 Haruhiko Fuwa
Haruhiko Fuwa Ryo Fukazawa
Ryo Fukazawa Makoto Sasaki
Makoto Sasaki- Graduate School of Life Sciences, Tohoku University, Sendai, Japan
Gambieric acid A (GAA) and its congeners belong to the family of marine polycyclic ether natural products. Their highly complex molecular architecture and unique biological activities have been of intense interest within the synthetic community. We have previously reported the first total synthesis, stereochemical reassignment, and preliminary structure–activity relationships of GAA. Here we disclose a concise synthesis of the A/BCD-ring fragment of GAA. The synthesis started from our previously reported synthetic intermediate that represents the A/B-ring. The C-ring was synthesized via an oxiranyl anion coupling and a 6-endo cyclization, and the D-ring was forged by means of an oxidative lactonization and subsequent palladium-catalyzed functionalization of the lactone ring. In this manner, the number of linear synthetic steps required for the construction of the C- and D-rings was reduced from 22 to 11.
Introduction
In 1992, Nagai, Yasumoto, and co-workers reported the isolation of gambieric acid A (GAA, 1) and its natural congeners, gambieric acids B–D (GAB–GAD, Figure 1) (Nagai et al., 1992a,b). Gambieric acids (GAs) are the secondary metabolites of the ciguatera causative dinoflagellate Gambierdiscus toxicus and belong to the family of marine polycyclic ether natural products (Yasumoto and Murata, 1993; Murata and Yasumoto, 2000). The gross structure and the relative configuration of the polycyclic ether region of GAs were determined on the basis of extensive 2D NMR experiments. The complete stereochemical assignment of GAs was subsequently made through conformational analysis of GAB on the basis of nuclear Overhauser effect (NOE) correlations coupled with 3JH,H values, application of chiral anisotropic reagents, and chiral HPLC analysis of degradation products (Morohashi et al., 2000). However, our synthesis and NMR spectroscopic analysis of a series of suitably designed A/B-ring model compounds of GAs strongly indicated that the absolute configuration of the polycyclic ether domain of GAs needs to be unambiguously established through total synthesis (Fuwa et al., 2008a, 2009a). The trans-fused polycyclic ether backbone of GAs is the common structural characteristic shared among the family of marine polycyclic ether neurotoxins, e.g., brevetoxins, ciguatoxins, and gambierol. Nonetheless, it has been reported that GAA shows only moderate toxicity against mice or cultured mammalian cells (Nagai et al., 1992b) and only weakly displaces binding of tritiated dihydrobrevetoxin B ([3H]-PbTx-3) to voltage-gated sodium channels (Inoue et al., 2003). Instead, GAs are known to impart extraordinary potent antifungal activity against Aspergillus niger, which is approximately 2000 times greater than that of amphotericin B (Nagai et al., 1993). In addition, it has been described that GAA is a possible endogenous growth-regulating factor of G. toxicus (Sakamoto et al., 1996). Unfortunately, the molecular basis for the biological activities of GAs has not been elucidated at all, partly due to the natural scarcity of these substances. The molecular complexity and intriguing biological activities of GAs have attracted the attention of the synthetic community (Kadota et al., 2001a,b; Clark et al., 2004, 2005; Sato and Sasaki, 2005, 2007; Fuwa et al., 2007, 2008a, 2009a,b, 2010; Roberts and Rainier, 2007; Saito and Nakata, 2009; Tsubone et al., 2011a,b).
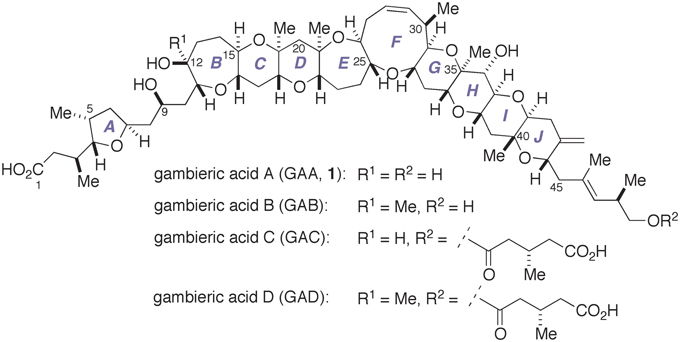
Figure 1. Structures of gambieric acids A–D.
We have recently completed the first total synthesis of GAA to establish its absolute configuration as that shown by 1 (Fuwa et al., 2012; Ishigai et al., 2013; Sasaki and Fuwa, 2014). Our synthesis entailed convergent assembly of the A/BCD- and F′GHIJ-ring fragments, i.e., 2 and 3, respectively, by means of Suzuki–Miyaura coupling (Miyaura and Suzuki, 1995; Sasaki and Fuwa, 2008; Suzuki, 2011) to give the endocyclic enol ether 4, followed by closure of the E- and F-rings via a stereoselective allylation of a thioacetal (Suga et al., 2014) and a ring-closing metathesis (Hoveyda and Zhugralin, 2007), respectively, to construct the nonacyclic polyether core 5 (Figure 2). Moreover, we have prepared several synthetic analogs of GAA by diversifying the synthetic route from the nonacyclic ether 5 and investigated the structure–activity relationships (SARs) of the peripheral substituents on the polycyclic ether skeleton (Ishigai et al., 2013). Toward the elucidation of the SARs of GAA in greater detail, however, it deemed indispensable to improve the synthetic availability of 2 and 3. Here we describe a concise synthesis of the A/BCD-ring fragment 2 of GAA, wherein the C-ring was constructed by using an oxiranyl anion coupling/6-endo cyclization sequence (Mori et al., 1997a,b, 1998) and the D-ring was forged via an oxidative lactonization and subsequent palladium-catalyzed functionalization of the derived lactone.
