2020.03.006
3. Karyadi E, Reddy JC, Dearden KA, Purwanti T, Mardewi Asri E, Roquero LB, et al. Antenatal care is associated with adherence to iron supplementation among pregnant women in selected low-middle-income-countries of Asia, Africa, and Latin America & the Caribbean regions: Insights from Demographic and Health Surveys. Matern Child Nutr. (2023) 19:e13477. doi: 10.1111/mcn.13477
4. Engidaw MT, Lee P, Fekadu G, Mondal P, Ahmed F. Effect of nutrition education during pregnancy on iron-folic acid supplementation compliance and anemia in low- and middle-income countries: a systematic review and meta-analysis. Nutr Rev. (2025) 83:e1472–87. doi: 10.1093/nutrit/nuae170
5. Wang M, Gao H, Wang J, Cao C, Ying X, Wei Y, et al. Global burden and inequality of iron deficiency: findings from the Global Burden of Disease datasets 1990-2017. Nutr J. (2022) 21:16. doi: 10.1186/s12937-022-00771-3
6. Hasan MM, Soares Magalhaes RJ, Garnett SP, Fatima Y, Tariqujjaman M, Pervin S, et al. Anaemia in women of reproductive age in low- and middle-income countries: progress towards the 2025 global nutrition target. Bull World Health Organ. (2022) 100:196–204. doi: 10.2471/BLT.20.280180
7. Yismaw AE, Tulu HB, Kassie FY, Araya BM. Iron-folic acid adherence and associated factors among pregnant women attending antenatal care at Metema District, Northwest Ethiopia. Front Public Health. (2022) 10:978084. doi: 10.3389/fpubh.2022.978084
8. Sanin KI, Alam Shaun M, Rita RS, Hasan MK, Khanam M, Haque MA. What makes Bangladeshi pregnant women more compliant to iron-folic acid supplementation: a nationally representative cross-sectional survey result. Nutrients. (2023) 15:1512. doi: 10.3390/nu15061512
9. Salifu Y, Agyeman YN, Lasong J. Adherence to and predictors of iron-folate acid supplementation among pregnant women in a pastoral population in Ghana: a community-based cross-sectional study. Reprod Health. (2024) 21:165. doi: 10.1186/s12978-024-01877-z
10. Felipe-Dimog EB, Yu CH, Ho CH, Liang FW. Factors influencing the compliance of pregnant women with iron and folic acid supplementation in the Philippines: 2017 Philippine demographic and health survey analysis. Nutrients. (2021) 13:3060. doi: 10.3390/nu13093060
11. Teichman J, Nisenbaum R, Lausman A, Sholzberg M. Suboptimal iron deficiency screening in pregnancy and the impact of socioeconomic status in a high-resource setting. Blood Adv. (2021) 5:4666–73. doi: 10.1182/bloodadvances.2021004352
12. Benson AE, Lo JO, Achebe MO, Aslan JS, Auerbach M, Bannow BTS, et al. Management of iron deficiency in children, adults, and pregnant individuals: evidence-based and expert consensus recommendations. Lancet Haematol. (2025) 12:e376–88. doi: 10.1016/S2352-3026(25)00038-9
13. Jin J. Screening for iron deficiency and iron deficiency anemia during pregnancy. JAMA. (2024) 332:942. doi: 10.1001/jama.2024.14791
14. Pai RD, Chong YS, Clemente-Chua LR, Irwinda R, Huynh TNK, Wibowo N, et al. Prevention and management of iron deficiency/iron-deficiency anemia in women: an Asian Expert Consensus. Nutrients. (2023) 15:125. doi: 10.3390/nu15143125
15. Agarwal AM, Rets A. Laboratory approach to investigation of anemia in pregnancy. Int J Lab Hematol. (2021) 43(Suppl 1):65–70. doi: 10.1111/ijlh.13551
16. Muckenthaler MU, Rivella S, Hentze MW, Galy B. A red carpet for iron metabolism. Cell. (2017) 168:344–61. doi: 10.1016/j.cell.2016.12.034
17. Zeidan RS, Han SM, Leeuwenburgh C, Xiao R. Iron homeostasis and organismal aging. Ageing Res Rev. (2021) 72:101510. doi: 10.1016/j.arr.2021.101510
18. Fisher AL, Nemeth E. Iron homeostasis during pregnancy. Am J Clin Nutr. (2017) 106:1567S−74S. doi: 10.3945/ajcn.117.155812
19. Cao C, O'Brien KO. Pregnancy and iron homeostasis: an update. Nutr Rev. (2013) 71:35–51. doi: 10.1111/j.1753-4887.2012.00550.x
20. McArdle HJ, Gambling L, Kennedy C. Iron deficiency during pregnancy: the consequences for placental function and fetal outcome. Proc Nutr Soc. (2014) 73:9–15. doi: 10.1017/S0029665113003637
21. Lozoff B, Georgieff MK. Iron deficiency and brain development. Semin Pediatr Neurol. (2006) 13:158–65. doi: 10.1016/j.spen.2006.08.004
22. Tamura T, Goldenberg RL, Hou J, Johnston KE, Cliver SP, Ramey SL, et al. Cord serum ferritin concentrations and mental and psychomotor development of children at five years of age. J Pediatr. (2002) 140:165–70. doi: 10.1067/mpd.2002.120688
23. Gundacker A, Glat M, Wais J, Stoehrmann P, Pollak A, Pollak DD. Early-life iron deficiency persistently disrupts affective behaviour in mice. Ann Med. (2023) 55:1265–77. doi: 10.1080/07853890.2023.2191003
24. Geng F, Mai X, Zhan J, Xu L, Zhao Z, Georgieff M, et al. Impact of fetal-neonatal iron deficiency on recognition memory at 2 months of age. J Pediatr. (2015) 167:1226–32. doi: 10.1016/j.jpeds.2015.08.035
25. Bothwell TH. Iron requirements in pregnancy and strategies to meet them. Am J Clin Nutr. (2000) 72:257S−64S. doi: 10.1093/ajcn/72.1.257S
26. Donovan A, Lima CA, Pinkus JL, Pinkus GS, Zon LI, Robine S, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. (2005) 1:191–200. doi: 10.1016/j.cmet.2005.01.003
27. van Santen S, Kroot JJ, Zijderveld G, Wiegerinck ET, Spaanderman ME, Swinkels DW. The iron regulatory hormone hepcidin is decreased in pregnancy: a prospective longitudinal study. Clin Chem Lab Med. (2013) 51:1395–401. doi: 10.1515/cclm-2012-0576
28. Sangkhae V, Fisher AL, Ganz T, Nemeth E. Iron homeostasis during pregnancy: maternal, placental, and fetal regulatory mechanisms. Annu Rev Nutr. (2023) 43:279–300. doi: 10.1146/annurev-nutr-061021-030404
29. Ohgami RS, Campagna DR, McDonald A, Fleming MD. The Steap proteins are metalloreductases. Blood. (2006) 108:1388–94. doi: 10.1182/blood-2006-02-003681
30. Wang CY, Jenkitkasemwong S, Duarte S, Sparkman BK, Shawki A, Mackenzie B, et al. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J Biol Chem. (2012) 287:34032–43. doi: 10.1074/jbc.M112.367284
