 Rosalba Juarez-Mosqueda
Rosalba Juarez-Mosqueda Andreas Mavrandonakis
Andreas Mavrandonakis Agnieszka B. Kuc
Agnieszka B. Kuc Lars G. M. Pettersson
Lars G. M. Pettersson Thomas Heine
Thomas Heine- 1School of Engineering and Science, Jacobs University Bremen, Bremen, Germany
- 2Department of Physics, AlbaNova University Center, Stockholm University, Stockholm, Sweden
The spillover mechanism of molecular hydrogen on carbon nanotubes in the presence of catalytically active platinum clusters was critically and systematically investigated by using density-functional theory. Our simulation model includes a Pt4 cluster for the catalyst nanoparticle and curved and planar circumcoronene for two exemplary single-walled carbon nanotubes (CNT), the (10,10) CNT and one of large diameter, respectively. Our results show that the H2 molecule dissociates spontaneously on the Pt4 cluster. However, the dissociated H atoms have to overcome a barrier of more than 2 eV to migrate from the catalyst to the CNT, even if the Pt4 cluster is at full saturation with six adsorbed and dissociated hydrogen molecules. Previous investigations have shown that the mobility of hydrogen atoms on the CNT surface is hindered by a barrier. We find that instead the Pt4 catalyst may move along the outer surface of the CNT with activation energy of only 0.16 eV, and that this effect offers the possibility of full hydrogenation of the CNT. Thus, although we have not found a low-energy pathway to spillover onto the CNT, we suggest, based on our calculations and calculated data reported in the literature, that in the hydrogen-spillover process the observed saturation of the CNT at hydrogen background pressure occurs through mobile Pt nanoclusters, which move on the substrate more easily than the substrate-chemisorbed hydrogens, and deposit or reattach hydrogens in the process. Initial hydrogenation of the carbon substrate, however, is thermodynamically unfavoured, suggesting that defects should play a significant role.
Introduction
Hydrogen spillover has been proposed as a promising mechanism for hydrogen storage (Lueking and Yang, 2004; Marella and Tomaselli, 2006; Li and Yang, 2006a,b; Chen and Huang, 2007, 2008; Liu et al., 2007; Zacharia et al., 2007; Zieliñski et al., 2007; Bhowmick et al., 2011). Over the past 9 years, Yang and coworkers (Lachawiec et al., 2005; Yang et al., 2006; Li and Yang, 2006a,b; Yang and Wang, 2009; Chen and Yang, 2010) have reported that hydrogen spillover is a viable technique to achieve high hydrogen storage on different carbon-based materials, even at ambient conditions. Carbon-based materials, such as carbon nanofibres (Lueking and Yang, 2004; Marella and Tomaselli, 2006), graphite (Mitchell et al., 2003a,b), and carbon nanotubes (CNTs) (Lueking and Yang, 2003, 2004; Nikitin et al., 2005, 2008; Zacharia et al., 2007; Chen and Huang, 2008; Yang and Wang, 2009; Bhowmick et al., 2011), are some of the common materials proposed as storage substrates. In 2008, Nilsson and coworkers have claimed that fully reversible hydrogenation of single-wall CNTs (SWCNT), with a diameter around 2.0 nm, can be achieved through reaction with atomic hydrogen (Nikitin et al., 2008). However, the veracity of these results and the efficiency of such graphitic materials to store hydrogen are still questionable. In two comprehensive reviews and an experimental work, Becher et al. (2003), Meregalli and Parrinello (2001) and Tibbetts et al. (2001) criticized earlier reports of hydrogen storage capacities of carbon-based materials greater than 1 wt % at ambient temperature.
