 Hui Liu1
Hui Liu1 Pawel M. Kozlowski
Pawel M. Kozlowski- 1Department of Chemistry, University of Louisville, Louisville, KY, USA
- 2Department of Theoretical Chemistry, Institute of Chemistry, University of Silesia, Katowice, Poland
Coenzyme B12 (AdoCbl) is one of the most biologically active forms of vitamin B12, and continues to be a topic of active research interest. The mechanism of Co-C bond cleavage in AdoCbl, and the corresponding enzymatic reactions are however, not well understood at the molecular level. In this work, time-dependent density functional theory (TD-DFT) has been applied to investigate the photodissociation of coenzyme B12. To reduce computational cost, while retaining the major spectroscopic features of AdoCbl, a truncated model based on ribosylcobalamin (RibCbl) was used to simulate Co-C photodissociation. Equilibrium geometries of RibCbl were obtained by optimization at the DFT/BP86/TZVP level of theory, and low-lying excited states were calculated by TD-DFT using the same functional and basis set. The calculated singlet states, and absorption spectra were simulated in both the gas phase, and water, using the polarizable continuum model (PCM). Both spectra were in reasonable agreement with experimental data, and potential energy curves based on vertical excitations were plotted to explore the nature of Co-C bond dissociation. It was found that a repulsive 3(σCo−C → σ*Co−C) triplet state became dissociative at large Co-C bond distance, similar to a previous observation for methylcobalamin (MeCbl). Furthermore, potential energy surfaces (PESs) obtained as a function of both Co-CRib and Co-NIm distances, identify the S1 state as a key intermediate generated during photoexcitation of RibCbl, attributed to a mixture of a metal-to-ligand charge transfer (MLCT) and a σ bonding-ligand charge transfer (SBLCT) states.
Introduction
Vitamin B12 derivatives (Figure 1) are a group of organometallic complexes that act as biologically active cofactors in many enzymatic reactions (Dolphin et al., 1982; Banerjee, 1997, 1999, 2001, 2003; Ludwig and Matthews, 1997; Kräutler et al., 1998; Marzilli, 1999; Toraya, 2000; Matthews, 2001; Banerjee and Ragsdale, 2003; Toraya, 2003; Brown, 2005; Randaccio et al., 2006, 2007). In addition to their pivotal roles in a variety of enzymatic processes, the B12 derivatives possess complex photophysical and photochemical properties (Endicott and Ferraudi, 1977; Endicott and Netzel, 1979; Rao and Symons, 1982; Chen and Chance, 1990, 1993; Sakaguchi et al., 1990; Chagovetz and Grissom, 1993; Grissom and Chagovetz, 1993; Lott et al., 1995; Natarajan and Grissom, 1996; Kruppa et al., 1997; Walker et al., 1998a,b; Shiang et al., 1999, 2006; Yoder et al., 2001; Cole et al., 2002; Sension et al., 2004, 2005a,b; Harris et al., 2007). The relatively weak organometallic Co-R bond (R = alkyl, hydroxyl, water, cyanide) in these compounds can undergo photodissociation under conditions of simple photon excitation, depending on the nature of the upper axial ligand. Cobalamin complexes with the alkyl axial ligands, such as enzymatically competent methylcobalamin (MeCbl) and adenosylcobalamin (AdoCbl), and their analogs, ethylcobalamin (EtCbl) and propylcobalamin (PropCbl), undergo photodissociation upon exposure to light (Walker et al., 1998a,b; Shiang et al., 1999; Yoder et al., 2001; Cole et al., 2002; Sension et al., 2004, 2005a,b; Harris et al., 2007). In certain cases, the nature of events following photon excitation is additionally influenced by a wavelength of exciting light, as found for MeCbl (Harris et al., 2007). On the other hand, the Co-R bond in cyannocobalimin (CNCbl) and non-alkylcobalamin analogs, such as azidocobalamin (N3Cbl) and aquocobalamin (H2OCbl+), are rather photostable (Shiang et al., 2006).
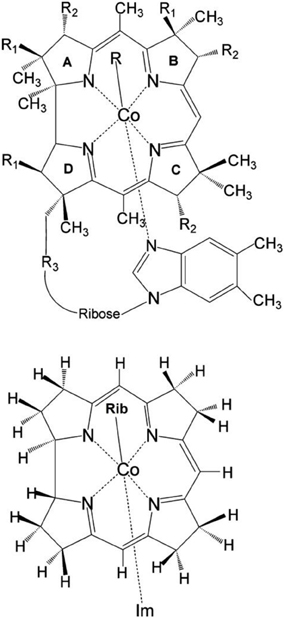
Figure 1. Upper: Molecular structure of vitamin B12 derivatives where R = Me, Ado, Et, Prop, CN, OH or N3 where R1 =CH2CONH2, R2 = CH2CH2CONH2, and R3 = (CH2)2CONHCH2CH(CH3)OPO−3. Lower: Structural model of RibCbl employed in present work (Rib refers to ribosyl with 5-hydroxyl group substituted by H atom). Reprinted (adapted) with permission from Jaworska et al. (2007). Copyright (2014) American Chemical Society.
The photochemistry of cobalt corrinoids has been probed using various experimental techniques such as laser flash photolysis (Endicott and Ferraudi, 1977; Endicott and Netzel, 1979; Chen and Chance, 1990; Chagovetz and Grissom, 1993; Lott et al., 1995), continuous wave (CW) photolysis (Chen and Chance, 1993), kinetic magnetic field effect (MFE) (Grissom and Chagovetz, 1993; Natarajan and Grissom, 1996), chemically induced dynamic electron polarization (CIDEP) (Rao and Symons, 1982; Sakaguchi et al., 1990) as well as nuclear polarization (CIDNP) (Kruppa et al., 1997). Systematic studies of photolysis in B12 alkyl-derivatives (MeCbl, EtCbl, PropCbl, and AdoCbl) using pump-probe transient absorption spectroscopy found that photoproduct yields for alkylcobalamins are different when excited at 400, or 520/530 nm (Walker et al., 1998a,b; Shiang et al., 1999; Yoder et al., 2001; Cole et al., 2002; Sension et al., 2004, 2005a,b; Harris et al., 2007). MeCbl for example, produces a mixture of 27% cob(II)alamin and 73% cob(III)alamin when excited at 400 nm as a result of partitioning between Co-C homolysis and heterolytic cleavage. Heterolytic bond cleavage products were identified as a metal-to-ligand charge transfer (MLCT) state upon excitation at 400 nm (Cole et al., 2002), and only metastable cob(III)alamin for 520 nm (Shiang et al., 1999). In contrast, photoexcitation of AdoCbl produces wavelength independent photoproducts at 400 and 520 nm, that correspond to an intermediate state observed as decay over a 14 ps time scale, of two relaxed radical pairs. These radical pairs were characterized as cob(II)alamin and an adenosyl-based radical. This observation corresponds to Co-C homolysis, or a trapped excited state with a weak Co-C bond (Shiang et al., 1999). Furthermore, although the photoproducts of AdoCbl were found to be insensitive to excitation wavelength, environmental effects have been shown to influence its photophysics (Yoder et al., 2001).
The photodissociation of Co-C in cobalamins has also been investigated computationally by means of time-dependent density functional theory (TD-DFT) (Jaworska et al., 2007; Lodowski et al., 2009, 2011; Kumar and Kozlowski, 2012). In the case of MeCbl, the key metastable photoproduct present during the photolysis process was identified as an S1 state with MLCT character (Jaworska et al., 2007), in accordance with experiment. Further analysis of the electron density map of S1 and S0 states indicated an additional contribution from σ bond, revealing that σ bonding-ligand charge transfer (SBLCT) character was mixed with MLCT (Lodowski et al., 2009). To explain the nature of the Co-C photo-scission in MeCbl, the presence of a repulsive 3(σCo−C → σ*Co−C) triplet state was proposed based on TD-DFT analysis of electronically excited states along the stretched Co-C bond. The same approach has also been applied to EtCbl, and the presence of a repulsive triplet state was used to explain the energetic differences between MeCbl and EtCbl, as well as why the photolysis mechanism is wavelength dependent in the case of MeCbl (Lodowski et al., 2009).