31. Georgieff MK, Wobken JK, Welle J, Burdo JR, Connor JR. Identification and localization of divalent metal transporter-1 (DMT-1) in term human placenta. Placenta. (2000) 21:799–804. doi: 10.1053/plac.2000.0566
32. Guller S, Buhimschi CS, Ma YY, Huang ST, Yang L, Kuczynski E, et al. Placental expression of ceruloplasmin in pregnancies complicated by severe preeclampsia. Lab Invest. (2008) 88:1057–67. doi: 10.1038/labinvest.2008.74
33. Fuqua BK, Lu Y, Darshan D, Frazer DM, Wilkins SJ, Wolkow N, et al. The multicopper ferroxidase hephaestin enhances intestinal iron absorption in mice. PLoS ONE. (2014) 9:e98792. doi: 10.1371/journal.pone.0098792
34. Chen H, Attieh ZK, Syed BA, Kuo YM, Stevens V, Fuqua BK, et al. Identification of zyklopen, a new member of the vertebrate multicopper ferroxidase family, and characterization in rodents and human cells. J Nutr. (2010) 140:1728–35. doi: 10.3945/jn.109.117531
35. Sangkhae V, Fisher AL, Wong S, Koenig MD, Tussing-Humphreys L, Chu A, et al. Effects of maternal iron status on placental and fetal iron homeostasis. J Clin Invest. (2020) 130:625–40. doi: 10.1172/JCI127341
36. Siddappa AJ, Rao RB, Wobken JD, Casperson K, Leibold EA, Connor JR, et al. Iron deficiency alters iron regulatory protein and iron transport protein expression in the perinatal rat brain. Pediatr Res. (2003) 53:800–7. doi: 10.1203/01.PDR.0000058922.67035.D5
37. Finkelstein JL, Cuthbert A, Weeks J, Venkatramanan S, Larvie DY, De-Regil LM, et al. Daily oral iron supplementation during pregnancy. Cochrane Database Syst Rev. (2024) 8:CD004736. doi: 10.1002/14651858.CD004736.pub6
38. Naveed K, Goldberg N, Shore E, Dhoot A, Gabrielson D, Goodarzi Z, et al. Defining ferritin clinical decision limits to improve diagnosis and treatment of iron deficiency: a modified Delphi study. Int J Lab Hematol. (2023) 45:377–86. doi: 10.1111/ijlh.14016
39. Jaime-Perez JC, Herrera-Garza JL, Gomez-Almaguer D. Sub-optimal fetal iron acquisition under a maternal environment. Arch Med Res. (2005) 36:598–602. doi: 10.1016/j.arcmed.2005.03.023
40. Kumar A, Rai AK, Basu S, Dash D, Singh JS. Cord blood and breast milk iron status in maternal anemia. Pediatrics. (2008) 121:e673–7. doi: 10.1542/peds.2007-1986
41. Bahr TM, Benson AE, Kling PJ, Ohls RK, Ward DM, Christensen RD. Maternal obesity and impaired offspring neurodevelopment: could fetal iron deficiency be a pathogenic link? J Perinatol. (2021) 41:1199–200. doi: 10.1038/s41372-021-00951-9
42. Jones AD, Zhao G, Jiang YP, Zhou M, Xu G, Kaciroti N, et al. Maternal obesity during pregnancy is negatively associated with maternal and neonatal iron status. Eur J Clin Nutr. (2016) 70:918–24. doi: 10.1038/ejcn.2015.229
43. Dao MC, Sen S, Iyer C, Klebenov D, Meydani SN. Obesity during pregnancy and fetal iron status: is Hepcidin the link? J Perinatol. (2013) 33:177–81. doi: 10.1038/jp.2012.81
44. Ganz T. Hepcidin and iron regulation, 10 years later. Blood. (2011) 117:4425–33. doi: 10.1182/blood-2011-01-258467
45. Flores-Quijano ME, Montalvo-Velarde I, Vital-Reyes VS, Rodríguez-Cruz M, Rendón-Macías ME, López-Alarcón M. Longitudinal analysis of the interaction between obesity and pregnancy on iron homeostasis: role of Hepcidin. Arch Med Res. (2016) 47:550–6. doi: 10.1016/j.arcmed.2016.11.011
46. Liang T, Jinglong X, Shusheng D, Aiyou W. Maternal obesity stimulates lipotoxicity and up-regulates inflammatory signaling pathways in the full-term swine placenta. Anim Sci J. (2018) 89:1310–22. doi: 10.1111/asj.13064
47. Carbia-Nagashima A, Arzt E. Intracellular proteins and mechanisms involved in the control of gp130/JAK/STAT cytokine signaling. IUBMB Life. (2004) 56:83–8. doi: 10.1080/15216540410001668064
48. Banerjee S, Katiyar P, Kumar L, Kumar V, Saini SS, Krishnan V, et al. Black pepper prevents anemia of inflammation by inhibiting hepcidin over-expression through BMP6-SMAD1/IL6-STAT3 signaling pathway. Free Radic Biol Med. (2021) 168:189–202. doi: 10.1016/j.freeradbiomed.2021.03.019
49. Dao MC, Meydani SN. Iron biology, immunology, aging, and obesity: four fields connected by the small peptide hormone hepcidin. Adv Nutr. (2013) 4:602–17. doi: 10.3945/an.113.004424
50. Flynn AC, Begum S, White SL, Dalrymple K, Gill C, Alwan NA, et al. Relationships between maternal obesity and maternal and neonatal iron status. Nutrients. (2018) 10:1000. doi: 10.3390/nu10081000
51. Cao C, Pressman EK, Cooper EM, Guillet R, Westerman M, O'Brien KO. Prepregnancy body mass index and gestational weight gain have no negative impact on maternal or neonatal iron status. Reprod Sci. (2016) 23:613–22. doi: 10.1177/1933719115607976
52. Verner AM, Manderson J, Lappin TR, McCance DR, Halliday HL, Sweet DG. Influence of maternal diabetes mellitus on fetal iron status. Arch Dis Child Fetal Neonatal Ed. (2007) 92:F399–401. doi: 10.1136/adc.2006.097279
53. Petry CD, Eaton MA, Wobken JD, Mills MM, Johnson DE, Georgieff MK. Iron deficiency of liver, heart, and brain in newborn infants of diabetic mothers. J Pediatr. (1992) 121:109–14. doi: 10.1016/S0022-3476(05)82554-5
54. Georgieff MK, Petry CD, Mills MM, McKay H, Wobken JD. Increased N-glycosylation and reduced transferrin-binding capacity of transferrin receptor isolated from placentae of diabetic women. Placenta. (1997) 18:563–8. doi: 10.1016/0143-4004(77)90011-X
55. Kato J, Kobune M, Ohkubo S, Fujikawa K, Tanaka M, Takimoto R, et al. Iron/IRP-1-dependent regulation of mRNA expression for transferrin receptor, DMT1 and ferritin during human erythroid differentiation. Exp Hematol. (2007) 35:879–87. doi: 10.1016/j.exphem.2007.03.005
56. Georgieff MK, Berry SA, Wobken JD, Leibold EA. Increased placental iron regulatory protein-1 expression in diabetic pregnancies complicated by fetal iron deficiency. Placenta. (1999) 20:87–93. doi: 10.1053/plac.1998.0339