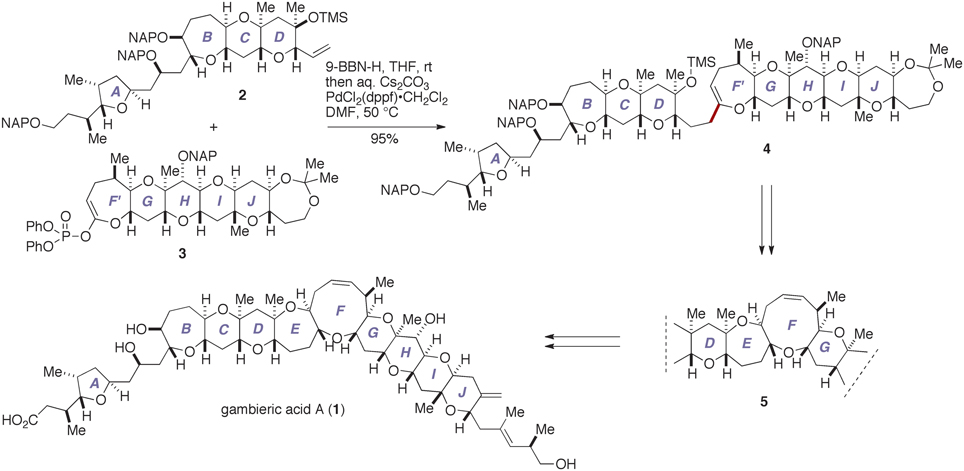
Figure 2. Outline of our total synthesis of gambieric acid A. aq., aqueous; 9-BBN-H, 9-borabicyclo[3.3.1]nonane; DMF, N,N-dimethylformamide; dppf, 1,1′-bis(diphenylphosphino)ferrocene; NAP, 2-naphthylmethyl; rt, room temperature; THF, tetrahydrofuran; TMS, trimethylsilyl.
Materials and Methods
Detailed experimental procedure and compound characterization data are furnished in the Supplementary Material.
Results and Discussion
As delineated in Figure 3, our previous synthesis of 2 (Fuwa et al., 2012; Ishigai et al., 2013) relied upon Suzuki–Miyaura coupling of an alkylborane prepared in situ from the A/B-ring exocyclic enol ether 6 with the enol phosphate 7, followed by ring-closing metathesis of the derived enol ether (Fuwa and Sasaki, 2008b). The closure of the C-ring was achieved by means of stereoselective methylation of the thioacetal 9 (Nicolaou et al., 1989; Fuwa et al., 2001), and subsequent elaboration of the D-ring completed the synthesis of 2. Although sufficient quantities of 2 for the total synthesis could actually be prepared, the synthetic sequence from 6 to 2 was rather lengthy (19 steps), partly because multiple steps were required for the introduction of the 1,3-diaxial methyl groups onto the D-ring.
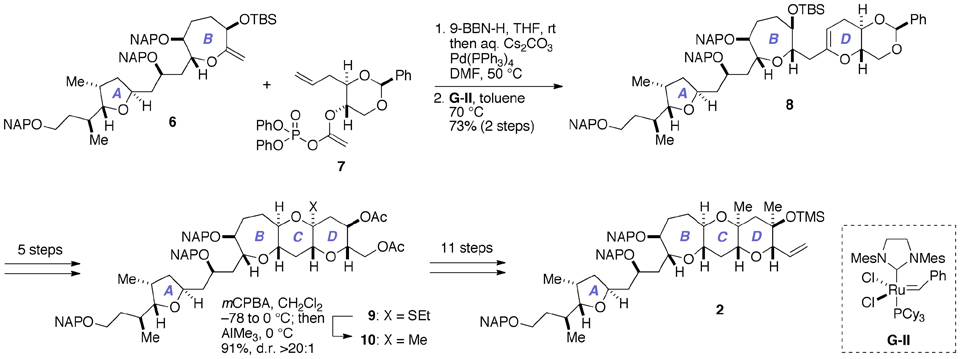
Figure 3. Outline of our previous synthesis of the A/BCD-ring fragment 2 of gambieric acid A. Cy, cyclohexyl; mCPBA, m-chloroperoxybenzoic acid; d.r., diastereomer ratio; Mes, 2,4,6-trimethylphenyl (mesityl); TBS, t-butyldimethylsilyl.
