 Gaëtan Maertens
Gaëtan Maertens Chloé L'Homme
Chloé L'Homme Sylvain Canesi
Sylvain Canesi- Laboratoire de Méthodologie et Synthèse de Produits Naturels, Département de Chimie, Université du Québec à Montréal, Montréal, QC, Canada
We present a review of natural product syntheses accomplished in our laboratory during the last 5 years. Each synthetic route features a phenol dearomatization promoted by an environmentally benign hypervalent iodine reagent. The dearomatizations demonstrate the “aromatic ring umpolung” concept, and involve stereoselective remodeling of the inert unsaturations of a phenol into a highly functionalized key intermediate that may contain a quaternary carbon center and a prochiral dienone system. Several new oxidative strategies were employed, including transpositions (1,3-alkyl shift and Prins-pinacol), a polycyclization, an ipso rearrangement, and direct nucleophilic additions at the phenol para position. Several alkaloids, heterocyclic compounds, and a polycyclic core have been achieved, including sceletenone (a serotonin reuptake inhibitor), acetylaspidoalbidine (an antitumor agent), fortucine (antiviral and antitumor), erysotramidine (curare-like effect), platensimycin (an antibiotic), and the main core of a kaurane diterpene (immunosuppressive agent and stimulator of apoptosis). These concise and in some cases enantioselective syntheses effectively demonstrate the importance of hypervalent iodine reagents in the total synthesis of bioactive natural products.
Introduction
Natural product synthesis is a subject of fascination for many chemists because of the inherent elegance of the synthetic procedure and because many of these compounds have demonstrated important bioactivities, including the noteworthy examples taxol and paclitaxel (Goodman and Walsh, 2001). Indeed, several of the natural products generated by Nature result from a long evolution of plants and microorganisms to develop defenses against pathogens and hostile environments. This long evolution is probably the key to the diverse bioactivity displayed by these compounds and the reason why many pharmaceutical compounds are isolated, inspired, or derived from natural sources. However, the total synthesis of natural products remains a challenge despite the powerful methodologies developed during the last century, and efficient new tools are still a necessity, not only for optimization of the synthetic process in terms of number of steps, yield, atom economy, and reduced environmental burden but in many cases to merely produce the desired architecture at all. Total synthesis has been the focus of many groups, enabling the advancement of basic knowledge as well as the development of new drugs and the creation of efficient tools and strategies to produce complex natural architectures. In this present article, we describe several of our own natural product syntheses accomplished during the past 5 years. The methods are based on phenol dearomatization mediated by hypervalent iodine reagents, and the target molecules are divided into two classes. The first class consists of molecules containing at least one quaternary carbon center obtained using methodologies developed in our laboratory and based on the phenol dearomatization process. Successful formation of a quaternary carbon involves a transposition (1,3-alkyl shift or Prins-pinacol), a polycyclization, or an ipso rearrangement. The second class represents molecules containing a trisubstituted alkene moiety obtained indirectly through hetero-nucleophilic addition followed by elimination. The strategies presented here have enabled us to synthesize alkaloids such as sceletenone (a serotonin reuptake inhibitor), acetylaspidoalbidine (an antitumor agent), fortucine (antiviral and antitumor), erysotramidine (curare-like effect), the heterocyclic antibiotic platensimycin, and the main tetracyclic core of a kaurane diterpene. Depending on the strategy employed, the molecules obtained may be racemic or enantiopure.
The Aromatic Ring Umpolung Concept
The central theme of these syntheses is what may be termed “aromatic ring umpolung” (Guérard et al., 2010). While an electron-rich aromatic nucleus normally reacts as a nucleophile in an electrophilic pathway, suitable oxidative activation can convert it to a reactive electrophilic intermediate 3. We assume that the formation of 3, often described as a phenoxonium ion, is mediated by a Single-Electron Transfer (SET). It has been demonstrated that in bimolecular processes, the electrophilic species can be intercepted at two different positions, depending on the nature of the nucleophile. When a heteronucleophile is involved, the pathway b is observed, resulting from the attack of the unhindered heteroatom on the site bearing the larger LUMO coefficient. However, when a more hindered carbon based nucleophile is used, the pathway a is preferred, due to steric interactions. In this case, a more substituted aromatic compound 4a is obtained. Finally, an intramolecular pathway remains an efficient strategy to force the nucleophile to interact at the desired position. It should be noted that when nucleophilic attack proceeds at the para-position (pathway b), a prochiral dienone system is rapidly obtained. This system may be used as a key intermediate in the total synthesis of several natural products. Indeed, this strategy provides a one-step route from an inexpensive phenol to the polyfunctionalized dienone core 4b. Furthermore, in the presence of a carbon-based nucleophile the formation of a quaternary carbon center is observed (Figure 1).
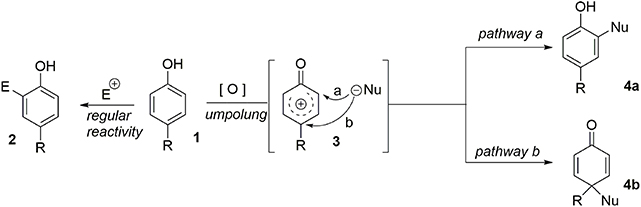
Figure 1. Aromatic ring umpolung concept.
While several oxidizing agents may be used to initiate formation of species 3 as first demonstrated in the remarkable work of Kita and coworkers (Tamura et al., 1987; Dohi et al., 2009, 2010, 2013; Ito et al., 2013), hypervalent iodine reagents (Quideau et al., 1999; Pouységu et al., 2008; Andrez et al., 2010; Liang and Ciufolini, 2010, 2011; Traoré et al., 2010; Desjardins et al., 2011; Yoshimura et al., 2012, 2013; Farid et al., 2013; Jacquemot et al., 2013) such as (diacetoxyiodo)benzene (DIB) or bis(trifluoroacetoxy)iodobenzene (PIFA) have emerged as the reagents of choice. These and similar approaches have been in use for several years, demonstrating the importance of this strategy in the total synthesis of natural products.
Syntheses of Natural Compounds Bearing a Quaternary Carbon Center
This section describes the synthesis of four compounds (two alkaloids, one heterocycle, and a diterpene) containing at least one quaternary carbon. It should be noted that the formation of such a subunit remains a challenge, particularly when stereocontrol of the quaternary center is desired. Oxidative methodologies based on phenol dearomatization mediated by an environmentally benign hypervalent iodine reagent have enabled the rapid formation of the main cores of our targets. These oxidative processes are examples of the “aromatic ring umpolung” concept, enabling the extension of several well-known aliphatic transformations involving an electrophilic intermediate to aromatic substrates. These methodologies promote the transformation of simple and inexpensive phenols into key intermediates containing a prochiral dienone system and a quaternary carbon center. For instance, a 1-3 alkyl shift process enabled synthesis of the hexacyclic alkaloid acetylaspidoalbidine, a Prins-pinacol rearrangement was a key step in formation of the main core of (−)-platensimycin, an oxidative polycyclization was used to construct the asymmetric main core of a kaurane diterpene, and the small alkaloid sceletenone was obtained through an oxidative ipso rearrangement (Figure 2).
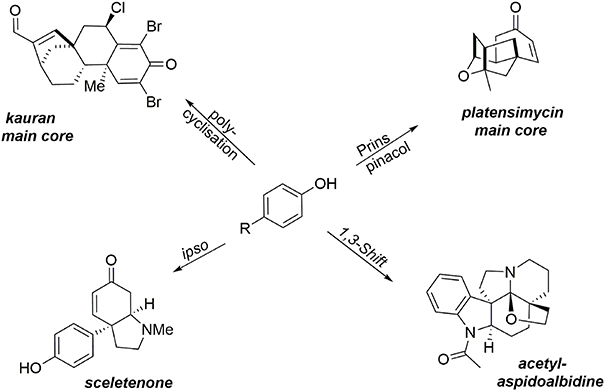
Figure 2. Total syntheses of natural products bearing at least one quaternary carbon center.
Synthesis of Acetylaspidoalbidine
Acetylaspidoalbidine (Walser and Djerassi, 1965; Hesse, 1968; Ban et al., 1975; Honma et al., 1976; Yoshida et al., 1987; Overman et al., 1991; Campbell et al., 2010; Zhang et al., 2013) 14 is a natural alkaloid isolated from the Venezuelan tree species aspidosperma fendleri woodson and aspidosperma rhombeosignatum markgraf, and the molecule belongs to a large class of complex and bioactive alkaloids known as the aspidosperma family (Hesse, 1968). Our retrosynthesis (Guérard et al., 2012) of this molecule from the simple and inexpensive phenol derivative 5 is depicted in Figure 3.
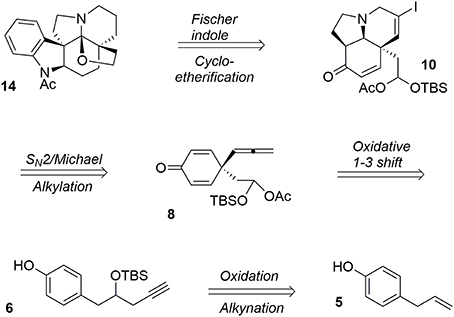
Figure 3. Retrosynthesis of acetylaspidoalbidine.