57. Yang A, Zhao J, Lu M, Gu Y, Zhu Y, Chen D, et al. Expression of hepcidin and ferroportin in the placenta, and ferritin and transferrin receptor 1 levels in maternal and umbilical cord blood in pregnant women with and without gestational diabetes. Int J Environ Res Public Health. (2016) 13:766. doi: 10.3390/ijerph13080766
58. Zimmermann P, Antonelli MC, Sharma R, Müller A, Zelgert C, Fabre B, et al. Prenatal stress perturbs fetal iron homeostasis in a sex specific manner Sci Rep. (2022) 12:9341. doi: 10.1038/s41598-022-13633-z
59. Rendina DN, Blohowiak SE, Coe CL, Kling PJ. Maternal perceived stress during pregnancy increases risk for low neonatal iron at delivery and depletion of storage iron at one year. J Pediatr. (2018) 200:166–173 e2. doi: 10.1016/j.jpeds.2018.04.040
60. Campbell RK, Tamayo-Ortiz M, Cantoral A, Schnaas L, Osorio-Valencia E, Wright RJ, et al. Maternal prenatal psychosocial stress and prepregnancy BMI associations with fetal iron status. Curr Dev Nutr. (2020) 4:nzaa018. doi: 10.1093/cdn/nzaa018
61. Coe CL, Lubach GR, Shirtcliff EA. Maternal stress during pregnancy predisposes for iron deficiency in infant monkeys impacting innate immunity. Pediatr Res. (2007) 61:520–4. doi: 10.1203/pdr.0b013e318045be53
62. Armony-Sivan R, Aviner S, Cojocaru L, Fytlovitch S, Ben-Alon D, Eliassy A, et al. Prenatal maternal stress predicts cord-blood ferritin concentration. J Perinat Med. (2013) 41:259–65. doi: 10.1515/jpm-2012-0125
63. Alyamani RAS, Murgatroyd C. Epigenetic programming by early-life stress. Prog Mol Biol Transl Sci. (2018) 157:133–50. doi: 10.1016/bs.pmbts.2018.01.004
64. Cortes M, Cao M, Liu HL, Moore CS, Durosier LD, Burns P, et al. alpha7 nicotinic acetylcholine receptor signaling modulates the inflammatory phenotype of fetal brain microglia: first evidence of interference by iron homeostasis. Sci Rep. (2017) 7:10645. doi: 10.1038/s41598-017-09439-z
65. Shaked I, Meerson A, Wolf Y, Avni R, Greenberg D, Gilboa-Geffen A, et al. MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity. (2009) 31:965–73. doi: 10.1016/j.immuni.2009.09.019
66. Sazak S, Kayiran SM, Paksoy Y. Umbilical cord serum erythropoietin levels and maternal smoking in pregnancy. ScientificWorldJournal. (2012) 2012:420763. doi: 10.1100/2012/420763
67. Chełchowska M, Maciejewski TM, Mazur J, Gajewska J, Zasimovich A, Ołtarzewski M, et al. Active tobacco smoke exposure in utero and concentrations of hepcidin and selected iron parameters in newborns. Int J Environ Res Public Health. (2019) 16:1996. doi: 10.3390/ijerph16111996
68. Sekovanić A, Jurasović J, Piasek M, Pašalić D, Orct T, Grgec AS, et al. Metallothionein 2A gene polymorphism and trace elements in mother-newborn pairs in the Croatian population. J Trace Elem Med Biol. (2018) 45:163–70. doi: 10.1016/j.jtemb.2017.10.011
69. Rao SS, Agadi R, Shetty S, Rao R, Shenoy RD. Smokeless tobacco exposure and fetal iron status: an analytical study. Indian J Community Med. (2022) 47:87–91. doi: 10.4103/ijcm.ijcm_1136_21
70. Carter RC, Dodge NC, Molteno CD, Meintjes EM, Jacobson JL, Jacobson SW. Mediating and moderating effects of iron homeostasis alterations on fetal alcohol-related growth and neurobehavioral deficits. Nutrients. (2022) 14:4332. doi: 10.3390/nu14204432
71. Bradley R, Lakpa KL, Burd M, Mehta S, Katusic MZ, Greenmyer JR. Fetal alcohol spectrum disorder and iron homeostasis. Nutrients. (2022) 14:4223. doi: 10.3390/nu14204223
72. Saini N, Helfrich KK, Kwan STC, Huebner SM, Abazi J, Flentke GR, et al. Alcohol's dysregulation of maternal-fetal IL-6 and p-STAT3 is a function of maternal iron status. Alcohol Clin Exp Res. (2019) 43:2332–43. doi: 10.1111/acer.14200
73. Huebner SM, Blohowiak SE, Kling PJ, Smith SM. Prenatal alcohol exposure alters fetal iron distribution and elevates hepatic hepcidin in a rat model of fetal alcohol spectrum disorders. J Nutr. (2016) 146:1180–8. doi: 10.3945/jn.115.227983
74. Masehi-Lano JJ, Deyssenroth M, Jacobson SW, Jacobson JL, Molteno CD, Dodge NC, et al. Alterations in placental inflammation-related gene expression partially mediate the effects of prenatal alcohol consumption on maternal iron homeostasis. Nutrients. (2023) 15:4105. doi: 10.3390/nu15194105
75. Andersen CBF, Stødkilde K, Sæderup KL, Kuhlee A, Raunser S, Graversen JH, et al. Haptoglobin. Antioxid Redox Signal. (2017) 26:814–31. doi: 10.1089/ars.2016.6793
76. Langlois MR, Delanghe JR. Biological and clinical significance of haptoglobin polymorphism in humans. Clin Chem. (1996) 42:1589–600. doi: 10.1093/clinchem/42.10.1589
77. Hu TY, Mayasari NR, Cheng TM, Bai CH, Chao JC, Huang YL, et al. Polymorphisms of haptoglobin modify the relationship between dietary iron and the risk of gestational iron-deficiency anemia. Eur J Nutr. (2023) 62:299–309. doi: 10.1007/s00394-022-02987-9
78. Ranade SC, Nawaz S, Chakrabarti A, Gressens P, Mani S. Spatial memory deficits in maternal iron deficiency paradigms are associated with altered glucocorticoid levels. Horm Behav. (2013) 64:26–36. doi: 10.1016/j.yhbeh.2013.04.005
79. Amin SB, Orlando M, Wang H. Latent iron deficiency in utero is associated with abnormal auditory neural myelination in >/= 35 weeks gestational age infants. J Pediatr. (2013) 163:1267–71. doi: 10.1016/j.jpeds.2013.06.020
80. Isasi E, Figares M, Abudara V, Olivera-Bravo S. Gestational and lactational iron deficiency anemia impairs myelination and the neurovascular unit in infant rats. Mol Neurobiol. (2022) 59:3738–54. doi: 10.1007/s12035-022-02798-3
81. Wu LL, Zhang L, Shao J, Qin YF, Yang RW, Zhao ZY. Effect of perinatal iron deficiency on myelination and associated behaviors in rat pups. Behav Brain Res. (2008) 188:263–70. doi: 10.1016/j.bbr.2007.11.003
82. Reinert A, Morawski M, Seeger J, Arendt T, Reinert T. Iron concentrations in neurons and glial cells with estimates on ferritin concentrations. BMC Neurosci. (2019) 20:25. doi: 10.1186/s12868-019-0507-7