A similar study was performed for vitamin B12 (CNCbl) to explain its photostability and the nature of its low-lying excited states (Lodowski et al., 2011). Potential energy curves for low-lying excited states along the Co-CN coordinate reveal that CNCbl has a repulsive triplet state, but is not dissociative. It was found that the CNCbl potential energy surface (PES) of the S1 state, when represented as a function of axial distances, had two energy minima. The first minima was located above the S0 minimum as an excitation with mixed π → π*/MLCT/SBLCT character, similar to the alkylcobalamin S1 state. The second minimum was found at longer Co-NIm and Co-CN bond lengths, and was characterized as ligand-to-metal charge-transfer (LMCT) excitation.
Interestingly, the Co-OH bond in hydroxycobalamin (HOCbl) is dissociative upon light exposure above 300 nm (Shell and Lawrence, 2011), although it is among the non-alkylcobalamins. Photolysis of HOCbl has also been studied by TD-DFT, by focusing on the Co-OH cleavage (Kumar and Kozlowski, 2012). TD-DFT results suggest that the photoactivity of HOCbl is mediated by the repulsive 1(n+dCo→ σ*Co−OH) singlet state whose energy drops with Co-OH bond stretching, to yield the cob(II)alamin and hydroxyl radical.
Although spectroscopic techniques have been extensively applied to alkyl- and non-alkylcobalamins, the mechanism of Co-C bond dissociation is still not well understood. The aim of this study is to provide the further insight into the photodissociation mechanism of the Co-C bond in the coenzyme B12 (AdoCbl, Figure 1) by TD-DFT computations. To accomplish this we employed the simplified structural model of AdoCbl, (RibCbl, Figure 1), in which the Ado group was simplified to a ribosyl (Rib) moiety. The intermediates involved in the photodissociation process were identified by systematically analyzing the nature of low-lying excited states as well as their changes along stretched Co-CRib coordinate. Finally, in order to provide a more accurate picture of key intermediates, the PESs of ground state and low-lying singlet states were analyzed as a function of both Co-CRib and Co-NIm axial bond distances.
Computational Details
All calculations reported in this work were based on nonlocal DFT with the gradient-corrected Becke-Perdew (BP86) (Perdew, 1986; Becke, 1993) functional and the TZVP basis set, as implemented in the Gaussian 09 (Frisch et al., 2009) or TURBOMOLE (Ahlrichs et al., 1989; Treutler and Ahlrichs, 1995; Furche and Ahlrichs, 2002; Furche and Rappoport, 2005; TURBOMOLE1) suites of programs for electronic structure calculations. The BP86 functional has been previously selected as the most appropriate for predicting both the structural and electronic properties of B12 cofactors, including Co-C bond dissociation energy (BDE) (Jensen and Ryde, 2003; Rovira et al., 2004; Kuta et al., 2006; Kozlowski et al., 2007; Rovira and Kozlowski, 2007; Galezowski et al., 2008; Lodowski et al., 2009, 2011; Kornobis et al., 2011, 2013). To account for environmental effects on geometries and electronic properties, ground states of RibCbl model (Im-[CoIII(corrin)]-Rib+, see Figure S1) were computed in the gas phase as well as in water via the Polarizable Continuum Model (PCM) (Miertuš et al., 1981; Cammi and Tomasi, 1995) as implemented in Gaussian 09.
Vertical excitations were obtained in both the gas phase and water solvent. Low-lying excited states were calculated from their corrdepsonding ground state geometries by TD-DFT, and both singlet and triplet excitations have been calculated to forty states (Tables S1–S4). To simulate the photodissociation process of RibCbl, the Co-CRib bond was stretched with a step size of 0.05 Å and the DFT/BP86/TZVP optimization was repeated at each point, followed by TD-DFT calculations at the BP86/TZVP level of theory. In addition, PESs of the ground state and key singlet states were plotted by stretching axial bonds in step of 0.05 Å. At each grid point, the geometry was optimized by DFT/BP86/TZVP, and the vertical excitation was obtained for S1 and S2 states.
Results and Discussion
Structural Model of RibCbl
The initial molecular structure of RibCbl was obtained from the high-resolution crystal structure of AdoCbl (Ouyang et al., 2004). The full structure of AdoCbl (Figure 1) consists of CoIII equatorially cooridinated via four nitrogen atoms to the corrin ring. The lower axial ligand, dimethylbenzimidazole (DBI) coordinates to the central Co atom via Nax, with the other N atom bound to the side of the corrin plane by a nucleotide loop. In the actual AdoCbl structure, the upper Ado ligand is 5′-deoxy-5′-adenosine.
To reduce computational cost, calculations were performed using the simplified structural model of RibCbl, denoted Im-[CoIII(corrin)]-Rib+for consistency with previous studies (Jaworska et al., 2007; Lodowski et al., 2009, 2011). In the RibCbl model (see Figure 1), the Ado group was simplified to ribosyl (Rib) by omitting the purine ring, leaving only ribose in the upper corrin plane. The corrin ring was truncated by replacing the side chains with hydrogen atoms, and DBI with imidazole (Im). The resulting overall charge of the system was +1 due to removal of the PO−4 nucleotide loop from below the plane. Previous studies have demonstrated that such simplified structural models accurately reproduce the electronic and spectroscopic properties of the full B12 structure (Jaworska and Lodowski, 2003; Jensen and Ryde, 2003, 2009; Stich et al., 2003, 2004; Jaworska et al., 2005, 2007; Kuta et al., 2006, 2009; Liptak and Brunold, 2006; Kozlowski et al., 2007, 2012; Rovira and Kozlowski, 2007; Galezowski et al., 2008; Lodowski et al., 2009, 2011). More inclusive structural models of coenzyme B12 will be employed in future studies utilizing multiple levels of theory.
Optimized RibCbl structures correspond to equilibrium geometries where Co-CRib = 2.02 Å and Co-NIm = 2.21 Å in the gas phase, and Co-CRib = 2.02 Å and Co-NIm = 2.19 Å in water (PCM). Optimized axial bond lengths are in reasonable agreement with X-ray data of Randaccio et al. (Ouyang et al., 2004) with corresponding experimental bond lengths of 2.04 Å for Co-CAdo and 2.23 Å for Co-NDBI, respectively. Molecular orbital (MO) energies and fragment contributions of RibCbl are collected in Table S5 for the gas phase, and Table S6 for water (PCM). The relevant MO diagram can be found in supporting information as Figure S2.
Frontier orbitals of RibCbl were determined by fragment analysis to be mainly corrin π, π*, and metal d orbitals. In particular, HOMO, HOMO−2, −4, −6, and LUMO+1, +2, +3 show major contributions from dCo and π* while HOMO−3, −5, −8, −9, −10, and LUMO+3 have increased weights from the Rib group. On the other hand, LUMO and LUMO+4 are composed of nearly pure π orbitals of the corrin ring. Finally, HOMO-7 and LUMO+5 are both composed of excitations from the imidazole group. The change of environment from gas phase to water solution (Table S6) did not influence the character of the MOs significantly, differences being attributed to reversion of energies between the HOMO-5 and −7 in the gas, and HOMO-3 and −4 in the PCM model.
Electronically Excited States of RibCbl
Simulated absorption (Abs) spectra generated from TD-DFT/BP86/TVZP vertical excitation energies with transition dipole moments, using corresponding optimized RibCbl geometries in the gas phase and in water, are shown in Figures 2, 3 respectively. Since hybrid B3LYP functional underestimates BDE as well as produce low-lying states as pure πcorrin → π*corrin excitations, we employed BP86 throughout this study which has been proved to produce correct BDE for both MeCbl and AdoCbl (Kuta et al., 2006; Kozlowski et al., 2007, 2012; Galezowski et al., 2008; Kornobis et al., 2011, 2013; Kumar et al., 2013). BP86 functional was also frequently applied in QM/MM calculations for the enzyme-active cofactors (Kumar et al., 2012; Kumar and Kozlowski, 2013). In both the gas phase and solvent-based calculations, forty excited states were calculated to cover a range appropriate for electronic states potentially involved in coenzyme B12 photolysis. The lowest ten singlet states are listed in Table 1, and the lowest ten triplet states are listed in Table 2 for the gas phase. In addition, the corresponding solvent-based calculations are listed in Table 3 for singlet states and Table 4 for triplet states. Complete lists of the forty excited states for each system can be found in the supporting information (Tables S1–S4). Taking into account that light exposure used in experimental studies was no shorter than 400 nm to induce Co-C cleavage (Walker et al., 1998a,b; Shiang et al., 1999; Yoder et al., 2001; Cole et al., 2002; Sension et al., 2004, 2005a,b; Harris et al., 2007), in further discussion we will focus on the low-energy region.