One particular advantage of the umpolung activation process is its ability to adapt aliphatic transformations to aromatic systems. We have demonstrated the possibility of exploiting oxidative variations of alkyl-shift processes (Guérard et al., 2009) in particular a 1,3-propargyl migration (Guérard et al., 2012) to produce a poly-functionalized prochiral dienone 7 from a phenol derivative 6. This approach established the first quaternary carbon center of the target as well as forming an allenyl moiety and a mixed acetal as a masked aldehyde that was used later in the synthesis (Scheme 1).
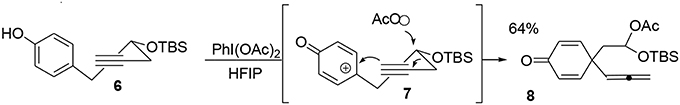
Scheme 1. First oxidative dearomatization process.
Once the central dienone core was obtained, the tricyclic system of the target molecule was obtained through more typical transformations such as iodination of the allenyl moiety and subsequent treatment with ethanolamine to yield the bicycle 9 via a SN2-Michael tandem process. Further activation of the alcohol functionality in the presence of mesyl chloride followed by treatment with potassium tert-butoxide led to the subunit 10. The remaining unsaturations and iodine were quantitatively removed during a final hydrogenation in the presence of palladium to produce 11, Scheme 2.
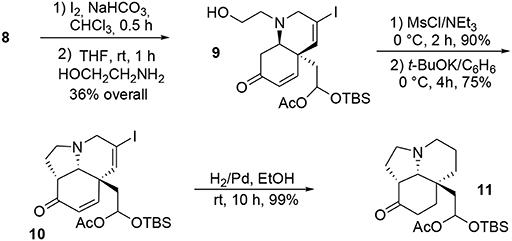
Scheme 2. Formation of the tricyclic core of acetylaspidoalbidine.
The final portion of the synthesis was inspired by the work of Stork and Dolfini, who demonstrated that a Fischer indole-like process could be used to produce the indoline moiety of the aspidosperma family (Stork and Dolfini, 1963). Initial deprotection of the acetal moiety followed by selective reduction using a hindered hydride led to the primary alcohol 12. Addition of phenylhydrazine (Fischer process) and further treatment with LiAlH4 afforded the pentacyclic system. Selective acetylation of the indoline nitrogen followed by oxidative cyclo-etherification (Ban et al., 1975) yielded acetylaspidoalbidine 14, Scheme 3.
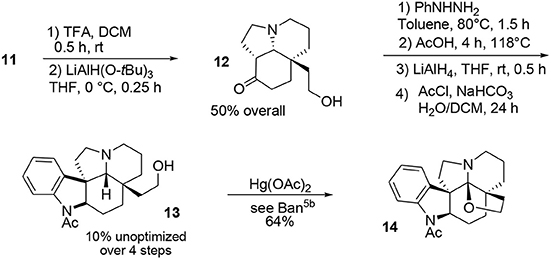
Scheme 3. Final steps in synthesis of acetylaspidoalbidine.
Synthesis of (−)-Platensimycin
Alkyl shifts are important transformations enabling rapid and stereoselective redesign of simple architectures into polyfunctionalized scaffolds, as illustrated by the synthesis of acetylaspidoalbidine. Another important transposition resulting in formation of a complex structure is the famous Prins-pinacol process developed by Overman and coworkers (Hirst et al., 1993; MacMillan and Overman, 1995; Overman and Pennington, 2000, 2003; Lebsack et al., 2001; Lavigne et al., 2005; Overman and Velthuisen, 2006; Armstrong et al., 2007; Tang et al., 2007). Oxidative extension of this process to aromatic systems opens interesting avenues in total synthesis. In 2010, we proposed an asymmetric formal synthesis of (−)-platensimycin 25 (Beaulieu et al., 2010, 2011), a natural antibiotic isolated from a strain of Streptomyces platensis by Wang and Soisson in 2006 and acting as a FabF inhibitor (Wang et al., 2006). The intriguing structure and bioactivity of this compound have evoked a great deal of interest in the synthetic arena (Nicolaou et al., 2006, 2007a,b, 2008, 2009a; Lalic and Corey, 2007; Li et al., 2007; Tiefenbacher and Mulzer, 2007, 2008; Zou et al., 2007; Kim et al., 2008; Matsuo et al., 2008; Ghosh and Xi, 2009; McGrath et al., 2009; Yun et al., 2009; Eey and Lear, 2010, 2014; Palanichamy and Kaliappan, 2010; Tiefenbacher et al., 2010; Xing et al., 2010; Hirai and Nakada, 2011; Ueda et al., 2011; Zheng et al., 2011; Horii et al., 2013). As illustrated in Figure 4, we targeted structure 24, an advanced intermediate in Nicolaou's synthesis (Nicolaou et al., 2009b) containing the main tetracyclic core of (−)-platensimycin. We envisaged producing this intermediate via a Schreiber fragmentation followed by Michael addition to dienone 21. The latter molecule contains the desired spiranic quaternary carbon center and may be obtained from phenol 18 through an oxidative Prins-pinacol tandem process (Beaulieu et al., 2010, 2011).
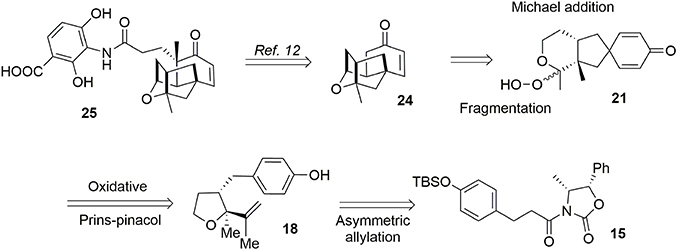
Figure 4. Retrosynthesis of (−)-platensimycin main core.
Preparation of the phenolic precursor 18 was achieved in nine simple steps including an Evans asymmetric allylation, an ozonolysis, and a cycloetherification under acidic conditions, Scheme 4.
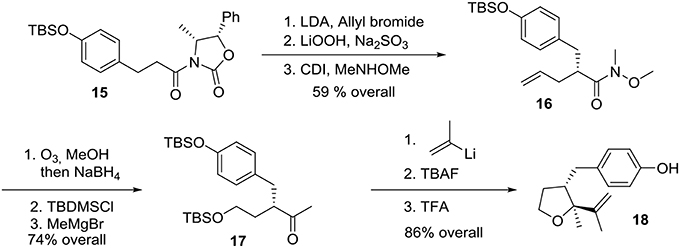
Scheme 4. Formation of phenol precursor.
Oxidation of phenol 18 using PhI(OAc)2 in a mixture of HFIP/DCM followed by addition of hydrogen peroxide furnished the hydroperoxyketal 21 in 64% yield as an unassigned 3:1 diastereomeric mixture (Scheme 5). During the process, the phenoxonium species underwent a stereoselective Prins-like reaction followed by a pinacol transposition through transition state 19, leading to the intermediate 20. This in turn reacted with hydrogen peroxide to produce the desired hydroperoxyketal 21. It should be noted that this oxidative process generated two quaternary carbon centers in a stereoselective manner. The hydroperoxyketal was treated with Fenton reagent (Schreiber, 1980; Schreiber and Liew, 1985) (FeSO4) and Cu(OAc)2 in methanol, and subsequent addition of K2CO3 produced the primary alcohol. The alcohol was oxidized to the corresponding aldehyde using PCC. Aldehydes 22 were obtained as a 3:1 mixture of exo- and endocyclic alkenes; however, both of these compounds may be used as synthetic precursors of (−)-platensimycin, Scheme 5.
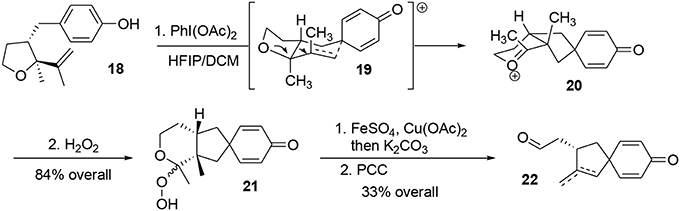
Scheme 5. Oxidative Prins-pinacol key step.
The mixture of aldehydes was subjected to Kagan's reagent (Molander, 1992; Nicolaou et al., 2009c) for stereoselective cyclization, yielding the alcohols 23. Final treatment with trifluoroacetic acid resulted in cycloetherification to complete the formal asymmetric synthesis of (−)-platensimycin, Scheme 6.
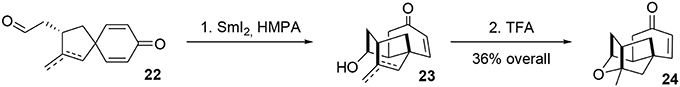
Scheme 6. Final steps of (−)-platensimycin main core synthesis.
Stereoselective Synthesis of Kaurane Main Core
In 2014, we reported an unprecedented tandem oxidative polycyclization-pinacol process (Desjardins et al., 2014) as a means of rapidly accessing the main core of kaurane diterpenes. These compounds belong to a large family of bioactive natural products including the examples in Figure 5. They are used in traditional Chinese medicines and exhibit bioactivities such as immunosuppression, induction of apoptosis, reduction of platelet aggregation, and anti-spasmodic effects.