83. Stephenson E, Nathoo N, Mahjoub Y, Dunn JF, Yong VW. Iron in multiple sclerosis: roles in neurodegeneration and repair. Nat Rev Neurol. (2014) 10:459–68. doi: 10.1038/nrneurol.2014.118
84. Cheli VT, Santiago González DA, Wan R, Rosenblum SL, Denaroso GE, Angeliu CG, et al. Transferrin receptor is necessary for proper oligodendrocyte iron homeostasis and development. J Neurosci. (2023) 43:3614–29. doi: 10.1523/JNEUROSCI.1383-22.2023
85. Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O'Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. (2014) 34:11929–47. doi: 10.1523/JNEUROSCI.1860-14.2014
86. Li Y, Guan Q, Chen Y, Han H, Liu W, Nie Z. Transferrin receptor and ferritin-H are developmentally regulated in oligodendrocyte lineage cells. Neural Regen Res. (2013) 8:6–12. doi: 10.4103/1673-5374.126960
87. Cheli VT, Correale J, Paez PM, Pasquini JM. Iron metabolism in oligodendrocytes and astrocytes, implications for myelination and remyelination. ASN Neuro. (2020) 12:1759091420962681. doi: 10.1177/1759091420962681
88. Badaracco ME, Ortiz EH, Soto EF, Connor J, Pasquini JM. Effect of transferrin on hypomyelination induced by iron deficiency. J Neurosci Res. (2008) 86:2663–73. doi: 10.1002/jnr.21709
89. Aparicio E, Mathieu P, Pereira Luppi M, Almeira Gubiani MF, Adamo AM. The Notch signaling pathway: its role in focal CNS demyelination and apotransferrin-induced remyelination. J Neurochem. (2013) 127:819–36. doi: 10.1111/jnc.12440
90. Perez MJ, Fernandez N, Pasquini JM. Oligodendrocyte differentiation and signaling after transferrin internalization: a mechanism of action. Exp Neurol. (2013) 248:262–74. doi: 10.1016/j.expneurol.2013.06.014
91. Perez MJ, Ortiz EH, Roffé M, Soto EF, Pasquini JM. Fyn kinase is involved in oligodendroglial cell differentiation induced by apotransferrin. J Neurosci Res. (2009) 87:3378–89. doi: 10.1002/jnr.21962
92. Su Y, Zhu W, Su T, Huang L, Qin M, Wang Q, et al. Endothelial TREM-1 mediates sepsis-induced blood–brain barrier disruption and cognitive impairment via the PI3K/Akt pathway. J Neuroinflammation. (2025) 22:142. doi: 10.1186/s12974-025-03469-5
93. Gonzalez R, Reinberg D. The Notch pathway: a guardian of cell fate during neurogenesis. Curr Opin Cell Biol. (2025) 95:102543. doi: 10.1016/j.ceb.2025.102543
94. Sun ZY, Ma DL, Gu LH, Chen X, Zhang L, Li L. DHF-7 ameliorates behavioral disorders and white matter lesions by regulating BDNF and Fyn in a mouse model of schizophrenia induced by cuprizone and MK-801. Int J Neuropsychopharmacol. (2022) 25:600–12. doi: 10.1093/ijnp/pyac022
95. Mukherjee C, Kling T, Russo B, Miebach K, Kess E, Schifferer M, et al. Oligodendrocytes provide antioxidant defense function for neurons by secreting ferritin heavy chain. Cell Metab. (2020) 32:259–72 e10. doi: 10.1016/j.cmet.2020.05.019
96. Rosato-Siri MV, Marziali L, Guitart ME, Badaracco ME, Puntel M, Pitossi F, et al. Iron availability compromises not only oligodendrocytes but also astrocytes and microglial. Cells Mol Neurobiol. (2018) 55:1068–81. doi: 10.1007/s12035-016-0369-2
97. Mason JL, Suzuki K, Chaplin DD, Matsushima GK. Interleukin-1beta promotes repair of the CNS. J Neurosci. (2001) 21:7046–52. doi: 10.1523/JNEUROSCI.21-18-07046.2001
98. Correale J, Farez MF. The role of astrocytes in multiple sclerosis progression. Front Neurol. (2015) 6:180. doi: 10.3389/fneur.2015.00180
99. Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci. (2001) 4:1116–22. doi: 10.1038/nn738
100. Rajendran R, Böttiger G, Stadelmann C, Karnati S, Berghoff M. FGF/FGFR pathways in multiple sclerosis and in its disease models. Cells. (2021) 10:884. doi: 10.3390/cells10040884
101. Zarruk JG, Berard JL, Passos dos Santos R, Kroner A, Lee J, Arosio P, et al. Expression of iron homeostasis proteins in the spinal cord in experimental autoimmune encephalomyelitis and their implications for iron accumulation. Neurobiol Dis. (2015) 81:93–107. doi: 10.1016/j.nbd.2015.02.001
102. Schulz K, Kroner A, David S. Iron efflux from astrocytes plays a role in remyelination. J Neurosci. (2012) 32:4841–7. doi: 10.1523/JNEUROSCI.5328-11.2012
103. Chen Z, Jiang R, Chen M, Zheng J, Chen M, Braidy N, et al. Multi-copper ferroxidase deficiency leads to iron accumulation and oxidative damage in astrocytes and oligodendrocytes. Sci Rep. (2019) 9:9437. doi: 10.1038/s41598-019-46019-9
104. Cramer SC, Sur M, Dobkin BH, O'Brien C, Sanger TD, Trojanowski JQ, et al. Harnessing neuroplasticity for clinical applications. Brain. (2011) 134(Pt 6):1591–609. doi: 10.1093/brain/awr039
105. Monk C, Georgieff MK, Xu D, Hao X, Bansal R, Gustafsson H, et al. Maternal prenatal iron status and tissue organization in the neonatal brain. Pediatr Res. (2016) 79:482–8. doi: 10.1038/pr.2015.248
106. Jorgenson LA, Wobken JD, Georgieff MK. Perinatal iron deficiency alters apical dendritic growth in hippocampal CA1 pyramidal neurons. Dev Neurosci. (2003) 25:412–20. doi: 10.1159/000075667
107. Greminger AR, Lee DL, Shrager P, Mayer-Pröschel M. Gestational iron deficiency differentially alters the structure and function of white and gray matter brain regions of developing rats. J Nutr. (2014) 144:1058–66. doi: 10.3945/jn.113.187732
108. Brunette KE, Tran PV, Wobken JD, Carlson ES, Georgieff MK. Gestational and neonatal iron deficiency alters apical dendrite structure of CA1 pyramidal neurons in adult rat hippocampus. Dev Neurosci. (2010) 32:238–48. doi: 10.1159/000314341
109. Jorgenson LA, Sun M, O'Connor M, Georgieff MK. Fetal iron deficiency disrupts the maturation of synaptic function and efficacy in area CA1 of the developing rat hippocampus. Hippocampus. (2005) 15:1094–102. doi: 10.1002/hipo.20128