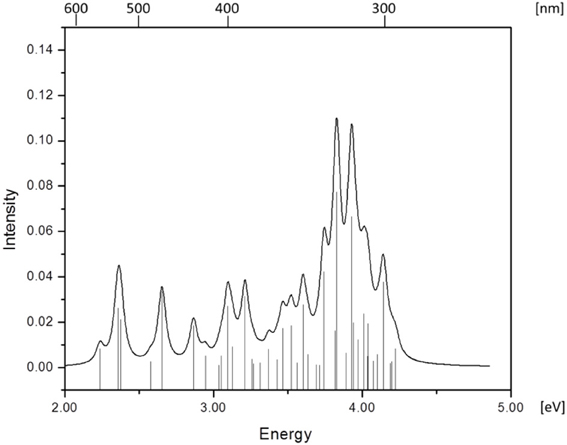
Figure 2. Absorption spectrum of RibCbl calculated in gas phase with BP86/TZVP.
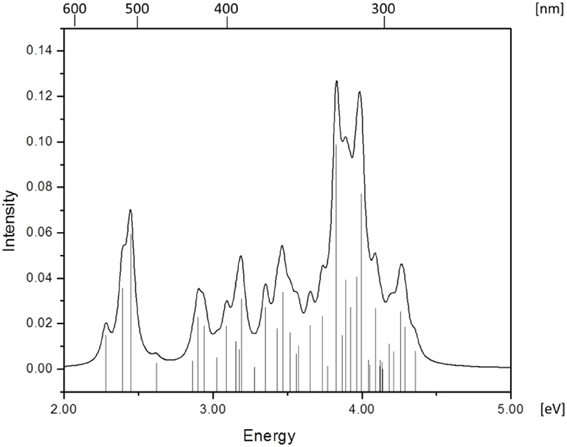
Figure 3. Absorption spectrum of RibCbl calculated in water solution with BP86/TZVP.
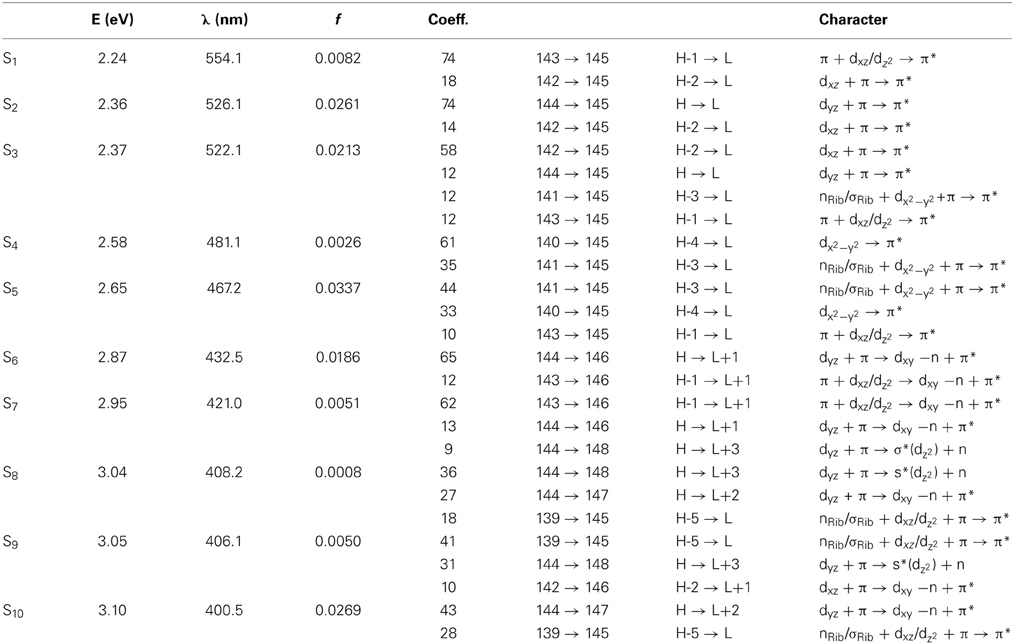
Table 1. The lowest ten singlet states for RibCbl received from TDDFT/TZVP gas phase calculations.
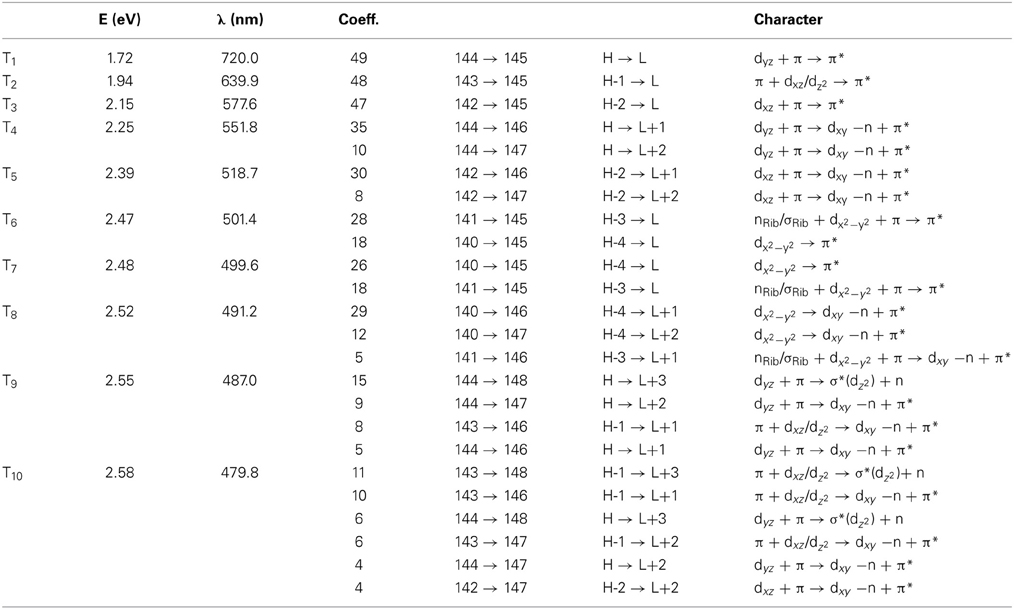
Table 2. The lowest ten triplet states for RibCbl received from TDDFT/TZVP gas phase calculations.
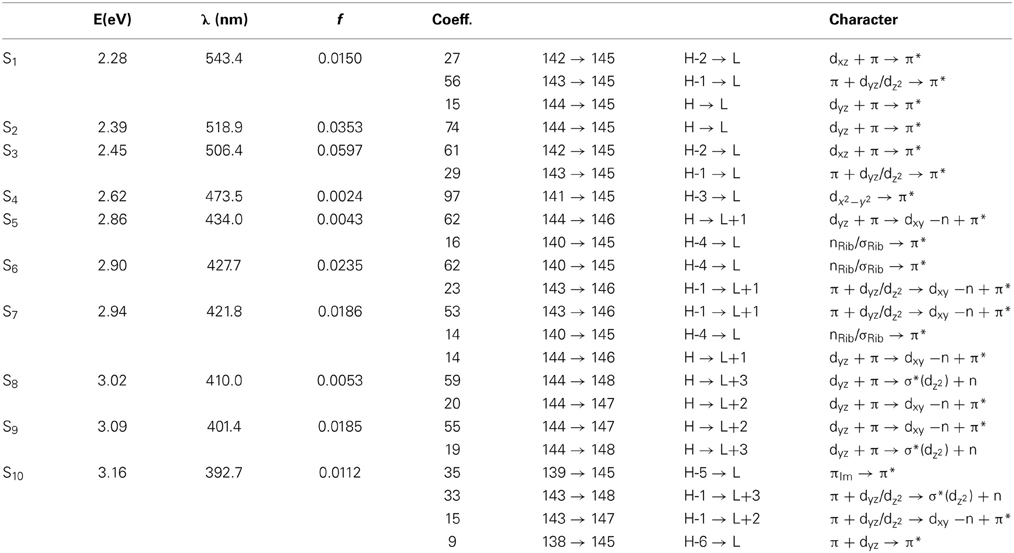
Table 3. The lowest ten singlet states for RibCbl received from TDDFT/TZVP PCM (water) calculations.