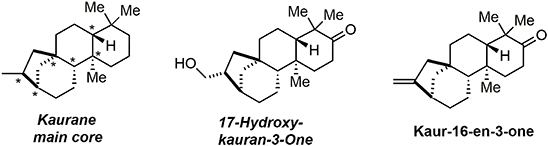
Figure 5. Kauran diterpen members.
The key transformation (Scheme 7) consisted of converting the phenol 36 into the elaborated tetracyclic compound 38 in a stereoselective manner. The proposed mechanism for this reaction involved formation of the electrophilic species 37, triggering a polycyclization cascade that concluded with a pinacol transposition and loss of the TIPS group to produce the aldehyde. The presence of the benzylic chlorine atom appears to be crucial for controlling the stereoselectivity of the polycyclization. Equatorial placement of the chlorine atom forces the side chain to react on the top face as represented in Scheme 7. Ortho protection of phenol 36 with bromine atoms prevented carbocation formation at these positions during oxidative activation, thus directing nucleophilic attack to the former para position. In this way, we were able to produce a compact and elaborated tetracyclic structure containing two quaternary carbon centers and five asymmetric carbons with total control of selectivity in one step and in 30% yield.
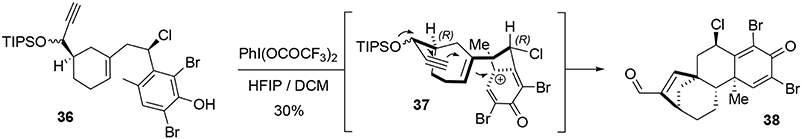
Scheme 7. Oxidative polycyclization-pinacol process.
The envisaged retrosynthetic approach to phenol 36 is presented in Figure 6. We expected that compound 36 could be produced through ring closure metathesis of diene 32. An asymmetric hydrocyanation would permit introduction of our second asymmetric center from Michael acceptor 29. This enone could be easily obtained from benzylic alcohol 27 through ozonolysis followed by simple further transformations. The first asymmetric center could be generated through an asymmetric Yamamoto allylation performed on aldehyde 26.
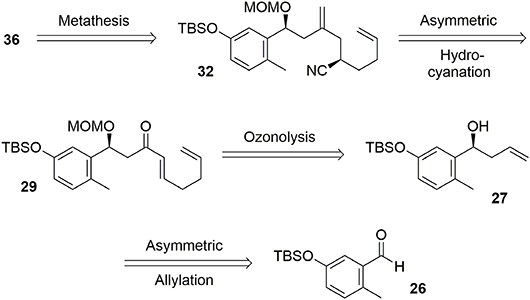
Figure 6. Kaurane main core retrosynthesis.
The synthesis of the phenolic precursor was achieved in 18 chemical steps. Initial asymmetric Yamamoto allylation of aldehyde 26 furnished the benzylic alcohol 27 in 87% yield and with good enantioselectivity (95% ee), allowing us to introduce our first asymmetric center (Scheme 8). Aldehyde 28 was obtained through ozonolysis after protection of the benzylic hydroxyl group with MOM. Michael acceptor 29 was quickly obtained through nucleophilic attack at the aldehyde followed by stereoselective reduction of the triple bond using LiAlH4 and oxidation of the resulting allylic alcohol using DMP.
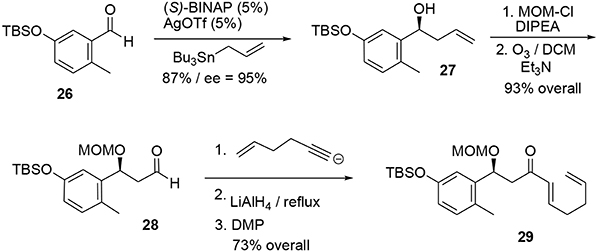
Scheme 8. Formation of first stereocenter.
A further asymmetric hydrocyanation using Ohkuma's ruthenium-based catalyst (Kurono et al., 2011) 30 followed by a Peterson reaction under acidic conditions led to the corresponding diene 32 (Scheme 9). At this stage, the bromine atoms were introduced through classic electrophilic aromatic substitution and the cyclohexene moiety was generated during a ring closure metathesis reaction in the presence of a catalytic amount of Grubbs Hoveyda II. Subsequent treatment with DIBAL-H afforded the enantiopure aldehyde 34.
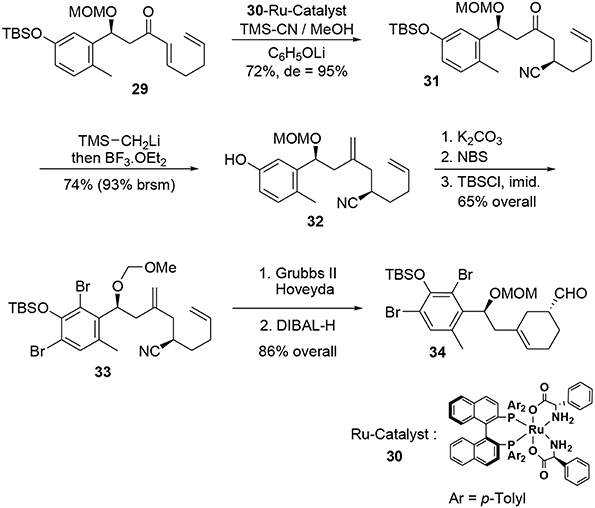
Scheme 9. Formation of second stereocenter.
The aldehyde was trapped by addition of the lithiated anion of trimethylsilylacetylene to produce a propargylic alcohol, which was protected with a TIPS group to avoid side reactions during the polycyclization (Scheme 10). After removal of the other protecting groups, the chlorine atom was introduced by treatment of benzylic alcohol 35 with thionyl chloride.
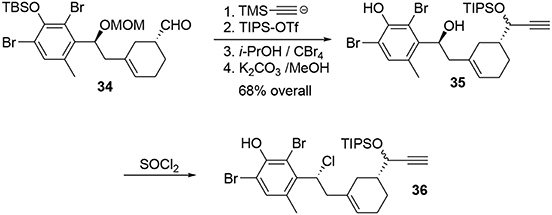
Scheme 10. Elaboration of phenol precursor.
Synthesis of Sceletenone
While transposition and polycyclization processes are eye-catching ways of producing a quaternary carbon center due to the spectacular rearrangements observed, a more direct way would involve a simple nucleophilic addition. However, in a bimolecular mode and in the presence of a hindered carbon-based nucleophile, attack on the phenoxonium ion 3 is favored at the ortho position (Guérard et al., 2010) (Figure 1, pathway a). In order to force the addition to occur at the desired para position, the phenol lateral chain may be used as a tether in a six-membered ring transition state mediated by an ipso-type rearrangement (Jacquemot and Canesi, 2012). As an illustration of this possibility, we synthesized sceletenone 45 (Jeffs et al., 1974, 1978; Sanchez et al., 1983), a small alkaloid belonging to the amaryllidaceae family. This molecule is isolated from the phenolic alkaloid fraction of Sceletium strictum and is a potent serotonin reuptake inhibitor. The natural product is isolated in racemic form, probably due to a potential aza-Michael/retro-Michael equilibrium. A retrosynthesis of this molecule is described in Figure 7.
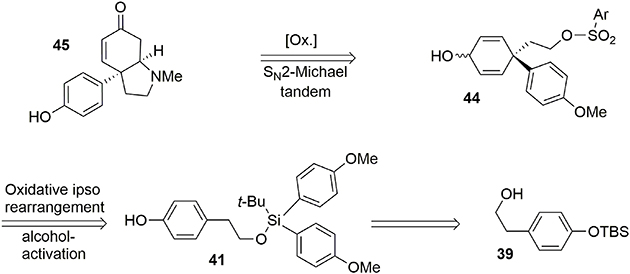
Figure 7. Retrosynthesis of sceletenone.
Our synthesis began with the known alcohol 39 (Nakajima et al., 2005), in which the hydroxyl group was used as a tether to introduce a carbon-based nucleophile on the lateral chain to force an intramolecular para attack, enabling formation of the quaternary carbon center required in the target 45. The modified silyl group was not only used as a protecting group during the synthesis but also acted as a linker to enable the formation of an important part of the target. The modified tert-butyldiarylsilyl chloride 40 bearing anisole groups was prepared by addition of two equivalents of aryl-organolithium reagent to commercially available tert-butyltrichlorosilane. Once the silyl reagent was obtained, an oxidative ipso-rearrangement was performed to produce the dienone 42 containing a quaternary carbon center connected to three sp2 centers in 74% yield. Interestingly, an asymmetric silicon center was obtained by substitution of one aryl group with hexafluoropropanol, which was used as a solvent, Scheme 11.
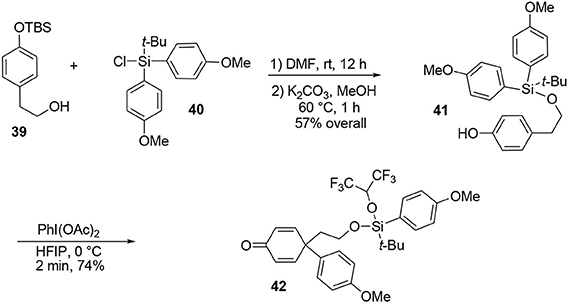
Scheme 11. Quaternary carbon center formation by oxidative ipso rearrangement.