With our previous synthesis in mind, we devised an improved synthesis of 2, which is outlined in Figure 4. Currently, a number of synthetic methods are available for the synthesis of tetrahydropyran derivatives (Nasir et al., 2014). We envisioned that the C-ring could be efficiently constructed in a concise manner by exploiting the chemistry developed by Mori et al. (1997a,b, 1998). Thus, a coupling of the triflate 11, which represents the A/B-ring, with an oxiranyl anion generated from the epoxy sulfone 12, followed by acid-catalyzed cleavage of the silyl ether and spontaneous 6-endo cyclization would directly afford the A/BC-ring tricycle 13. Meanwhile, the oxiranyl anion chemistry cannot be directly applied to the D-ring with 1,3-diaxial methyl groups. Accordingly, we planned to construct the D-ring via the lactone 14. Functionalization of lactones is a versatile means for the synthesis of cyclic ethers (e.g., Nicolaou et al., 1997; Suga et al., 2014). A palladium-catalyzed vinylation of an enol phosphate or triflate derived from 14 would give the diene 15. Chemo- and stereoselective epoxidation of 15 and subsequent stereoselective reduction of the resultant epoxide would allow a rapid access to the targeted 2.
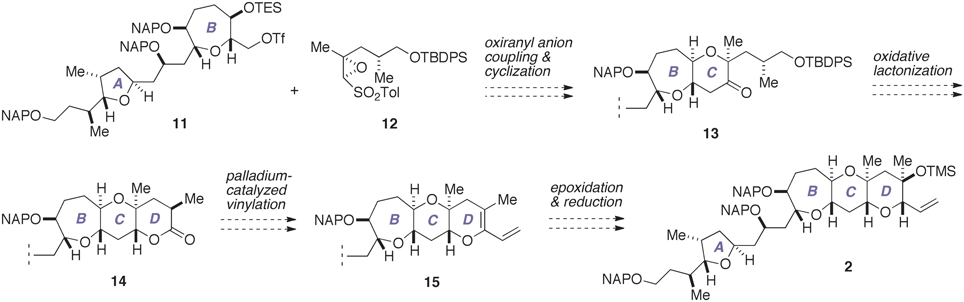
Figure 4. Improved synthesis plan toward 2. TBDPS, t-butyldiphenylsilyl; TES, triethylsilyl; Tf, trifluoromethanesulfonate (triflate); Tol, p-tolyl.
Initially, we prepared the epoxy sulfone 12 and examined its use in a model system (Figure 5). The synthesis of 12 started with the known methyl ketone 16 (Edmunds et al., 1997). Coupling of 16 with a lithiated sulfoxide generated in situ from 17 (Satoh et al., 1989; Mori et al., 1998) provided the chlorohydrins 18a (36%) and 18b (46%) as a separable mixture. The minor diastereomer 18a was treated with a base and then oxidized with mCPBA to afford the epoxy sulfone 12 (89%, two steps). At this stage, however, we were unable to establish the absolute configuration of the newly introduced stereogenic centers of 12. Accordingly, we reacted an oxiranyl anion prepared from 12 with the triflate 21 as a model experiment. The triflate 21 was readily prepared from the known alcohol 19 (Inoue et al., 1999) in three steps, including silylation, ozonolysis/NaBH4 reduction, and triflation. Treatment of a mixture of 12 and 21 with n-BuLi in THF/HMPA at −100°C cleanly provided the desired coupling product 22 (95%). Exposure of 22 to TsOH·H2O in CHCl3 at 55°C resulted in cleavage of the TES ether and spontaneous 6-endo cyclization, as expected, to afford the ketone 23 in 93% yield as a single stereoisomer (d.r. >20:1). Here we were able to establish the stereostructure of 23 by an NOE experiment as shown, thus confirmed the absolute configuration of the epoxy sulfone 12.
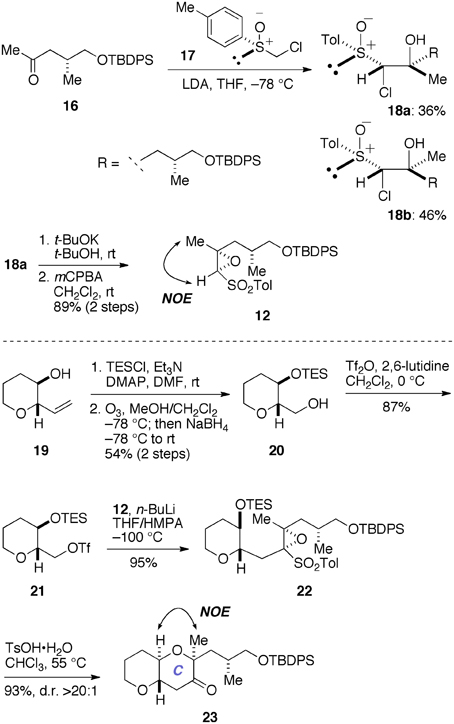
Figure 5. Synthesis of epoxy sulfone 12 and a model study for the construction of the C-ring. DMAP, 4-(dimethylamino)pyridine; HMPA, hexamethylphosphoramide; LDA, lithium diisopropylamide; NOE, nuclear Overhauser effect; TsOH, p-toluenesulfonic acid.
With the requisite epoxy sulfone 12 available, we proceeded to construct the C-ring in the real system, as shown in Figure 6. Sequential triflation/silylation (Mori et al., 1997a) of the AB-ring diol 24 (Fuwa et al., 2012; Ishigai et al., 2013) gave the triflate 11. This was immediately coupled with an oxiranyl anion generated from 12 under the same conditions employed above (n-BuLi, THF/HMPA, −100°C) to afford the coupling product 25. Subsequent treatment of 25 with TsOH·H2O in CHCl3 at 0°C led to the ketone 13 in 76% overall yield from 24. Stereoselective reduction of 13 with NaBH4 afforded the alcohol 26 (96%, d.r. >20:1). The absolute configuration of the C18 and C19 stereogenic centers was confirmed by NOE experiments, as shown. Thus, we successfully elaborated the C-ring in only four steps from 24.