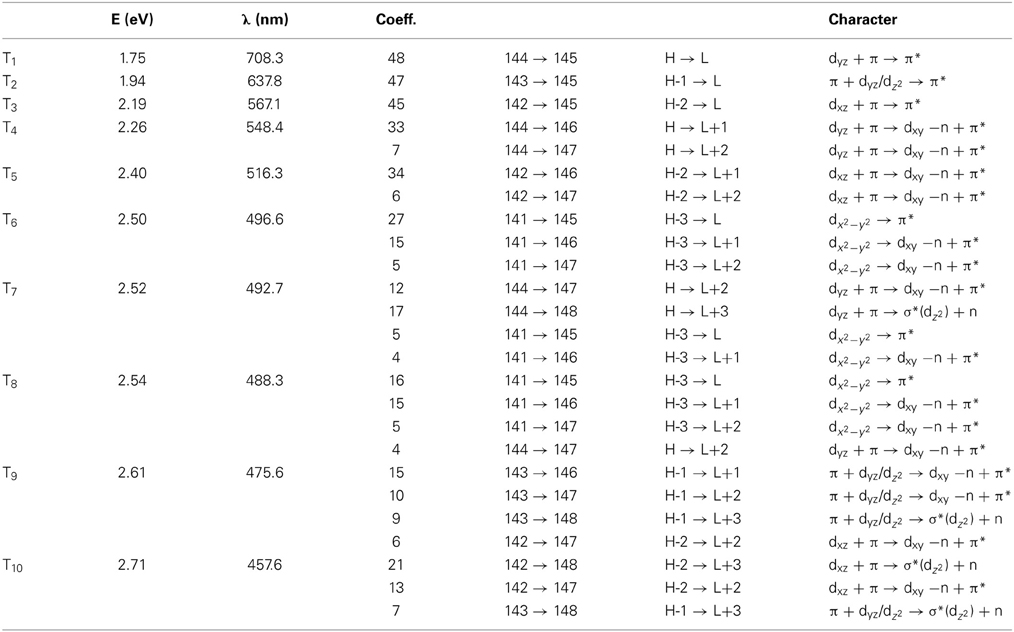
Table 4. The lowest ten triplet states for RibCbl received from TDDFT/TZVP PCM (water) calculations.
Since the studied RibCbl model is a truncated form of full coenzyme B12, no direct comparison to the experimental spectrum can be made. However, the predicted spectra can be correlated to that of AdoCbl with good agreement (Shiang et al., 1999). Several calculated electronic transitions can be assigned to experimental spectral bands. Specifically, the S2 at 2.36 eV and the S3 at 2.37 eV agree well with the experimental transition at 2.35 eV (528 nm), the S6 at 2.87 eV agrees with the experimental transition at 2.90 eV (428 nm), and the S10 state at 3.10 eV can be assigned to absorption observed at 3.07 eV (404 nm). The same trend applies to PCM geometries (Table 3). Namely, the S2, S3, S6, and S9 can all be correlated with experimental absorption bands for AdoCbl observed at 2.35 eV (528 nm), 2.90 eV (428 nm), and 3.07 eV (404 nm), respectively. Furthermore, the simulated RibCbl Abs spectra are also in reasonable agreement with the simulated AdoCbl spectra published by Andruniow et al. (2009) further supporting the ability of the truncated AdoCbl model, (i.e., RibCbl) to accurately reproduce the experimental spectroscopic features of AdoCbl.
The absorption spectrum of RibCbl calculated in the gas phase at the BP86/TZVP level is shown in Figure 2, and assignments for the lowest forty singlet states are collected in Table S1. Based on these results it can be deduced that the α/β bands are located in the low energy region from 2.00 to 2.80 eV, covering first five calculated singlet transitions, the D/E band ranges from 2.80 to 3.30 eV and is composed of multiple excitations, and the high-energy region, or so-called γ band, has significant intensity covering several transitions from 3.30 to 4.40 eV. Based on the nature of the calculated electronic excited states (Table 1), the α/β bands involve primarily dCo/π → π* contributions, the D/E bands result from various electronic transitions with more contributions from the Rib group, and the same tendency is observed in the γ band, where oscillator strength increases significantly. It should be noted that ribose increases its role in photoelectronic transitions in the high energy region, i.e., the D/E and γ bands, respectively.
Table 1 summarizes the ten lowest calculated excited singlet states of RibCbl in the gas phase. As shown in Table 1, the S1 excitation present at 2.24 eV, carries only a small oscillator strength (0.008). The major contribution from the S1 state is an excitation from HOMO-1 to LUMO, in which π + dxz/dz2 → π* is dominant. The MO diagram (Figure S2), shows that S1 excitation also involves a transition from a σ orbital of axial bonding to the π* of the corrin ring, and is identified as SBLCT (Lodowski et al., 2009). The second noticeable contribution from the S1 state is the transition from HOMO-2 to LUMO, identified as π + dxz → π*. Based on the MO diagram it is apparent that LUMO is a pure corrin π* orbital, to which all the excitations in the α/β band generally take place. The next two states, the S2 and S3, are adjacent closely in energy. The most significant components of the S2 and S3 states are HOMO → LUMO and HOMO-2 → LUMO, where HOMO is a mixture of π and dyz and HOMO-2 refers to π and dxz. Therefore, the d to π* transition is dominant in this region, and should be described as MLCT transition (Cole et al., 2002; Jaworska et al., 2007; Lodowski et al., 2009, 2011).
The S4 state has even less oscillator strength intensity compared to that of S1, in which both HOMO-3 and HOMO-4 contribute almost evenly to excitation at 2.58 eV. HOMO-3 derives from ribose, according to MO diagram, while HOMO-4 is composed from the Co dx2 −y2 orbital. Coincidently, the S5 involves the same set of orbitals which contribute to the S4 state. The only difference, considering their specific characters, is that the S5 state mixes a minor contribution from the HOMO-1→ LUMO transition. However, the larger oscillator strength of the S5 state makes it the strongest component of the α/β band.
Electronic transitions in the D/E band are more complex than those in the α/β band, and the oscillator strengths are noticeably smaller. This region can be represented by electronic transitions from the S6 to S14 with three strong excitations occurring at 2.87 (S6), 3.10 (S10), and 3.21 eV (S12) (Figure 2). The S6 and S10 have dCo/π → π* character while the S12 shows a dCo/π → σ*Co−C nature. The other small intensity transitions in this region are of similar character.
More than twenty singlet states can be associated with γ band ranging from 3.31 (S15) to 4.23 eV (S40) (Table S1). Among them, the S25 and S27 have the largest transition dipole moment and are composed of nRib/σRib + dxz/dz2 + π →dxy −n + π* transitions with a minor dCo/ π → π* contribution. The S29 is dominated by dCo/π → π* transitions, while the S32 has dCo/π → π*Im character. Near the end of the simulated spectrum, another noticeable transition of nRib/σRib + dxz/dz2 + π → σ*(dz2) + n type is found at 4.14 eV corresponding to the S37 state (Table S1).
Since triplet excitations have zero transition dipole moments, we will not take them into account for spectral analysis. The first three triplet states are energetically below the S1 (2.24 eV), where the T1 occurs at 1.72 eV from an electronic transition of HOMO→LUMO, while T2 and T3 are transitions at 1.94 and 2.15 eV, originating from HOMO-1→LUMO and HOMO-2→LUMO, respectively. All three electronic transitions can be described as dCo/π → π* type (see Table 2 and Table S2).