At this stage the formation of the pyrrolidine moiety was undertaken. A 1,2-reduction of the dienone mediated by DIBAL-H led stereoselectively to alcohol 43. Subsequent deprotection of the silyl ether using TBAF produced the free alcohol in 85% yield. Selective activation of the primary alcohol promoted by ortho-nosyl chloride furnished 44 in 40% yield. Further oxidation with TPAP/NMO led to a dienone core which was treated with methylamine to afford the pyrrolidine core of sceletenone through a SN2-aza-Michael tandem process in 76% yield. The synthesis of sceletenone 45 was concluded by a final treatment with BBr3, Scheme 12.
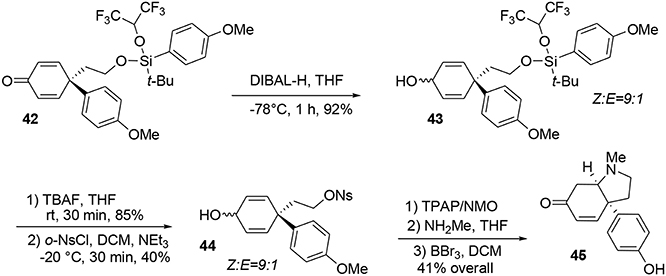
Scheme 12. Final steps of sceletenone synthesis.
Syntheses ok Alkaloids Bearing a Cyclic Trisubstituted Alkene
One characteristic of our methodologies employing the phenol dearomatization process is the production of a carbon center at the para position of the original phenol that is not connected to any hydrogen atoms, either in the form of a quaternary carbon center or a substituted olefin. The second portion of this review highlights syntheses of the alkaloids erysotramidine and (−)-fortucine. Elaboration of these molecules is mediated by a two-step oxidative elimination process in which an oxygen-based nucleophile is introduced at the para-position to enable the dearomatization process and block the para position, followed by elimination of this ether moiety to insert the required polysubstituted double bond at the former para position (Figure 8).
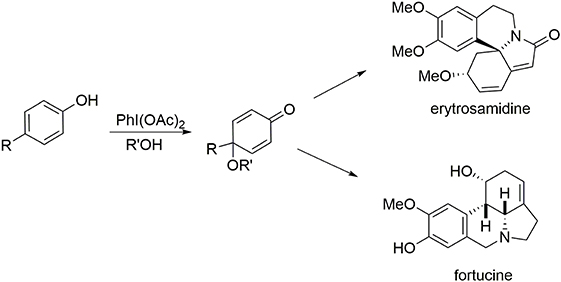
Figure 8. Alkaloids bearing a cyclic trisubstituted alkene.
Synthesis of Erysotramidine
Erysotramidine belongs to the erythrina alkaloid family and was isolated from tropical plants of the erythrina genus (Tsuda and Sano, 1996). These compounds contain an aza-spiran ring and exhibit several biological activities including hypotensive, sedative, anticonvulsant, and curare-like effects (Boekelheide, 1960; Hill, 1967; Dyke and Quessy, 1981; Chawla and Jackson, 1984). While some erythrina alkaloids have nonaromatic structures, others such as erysotramidine and erysotrine contain an aromatic subunit, Figure 9.
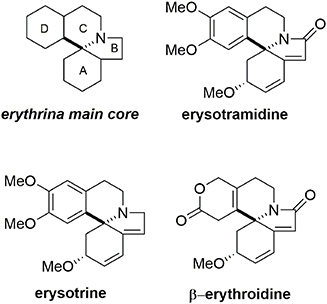
Figure 9. Erythrina alkaloid members.
Our synthesis of erysotramidine was achieved in eight steps (L'Homme et al., 2014). A retrosynthetic description is provided in Figure 10. The key steps included a stereoselective ketone reduction, a Pictet-Spengler cyclization to produce the main tetracyclic system, a novel tandem aza-Michael-rearomatization, and two oxidative dearomatization processes mediated by a hypervalent iodine reagent.
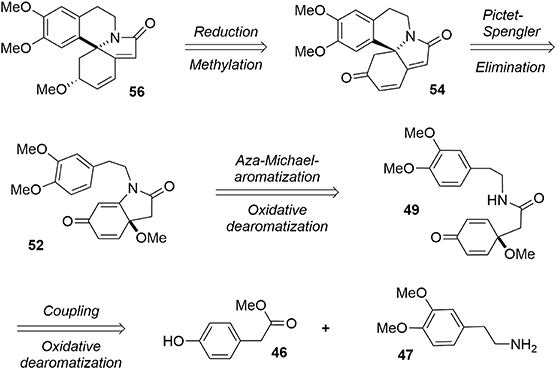
Figure 10. Retrosynthesis of erysotramidine.
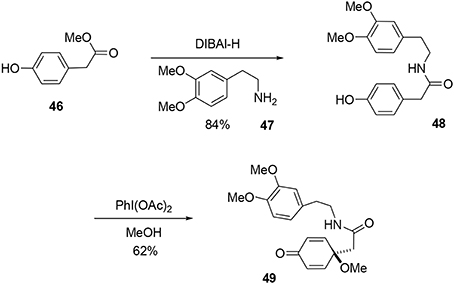
Our synthesis began with a coupling reaction between phenol 46 and amine 47 in the presence of DIBAL-H to produce the corresponding amide 48. An oxidative dearomatization using PhI(OAc)2 in methanol was then undertaken to transform phenol 48 into prochiral dienone 49 in 62% yield (Scheme 13).
Scheme 13. Elaboration of the bicyclic precursor.
The bicycle 51 was subsequently obtained through a new tandem aza-Michael rearomatization process. Dienone 49 was treated with TMS-OTf in the presence of triethylamine to generate intermediate 50. We expect the reaction first occurs through formation of an imino-ether, followed by activation of the enone moiety by the siliconium Lewis acid enabling an aza-Michael addition to yield the enol ether 50. The methoxy group was removed by addition of BF3·Et2O in the same pot, leading to a rearomatization furnishing 51. The phenol 51 was re-oxidized with PIFA in methanol to generate another dienone 52. However, we observed the formation of a byproduct 53 containing two methoxy groups; this by-product was probably formed through a double activation mediated by the hypervalent iodine reagent. The desired dienone 52 was obtained in 53% yield, Scheme 14.
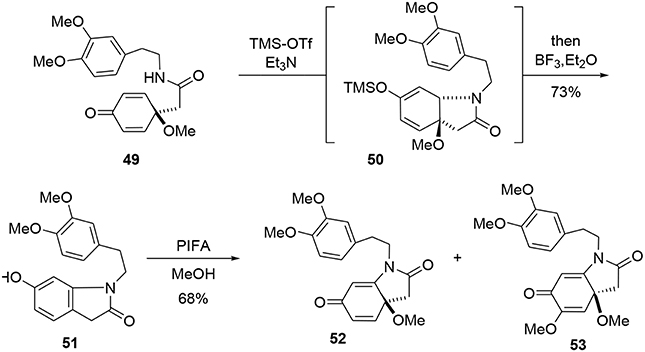
Scheme 14. Formation of main bicyclic system of erysotramidine.
The tetracyclic structure 54 was produced in a Pictet-Spengler type reaction. Under acidic conditions the aromatic moiety attacked the Michael acceptor, producing the aza-spiran in 71% yield (Maryanoff et al., 2004). At this stage, the tetracycle was treated with KHMDS to eliminate the methoxy group through an E1cB mechanism to form the polyconjugated product 55 in 73% yield, Scheme 15. The ketone functionality was reduced under Luche conditions to yield the desired alcohol with good stereoselectivity (9:1). Erysotramidine 56 was quantitatively obtained by methylation of the alcohol using Ag2O and iodomethane.
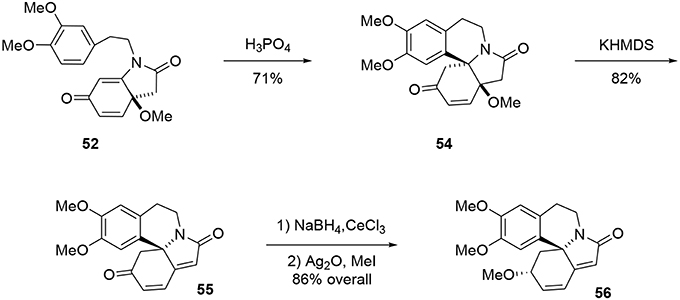
Scheme 15. Final steps of erysotramidine synthesis.
Stereoselective Synthesis of Fortucine
Fortucine belongs to the lycorine alkaloid family (Dalton, 1979) and was isolated from a fortune variety of narcissus. Lycorine alkaloids have demonstrated antitumor and antiviral activities (De Leo et al., 1973; Chattopadhyay et al., 1984; Ghosal et al., 1985; Martin, 1987). They all contain a tetracyclic pyrrolo[d,e]phenanthridine core and most (including (−)-lycorine) (Fales et al., 1955; Takagi et al., 1955; Yamada et al., 2009) present a trans B/C-ring junction; however, (+)-fortucine (Gorbunova et al., 1984; Tokhtabaeva et al., 1987), (+)-kirkine (Bastida et al., 1995), and (−)-siculinine (Richomme et al., 1989) contain a cis ring junction (Figure 11).