110. Carlson ES, Stead JD, Neal CR, Petryk A, Georgieff MK. Perinatal iron deficiency results in altered developmental expression of genes mediating energy metabolism and neuronal morphogenesis in hippocampus. Hippocampus. (2007) 17:679–91. doi: 10.1002/hipo.20307
111. Wang Y, Gu C, Ewing AG. Single-vesicle electrochemistry following repetitive stimulation reveals a mechanism for plasticity changes with iron deficiency. Angew Chem Int Ed Engl. (2022) 61:e202200716. doi: 10.1002/anie.202200716
112. Radlowski EC, Johnson RW. Perinatal iron deficiency and neurocognitive development. Front Hum Neurosci. (2013) 7:585. doi: 10.3389/fnhum.2013.00585
113. Aguilar-Valles A, Flores C, Luheshi GN. Prenatal inflammation-induced hypoferremia alters dopamine function in the adult offspring in rat: relevance for schizophrenia. PLoS ONE. (2010) 5:e10967. doi: 10.1371/journal.pone.0010967
114. Beard JL, Connor JR. Iron status and neural functioning. Annu Rev Nutr. (2003) 23:41–58. doi: 10.1146/annurev.nutr.23.020102.075739
115. Erikson KM, Jones BC, Beard JL. Iron deficiency alters dopamine transporter functioning in rat striatum. J Nutr. (2000) 130:2831–7. doi: 10.1093/jn/130.11.2831
116. Erikson KM, Jones BC, Hess EJ, Zhang Q, Beard JL. Iron deficiency decreases dopamine D1 and D2 receptors in rat brain. Pharmacol Biochem Behav. (2001) 69:409–18. doi: 10.1016/S0091-3057(01)00563-9
117. Unger EL, Bianco LE, Jones BC, Allen RP, Earley CJ. Low brain iron effects and reversibility on striatal dopamine dynamics. Exp Neurol. (2014) 261:462–8. doi: 10.1016/j.expneurol.2014.06.023
118. Wang X, Wiesinger J, Beard J, Felt B, Menzies S, Earley C, et al. Thy1 expression in the brain is affected by iron and is decreased in Restless Legs Syndrome. J Neurol Sci. (2004) 220:59–66. doi: 10.1016/j.jns.2004.02.004
119. Beard JL, Felt B, Schallert T, Burhans M, Connor JR, Georgieff MK. Moderate iron deficiency in infancy: biology and behavior in young rats. Behav Brain Res. (2006) 170:224–32. doi: 10.1016/j.bbr.2006.02.024
120. Unger EL, Hurst AR, Georgieff MK, Schallert T, Rao R, Connor JR, et al. Behavior and monoamine deficits in prenatal and perinatal iron deficiency are not corrected by early postnatal moderate-iron or high-iron diets in rats. J Nutr. (2012) 142:2040–9. doi: 10.3945/jn.112.162198
121. Burhans MS, Dailey C, Beard Z, Wiesinger J, Murray-Kolb L, Jones BC, et al. Iron deficiency: differential effects on monoamine transporters. Nutr Neurosci. (2005) 8:31–8. doi: 10.1080/10284150500047070
122. Carter AM. Animal models of human pregnancy and placentation: alternatives to the mouse. Reproduction. (2020) 160:R129–43. doi: 10.1530/REP-20-0354
123. Carter AM. Animal models of human placentation–a review. Placenta. (2007) 28(Suppl A):S41–7. doi: 10.1016/j.placenta.2006.11.002
124. Morrison JL, Botting KJ, Darby JRT, David AL, Dyson RM, Gatford KL, et al. Guinea pig models for translation of the developmental origins of health and disease hypothesis into the clinic. J Physiol. (2018) 596:5535–69. doi: 10.1113/JP274948
125. Garcia Y, Diaz-Castro J. Advantages and disadvantages of the animal models v. in vitro studies in iron metabolism: a review. Animal. (2013) 7:1651–8. doi: 10.1017/S1751731113001134
126. Schmidt AT, Alvarez GC, Grove WM, Rao R, Georgieff MK. Early iron deficiency enhances stimulus-response learning of adult rats in the context of competing spatial information. Dev Cogn Neurosci. (2012) 2:174–80. doi: 10.1016/j.dcn.2011.07.014
127. McEchron MD, Cheng AY, Liu H, Connor JR, Gilmartin MR. Perinatal nutritional iron deficiency permanently impairs hippocampus-dependent trace fear conditioning in rats. Nutr Neurosci. (2005) 8:195–206. doi: 10.1080/10284150500162952
128. McEchron MD, Alexander DN, Gilmartin MR, Paronish MD. Perinatal nutritional iron deficiency impairs hippocampus-dependent trace eyeblink conditioning in rats. Dev Neurosci. (2008) 30:243–54. doi: 10.1159/000110502
129. Carlson ES, Tkac I, Magid R, O'Connor MB, Andrews NC, Schallert T, et al. Iron is essential for neuron development and memory function in mouse hippocampus. J Nutr. (2009) 139:672–9. doi: 10.3945/jn.108.096354
130. Shero N, Fiset S, Blakley B, Jougleux JL, Surette ME, Thabet M, et al. Impact of maternal iron deficiency on the auditory functions in the young and adult guinea pig. Nutr Neurosci. (2019) 22:444–52. doi: 10.1080/1028415X.2017.1408946
131. Jougleux JL, Rioux FM, Church MW, Fiset S, Surette ME. Mild iron deficiency anaemia during pregnancy and lactation in guinea pigs alters amplitudes and auditory nerve velocity but not brainstem transmission times in the offspring's auditory brainstem response. Nutr Neurosci. (2014) 17:37–47. doi: 10.1179/1476830513Y.0000000067
132. Fiset C, Rioux FM, Surette ME, Fiset S. Prenatal iron deficiency in guinea pigs increases locomotor activity but does not influence learning and memory. PLoS ONE. (2015) 10:e0133168. doi: 10.1371/journal.pone.0133168
133. Golub MS, Hogrefe CE, Tarantal AF, Germann SL, Beard JL, Georgieff MK, et al. Diet-induced iron deficiency anemia and pregnancy outcome in rhesus monkeys. Am J Clin Nutr. (2006) 83:647–56. doi: 10.1093/ajcn.83.3.647
134. Golub MS, Hogrefe CE, Germann SL, Capitanio JP, Lozoff B. Behavioral consequences of developmental iron deficiency in infant rhesus monkeys. Neurotoxicol Teratol. (2006) 28:3–17. doi: 10.1016/j.ntt.2005.10.005
135. Golub MS, Hogrefe CE, Germann SL. Iron deprivation during fetal development changes the behavior of juvenile rhesus monkeys. J Nutr. (2007) 137:979–84. doi: 10.1093/jn/137.4.979
136. Cortes RA, Green AE, Barr RF, Ryan RM. Fine motor skills during early childhood predict visuospatial deductive reasoning in adolescence. Dev Psychol. (2022) 58:1264–76. doi: 10.1037/dev0001354
137. Zhao G, Bian Y, Li M. Impact of passing items above the ceiling on the assessment results of Peabody developmental motor scales. Beijing Da Xue Xue Bao Yi Xue Ban. (2013) 45:928–32.