The Abs spectrum of RibCbl calculated in water solution using the same level of theory was plotted in Figure 3. The energy of the γ band remains unchanged in comparison to gas phase calculation. However, the α/β band is narrowed due to the blue shift of the S5 to the D/E region. The S1, S2, and S3 are assigned to dCo/π → π* transition. A significant contribution to S1 state is composed of a π + dxz/dz2 → π* excitation and, like in the gas phase, is characterized as SBLCT. The S4 has pure dx2 −y2 → π* character. The D/E band covers states S5–S13. The states S5 and S7 are identified as d/π → π*/d−n excitations with a small admixture of long range CT (LRCT) type transitions (nRib/σRib → π*) (Andruniow et al., 2009). The S6 state at 2.90 eV is a mixture of a dominant LRCT, and minor d/π → π*/d−n character. In contrast, the S7 state is mainly composed of a dCo/π → π* transition, with minor contributions from SBLCT. The other intense singlet transitions in this region are assigned to the dCo/π → π* transition, which is consistent with singlet excited states observed in the gas phase calculation. The complete assignment of electronic transitions is presented in Table S3. In water solvated system, the T1–T4 states are below the S1 state (Table 4), and all possess contributions from the dCo/π → π* transition.
Mechanism of Photolysis
In order to simulate the photodissociation of AdoCbl, the Co-CRib bond in RibCbl was elongated to mimic the light induced bond scission. The length of the Co-CRib bond was systematically stretched with the increment of 0.05 Å in a range from 1.90 to 2.65 Å. For each constrained distance, the ground state geometry was optimized at the BP86/TZVP level, followed by TD-DFT calculations for both singlet and triplet states. Figure 4 depicts the potential energy curves of a manifold of excited states computed as a function of Co-CRib bond length.
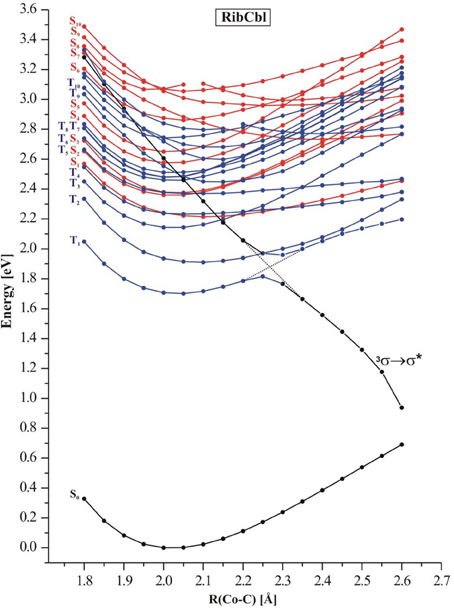
Figure 4. Potential energy curves of the lowest-excited singlet (red) and triplet (blue) states of the RibCbl model complex along the Co-C bond stretch computed at TD-DFT/BP86/TZVP. The triplet repulsive state is denoted as 3σ → σ*.
The S1–S5 singlet states, which belong to α/β band, have non-repulsive character, and the gap between the S1 and higher states gets larger when the bond length increases. States denoted as S2 and S3 are parallel at short Co-CRib distance but cross at ~2.50 Å. On the other hand the S4 and S5 keep similar energy but visibly separate from the first group of transitions (S1–S3). However, at ~2.30 Å these two states show avoiding crossing, and the S4 approaches the S2 at long length bond region. The excited states higher in energy (S6–S10), display multiple crossings at even short Co-C distances. According to the TD-DFT data, low-lying singlet states do not participate in photodissociation because there is no 1(σCo−C → σ*Co−C) dominant. Most likely such a transition requires an energetically high excitation and none of singlet states with predominant 1(σCo−C → σ*Co−C) character are in a reasonable transition region. Additionally, it is expected that such states would not dissociate to the cob(II)alamin and ribosyl radicals, but rather to ionic fragments. However, the low-lying triplet states appear to have σCo−C → σ*Co−C character in the achievable energy region. By connecting the energy points of σCo−C-featured triplet states at different dissociation distances, the 3(σCo−C → σ*Co−C) state with repulsive character can be located (black curve in Figure 4). At 2.35 Å, this state becomes the lowest level in terms of energy, and displays an avoided crossing with T1 state, that typically accounts for dCo/π → π* excitation. Together with an increasing Co-CRib distance, the energy of the triplet state drops dramatically above a Co-C bond distance of 2.40 Å, indicating that dissociation may not be properly described with a single-determinant wavefunction approximation at long Co-CRib distances (Kumar et al., 2011a,b). It is also reasonable to postulate that there is a conical intersection existing between the two states. Although the current study has limitations on the longer bond distances, it is expected that the photolysis happens at much shorter bond lengths, where 3(σCo−C → σ*Co−C) excitation is described properly. Further multi-configuration wave functions would be required to obtain a more detailed picture.
Potential Energy Surfaces of Low-Lying Excited States
Since the photoexcitation of B12 derivatives involves primarily the structural changes of the axial ligand involved in N-Co-C bonding, this investigation was extended to the PESs computed as a function of both Co-CRib and Co-NIm distances. The S1 and S2 PESs were constructed from the equilibrium structure of the RibCbl (S0 state), and systematically stretching the two axial bonds with an increment of 0.05 Å. Geometries at each grid point were optimized by BP86/TZVP, and the vertical excitations were calculated by TD-DFT at the BP86/TZVP level of theory (Figures 5, 6).
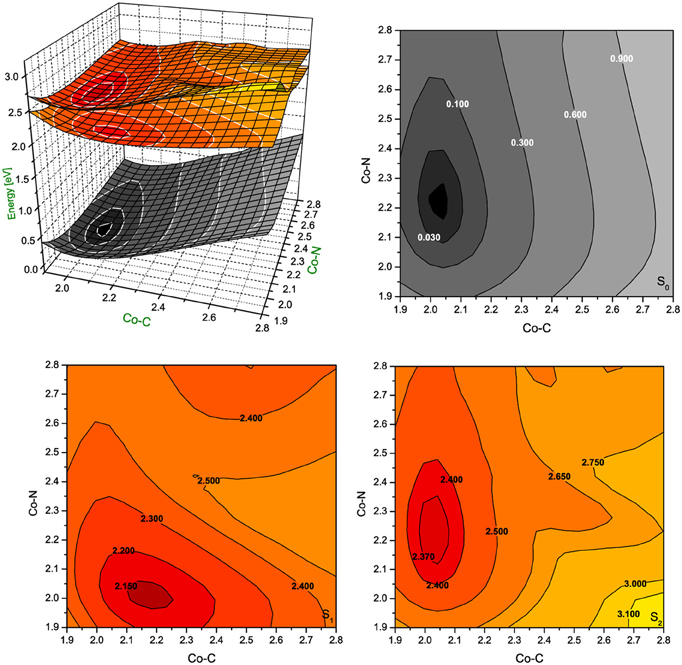
Figure 5. Potential energy surfaces for singlet ground state and two lowest singlet excited states of RibCbl together with their vertical projections plotted as a function of axial bond lengths (expressed in Å) calculated in gas phase with BP86/TZVP.
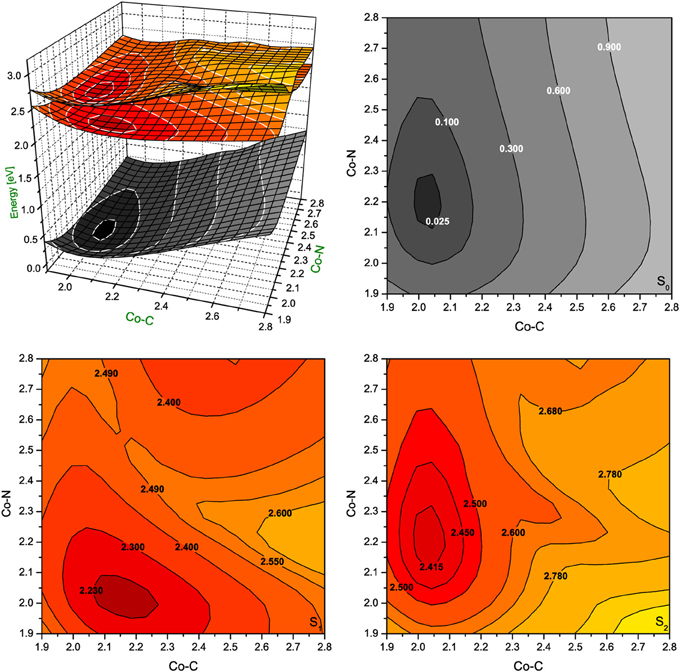
Figure 6. Potential energy surfaces for singlet ground state and two lowest singlet excited states of RibCbl together with their vertical projections plotted as a function of axial bond lengths (expressed in Å) calculated in water solution (PCM) with BP86/TZVP.