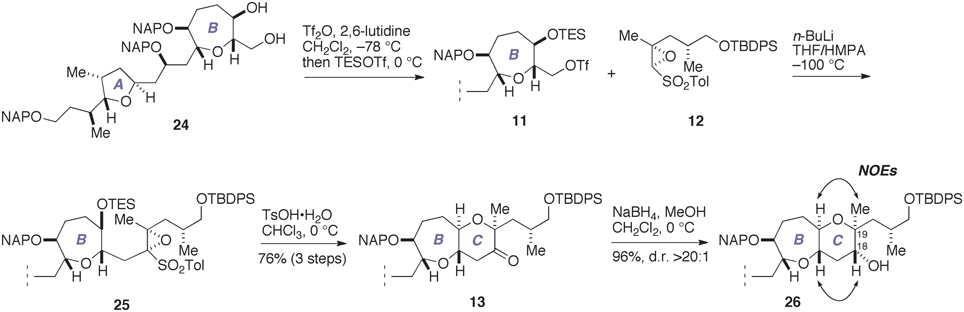
Figure 6. Construction of the C-ring.
Next, we investigated the construction of the D-ring, as shown in Figure 7. Removal of the silyl group from 26 with TBAF gave the diol 27 (92%), which was oxidized with TEMPO/PhI(OAc)2 (Hansen et al., 2003) to directly afford the lactone 14 (92%).
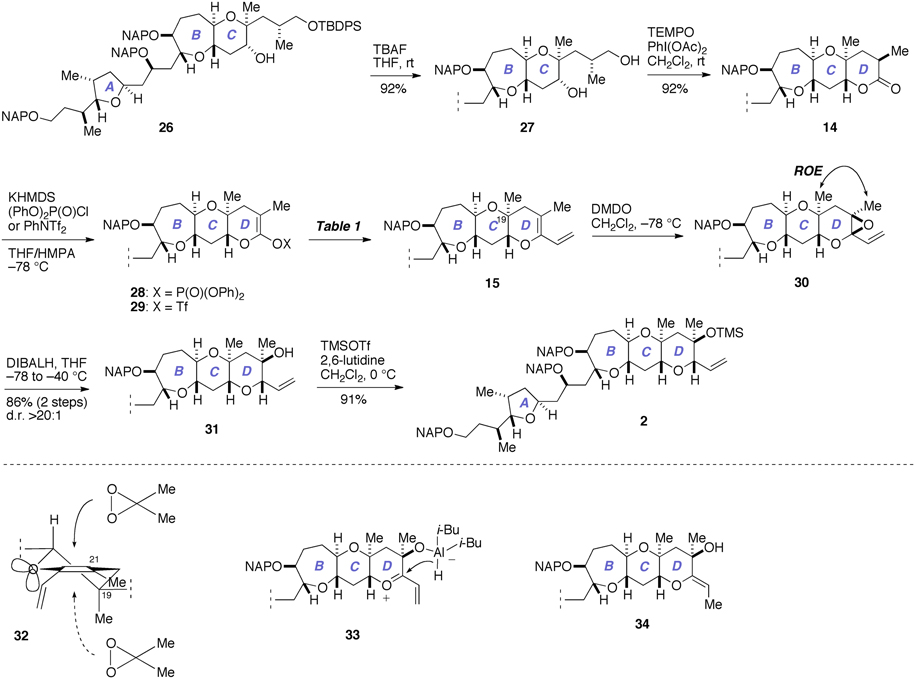
Figure 7. Construction of the D-ring and completion of the synthesis of 2. DIBALH, diisobutylaluminum hydride; DMDO, dimethyldioxirane; KHMDS, potassium bis(trimethylsilyl)amide; ROE, rotating-frame Overhauser effect; TBAF, tetra-n-butylammonium fluoride; TEMPO, 2,2,6,6-tetramethylpiperidin-1-oxyl.
We investigated the functionalization of the lactone ring of 14 to elaborate the D-ring. Exposure of 14 to KHMDS in the presence of (PhO)2P(O)Cl smoothly provided the enol phosphate 28 (Nicolaou et al., 1997). Initially, we examined the palladium-catalyzed vinylation of 28 under Suzuki–Miyaura conditions (Miyaura and Suzuki, 1995; Suzuki, 2011), as summarized in Table 1. Treatment of 28 with vinylboronic acid pinacol ester under the influence of aqueous Cs2CO3 solution and PdCl2(dppf)·CH2Cl2 catalyst, however, did not give the diene 15 at all and only returned the enol phosphate 28 (entry 1). Changing the catalyst to Pd(PPh3)4 was also ineffective (entry 2). We suspected that the low reactivity of the enol phosphate 28 would stem from the steric bulk of the α-methyl group (e.g., Nicolaou et al., 1997). Thus, we also prepared the enol triflate 29 (Tsushima et al., 1989) as a more reactive surrogate. Because our previous studies have shown that highly reactive enol triflates favor palladium catalyst with electron deficient supporting ligands (Sasaki et al., 1998, 2002), we examined Suzuki–Miyaura coupling of 29 with vinylboronic acid pinacol ester under the influence of the Pd2(dba)3/Ph3As catalyst system (entries 3 and 4). To our dismay, we isolated 15 in only moderate yields under these conditions. These unsatisfactory results could be ascribed to undesirable hydrolysis of 29 under alkaline conditions. Accordingly, we turned our attention to Stille coupling of 29 with vinyl(tri-n-butyl)stannane by the action of Pd(PPh3)4 catalyst and LiCl in 1,4-dioxane at 80°C (Scott and Stille, 1986) (entry 5). Under these conditions, we were able to isolate the diene 15 in 63% overall yield from 14. Here it was necessary to purify the diene 15 by aqueous 20% KF and DL-serine workup and by flash column chromatography using potassium carbonate–silica gel to scavenge organotin byproducts and palladium salts (Leibner and Jacobus, 1979; Harrowven et al., 2010; Yoshimura et al., 2011), as traces of these weakly Lewis acidic contaminants were found to adversely affect the outcome of subsequent epoxidation process.