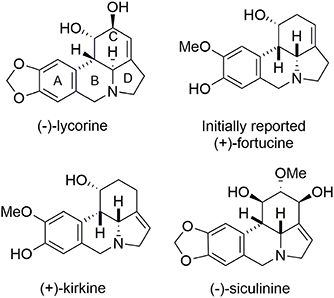
Figure 11. Lycorine alkaloids.
A racemic synthesis of fortucine was reported by Zard and coworkers (Biechy et al., 2008, 2009) and involved a noteworthy radical cascade reaction for formation of the main core of the target. We envisaged an enantioselective synthesis of fortucine based on the retrosynthesis presented in Figure 12. The tetracyclic structure 68 could be assembled by stereoselective carbo-palladation and decarboxylation of enol ether 63. The latter could be obtained from a diastereoselective oxidative dearomatization – aza-Michael addition sequence applied to substrate 61 in an elegant approach first developed by Wipf and coworkers (Wipf and Kim, 1992; Wipf et al., 1995; Wipf and Li, 1999; Wipf and Mareska, 2000; Wipf and Methot, 2000). Finally, phenol 61 could be produced through a Schotten-Baumann coupling between l-tyrosine methyl ester and acyl chloride 59, which is derived from the commercially available aldehyde 57.
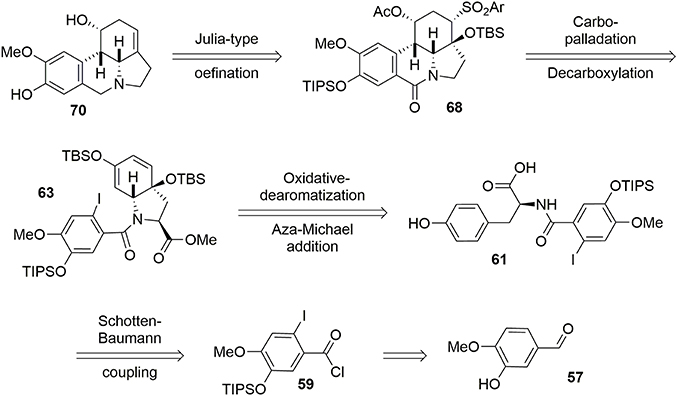
Figure 12. Fortucine retrosynthesis.
The synthesis of intermediate 59 was achieved in four steps from commercial 3-hydroxy-4-methoxybenzaldehyde 57. After protection of the hydroxy group using TIPSCl, the iodine atom was introduced by electrophilic aromatic substitution in the presence of I2 and silver nitrate. Aldehyde 58 was oxidized to the corresponding carboxylic acid (Das and Chakraborty, 2011) using tert-BuOOH in the presence of CuCl2, and treatment with SOCl2 catalyzed by DMF yielded the acyl chloride 59 in 55% yield over the four steps, Scheme 16.
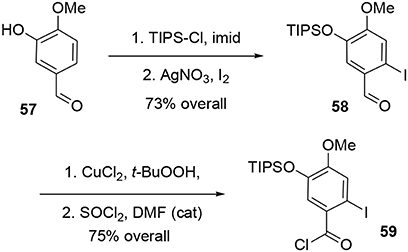
Scheme 16. Synthesis of aromatic moiety.
Acyl chloride 59 was coupled with l-tyrosine methyl ester 60 in the presence of NaHCO3 to generate the corresponding amide, which was subjected to mild Krapcho-like conditions to produce the intermediate 61. At this stage, we applied the methodology developed by Wipf and coworkers (Wipf and Kim, 1992; Wipf et al., 1995; Wipf and Li, 1999; Wipf and Mareska, 2000; Wipf and Methot, 2000). This strategy consisted of activating the phenol with PhI(OAc)2to form the phenoxonium ion, which was trapped by the acid function to yield the spiro-lactone 62. This was treated with KOH in methanol to cleave the lactone and promote conjugated addition of the amino group to the Michael acceptor, leading to the bicyclic enone 63. It should be noted that the stereochemistry of that reaction was governed by the chiral center of the tyrosine (Tomioka et al., 1977), allowing us to obtain compound 63 with high stereoselectivity, Scheme 17.
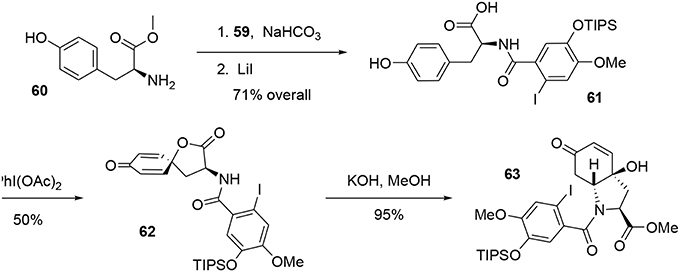
Scheme 17. Formation of heterocyclic system of fortucine.
Silylation of enone 63 followed by a Heck-type carbopalladation in the presence of Pd(PPh3)4 produced the cis tetracyclic pyrrolo[d,e]phenanthridine 64 in 82% over two steps. We assume that the observed cis ring junction was a consequence of the planar geometry of the lactam moiety. It should be stressed that the tetracyclic compound 64 not only represents the main core of fortucine but also the central architecture of siculinine and kirkine. A kinetic Michael addition of 2-methoxy benzenethiol in the presence of Et3N formed the thioether 65. This functionality was necessary for the planned Julia-type reaction at the end of the synthesis. An enantiopure protected alcohol was obtained by reduction of ketone 65 with LiBH4 followed by acetylation of the hydroxyl group under classic conditions. Methyl ester 66 was subsequently converted to a carboxylic acid under mild Krapcho-like conditions and the thioether was oxidized to the corresponding sulfone 67 by treatment with m-CPBA, Scheme 18.
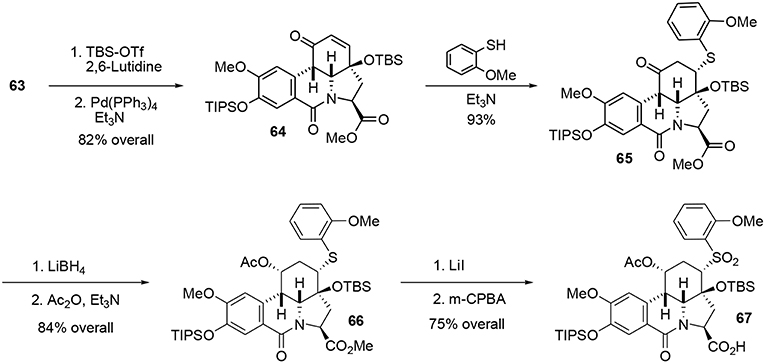
Scheme 18. Formation of tetracyclic portion.
Compound 67 was decarboxylated using a photochemical process developed by Boto et al. (2000, 2001) and involving a hypervalent iodine reagent in the presence of molecular iodine. The resulting iminium ion was trapped using Et3SiH to yield the lactam 68. The latter was reduced with DIBAL-H, and the TIPS and acetate groups were selectively removed. Finally, a Julia-type reaction (Julia and Paris, 1973) in the presence of Li/naphthalene resulted in formation of the desired unsaturation in 65% yield. Although the NMR and mass spectrometric data for our synthetic fortucine were in accordance with the literature (Bastida et al., 1995), we observed that the optical rotation had the opposite sign from natural fortucine. We also observed that our compound and natural fortucine had opposite Cotton effects at 285 nm. A crystallographic study of substrate 69 confirmed that we had synthesized the reported enantiomer of fortucine (Beaulieu et al., 2014), meaning that the natural product is actually the mirror image of the structure that had previously been described, Scheme 19.
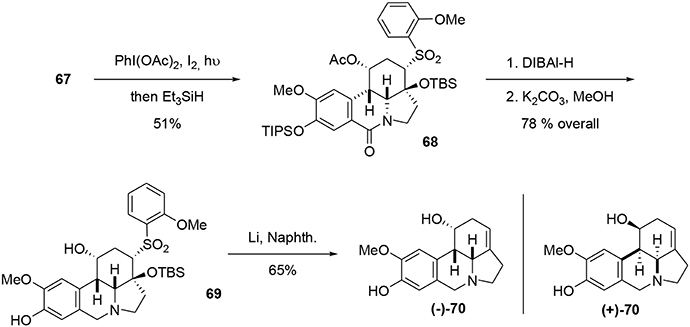
Scheme 19. End of synthesis and correction of absolute configuration.
Conclusion
We have summarized several total natural product syntheses developed by our group. All of the syntheses highlight the importance of hypervalent iodine reagents as well as the utility of the “aromatic ring umpolung” concept. Indeed, one to two key steps in each synthesis involved hypervalent iodine reagents. A main challenge that requires to be addressed in a near future is the development of an efficient stereoselective methodology enabling to desymmetrize prochiral systems into enantiopur precursors for asymmetric synthesis. Hypervalent iodine chemistry is in constant growth, and these strategies are only examples of the expanding literature on the applications of hypervalent iodine reagents in the synthesis of natural products.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are very grateful to the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Foundation for Innovation (CFI), and the provincial government of Quebec (FQRNT and CCVC) for their precious financial support in this research.