In gas phase PES contours (Figure 5), the S0 shows an equilibrium geometry with the lowest energy at 2.02 Å for Co-CRib and 2.21 Å for Co-NIm, while for the S1 PES, the minimum occurs at the longer Co-CRib bond (~2.17 Å) but shorter Co-NIm (~2.00 Å) distance. According to pump-probe time resolved experiments, an intermediate state of AdoCbl is populated through photoelectronic excitation, leading to a partial mixture of adenosyl radical and cob(II)alamin. It is therefore reasonable to postulate that such an intermediate state can be identified as the S1, which according to TD-DFT calculations has the Co-CRib bond elongated and Co-NIm shortened due to an electron density shift from the metal to the equatorial bonds of the corrin ring. The PES plot of the S2 is nearly parallel to the S0 surface. Estimates based on PES projections (Figure 5) predict the energy minimum of the S2 surface is located at Co-CRib ≈ 2.04 Å, and Co-NIm ≈ 2.23 Å, ± 0.02 Å, compared to the equilibrium geometry. This indicates no significant change of axial bond distances occur from the S0 to S2 transition. If this is true, the energy difference between the S0 and S2 states are mainly attributed the geometry changes of the corrin ring. At this point, the distortion of the corrin ring, mainly due to π→π* electronic transitions, increases the potential energy of the system. Since the geometry change in the S1 state involves Co-CRib bond stretching, it is likely that the S1 state is the most involved in the photolytic cleavage of Co-C, and thus the formation of the radical pair generated when the Co-C bond is stretched.
The PES of RibCbl in water retains nearly all of the general features of the gas phase PES (Figure 6). However, although the equilibrium geometry of RibCbl in water has similar axial bond lengths to those obtained in the gas phase calculation (with the exception of a slightly reduced Co-N bond distance of 0.02 Å), there remain subtle differences in energies between the S0 and S1, and S0 and S2 states. The energy gaps between S0 and S1, as well as S0 and S2, are larger for the solvated geometry than those of the gas phase. The vertical energy gaps between the minimum point of solvated ground state and the lowest two excited states are ~2.30 eV (S0–S1), and ~2.40 eV (S0–S2) respectively, while the vertical energy gaps in the gas phase calculation are ~2.20 eV (S0–S1) and ~2.35 eV (S0–S2). It can be reasoned that the larger vertical energy gaps from the ground state to the first two excited states in the PCM calculation, that the polar effect of solvent tends to increase the energies required for RibCbl excitation.
Summary and Conclusions
The purpose of this study was to explore the mechanisms of Co-C bond scission in AdoCbl upon light exposure using the TD-DFT method. To accomplish this we used a simplified RibCbl model to mimic the molecular features of photoexcited AdoCbl. The optimized RibCbl geometry at the DFT/BP86/TVZP level of theory was very close to the X-ray structure of AdoCbl with respect to the Co-C and Co-Nax bond distances, within the deviation of 0.02 Å. Electronically excited states were calculated with TD-DFT, and both low-lying singlet and triplet states were analyzed. Calculated singlet states of RibCbl were used to generate electronic spectra that agree well with the experimental UV-Vis for AdoCbl, especially considering the characteristic low energy α/β band. In addition, PESs were generated as a function of both Co-CRib and Co-NIm distances for singlet excited states using TD-DFT. The S1 state was identified as the key state of RibCbl photoexcitation, with contributions from both MLCT and SBLCT. Similar to the analysis of low-lying excited states of MeCbl, a repulsive 3(σCo−C → σ*Co−C) triplet state was found to facilitate photon induced dissociation of the Co-C bond in RibCbl.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to acknowledge the Cardinal Research Cluster (Supercomputing Facilities at the University of Louisville) for providing computational resources. This work has been supported by the National Science Centre, Poland under Grant No. 2013/09/B/ST4/03014.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fchem.2013.00041/abstract
Footnotes
1. ^TURBOMOLE has been designed by the QuantumChemistryGroup, University of Karlsruhe, Germany, since 1988. The following members of the group have made contributions: Reinhart Ahlrichs, Michael Bär, Hans-Peter Baron, Rüdiger Bauernschmitt, Stephan Böcker, Nathan Crawford, Peter Deglmann, Michael Ehrig, Karin Eichkorn, Simon Elliott, Filipp Furche, Frank Haase, Marco Häser, Christof Hättig, Arnim Hellweg, Hans Horn, Christian Huber, Uwe Huniar, Marco Kattannek, Andreas Köhn, Christoph Kölmel, Markus Kollwitz, Klaus May, Paola Nava, Christian Ochsenfeld, Holger Öhm, Holger Patzelt, Dmitrij Rappoport, Oliver Rubner, Ansgar Schäfer, Uwe Schneider, Marek Sierka, Oliver Treutler, Barbara Unterreiner, Malte von Arnim, Florian Weigend, Patrick Weis, Horst Weiss. Available online at: http://www.turbomole.com.
References
Ahlrichs, R., Bär, M., Häser, M., Horn, H., and Kölmel, C. (1989). Electronic structure calculations on workstation computers: the program system turbomole. Chem. Phys. Lett. 162, 165–169. doi: 10.1016/0009-2614(89)85118-8
Andruniow, T., Jaworska, M., Lodowski, P., Zgierski, M. Z., Dreos, R., Randaccio, L., et al. (2009). Time-dependent density functional theory study of cobalt corrinoids: electronically excited states of coenzyme B12. J. Chem. Phys. 131, 105105. doi: 10.1063/1.3190326
Banerjee, R. (1997). The Yin-Yang of cobalamin biochemistry. Chem. Biol. 4, 175–186. doi: 10.1016/S1074-5521(97)90286-6
Banerjee, R. (2001). Radical peregrinations catalyzed by coenzyme B12-dependent enzymes. Biochemistry 40, 6191–6198. doi: 10.1021/bi0104423
Banerjee, R. (2003). Radical carbon skeleton rearrangements: catalysis by coenzyme b12-dependent mutases. Chem. Rev. 103, 2083–2094. doi: 10.1021/cr0204395
Banerjee, R., and Ragsdale, S. W. (2003). The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu. Rev. Biochem. 72, 209–247. doi: 10.1146/annurev.biochem.72.121801.161828
Becke, A. D. (1993). Density–functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. doi: 10.1063/1.464913
Brown, K. L. (2005). Chemistry and enzymology of vitamin B12. Chem. Rev. 105, 2075–2149. doi: 10.1021/cr030720z
Cammi, R., and Tomasi, J. (1995). Remarks on the use of the apparent surface charges (ASC) methods in solvation problems: iterative versus matrix-inversion procedures and the renormalization of the apparent charges. J. Comput. Chem. 16, 1449–1458. doi: 10.1002/jcc.540161202
Chagovetz, A. M., and Grissom, C. B. (1993). Magnetic field effects in adenosylcob(III)alamin photolysis: relevance to B12 enzymes. J. Am. Chem. Soc. 115, 12152–12157. doi: 10.1021/ja00078a063
Chen, E., and Chance, M. R. (1990). Nanosecond transient absorption spectroscopy of coenzyme B12. Quantum yields and spectral dynamics. J. Biol. Chem. 265, 12987–12994.