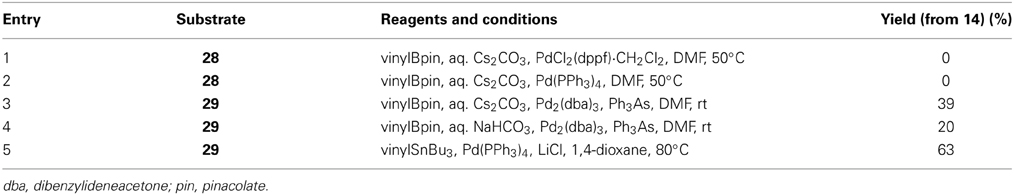
Table 1. Examination of palladium-catalyzed vinylation of enol phosphate 28 and triflate 29.
Our final task was to elaborate the diene 15 to the A/BCD-ring fragment 2 via chemo- and stereoselective epoxidation of 15 and subsequent reductive opening of the derived epoxide 30 (Figure 7). Thus, treatment of 15 with DMDO in CH2Cl2 at −78°C provided the epoxide 30 as a single stereoisomer (d.r. >20:1, judged by 1H NMR analysis). This epoxide was isolated by aqueous workup and immediately reduced with DIBALH in THF at −78 to −40°C to afford the tertiary alcohol 31 in 86% yield (two steps). The chemoselectivity of the epoxidation of 15 was secured by the differential reactivity of the enol ether and the terminal olefin (Fujiwara et al., 1999; Clark et al., 2007). The stereochemical outcome of the epoxidation of 15 with DMDO was in accordance with that of glycal derivatives (Halcomb and Danishefsky, 1989; Allwein et al., 2002) and could be reasoned by considering stereoelectronic effect as well as the steric bulk of the axial methyl group at the C19 position (e.g., 32). The purity of the diene 15 was crucial for the success of the epoxidation; when 15 containing traces of organotin byproducts and/or palladium salts was used, in situ hydrolysis of the epoxide 30 with traces of adventitious H2O occurred as a serious side reaction. Meanwhile, the stereoselectivity of the DIBALH reduction of the epoxide 30 could be explained by considering the aluminum ate complex 33 as the intermediate, as previously proposed by Majumder et al. (2006). Our initial attempts to reduce 30 with DIBALH in CH2Cl2 at −78°C resulted in only 19% yield of the tertiary alcohol 31 and the exocyclic enol ether 34 was isolated alongside in 44% yield. The undesired product 34 might arise from an SN2′-type reduction of 33. Consequently, we chose to perform the reduction in THF to reduce the Lewis acidity of DIBALH as well as to solvate the presumed oxocarbenium ion intermediate 33. Other reducing conditions, such as Et3SiH/BF3·OEt2 (Clark et al., 2007) or NaBH3CN (Zimmermann et al., 2000), gave unsatisfactory results. Finally, silylation of 31 with TMSOTf/2,6-lutidine afforded the A/BCD-ring fragment 2 in 91% yield.