138. Gaul D, Issartel J. Fine motor skill proficiency in typically developing children: on or off the maturation track? Hum Mov Sci. (2016) 46:78–85. doi: 10.1016/j.humov.2015.12.011
139. Mireku MO, Davidson LL, Koura GK, Ouédraogo S, Boivin MJ, Xiong X, et al. Prenatal hemoglobin levels and early cognitive and motor functions of one-year-old children. Pediatrics. (2015) 136:e76–83. doi: 10.1542/peds.2015-0491
140. Ouyang J, Cai W, Wu P, Tong J, Gao G, Yan S, et al. Association between dietary patterns during pregnancy and children's neurodevelopment: a birth cohort study. Nutrients. (2024) 16:1530. doi: 10.3390/nu16101530
141. Santos DCC, Angulo-Barroso RM, Li M, Bian Y, Sturza J, Richards B, et al. Timing, duration, and severity of iron deficiency in early development and motor outcomes at 9 months. Eur J Clin Nutr. (2018) 72:332–41. doi: 10.1038/s41430-017-0015-8
142. Mireku MO, Davidson LL, Boivin MJ, Zoumenou R, Massougbodji A, Cot M, et al. Prenatal iron deficiency, neonatal ferritin, and infant cognitive function. Pediatrics. (2016) 138:e20161319. doi: 10.1542/peds.2016-1319
143. Hanieh S, Ha TT, Simpson JA, Casey GJ, Khuong NC, Thoang DD, et al. The effect of intermittent antenatal iron supplementation on maternal and infant outcomes in rural Viet Nam: a cluster randomised trial. PLoS Med. (2013) 10:e1001470. doi: 10.1371/journal.pmed.1001470
144. Tran TD, Biggs BA, Tran T, Simpson JA, Hanieh S, Dwyer T, et al. Impact on infants' cognitive development of antenatal exposure to iron deficiency disorder and common mental disorders. PLoS ONE. (2013) 8:e74876. doi: 10.1371/journal.pone.0074876
145. Fararouei M, Robertson C, Whittaker J, Sovio U, Ruokonen A, Pouta A, et al. Maternal Hb during pregnancy and offspring's educational achievement: a prospective cohort study over 30 years. Br J Nutr. (2010) 104:1363–8. doi: 10.1017/S0007114510002175
146. Chang S, Zeng L, Brouwer ID, Kok FJ, Yan H. Effect of iron deficiency anemia in pregnancy on child mental development in rural China. Pediatrics. (2013) 131:e755–63. doi: 10.1542/peds.2011-3513
147. Squire LR. Memory systems of the brain: a brief history and current perspective. Neurobiol Learn Mem. (2004) 82:171–7. doi: 10.1016/j.nlm.2004.06.005
148. Riggins T, Miller NC, Bauer PJ, Georgieff MK, Nelson CA. Consequences of low neonatal iron status due to maternal diabetes mellitus on explicit memory performance in childhood. Dev Neuropsychol. (2009) 34:762–79. doi: 10.1080/87565640903265145
149. Siddappa AM, Georgieff MK, Wewerka S, Worwa C, Nelson CA, Deregnier RA. Iron deficiency alters auditory recognition memory in newborn infants of diabetic mothers. Pediatr Res. (2004) 55:1034–41. doi: 10.1203/01.pdr.0000127021.38207.62
150. Geng F, Mai X, Zhan J, Xu L, Georgieff M, Shao J, et al. Timing of iron deficiency and recognition memory in infancy. Nutr Neurosci. (2022) 25:1–10. doi: 10.1080/1028415X.2019.1704991
151. Harvey L, Boksa P. Additive effects of maternal iron deficiency and prenatal immune activation on adult behaviors in rat offspring. Brain Behav Immun. (2014) 40:27–37. doi: 10.1016/j.bbi.2014.06.005
152. Schmidt RJ, Tancredi DJ, Krakowiak P, Hansen RL, Ozonoff S. Maternal intake of supplemental iron and risk of autism spectrum disorder. Am J Epidemiol. (2014) 180:890–900. doi: 10.1093/aje/kwu208
153. Brynge M, Gardner R, Sjöqvist H, Karlsson H, Dalman C. Maternal levels of acute phase proteins in early pregnancy and risk of autism spectrum disorders in offspring. Transl Psychiatry. (2022) 12:148. doi: 10.1038/s41398-022-01907-z
154. Sørensen HJ, Nielsen PR, Pedersen CB, Mortensen PB. Association between prepartum maternal iron deficiency and offspring risk of schizophrenia: population-based cohort study with linkage of Danish national registers. Schizophr Bull. (2011) 37:982–7. doi: 10.1093/schbul/sbp167
155. Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. (2007) 128:669–81. doi: 10.1016/j.cell.2007.01.033
156. Boland MJ, Nazor KL, Loring JF. Epigenetic regulation of pluripotency and differentiation. Circ Res. (2014) 115:311–24. doi: 10.1161/CIRCRESAHA.115.301517
157. Lien YC, Condon DE, Georgieff MK, Simmons RA, Tran PV. Dysregulation of neuronal genes by fetal-neonatal iron deficiency anemia is associated with altered DNA methylation in the rat hippocampus. Nutrients. (2019) 11:1191. doi: 10.3390/nu11051191
158. Taeubert MJ, de Prado-Bert P, Geurtsen ML, Mancano G, Vermeulen MJ, Reiss IKM, et al. Maternal iron status in early pregnancy and DNA methylation in offspring: an epigenome-wide meta-analysis. Clin Epigenetics. (2022) 14:59. doi: 10.1186/s13148-022-01276-w
159. Wang RN, Green J, Wang Z, Deng Y, Qiao M, Peabody M, et al. Bone morphogenetic protein (BMP) signaling in development and human diseases. Genes Dis. (2014) 1:87–105. doi: 10.1016/j.gendis.2014.07.005
160. Barks AK, Liu SX, Georgieff MK, Hallstrom TC, Tran PV. Early-life iron deficiency anemia programs the hippocampal epigenomic landscape. Nutrients. (2021) 13:3857. doi: 10.3390/nu13113857
161. Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. (2014) 156:45–68. doi: 10.1016/j.cell.2013.12.019
162. Hahn MA, Qiu R, Wu X, Li AX, Zhang H, Wang J, et al. Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell Rep. (2013) 3:291–300. doi: 10.1016/j.celrep.2013.01.011