References
Andrez, J. A., Giroux, M. A., Lucien, J., and Canesi, S. (2010). Rapid formation of hindered cores using an oxidative prins process. Org. Lett. 12, 4368–4371. doi: 10.1021/ol101851z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Armstrong, A., Bhonoah, Y., and Shanahan, S. E. (2007). Aza-Prins-pinacol approach to 7-azabicyclo[2.2.1]heptanes: syntheses of (±)-epibatidine and (±)-epiboxidine. J. Org. Chem. 72, 8019–8024. doi: 10.1021/jo701536a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ban, Y., Ohnuma, T., Seki, K., and Oishi, T. (1975). The total synthesis of the alkaloid (±)-1-acetylaspidoalbidine. Tetrahedron Lett. 16, 727–730. doi: 10.1016/S0040-4039(00)71968-X
Bastida, J., Codina, C., Peeters, P., Rubiralta, M., Orozco, M., Luque, F. J., et al. (1995). Alkaloids from crinum kirkii. Phytochemistry 40, 1291–1293. doi: 10.1016/0031-9422(95)00391-J
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beaulieu, M. A., Guérard, K. C., Maertens, G., Sabot, C., and Canesi, S. (2011). Oxidative prins-pinacol tandem process mediated by a hypervalent iodine reagent: scope, limitations and applications. J. Org. Chem. 76, 9460–9471. doi: 10.1021/jo2019027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beaulieu, M. A., Ottenwaelder, X., and Canesi, S. (2014). Asymmetric synthesis of fortucine and reassignment of its absolute configuration. Chem. Eur. J. 20, 7581–7584. doi: 10.1002/chem.201402323
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beaulieu, M. A., Sabot, C., Achache, N., Guérard, K. C., and Canesi, S. (2010). An oxidative prins-pinacol tandem process and its application to the synthesis of (−)-platensimycin. Chem. Eur. J. 16, 11224–11228. doi: 10.1002/chem.201001813
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Biechy, A., Hachisu, S., Quiclet-Sire, B., Ricard, L., and Zard, S. Z. (2008). The total synthesis of (±)-fortucine and a revision of the structure of kirkine. Angew. Chem. Int. Ed. 47, 1436–1438. doi: 10.1002/anie.200704996
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Biechy, A., Hachisu, S., Quiclet-Sire, B., Ricard, L., and Zard, S. Z. (2009). Application of an amidyl radical cascade to the total synthesis of (±)-fortucine leading to the structural revision of kirkine. Tetrahedron 65, 6730–6738. doi: 10.1016/j.tet.2009.04.027
Boto, A., Hernandez, R., De Leon, Y., and Suarez, E (2001). Synthesis of 2,3-disubstituted pyrrolidines and piperidines via one-pot oxidative decarboxylation-β-iodination of amino acids. J. Org. Chem. 66, 7796–7803. doi: 10.1021/jo015877a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boto, A., Hernandez, R., and Suarez, E. (2000). Tandem radical decarboxylation-oxidation of amino acids: a mild and efficient method for the generation of N-acyliminium ions and their nucleophilic trapping. J. Org. Chem. 64, 4930–4937. doi: 10.1021/jo000356t
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Campbell, E. L., Zuhl, A. M., Liu, C. M., and Boger, D. L. (2010). Total synthesis of (+)-fendleridine (aspidoalbidine) and (+)-1-acetylaspidoalbidine. J. Am. Chem. Soc. 132, 3009–3012. doi: 10.1021/ja908819q
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chattopadhyay, U., Chaudhuri, L., Das, S., Kumar, Y., and Ghosal, S. (1984). Activation of lymphocytes by lycorine-1-O-β-D-glucoside. Pharmazie 39, 855–856.
Chawla, A. S., and Jackson, A. H. (1984). Erthrina and related alkaloids. Nat. Prod. Rep. 1, 371–373. doi: 10.1039/np9840100371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dalton, D. R. (1979). The Alkaloids: The Fundamental Chemistry - A Biogenetic Approach, Vol. 7, ed M. Dekker (New York, NY: CRC Press).
Das, R., and Chakraborty, D. (2011). Cu(II) bromide catalyzed oxidation of aldehydes and alcohols. Appl. Organomet. Chem. 25, 437–442. doi: 10.1002/aoc.1783
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Leo, P., Dalessandro, G., De Santis, A., and Arigoni, O. (1973). Inhibitory effect of lycorine on cell division and cell elongation. Plant Cell Physiol. 14, 487–496.
Desjardins, S., Andrez, J. A., and Canesi, S. (2011). A stereoselective oxidative polycyclization process mediated by a hypervalent iodine reagent. Org. Lett. 13, 3406–3409. doi: 10.1021/ol201149u
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Desjardins, S., Maertens, G., and Canesi, S. (2014). Asymmetric synthesis of the main core of kaurane family members triggered by an oxidative polycyclization-pinacol tandem process. Org. Lett. 16, 4928–4931. doi: 10.1021/ol5024486
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dohi, T., Ito, M., Yamaoka, N., Morimoto, K., Fujioka, H., and Kita, Y. (2009). Hypervalent iodine(III): selective and efficient single-electron-transfer (SET) oxidizing agent. Tetrahedron 65, 10797–10815. doi: 10.1016/j.tet.2009.10.040
Dohi, T., Takenaga, N., Nakae, T., Toyoda, Y., Yamasaki, M., Shiro, M., et al. (2013). Asymmetric dearomatizing spirolactonization of naphthols catalyzed by spirobiindane-based chiral hypervalent iodine species. J. Am. Chem. Soc. 135, 4558–4566. doi: 10.1021/ja401074u
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dohi, T., Yamaoka, N., and Kita, Y. (2010). Fluoroalcohols: versatile solvents in hypervalent iodine chemistry and syntheses of diaryliodonium(III) salts. Tetrahedron. 66, 5775–5785. doi: 10.1016/j.tet.2010.04.116
Eey, S. T. C., and Lear, M. (2010). A bismuth(III)-catalyzed friedel-crafts cyclization and stereocontrolled organocatalytic approach to (–)-platensimycin. J. Org. Lett. 12, 5510–5513. doi: 10.1021/ol102390t
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eey, S. T. C., and Lear, M. J. (2014). Total Synthesis of (–)-Platensimycin by advancing oxocarbenium- and iminium-mediated catalytic methods. Chem. Eur. J. 20, 11556–11573. doi: 10.1002/chem.201400131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fales, H., Warnhoff, E. M., and Wildman, W. C. (1955). Alkaloids of the amaryllidaceae. VI. the action of oxidizing agents on lycorine and caranine. J. Am. Chem. Soc. 77, 5885–5890. doi: 10.1021/ja01627a032
Farid, U., Malmedy, F., Claveau, R., Albers, L., and Wirth, T. (2013). Stereoselective rearrangements with chiral hypervalent iodine reagents. Angew. Chem. Int. Ed. 52, 7018–7022. doi: 10.1002/anie.201302358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghosal, S., Saini, K. S., and Razdan, S. (1985). Crinum alkaloids: their chemistry and biology. Phytochemistry 24, 2141–2156. doi: 10.1016/S0031-9422(00)83001-0
Ghosh, A. K., and Xi, K. (2009). Total synthesis of (–)-platensimycin, a novel antibacterial agent. J. Org. Chem. 74, 1163–1170. doi: 10.1021/jo802261f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Goodman, J., and Walsh, V. (2001). The Story of Taxol: Nature and Politics in the Pursuit of an Anti-Cancer Drug. New York, NY: Cambridge University Press
Gorbunova, G. M., Sheichenko, V. I., and Tolkachev, O. N. (1984). Alkaloids of narcissus fortune. Chem. Nat. Compd. 20, 800–801.