Conclusions
In this paper, we described a concise synthesis of the A/BCD-ring fragment 2 of GAA, which is significantly improved over our previous synthesis in terms of “step economy” (Wender et al., 2008). Starting from the A/B-ring diol 24, the C-ring was rapidly constructed by means of an oxiranyl anion coupling and subsequent 6-endo cyclization. The D-ring was first forged as a six-membered lactone and further elaborated via a Stille coupling. The present synthesis minimized the use of protecting group chemistry and enabled rapid synthesis of 2 from 24 in just 11 linear steps, which compares favorably with our previously reported synthesis (22 linear steps from 24).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research (A) (No. 21241050), a Grant-in-Aid for Young Scientists (A) (No. 23681045), and a Grant-in-Aid for Scientific Research on Innovative Areas (No. 26102708) from the Japan Society for Promotion of Science (JSPS).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fchem.2014.00116/abstract
References
Allwein, S. P., Cox, J. M., Howard, B. E., Johnson, H. W. B., and Rainier, J. D. (2002). C-Glycosides to fused polycyclic ethers. Tetrahedron 58, 1997–2009. doi: 10.1016/S0040-4020(02)00057-1
Clark, J. S., Conroy, J., and Blake, A. J. (2007). Rapid synthesis of the A–E fragment of ciguatoxin CTX3C. Org. Lett. 9, 2091–2094. doi: 10.1021/ol0706096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clark, J. S., Fessard, T. C., and Wilson, C. (2004). A concise and stereoselective synthesis of the A-ring fragment of gambieric acids. Org. Lett. 6, 1773–1776. doi: 10.1021/ol049483s
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Clark, J. S., Kimber, M. C., Robertson, J., McErlean, C. S., and Wilson, C. (2005). Rapid two-directional synthesis of the F–J fragment of gambieric acids by iterative double ring-closing metathesis. Angew. Chem. Int. Ed. Engl. 44, 6157–6162. doi: 10.1002/anie.200501925
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edmunds, A. J. F., Trueb, W., Oppolzer, W., and Cowley, P. (1997). Herboxidiene: determination of absolute configuration by degradation and synthetic studies. Tetrahedron 53, 2785-2802. doi: 10.1016/S0040-4020(97)00021-5
Fujiwara, K., Awakura, D., Tsunashima, M., Nakamura, A., Honma, T., and Murai, A. (1999). Total synthesis of (+)-obtusenyne. J. Org. Chem. 64, 2616–2617. doi: 10.1021/jo990212i
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fuwa, H., Goto, T., and Sasaki, M. (2008a). Stereocontrolled synthesis of the A/B-ring fragment of gambieric acid B: reassignment of the absolute configuration of the polycyclic ether region. Org. Lett. 10, 2211–2214. doi: 10.1021/ol800642t
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fuwa, H., Ishigai, K., Goto, T., Suzuki, A., and Sasaki, M. (2009a). Synthetic studies on gambieric acids, potent antifungal polycyclic ether natural products: reassignment of the absolute configuration of the nonacyclic polyether core by NMR analysis of model compounds. J. Org. Chem. 74, 4024–4040. doi: 10.1021/jo900332q
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fuwa, H., Ishigai, K., Hashizume, K., and Sasaki, M. (2012). Total synthesis and complete stereostructure of gambieric acid A. J. Am. Chem. Soc. 134, 11984–11987. doi: 10.1021/ja305864z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fuwa, H., Noji, S., and Sasaki, M. (2009b). Stereocontrolled synthesis of the DEFG-ring skeleton of gambieric acids. Chem. Lett. 38, 866–867. doi: 10.1246/cl.2009.866
Fuwa, H., Noji, S., and Sasaki, M. (2010). Studies toward the total synthesis of gambieric acids: stereocontrolled synthesis of a DEFG-ring model compound. J. Org. Chem. 75, 5072–5082. doi: 10.1021/jo1008146
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fuwa, H., and Sasaki, M. (2008b). An efficient strategy for the synthesis of endocyclic enol ethers and its application to the synthesis of spiroacetals. Org. Lett. 10, 2549–2552. doi: 10.1021/ol800815t
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fuwa, H., Sasaki, M., and Tachibana, K. (2001). Synthetic studies on a marine polyether toxin, gambierol: stereoselective synthesis of the EFGH ring system via B-alkyl Suzuki coupling. Tetrahedron 57, 3019–3033. doi: 10.1016/S0040-4020(01)00164-8
Fuwa, H., Suzuki, A., Sato, K., and Sasaki, M. (2007). Stereoselective synthesis of the AB-ring fragment of gambieric acid A. Heterocycles 72, 139–144. doi: 10.3987/COM-06-S(K)30
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Halcomb, R. L., and Danishefsky, S. J. (1989). On the direct epoxidation of glycals: application of a reiterative strategy for the synthesis of β-linked oligosaccharides. J. Am. Chem. Soc. 111, 6661–6666. doi: 10.1021/ja00199a028
Hansen, T. M., Florence, G. J., Lugo-Mas, P., Chen, J., Abrams, J. N., and Forsyth, C. J. (2003). Highly chemoselective oxidation of 1,5-diols to δ-lactones with TEMPO/BAIB. Tetrahedron Lett. 44, 57–59. doi: 10.1016/S0040-4039(02)02489-9
Harrowven, D. C., Curran, D. P., Kostiuk, S. L., Wallis-Guy, I. L., Whiting, S., Stenning, K. J., et al. (2010). Potassium carbonate–silica: a highly effective stationary phase for the chromatographic removal of organotin impurities. Chem. Commun. 46, 6335–6337. doi: 10.1039/c0cc01328e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoveyda, A. H., and Zhugralin, A. R. (2007). The remarkable metal-catalysed olefin metathesis reaction. Nature 450, 243–251. doi: 10.1038/nature06351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inoue, M., Hirama, M., Satake, M., Sugiyama, K., and Yasumoto, T. (2003). Inhibition of brevetoxin binding to the voltage-gated sodium channel by gambierol and gambieric acid-A. Toxicon 41, 469–474. doi: 10.1016/S0041-0101(02)00369-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Inoue, M., Sasaki, M., and Tachibana, K. (1999). A convergent synthesis of the trans-fused hexahydrooxonine ring system and reproduction of conformational behavior shown by ring F of ciguatoxin. Tetrahedron 55, 10949–10970. doi: 10.1016/S0040-4020(99)00620-1
Ishigai, K., Fuwa, H., Hashizume, K., Fukazawa, R., Cho, Y., Yotsu-Yamashita, M., et al. (2013). Total synthesis and biological evaluation of (+)-gambieric acid A and its analogues. Chem. Eur. J. 19, 5276–5288. doi: 10.1002/chem.201204303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kadota, I., Oguro, N., and Yamamoto, Y. (2001a). Synthesis of the A ring segment of gambieric acid. Tetrahedron Lett. 42, 3645–3647. doi: 10.1016/S0040-4039(01)00529-9
Kadota, I., Takamura, H., and Yamamoto, Y. (2001b). Synthesis of the J ring segment of gambieric acid. Tetrahedron Lett. 42, 3649–3651. doi: 10.1016/S0040-4039(01)00530-5
Leibner, J. E., and Jacobus, J. (1979). Facile product isolation from organostannane reductions of organic halides. J. Org. Chem. 44, 449–450. doi: 10.1021/jo01317a032
Majumder, U., Cox, J. M., Johnson, H. W. B., and Rainier, J. D. (2006). Total synthesis of gambierol: the generation of the A–C and F–H subunits by using a C-glycoside centered strategy. Chem. Eur. J. 12, 1736–1746. doi: 10.1002/chem.200500993
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miyaura, N., and Suzuki, A. (1995). Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 95, 2457–2483. doi: 10.1021/cr00039a007
Mori, Y., Yaegashi, K., and Furukawa, H. (1997a). Oxiranyl anions in organic synthesis: application to the synthesis of hemibrevetoxin B. J. Am. Chem. Soc. 119, 4557–4558.