Chen, E., and Chance, M. R. (1993). Continuous-wave quantum yields of various cobalamins are influenced by competition between geminate recombination and cage escape. Biochemistry 32, 1480–1487. doi: 10.1021/bi00057a011
Cole, A. G., Yoder, L. M., Shiang, J. J., Anderson, N. A., Walker, L. A. II., Banaszak Holl, M. M., et al. (2002). Time-resolved spectroscopic studies of B12 coenzymes: a comparison of the primary photolysis mechanism in methyl-, ethyl-, n-propyl-, and 5′-deoxyadenosylcobalamin. J. Am. Chem. Soc. 124, 434–441. doi: 10.1021/ja011628s
Endicott, J. F., and Ferraudi, G. J. (1977). A flash photolytic investigation of low energy homolytic processes in methylcobalamin. J. Am. Chem. Soc. 99, 243–245. doi: 10.1021/ja00443a043
Endicott, J. F., and Netzel, T. L. (1979). Early events and transient chemistry in the photohomolysis of alkylcobalamins. J. Am. Chem. Soc. 101, 4000–4002. doi: 10.1021/ja00508a066
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09, Revision A.1. Wallingford, CT: Gaussian, Inc.
Furche, F., and Ahlrichs, R. (2002). Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 117, 7433–7447. doi: 10.1063/1.1508368
Furche, F., and Rappoport, D. (2005). “Density functional methods for excited states: equilibrium structure and electronic spectra computational photochemistry,” in Theoretical and Computational Chemistry, Vol. 16, Chapter III, ed M. Olivucci (Amsterdam: Elsevier), 93–128.
Galezowski, W., Kuta, J., and Kozlowski, P. M. (2008). DFT study of Co-C bond cleavage in the neutral and one-electron-reduced alkyl-cobalt(iii) phthalocyanines. J. Phys. Chem. B 112, 3177–3183. doi: 10.1021/jp0769678
Grissom, C. B., and Chagovetz, A. M. (1993). Magnetic field effects in model B12, enzymatic reactions: the photolysis of methyl cob(III)alamin. Z. Phys. Chem. 182, 181–188. doi: 10.1524/zpch.1993.182.Part_1_2.181
Harris, D. A., Stickrath, A. B., Carroll, E. C., and Sension, R. J. (2007). Influence of environment on the electronic structure of Cob(III)alamins: time-resolved absorption studies of the S1 state spectrum and dynamics. J. Am. Chem. Soc. 129, 7578–7585. doi: 10.1021/ja066197y
Jaworska, M., Kozibut, G., and Lodowski, P. (2005). Electronic spectrum of cobalt-free corrins calculated by TDDFT method. J. Phys. Chem. A 107, 1339–1347. doi: 10.1021/jp021261t
Jaworska, M., and Lodowski, P. (2003). Electronic spectrum of Co-corrin calculated with the TDDFT method. J. Mol. Struct. THEOCHEM 631, 209–223. doi: 10.1016/S0166-1280(03)00249-5
Jaworska, M., Lodowski, P., Andruniow, T., and Kozlowski, P. M. (2007). Photolysis of methylcobalamin: identification of the relevant excited states involved in Co-C bond scission. J. Phys. Chem. B. 111, 2419–2422. doi: 10.1021/jp0685840
Jensen, K., and Ryde, U. (2009). Cobalamins uncovered by modern electronic structure calculations. Coord. Chem. Rev. 253, 769–778. doi: 10.1016/j.ccr.2008.04.015
Jensen, K. P., and Ryde, U. (2003). Theoretical prediction of the Co-C bond strength in cobalamins. J. Phys. Chem. A 107, 7539–7545. doi: 10.1021/jp027566p
Kornobis, K., Kumar, N., Lodowski, P., Jaworska, M., Piecuch, P., Lutz, J. J., et al. (2013). Electronic structure of the S1 state in methylcobalamin: insight from CASSCF/MC-XQDPT2, EOM-CCSD, and TD-DFT calculations. J. Comput. Chem. 34, 987–1004. doi: 10.1002/jcc.23204
Kornobis, K., Kumar, N., Wong, B. M., Lodowski, P., Jaworska, M., Andruniow, T., et al. (2011). Electronically excited states of vitamin B12: benchmark calculations including time-dependent density functional theory and correlated ab initio methods. J. Phys. Chem. A 115, 1280–1292. doi: 10.1021/jp110914y
Kozlowski, P. M., Kumar, M., Piecuch, P., Li, W., Bauman, N. P., Hansen, J. A., et al. (2012). The cobalt–methyl bond dissociation in methylcobalamin: new benchmark analysis based on density functional theory and completely renormalized coupled-cluster calculations. J. Chem. Theor. Comput. 8, 1870–1894. doi: 10.1021/ct300170y
Kozlowski, P. M., Kuta, J., and Galezowski, W. (2007). Reductive cleavage mechanism of methylcobalamin: elementary steps of Co-C bond breaking. J. Phys. Chem. B 111, 7638–7645. doi: 10.1021/jp066972w
Kräutler, B., Arigoni, D., and Golding, B. T. (eds.). (1998). Vitamin B12 and B12 Proteins. New York, NY: Wiley-VCH. doi: 10.1002/9783527612192
Kruppa, A. I., Taraban, M. B., Leshina, T. V., Natarajan, E., and Grissom, C. B. (1997). CIDNP in the photolysis of coenzyme B12 model compounds suggesting that C-Co bond homolysis occurs from the singlet state. Inorg. Chem. 36, 758–759. doi: 10.1021/ic960562c
Kumar, M., and Kozlowski, P. M. (2012). Why hydroxocobalamin is photocatalytically active? Chem. Phys. Lett. 543, 133–136. doi: 10.1016/j.cplett.2012.06.007
Kumar, N., and Kozlowski, P. M. (2013). Mechanistic insights for formation of an organometallic co–C bond in the methyl transfer reaction catalyzed by methionine synthase. J. Chem. Phys. B 117, 16044–16057. doi: 10.1021/jp4093145
Kumar, N., Jaworska, M., Lodowski, P., Kumar, M., and Kozlowski, P. M. (2011a). Electronic structure of cofactor-substrate reactant complex involved in the methyl transfer reaction catalyzed by cobalamin-dependent methionine synthase. J. Phys. Chem. B 115, 6722–6731. doi: 10.1021/jp200945a
Kumar, N., Alfonso-Prieto, M., Rovira, C., Lodowski, P., Jaworska, M., and Kozlowski, P. M. (2011b). Role of the axial base in the modulation of the cob(I)alamin electronic properties: insight from QM/MM, DFT, and CASSCF calculations. J. Chem. Theory Comput. 7, 1541–1551. doi: 10.1021/ct200065s
Kumar, N., Kuta, J., Galezowski, W., and Kozlowski, P. M. (2013). Electronic structure of one-electron-oxidized form of the methylcobalamin cofactor: spin density distribution and pseudo-jahn–teller effect. Inorg. Chem. 52, 1762–1771. doi: 10.1021/ic3013443
Kumar, N., Liu, S., and Kozlowski, P. M. (2012). Charge separation propensity of the coenzyme B12–tyrosine complex in adenosylcobalamin-dependent methylmalonyl–CoA mutase enzyme. J. Phys. Chem. Lett. 3, 1035–1038. doi: 10.1021/jz300102s
Kuta, J., Patchkovskii, S., Zgierski, M. Z., and Kozlowski, P. M. (2006). Performance of DFT in modeling electronic and structural properties of cobalamins. J. Comput. Chem. 27, 1429–1437. doi: 10.1002/jcc.20454
Kuta, J., Wuerges, J., Randaccio, L., and Kozlowski, P. M. (2009). Axial bonding in alkylcobalamins: DFT analysis of the inverse versus normal trans influence. J. Phys. Chem. A 113, 11604–11612. doi: 10.1021/jp901397p
Liptak, M. D., and Brunold, T. C. (2006). Spectroscopic and computational studies of Co1+cobalamin: spectral and electronic properties of the “superreduced” B12 cofactor. J. Am. Chem. Soc. 128, 9144–9156. doi: 10.1021/ja061433q
Lodowski, P., Jaworska, M., Andruniow, T., Kumar, M., and Kozlowski, P. M. (2009). Photodissociation of Co-C bond in methyl- and ethylcobalamin: an insight from TD-DFT calculations. J. Phys. Chem. B. 113, 6898–6909. doi: 10.1021/jp810223h