163. Khare T, Pai S, Koncevicius K, Pal M, Kriukiene E, Liutkeviciute Z, et al. 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat Struct Mol Biol. (2012) 19:1037–43. doi: 10.1038/nsmb.2372
164. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. (2011) 21:381–95. doi: 10.1038/cr.2011.22
165. Blegen MB, Kennedy BC, Thibert KA, Gewirtz JC, Tran PV, Georgieff MK. Multigenerational effects of fetal-neonatal iron deficiency on hippocampal BDNF signaling. Physiol Rep. (2013) 1:e00096. doi: 10.1002/phy2.96
166. Liu SX, Barks AK, Lunos S, Gewirtz JC, Georgieff MK, Tran PV. Prenatal iron deficiency and choline supplementation interact to epigenetically regulate Jarid1b and Bdnf in the rat hippocampus into adulthood. Nutrients. (2021) 13:4527. doi: 10.3390/nu13124527
167. Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. (2006) 439:811–6. doi: 10.1038/nature04433
168. Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, et al. Structural insights into histone demethylation by JMJD2 family members. Cell. (2006) 125:691–702. doi: 10.1016/j.cell.2006.04.024
169. Vernimmen D, Lynch MD, De Gobbi M, Garrick D, Sharpe JA, Sloane-Stanley JA, et al. Polycomb eviction as a new distant enhancer function. Genes Dev. (2011) 25:1583–8. doi: 10.1101/gad.16985411
170. Pasini D, Cloos PA, Walfridsson J, Olsson L, Bukowski JP, Johansen JV, et al. JARID2 regulates binding of the Polycomb repressive complex 2 to target genes in ES cells. Nature. (2010) 464:306–10. doi: 10.1038/nature08788
171. Kouzarides T. Chromatin modifications and their function. Cell. (2007) 128:693–705. doi: 10.1016/j.cell.2007.02.005
172. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell. (2007) 129:823–37. doi: 10.1016/j.cell.2007.05.009
173. Tran PV, Kennedy BC, Lien YC, Simmons RA, Georgieff MK. Fetal iron deficiency induces chromatin remodeling at the Bdnf locus in adult rat hippocampus. Am J Physiol Regul Integr Comp Physiol. (2015) 308:R276–82. doi: 10.1152/ajpregu.00429.2014
174. Davison JM, Mellott TJ, Kovacheva VP, Blusztajn JK. Gestational choline supply regulates methylation of histone H3, expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a), and DNA methylation of their genes in rat fetal liver and brain. J Biol Chem. (2009) 284:1982–9. doi: 10.1074/jbc.M807651200
175. Correia de Sousa M, Gjorgjieva M, Dolicka D, Sobolewski C, Foti M. Deciphering miRNAs' Action through miRNA Editing. Int J Mol Sci. (2019) 20:6249. doi: 10.3390/ijms20246249
176. Gao Q, Zhou Y, Chen Y, Hu W, Jin W, Zhou C, et al. Role of iron in brain development, aging, and neurodegenerative diseases. Ann Med. (2025) 57:2472871. doi: 10.1080/07853890.2025.2472871
177. Bastian TW, von Hohenberg WC, Mickelson DJ, Lanier LM, Georgieff MK. Iron deficiency impairs developing hippocampal neuron gene expression, energy metabolism, and dendrite complexity. Dev Neurosci. (2016) 38:264–76. doi: 10.1159/000448514
178. Bastian TW, von Hohenberg WC, Georgieff MK, Lanier LM. Chronic energy depletion due to iron deficiency impairs dendritic mitochondrial motility during hippocampal neuron development. J Neurosci. (2019) 39:802–13. doi: 10.1523/JNEUROSCI.1504-18.2018
179. Silva CA, Yalnizyan-Carson A, Fernández Busch MV, van Zwieten M, Verhage M, Lohmann C. Activity-dependent regulation of mitochondrial motility in developing cortical dendrites. Elife. (2021) 10:e62091. doi: 10.7554/eLife.62091
180. Hirokawa N, Niwa S, Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. (2010) 68:610–38. doi: 10.1016/j.neuron.2010.09.039
181. Bastian TW. Potential mechanisms driving mitochondrial motility impairments in developing iron-deficient neurons. J Exp Neurosci. (2019) 13:1179069519858351. doi: 10.1177/1179069519858351
182. Pekkurnaz G, Trinidad JC, Wang X, Kong D, Schwarz TL. Glucose regulates mitochondrial motility via Milton modification by O-GlcNAc transferase. Cell. (2014) 158:54–68. doi: 10.1016/j.cell.2014.06.007
183. Mishra P, Carelli V, Manfredi G, Chan DC. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. (2014) 19:630–41. doi: 10.1016/j.cmet.2014.03.011
184. Baricault L, Ségui B, Guégand L, Olichon A, Valette A, Larminat F, et al. OPA1 cleavage depends on decreased mitochondrial ATP level and bivalent metals. Exp Cell Res. (2007) 313:3800–8. doi: 10.1016/j.yexcr.2007.08.008
185. Madalena KM, Lerch JK. Glucocorticoids and nervous system plasticity. Neural Regen Res. (2016) 11:37–41. doi: 10.4103/1673-5374.175039
186. Wei B, Shi H, Yu X, Shi Y, Zeng H, Zhao Y, et al. GR/Ahi1 regulates WDR68-DYRK1A binding and mediates cognitive impairment in prenatally stressed offspring. Cell Mol Life Sci. (2024) 81:20. doi: 10.1007/s00018-023-05075-1
187. Nemeroff CB, Vale WW. The neurobiology of depression: inroads to treatment and new drug discovery. J Clin Psychiatry. (2005) 66(Suppl 7):5–13.
188. Sinclair D, Fillman SG, Webster MJ, Weickert CS. Dysregulation of glucocorticoid receptor co-factors FKBP5, BAG1 and PTGES3 in prefrontal cortex in psychotic illness. Sci Rep. (2013) 3:3539. doi: 10.1038/srep03539
189. Ratajczak T, Cluning C, Ward BK. Steroid receptor-associated immunophilins: a gateway to steroid signalling. Clin Biochem Rev. (2015) 36:31–52.