Mori, Y., Yaegashi, K., and Furukawa, H. (1997b). Stereoselective synthesis of the 6,7,6- and 6,7,7-ring systems of polycyclic ethers by 6-endo cyclization and ring expansion. Tetrahedron 53, 12917–12932.
Mori, Y., Yaegashi, K., and Furukawa, H. (1998). Formal total synthesis of hemibrevetoxin B by an oxiranyl anion strategy. J. Org. Chem. 63, 6200–6209. doi: 10.1021/jo980320p
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morohashi, A., Satake, M., Nagai, H., Oshima, Y., and Yasumoto, T. (2000). The absolute configuration of gambieric acids A–D, potent antifungal polyethers, isolated from the marine dinoflagellate Gambierdiscus toxicus. Tetrahedron 56, 8995–9001. doi: 10.1016/S0040-4020(00)00753-5
Murata, M., and Yasumoto, T. (2000). The structure elucidation and biological activities of high molecular weight algal toxins: maitotoxin, prymnesins and zooxanthellatoxins. Nat. Prod. Rep. 17, 293–314. doi: 10.1039/a901979k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagai, H., Mikami, Y., Yazawa, K., Gonoi, T., and Yasumoto, T. (1993). Biological activities of novel polyether antifungals, gambieric acids A and B from a marine dinoflagellate Gambierdiscus toxicus. J. Antibiot. 46, 520–522. doi: 10.7164/antibiotics.46.520
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagai, H., Murata, M., Torigoe, K., Satake, M., and Yasumoto, T. (1992b). Gambieric acids, new potent antifungal substances with unprecedented polyether structures from a marine dinoflagellate Gambierdiscus toxicus. J. Org. Chem. 57, 5448–5453.
Nagai, H., Torigoe, K., Satake, M., Murata, M., Yasumoto, T., and Hirota, H. (1992a). Gambieric acids: unprecedented potent antifungal substances isolated from cultures of a marine dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 114, 1102–1103.
Nasir, N. M., Ermanis, K., and Clarke, P. A. (2014). Strategies for the construction of tetrahydropyran rings in the synthesis of natural products. Org. Biomol. Chem. 12, 3323–3335. doi: 10.1039/c4ob00423j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nicolaou, K. C., Prasad, C. V. C., Hwang, C. K., Duggan, M. E., and Veale, C. A. (1989). Cyclizations of hydroxy dithioketals. New synthetic technology for the construction of oxocenes and related medium-ring systems. J. Am. Chem. Soc. 111, 5321–5330. doi: 10.1021/ja00196a042
Nicolaou, K. C., Shi, G.-Q., Gunzner, J. L., Gärtner, P., and Yang, Z. (1997). Palladium-catalyzed functionalization of lactones via their cyclic ketene acetal phosphates. Efficient new synthetic technology for the construction of medium and large cyclic ethers. J. Am. Chem. Soc. 119, 5467–5468. doi: 10.1021/ja970619+
Roberts, S. W., and Rainier, J. D. (2007). Synthesis of an A–E gambieric acid subunit with use of a C-glycoside centered strategy. Org. Lett. 9, 2227–2230. doi: 10.1021/ol0707970
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saito, T., and Nakata, T. (2009). Stereoselective synthesis of trans-fused 7,6,6,7-membered tetracyclic ether, corresponding to the EFGH-ring of gambierol and the BCDE-ring of gambieric acids. Org. Lett. 11, 113–116. doi: 10.1021/ol8024555
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sakamoto, B., Nagai, H., and Hokama, Y. (1996). Stimulators of Gambierdiscus toxicus (Dinophyceae) growth: the possible role of gambieric acid-A as an endogenous growth enhancer. Phycologia 35, 350–353. doi: 10.2216/i0031-8884-35-4-350.1
Sasaki, M., and Fuwa, H. (2008). Convergent strategies for the total synthesis of polycyclic ether metabolites. Nat. Prod. Rep. 25, 401–426. doi: 10.1039/b705664h
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sasaki, M., and Fuwa, H. (2014). Total synthesis and complete structural assignment of gambieric acid A, a large polycyclic ether marine natural product. Chem. Rec. 14, 678–703. doi: 10.1002/tcr.201402052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sasaki, M., Fuwa, H., Inoue, M., and Tachibana, K. (1998). New strategy for convergent synthesis of trans-fused polyether frameworks based on palladium-catalyzed Suzuki cross-coupling reaction. Tetrahedron Lett. 39, 9027–9030. doi: 10.1016/S0040-4039(98)02025-5
Sasaki, M., Ishikawa, M., Fuwa, H., and Tachibana, K. (2002). A general strategy for the convergent synthesis of fused polycyclic ethers via B-alkyl Suzuki coupling: synthesis of the ABCD ring fragment of ciguatoxins. Tetrahedron 58, 1889–1911. doi: 10.1016/S0040-4020(02)00045-5