Guérard, K. C., Chapelle, C., Giroux, M. A., Sabot, C., Beaulieu, M. A., Achache, N., et al. (2009). An unprecedented oxidative wagner-meerwein transposition. Org. Lett. 11, 4756–4759. doi: 10.1021/ol902000j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guérard, K. C., Guerinot, A., Bouchard-Aubin, C., Ménard, M. A., Lepage, M., Beaulieu, M. A., et al. (2012). Oxidative 1,2- and 1,3-alkyl shift processes: developments and applications in synthesis. J. Org. Chem. 77, 2121–2133. doi: 10.1021/jo300169k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Guérard, K. C., Sabot, C., Beaulieu, M. A., Giroux, M. A., and Canesi, S. (2010). “Aromatic ring umpolung,” a rapid access to the main core of several natural products. Tetrahedron 66, 5893–5901. doi: 10.1016/j.tet.2010.03.096
Hirai, S., and Nakada, M. (2011). Enantioselective divergent approaches to both (–)-platensimycin and (–)-platencin. Tetrahedron 67, 518–530. doi: 10.1016/j.tet.2010.10.076
Hirst, G. C., Johnson, T. O., and Overman, L. E. (1993). First total synthesis of lycopodium alkaloids of the magellanane group. Enantioselective total syntheses of (–)-magellanine and (+)-magellaninone. J. Am. Chem. Soc. 115, 2992–2993. doi: 10.1021/ja00060a064
Honma, Y., Ohnuma, T., and Ban, Y. (1976). A total synthesis of (±)-fendleridine. Heterocycles 5, 47–51. doi: 10.3987/S-1976-01-0047
Horii, S., Torihata, M., Nagasawa, T., and Kuwahara, S. (2013). Stereoselective approach to the racemic oxatetracyclic core of platensimycin. J. Org. Chem. 78, 2798–2801. doi: 10.1021/jo302813y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ito, M., Kubo, H., Itani, I., Morimoto, K., Dohi, T., and Kita, Y. (2013). Organocatalytic C-H/C-H' cross-biaryl coupling: C-selective arylation of sulfonanilides with aromatic hydrocarbons. J. Am. Chem. Soc. 135, 14078–14081. doi: 10.1021/ja407944p
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jacquemot, G., and Canesi, S. (2012). Oxidative ipso-rearrangement performed by a hypervalent iodine reagent and its application. J. Org. Chem. 77, 7588–7594. doi: 10.1021/jo301408j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jacquemot, G., Menard, M. A., L'Homme, C., and Canesi, S. (2013). Oxidative cycloaddition and cross-coupling processes on unactivated benzene derivatives. Chem. Sci. 4, 1287–1292. doi: 10.1039/C2SC22318J
Jeffs, P. W., Karle, J. M., and Martin, N. H. (1978). Cinnamic acid intermediates as precursors to mesembrine and some observations on the late stages in the biosynthesis of the mesembrine alkaloids. Phytochemistry 17, 719–728. doi: 10.1016/S0031-9422(00)94215-8
Jeffs, W., Capps, T. M., Johnson, D. B., Karle, J. M., Martin, N. H., and Rauckman, B. (1974). Sceletium alkaloids. VI. minor alkaloids of S. namaquense and S. strictum. J. Org. Chem. 39, 2703–2710. doi: 10.1021/jo00932a008
Julia, M., and Paris, J. M. (1973). Synthèses à l'aide de (+)-sulfones–méthode de synthèse générale de doubles liaisons. Tetrahedron Lett. 14, 4833–4836. doi: 10.1016/S0040-4039(01)87348-2
Kim, C. H., Jang, K. P., Choi, S. Y., Chung, Y. K., and Lee, E. (2008). A carbonyl ylide cycloaddition approach to platensimycin. Angew. Chem. Int. Ed. 47, 4009–4011. doi: 10.1002/anie.200800568
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kurono, N., Nii, N., Sakaguchi, Y., Uemura, M., and Ohkuma, T. (2011). Asymmetric hydrocyanation of α,β-unsaturated ketones into β-cyano ketones with the [Ru(phgly)2(binap)]/C6H5OLi catalyst system. Angew. Chem. Int. Ed. 50, 5541–5544. doi: 10.1002/anie.201100939
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
L'Homme, C., Menard, M. A., and Canesi, S. (2014). Concise syntheses of the erythrina alkaloids erysotramidine and erysotrine. J. Org. Chem. 79, 8481–8485. doi: 10.1021/jo501583c
Lalic, G., and Corey, E. J. (2007). An effective enantioselective route to the platensimycin core. Org. Lett. 9, 4921–4923. doi: 10.1021/ol702323s
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lavigne, R. M. A., Riou, M., Girardin, M., Morency, L., and Barriault, L. (2005). Synthesis of highly functionalized bicyclo[m.n.1]alkanones via a cationic reaction cascade. Org. Lett. 7, 5921–5923. doi: 10.1021/ol0527072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lebsack, A. D., Overman, L. E., and Valentkovitch, R. J. (2001). Enantioselective total synthesis of shahamin K. J. Am. Chem. Soc. 123, 4851–4852. doi: 10.1021/ja015802o
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, P., Payette, J. N., and Yamamoto, H. (2007). Enantioselective route to platensimycin: an intramolecular Robinson annulation approach. J. Am. Chem. Soc. 129, 9534–9535. doi: 10.1021/ja073547n
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liang, H., and Ciufolini, M. A. (2010). Synthetic aspects of the oxidative amidation of phenols. Tetrahedron 66, 5884–5892. doi: 10.1016/j.tet.2010.05.020
Liang, H., and Ciufolini, M. A. (2011). Chiral hypervalent iodine reagents in asymmetric reactions. Angew. Chem. Int. Ed. 50, 11849–11851. doi: 10.1002/anie.201106127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
MacMillan, D. W. C., and Overman, L. E. (1995). Enantioselective total synthesis of (–)-7-deacetoxyalcyonin acetate. First synthesis of a eunicellin diterpene. J. Am. Chem. Soc. 117, 10391–10392. doi: 10.1021/ja00146a028
Maryanoff, B. E., Zhang, H. C., Cohen, J. H., Turchi, I. J., and Maryanoff, C. A. (2004). Cyclizations of N-acyliminium ions. Chem. Rev. 104, 1431–1628. doi: 10.1021/cr0306182
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Matsuo, J. I., Takeuchi, K., and Ishibashi, H. (2008). Stereocontrolled formal synthesis of (±)-platensimycin. Org. Lett. 10, 4049–4052. doi: 10.1021/ol801584r
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McGrath, N. A., Bartlett, E. S., Sittihan, S., and Njardarson, J. T. (2009). A concise ring-expansion route to the compact core of platensimycin. Angew. Chem. Int. Ed. 48, 8543–8546. doi: 10.1002/anie.200903347
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Molander, G. A. (1992). Application of lanthanide reagents in organic synthesis. Chem. Rev. 92, 29–68. doi: 10.1021/cr00009a002
Nakajima, R., Ono, M., Aiso, S., and Akita, H. (2005). Synthesis of methyl 1-0-(4-hydroxymethamphetaminyl)-α-D-glucopyranouronate. Chem. Pharm. Bull. 53, 684–687. doi: 10.1248/cpb.53.684
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicolaou, K. C., Chen, J. S., Edmonds, D. J., and Estrada, A. A. (2009a). Recent advances in the chemistry and biology of naturally occurring antibiotics. Angew. Chem. Int. Ed. 48, 660–719. doi: 10.1002/anie.200801695
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicolaou, K. C., Edmonds, D. J., Li, A., and Tria, G. S. (2007a). Asymmetric total syntheses of platensimycin. Angew. Chem. Int. Ed. 46, 3942–3945. doi: 10.1002/anie.200700586
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicolaou, K. C., Ellery, S. P., and Chen, J. S. (2009c). Samarium diiodide mediated reactions in total synthesis. Angew. Chem. Int. Ed. 48, 7140–7165. doi: 10.1002/anie.200902151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicolaou, K. C., Li, A., and Edmonds, D. J. (2006). Total synthesis of platensimycin. Angew. Chem. Int. Ed. 45, 7086–7090. doi: 10.1002/anie.200603892
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicolaou, K. C., Li, A., Edmonds, D. J., Tria, G. S., and Ellery, S. P. (2009b). Total synthesis of platensimycin and related natural products. J. Am. Chem. Soc. 131, 16905–16918. doi: 10.1021/ja9068003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicolaou, K. C., Pappo, D., Tsang, K. Y., Gibe, R., and Chen, D. Y. K. (2008). A chiral pool based synthesis of platensimycin. Angew. Chem. Int. Ed. 47, 944–946. doi: 10.1002/anie.200705080
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicolaou, K. C., Tang, Y., and Wang, J. (2007b). Formal synthesis of (±)-platensimycin. Chem. Commun. 1922–1923. doi: 10.1039/b704589a
Overman, L. E., and Pennington, L. D. (2000). A new strategy for synthesis of attached rings. Can. J. Chem. 78, 732–738. doi: 10.1139/v00-022
Overman, L. E., and Pennington, L. D. (2003). Strategic use of pinacol-terminated prins cyclizations in target-oriented total synthesis. J. Org. Chem. 68, 7143–7157. doi: 10.1021/jo034982c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Overman, L. E., Robertson, G. M., and Robichaud, A. J. (1991). Use of aza-cope rearrangement-mannich cyclization reactions to achieve a general entry to melodinus and aspidosperma alkaloids. stereocontrolled total syntheses of (±)-deoxoapodine, (±)-meloscine, and (±)-epimeloscine and a formal synthesis of (±)-1-acetylaspidoalbidine. J. Am. Chem. Soc. 113, 2598–2610. doi: 10.1021/ja00007a038
Overman, L. E., and Velthuisen, E. J. (2006). Scope and facial selectivity of the prins-pinacol synthesis of attached rings. J. Org. Chem. 71, 1581–1587. doi: 10.1021/jo0522862
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Palanichamy, K., and Kaliappan, K. P. (2010). Discovery and syntheses of “superbug challengers” – platensimycin and platencin. Chem. Asian. J. 5, 668–703. doi: 10.1002/asia.200900423