190. Maeng S, Hunsberger JG, Pearson B, Yuan P, Wang Y, Wei Y, et al. BAG1 plays a critical role in regulating recovery from both manic-like and depression-like behavioral impairments. Proc Natl Acad Sci USA. (2008) 105:8766–71. doi: 10.1073/pnas.0803736105
191. Zgajnar NR, De Leo SA, Lotufo CM, Erlejman AG, Piwien-Pilipuk G, Galigniana MD. Biological actions of the Hsp90-binding immunophilins FKBP51 and FKBP52. Biomolecules. (2019) 9:52. doi: 10.3390/biom9020052
192. Storer CL, Dickey CA, Galigniana MD, Rein T, Cox MB. FKBP51 and FKBP52 in signaling and disease. Trends Endocrinol Metab. (2011) 22:481–90. doi: 10.1016/j.tem.2011.08.001
193. Guidotti G, Calabrese F, Anacker C, Racagni G, Pariante CM, Riva MA. Glucocorticoid receptor and FKBP5 expression is altered following exposure to chronic stress: modulation by antidepressant treatment. Neuropsychopharmacology. (2013) 38:616–27. doi: 10.1038/npp.2012.225
194. Anacker C, Zunszain PA, Cattaneo A, Carvalho LA, Garabedian MJ, Thuret S, et al. Antidepressants increase human hippocampal neurogenesis by activating the glucocorticoid receptor. Mol Psychiatry. (2011) 16:738–50. doi: 10.1038/mp.2011.26
195. Zhang H, He L, Li S, Zhai M, Ma S, Jin G, et al. Cerebral iron deficiency may induce depression through downregulation of the hippocampal glucocorticoid-glucocorticoid receptor signaling pathway. J Affect Disord. (2023) 332:125–35. doi: 10.1016/j.jad.2023.03.085
196. Santa-Marina L, Lertxundi N, Andiarena A, Irizar A, Sunyer J, Molinuevo A, et al. Maternal ferritin levels during pregnancy and ADHD symptoms in 4-year-old children: results from the INMA-INfancia y Medio Ambiente (Environment and Childhood) Prospective Birth Cohort Study. Int J Environ Res Public Health. (2020) 17:7704. doi: 10.3390/ijerph17217704
197. Díaz-López A, Canals-Sans J, Julvez J, Fernandez-Barrés S, Llop S, Rebagliato M, et al. Maternal iron status during pregnancy and attention deficit/hyperactivity disorder symptoms in 7-year-old children: a prospective cohort study. Sci Rep. (2022) 12:20762. doi: 10.1038/s41598-022-23432-1
198. Rioux FM, Bélanger-Plourde J, Leblanc CP, Vigneau F. Relationship between maternal DHA and iron status and infants' cognitive performance. Can J Diet Pract Res. (2011) 72:76. doi: 10.3148/72.2.2011.e140
199. Lynch S, Pfeiffer CM, Georgieff MK, Brittenham G, Fairweather-Tait S, Hurrell RF, et al. Biomarkers of nutrition for development (BOND)-iron review. J Nutr. (2018) 148(suppl_1): 1001S−67S. doi: 10.1093/jn/nxx036
200. Tran PV, Kennedy BC, Pisansky MT, Won KJ, Gewirtz JC, Simmons RA, et al. Prenatal choline supplementation diminishes early-life iron deficiency-induced reprogramming of molecular networks associated with behavioral abnormalities in the adult rat hippocampus. J Nutr. (2016) 146:484–93. doi: 10.3945/jn.115.227561
201. Bastian TW, von Hohenberg WC, Kaus OR, Lanier LM, Georgieff MK. Choline supplementation partially restores dendrite structural complexity in developing iron-deficient mouse hippocampal neurons. J Nutr. (2022) 152:747–57. doi: 10.1093/jn/nxab429
202. Liu SX, Fredrickson TK, Calixto Mancipe N, Georgieff MK, Tran PV. Sex-specific effects of early-life iron deficiency and prenatal choline treatment on adult rat hippocampal transcriptome. Nutrients. (2023) 15:1316. doi: 10.3390/nu15061316
203. Bahnfleth CL, Strupp BJ, Caudill MA, Canfield RL. Prenatal choline supplementation improves child sustained attention: a 7-year follow-up of a randomized controlled feeding trial. FASEB J. (2022) 36:e22054. doi: 10.1096/fj.202101217R
204. Boeke CE, Gillman MW, Hughes MD, Rifas-Shiman SL, Villamor E, Oken E. Choline intake during pregnancy and child cognition at age 7 years. Am J Epidemiol. (2013) 177:1338–47. doi: 10.1093/aje/kws395
205. Irvine N, England-Mason G, Field CJ, Dewey D, Aghajafari F. Prenatal folate and choline levels and brain and cognitive development in children: a critical narrative review. Nutrients. (2022) 14:364. doi: 10.3390/nu14020364
206. Wu BT, Dyer RA, King DJ, Richardson KJ, Innis SM. Early second trimester maternal plasma choline and betaine are related to measures of early cognitive development in term infants. PLoS ONE. (2012) 7:e43448. doi: 10.1371/journal.pone.0043448
207. Arija V, Hernández-Martínez C, Tous M, Canals J, Guxens M, Fernández-Barrés S, et al. Association of iron status and intake during pregnancy with neuropsychological outcomes in children aged 7 years: the prospective birth cohort Infancia y Medio Ambiente (INMA) Study. Nutrients. (2019) 11:2999. doi: 10.3390/nu11122999
208. Jia SZ, Li Y, Xu XW, Huang YP, Deng XY, Zhang ZH, et al. Selenoprotein K confers protection against iron dyshomeostasis-related neurotoxicity by regulating the palmitoylation of TfR-1. J Agric Food Chem. (2025) 73:12233–46. doi: 10.1021/acs.jafc.4c08266
Keywords: iron deficiency, pregnancy, fetal origins of adult disease, neurologic development, offspring, behavior
Citation: Zhao Z, Shi Y, Sun M and Wang B (2025) Effects of prenatal iron deficiency on neurological development and related disorders in offspring. Front. Nutr. 12:1637398. doi: 10.3389/fnut.2025.1637398
Received: 29 May 2025; Accepted: 18 August 2025;
Published: 05 September 2025.
Edited by:
Patrick Noël Pallier, Queen Mary University of London, United KingdomReviewed by:
Jonas Wolf, Moinhos de Vento Hospital, BrazilFaruk Saudatu, Usman Danfodiyo University, Nigeria
Copyright © 2025 Zhao, Shi, Sun and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bin Wang, Ymlud2FuZzIyMzNAc3VkYS5lZHUuY24=; Miao Sun, bWlhb3N1bkBpYm1zLnB1bWMuZWR1LmNu
†These authors have contributed equally to this work