Sato, K., and Sasaki, M. (2005). Studies toward the total synthesis of gambieric acids, potent antifungal polycyclic ethers: convergent synthesis of the CDEFG-ring system. Org. Lett. 7, 2441–2444. doi: 10.1021/ol050760k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sato, K., and Sasaki, M. (2007). Studies toward the total synthesis of gambieric acids A and C: convergent assembly of the nonacyclic polyether skeleton. Angew. Chem. Int. Ed. Engl. 46, 2518–2522. doi: 10.1002/anie.200604625
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Satoh, T., Oohara, T., Ueda, Y., and Yamakawa, K. (1989). A novel approach to the asymmetric synthesis of epoxides, allylic alcohols, α-amino ketones, and α-amino aldehydes from carbonyl compounds through α,β-epoxy sulfoxides using the optically active p-tolylsulfinyl group to induce chirality. J. Org. Chem. 54, 3130–3136. doi: 10.1021/jo00274a032
Scott, W. J., and Stille, J. K. (1986). Palladium-catalyzed coupling of vinyl triflates with organostannanes. Synthetic and mechanistic studies. J. Am. Chem. Soc. 108, 3033–3040. doi: 10.1021/ja00271a037
Suga, Y., Fuwa, H., and Sasaki, M. (2014). Stereoselective synthesis of medium-sized cyclic ethers: application of C-glycosylation chemistry to seven- to nine-membered lactone-derived thioacetals and their sulfone counterparts. J. Org. Chem. 79, 1656–1682. doi: 10.1021/jo4025545
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Suzuki, A. (2011). Cross-coupling reactions of organoboranes. An easy way to construct C–C bonds (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 50, 6722–6737. doi: 10.1002/anie.201101379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsubone, K., Hashizume, K., Fuwa, H., and Sasaki, M. (2011a). Studies toward the total synthesis of gambieric acids: convergent synthesis of the GHIJ-ring fragment having a side chain. Tetrahedron Lett. 52, 548–551. doi: 10.1016/j.tetlet.2010.11.127
Tsubone, K., Hashizume, K., Fuwa, H., and Sasaki, M. (2011b). Studies toward the total synthesis of gambieric acids, potent antifungal polycyclic ethers: convergent synthesis of a fully elaborated GHIJ-ring fragment. Tetrahedron 67, 6600–6615. doi: 10.1016/j.tet.2011.05.082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsushima, K., Araki, K., and Murai, A. (1989). Conversion of lactones into substituted cyclic ethers. Chem. Lett. 1313–1316.
Wender, P. A., Verma, V. A., Paxton, T. J., and Pillow, T. H. (2008). Function-oriented synthesis, step economy, and drug design. Acc. Chem. Res. 41, 40–49. doi: 10.1021/ar700155p
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yasumoto, T., and Murata, M. (1993). Marine toxins. Chem. Rev. 93, 1897–1909. doi: 10.1021/cr00021a011
Yoshimura, F., Takahashi, Y., Tanino, K., and Miyashita, M. (2011). Synthetic studies of the zoanthamine alkaloids: total synthesis of zoanthenol based on an isoaromatization strategy. Chem. Asian J. 6, 922–931. doi: 10.1002/asia.201000552
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zimmermann, P. J., Blanarikova, I., and Jäger, V. (2000). A general approach to L-(+)-furanomycin and some stereoisomers and analogues using furoisooxazoline intermediates. Angew. Chem. Int. Ed. Engl. 39, 910–912. doi: 10.1002/(SICI)1521-3773(20000303)39:5<910::AID-ANIE910>3.0.CO;2-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: marine polycyclic ethers, oxiranyl anions, 6-endo cyclization, oxidative lactonization, palladium-catalyzed reactions
Citation: Fuwa H, Fukazawa R and Sasaki M (2015) Concise synthesis of the A/BCD-ring fragment of gambieric acid A. Front. Chem. 2:116. doi: 10.3389/fchem.2014.00116
Received: 17 November 2014; Paper pending published: 16 December 2014;
Accepted: 18 December 2014; Published online: 13 January 2015.
Edited by:
Bastien Nay, Centre National de la Recherche Scientifique, FranceReviewed by:
Irina Bakunina, Russian Academy of Sciences, RussiaBogdan Olenyuk, University of Southern California, USA
Copyright © 2015 Fuwa, Fukazawa and Sasaki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haruhiko Fuwa, Graduate School of Life Sciences, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan e-mail:aGZ1d2FAbS50b2hva3UuYWMuanA=