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pouységu, L., Marguerit, M., Gagnepain, J., Lyvinec, G., Eatherton, A. J., and Quideau, S. (2008). Total synthesis of wasabidienones B1 and B0 via SIBX-mediated hydroxylative phenol dearomatization. Org. Lett. 10, 5211–5214. doi: 10.1021/ol802183p
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Quideau, S., Looney, M. A., and Pouységu, L. (1999). Oxidized arenol intermediates in intermolecular carbon-carbon bond formation. naphthoid cyclohexa-2,4-dienones via oxidative nucleophilic substitution. Org. Lett. 1, 1651–1654. doi: 10.1021/ol9910363
Richomme, P., Pabuççuoglu, V., Gözler, T., Freyer, A. J., and Shamma, M. (1989). (–)-Siculinine: a lycorine-type alkaloid from sternbergia sicula. J. Nat. Prod. 52, 1150–1152. doi: 10.1021/np50065a040
Sanchez, I. H., Larraza, M. I., Flores, H. J., Rojas, I., and Alcala, R. (1983). Sceletium (aizoaceae) alkaloids: total synthesis of racemic mesembranone, mesembrenone, O-methylsceletenone and O-methyl dihydrosceletenone. Synth. Commun. 13, 35–41. doi: 10.1080/00397918308061956
Schreiber, S. L. (1980). Fragmentation reactions of α-alkoxy hydroperoxides and application to the synthesis of the macrolide (±)-recifeiolide. J. Am. Chem. Soc. 102, 6163–6165. doi: 10.1021/ja00539a041
Schreiber, S. L., and Liew, W. (1985). Iron/copper promoted fragmentation reactions of α-alkoxy hydroperoxides. The conversion of octalins into fourteen-membered ring macrolides. J. Am. Chem. Soc. 107, 2980–2982. doi: 10.1021/ja00296a029
Stork, G., and Dolfini, J. E. (1963). The total synthesis of dl-aspidospermine and of dl-quebrachamine. J. Am. Chem. Soc. 85, 2872–2873. doi: 10.1021/ja00901a061
Takagi, S., Taylor, W. I., Uyeo, S., and Yajima, H. (1955). The site of the hydroxyl groups in lycorine. J. Chem. Soc. 4003–4007. doi: 10.1039/jr9550004003
Tamura, Y., Yakura, T., Haruta, J., and Kita, Y. (1987). Hypervalent iodine oxidative of p-alkoxyphenols and related compounds: a general route to p-benzoquinone monoacetals and spiro lactones. J. Org. Chem. 52, 3927–3930. doi: 10.1021/jo00226a041
Tang, J. M., Bhunia, S., Sohel, S. M. A., Lin, M. Y., Liao, H. Y., Datta, S., et al. (2007). The skeletal rearrangement of gold- and platinium-catalyzed cycloisomerization of cis-4,6-dien-1-yn-3-ols: pinacol rearrangement and formation of bicyclo[4.1.0]heptenone and reorganized styrene derivatives. J. Am. Chem. Soc. 129, 15677–15683. doi: 10.1021/ja076175r
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tiefenbacher, K., and Mulzer, J. (2007). Protecting-group-free formal synthesis of platensimycin. Angew. Chem. Int. Ed. 46, 8074–8075. doi: 10.1002/anie.200702852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tiefenbacher, K., and Mulzer, J. (2008). Synthesis of platensimycin. Angew. Chem. Int. Ed. 47, 2548–2555. doi: 10.1002/anie.200705303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tiefenbacher, K., Tröndlin, L., Mulzer, J., and Pfaltz, A. (2010). An expeditious asymmetric formal synthesis of the antibiotic platensimycin. Tetrahedron 66, 6508–6513. doi: 10.1016/j.tet.2010.04.098
Tokhtabaeva, G. M., Sheichenko, V. I., Yartseva, I. V., and Tolkachev, O. N. (1987). Structure of a new alkaloid from the fortune variety of narcissus. Khim. Prir. Soedin. 23, 727–730.
Tomioka, K., Koga, K., and Yamada, S. (1977). Stereochemical studies L-reductive decyanization of α-amino nitriles with sodium in liquid ammonia. An alternate method for the application to the asymmetric synthesis of optically active natural products. Chem. Pharm. Bull. 25, 2689–2691. doi: 10.1248/cpb.25.2689
Traoré, M., Ahmed-Ali, S., Peuchmaur, M., and Wong, Y. S. (2010). Hypervalent iodine(III)-mediated tandem oxidative reactions: application for the synthesis of bioactive polyspirocyclohexa-2,5-dienones. Tetrahedron 66, 5863–5872. doi: 10.1016/j.tet.2010.04.135
Ueda, Y., Iwahashi, K., Iguchi, K., and Ito, H. (2011). Enantioselective synthesis of the tetracyclic core of platensimycin. Synthesis 28, 1532–1536. doi: 10.1055/s-0030-1260002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walser, A., and Djerassi, C. (1965). Alkaloid-studen LII. die alkaloide aus vallesia dichotoma RUIZ et PAV. Helv. Chim. Acta. 48, 391–404. doi: 10.1002/hlca.19650480220
Wang, J., Soisson, S. M., Young, K., Shoop, W., Kodali, S., Galgoci, A., et al. (2006). Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 441, 358–361. doi: 10.1038/nature04784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wipf, P., and Kim, Y. (1992). Studies on the synthesis of stemona alkaloids; stereoselective preparation of the hydroindole ring system by oxidative cyclization of tyrosine. Tetrahedron Lett. 33, 5477–5480. doi: 10.1016/S0040-4039(00)61121-8
Wipf, P., Kim, Y., and Goldstein, D. (1995). Asymmetric total synthesis of the stemona alkaloid (–)-stenine. J. Am. Chem. Soc. 117, 11106–11112. doi: 10.1021/ja00150a010
Wipf, P., and Li, W. (1999). Formation of nine-membered lactams by oxidative ring expansion of 4-hydroxyhydroindoles: a biomimetic approach toward the tuberostemonone ring system. J. Org. Chem. 64, 4576–4577. doi: 10.1021/jo9905064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Wipf, P., and Mareska, D. A. (2000). Radical cleavage of a β-hydroxy azide: a reversal of regioselectivity in the oxidative fragmentation of hydroindoles. Tetrahedron Lett. 41, 4723–4727. doi: 10.1016/S0040-4039(00)00706-1
Wipf, P., and Methot, J. L. (2000). Total synthesis and stereochemical revision of (+)-aeruginosin 298-A. Org. Lett. 2, 4213–4216. doi: 10.1021/ol006759x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xing, S., Pan, W., Liu, C., Ren, J., and Wang, Z. (2010). Efficient construction of oxa- and aza-[n.2.1] skeletons: lewis acid catalyzed intramolecular [3+2] cycloaddition of cyclopropane 1,1-diesters with carbonyls and imines. Angew. Chem. Int. Ed. 49, 3215–3218. doi: 10.1002/anie.201000563
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamada, K. I., Yamashita, M., Sumiyoshi, T., Nishimura, K., and Tomioka, K. (2009). Total synthesis of (–)-lycorine and (–)-2-epi-lycorine by asymmetric conjugate addition cascade. Org. Lett. 11, 1631–1633. doi: 10.1021/ol9003564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yoshida, K., Sakuma, Y., and Ban, Y. (1987). Synthetic studies on oxygenated aspidosperma alkaloids: facile syntheses of 1-acetylaspidoalbidine and deoxyaspidodispermine. Heterocycles 25, 47–50. doi: 10.3987/S-1987-01-0047
Yoshimura, A., Middleton, K. R., Luedtke, M. W., Zhu, C., and Zhdankin, V. V. (2012). Hypervalent iodine catalyzed Hofmann rearrangement of carboxamides using oxone as terminal oxidant. J. Org. Chem. 77, 11399–11404. doi: 10.1021/jo302375m
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yoshimura, A., Middleton, K. R., Todora, A. D., Kastern, B. J., Koski, S. R., Maskaev, A. V., et al. (2013). Hypervalent iodine catalyzed generation of nitrile oxides from oximes and their cycloaddition with alkenes or alkynes. Org. Lett. 15, 4010–4013. doi: 10.1021/ol401815n
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yun, S. Y., Zheng, J. C., and Lee, D. (2009). Stereoelectronic effect for the selectivity in C-H insertion of alkylidene carbenes and its application to the synthesis of platensimycin. J. Am. Chem. Soc. 131, 8413–8415. doi: 10.1021/ja903526g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, S. X., Shen, X. L., Li, Z. Q., Zou, L. W., Wang, F. Q., Zhang, H. B., et al. (2013). Enantioselective total synthesis of (–)-limaspermidine and formal synthesis of (–)-1-acetylaspidoalbidine. J. Org. Chem. 78, 11444–11449. doi: 10.1021/jo402004f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zheng, J. C., Yun, S. Y., Sun, C., Lee, N. K., and Lee, D. (2011). Selectivity control in alkylidene carbene-mediated C-H insertion and allene formation. J. Org. Chem. 76, 1086–1099. doi: 10.1021/jo102180f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zou, Y., Chen, C. H., Taylor, C. D., Foxman, B. M., and Snider, B. B. (2007). Formal synthesis of (±)-platensimycin. Org. Lett. 9, 1825–1828. doi: 10.1021/ol070563g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: total synthesis, hypervalent iodine, alkaloids, dienone, natural products
Citation: Maertens G, L'Homme C and Canesi S (2015) Total synthesis of natural products using hypervalent iodine reagents. Front. Chem. 2:115. doi: 10.3389/fchem.2014.00115
Received: 03 November 2014; Paper pending published: 01 December 2014;
Accepted: 10 December 2014; Published online: 05 January 2015.
Edited by:
Stellios Arseniyadis, Université Louis Pasteur, FranceCopyright © 2015 Maertens, L'Homme and Canesi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvain Canesi, Laboratoire de Méthodologie et Synthèse de Produits Naturels, Department of Chemistry, Université du Québec à Montréal, C.P. 8888, Succ. Centre-Ville, Montréal, QC H3C 3P8, Canada e-mail:Y2FuZXNpLnN5bHZhaW5AdXFhbS5jYQ==