Lodowski, P., Jaworska, M., Kornobis, K., Andruniow, T., and Kozlowski, P. M. (2011). Electronic and structural properties of low-lying excited states of vitamin B12. J. Phys. Chem. B. 115, 13304–13319. doi: 10.1021/jp200911y
Lott, W. B., Chagovetz, A. M., and Grissom, C. B. (1995). Alkyl radical geometry controls geminate cage recombination in alkylcobalamins. J. Am. Chem. Soc. 117, 12194–12201. doi: 10.1021/ja00154a020
Ludwig, M. L., and Matthews, R. G. (1997). Structure-based perspectives on B12-dependent enzymes. Annu. Rev. Biochem. 66, 269–313. doi: 10.1146/annurev.biochem.66.1.269
Marzilli, L. G. (1999). “The two B12 cofactors: Influence of the trans Nitrogen ligand on homolytic and heterolytic processes,” Bioinorganic Catalysis, eds J. Reedijk and E. Bouwman (New York, NY: Marcel Dekker), 423–468. doi: 10.1201/9780203908457.ch13
Matthews, R. G. (2001). Cobalamin-dependent methyltransferases. Acc. Chem. Res. 34, 681–689. doi: 10.1021/ar0000051
Miertuš, S., Scrocco, E., and Tomasi, J. (1981). Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 55, 117–129. doi: 10.1016/0301-0104(81)85090-2
Natarajan, E., and Grissom, C. B. (1996). The origin of magnetic field dependent recombination in alkylcobalamin radical pairs. Photochem. Photobiol. 64, 286–295. doi: 10.1111/j.1751-1097.1996.tb02460.x
Ouyang, L., Rulis, P., Ching, G., Nardin, L., and Randaccio, L. (2004). Accurate redetermination of the X-ray structure and electronic bonding in adenosylcobalamin. Inorg. Chem. 43, 1235–1241. doi: 10.1021/ic0348446
Perdew, J. P. (1986). Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 33, 8822–8824. doi: 10.1103/PhysRevB.33.8822
Randaccio, L., Geremia, S., Nardin, G., and Wuerges, J. (2006). X-ray structural chemistry of cobalamins. Coord. Chem. Rev. 250, 1332–1350. doi: 10.1016/j.ccr.2005.12.005
Randaccio, L., Geremia, S., and Wuerges, J. (2007). Crystallography of vitamin B12 proteins. J. Organomet. Chem. 692, 1198–1215. doi: 10.1016/j.jorganchem.2006.11.040
Rao, D. N. R., and Symons, M. C. R. (1982). Effects of ionizing radiation and photolysis on methyl-cobalamin in low temperature matrices: an e.s.r. study. J. Chem. Soc. Chem. Commun. 954–955. doi: 10.1039/C39820000954
Rovira, C., Biarnes, X., and Kunc, K. (2004). Structure-energy relations in methylcobalamin with and without bound axial base. Inorg. Chem. 43, 6628–6632. doi: 10.1021/ic049810s
Rovira, C., and Kozlowski, P. M. (2007). First principles study of coenzyme B12. Crystal packing forces effect on axial bond lengths. J. Phys. Chem. B 111, 3251–3257. doi: 10.1021/jp0660029
Sakaguchi, Y., Hayashi, H., and I'Haya, Y. J. (1990). Fast formation of methyl radical from methylaquocobaloxime as studied by time-resolved optical and ESR techniques. J. Phys. Chem. 94, 291–293. doi: 10.1021/j100364a048
Sension, R. J., Cole, A. G., Harris, A. D., Fox, C. C., Woodbury, N. W., Lin, S., et al. (2004). Photolysis and recombination of adenosylcobalamin bound to glutamate mutase. J. Am. Chem. Soc. 126, 1598–1599. doi: 10.1021/ja0396910
Sension, R. J., Harris, A. D., Stickrath, A., Cole, A. G., Fox, C. C., and Marsh, E. N. G. (2005a). Time-resolved measurements of the photolysis and recombination of adenosylcobalamin bound to glutamate mutase. J. Phys. Chem. B. 109, 18146–18152. doi: 10.1021/jp052492d
Sension, R. J., Harris, A. D., and Cole, A. G. (2005b). Time-resolved spectroscopic studies of B12 coenzymes: comparison of the influence of solvent on the primary photolysis mechanism and geminate recombination of methyl-, ethyl-, n-propyl-, and 5′-deoxyadenosylcobalamin. J. Phys. Chem. B. 109, 21954–21962. doi: 10.1021/jp053202w
Shell, T. A., and Lawrence, D. S. (2011). A new trick (hydroxyl radical generation) for an old vitamin (B12). J. Am. Chem. Soc. 133, 2148–2150. doi: 10.1021/ja111585c
Shiang, J. J., Cole, A. G., Sension, R. J., Hang, K., Weng, Y., Trommel, J. S., et al. (2006). Ultrafast excited-state dynamics in vitamin B12 and related cob(III)alamins. J. Am. Chem. Soc. 128, 801–808. doi: 10.1021/ja054374+
Shiang, J. J., Walker, L. A. II., Anderson, N. A., Cole, A. G., and Sension, R. J. (1999). Time-resolved spectroscopic studies of B12 coenzymes: the photolysis of methylcobalamin is wavelength dependent. J. Phys. Chem. B. 103, 10532–10539. doi: 10.1021/jp992358r
Stich, T. R., Brooks, A. J., Buan, N. R., and Brunold, T. C. (2003). Spectroscopic and computational studies of Co3+-corrinoids: spectral and electronic properties of the B12 cofactors and biologically relevant precursors. J. Am. Chem. Soc. 125, 5897–5914. doi: 10.1021/ja029328d
Stich, T. R., Buan, N. R., and Brunold, T. C. (2004). Spectroscopic and computational studies of Co2+corrinoids: spectral and electronic properties of the biologically relevant base-on and base-off forms of Co2+cobalamin. J. Am. Chem. Soc. 126, 9735–9749. doi: 10.1021/ja0481631
Toraya, T. (2000). Radical catalysis of B12 enzymes: structure, mechanism, inactivation, and reactivation of diol and glycerol dehydratases. Cell. Mol. Life Sci. 57, 106–127. doi: 10.1007/s000180050502
Toraya, T. (2003). Radical catalysis in coenzyme B12-dependent isomerization (eliminating) reactions. Chem. Rev. 103, 2095–2127. doi: 10.1021/cr020428b
Treutler, O., and Ahlrichs, R. (1995). Efficient molecular numerical integration schemes. J. Chem. Phys. 102, 346–354. doi: 10.1063/1.469408
Walker, L. A. II., Jarrett, J. T., Anderson, N. A., Pullen, S. H., Matthews, R. G., and Sension, R. J. (1998a). Time-resolved spectroscopic studies of B12 coenzymes: the identification of a metastable cob(III)alamin photoproduct in the photolysis of methylcobalamin. J. Am. Chem. Soc. 120, 3597–3603. doi: 10.1021/ja974024q
Walker, L. A. II., Shiang, J. J., Anderson, N. A., Pullen, S. H., and Sension, R. J. (1998b). Time-resolved spectroscopic studies of B12 coenzymes: the photolysis and geminate recombination of adenosylcobalamin. J. Am. Chem. Soc. 120, 7286–7292. doi: 10.1021/ja981029u
Keywords: coenzyme B12, Co-C bond, photodissociation, ribosylcobalamin, time-dependent density functional theory
Citation: Liu H, Kornobis K, Lodowski P, Jaworska M and Kozlowski PM (2014) TD-DFT insight into photodissociation of the Co-C bond in coenzyme B12. Front. Chem. 1:41. doi: 10.3389/fchem.2013.00041
Received: 20 August 2013; Accepted: 24 December 2013;
Published online: 05 February 2014.
Edited by:
Kazunari Yoshizawa, Kyushu University, JapanReviewed by:
Mathieu Linares, Linköping University, SwedenJean-Philip Piquemal, UPMC, Paris 6-Sorbonne Universités, France
Takashi Kamachi, Kyushu University, Japan
Copyright © 2014 Liu, Kornobis, Lodowski, Jaworska and Kozlowski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pawel M. Kozlowski, Department of Chemistry, University of Louisville, 2320 South Brook Street, Louisville, KY 40292, USA e-mail:cGF3ZWxAbG91aXN2aWxsZS5lZHU=