A hydrogen spillover mechanism should consist of several steps: In the first, molecular hydrogen is activated and dissociated on a transition-metal catalyst (e.g., platinum, palladium, ruthenium, or nickel, Somorjai, 1994) in close contact with the substrate (e.g., carbon-based substrates). Secondly, the migration of H atoms from the catalyst particles to the substrate should occur. The last two steps consist in the diffusion and recombination of H atoms on the substrate surface (Conner and Falconer, 1995; Cheng et al., 2008; Psofogiannakis and Froudakis, 2009). Many of these steps have already been studied independently by several groups, suggesting that the picture might not be as simple as it appears. Han et al. (2012) found on the basis of quantum simulations that hydrogen migration along a graphene-like surface is an impractical process at standard conditions since it requires high-temperature heating (several 100°C). Furthermore, Chen et al. reported that, for CNTs, the curvature also has an important effect hampering the H diffusion between adjacent C atoms (Chen et al., 2007a). The authors found endothermic activation energies of 1.43, 1.07, and 0.82 eV for the H diffusion on (5,5) and (9,9) SWCNTs and graphene, respectively (Chen et al., 2007a). In addition, the migration of H atoms from a saturated Pt cluster to the carbon surface has been reported by Wu et al. (2011) and Psofogiannakis and Froudakis (2009), showing that this process is rather endothermic.
In the present work, we study the viability of hydrogen spillover as a mechanism to hydrogen storage. For this purpose, we performed quantum mechanical calculations based on DFT, to determine the energy profiles of the aforementioned mechanistic steps. We used bent-C54H18 and planar-C54H18 structures as model systems for small and large diameter single-walled carbon nanotubes (SWCNT), respectively. Firstly, we calculate the interaction energy between H2 and the carbon substrate for the initial reaction on a finite CNT model H2 + C54H18 → H2@C54H18 without any catalyst. We found that to load the carbon structure with hydrogen from gas-phase is a rather endothermic process with reaction energies of 1.3–2.9 eV and 1.9–3.4 eV for small and large diameter SWCNTs, respectively. Furthermore, the barrier to H2 chemisorption is larger than 3.5 eV. On the other hand, the recombination of H atoms to form a H2 molecule, in the absence of any catalyst, requires overcoming an energy barrier of 0.4, 1.6 and 2.0 eV when the H pair is removed from the meta, para and ortho positions of large diameter SWCNTs, respectively. This barrier becomes higher [1.2 (meta), 2.9 (ortho) and 2.9 eV (para)] when H atoms are removed from the small diameter SWCNTs. However, the full dehydrogenation of CNT without catalyst support by heating up to 600°C has been reported (Nikitin et al., 2005).
To study the effect of the catalyst on loading and unloading of hydrogen, a Pt4 cluster was used as a model catalyst. In accordance with many reports (Zhou et al., 2007; Chen et al., 2009; Gomez et al., 2011), we found that H2 is split into H atoms spontaneously on the cluster, i.e., without any energy barrier. However, we found that the migration of H atoms from Pt4 to the carbon substrate defines the energetically most expensive step of the spillover mechanism with an energy barrier of about 2 eV which is 50% lower than that obtained for the non-catalyzed process. Furthermore, we have found relatively lower energy barriers for the Pt4 mobility on the substrate which is energetically favored over the diffusion of the H atoms (Chen et al., 2007a). Therefore, we propose that Pt4 mobility is the key to the hydrogenation of the SWCNT.
Methods and Computational Details
Bent and planar circumcoronene (C54H18) structures were used to model the (10,10) CNT and a large diameter CNT or, equivalently, graphene (Figure 1), respectively. We have recently shown in the case of inorganic nanotubes that the properties of the large-diameter species are well-described by planar models (Ghorbani-Asl et al., 2013). Both models were simulated using the experimental bond lengths of 1.44 Å (White and Todorov, 1998; Saito et al., 2000) as initial geometry and with the outermost atoms fixed in the structural optimizations while the central atoms (darker color in Figure 1) were free to move. In earlier works (Jeloaica and Sidis, 1999; Mitchell et al., 2003a,b; Bonfanti et al., 2007; Psofogiannakis and Froudakis, 2009), a smaller coronene system has been shown to be a good model of the (0001) graphitic surface. A small platinum cluster, namely Pt4 (Figure 1), with tetrahedral symmetry, was used as a catalyst to dissociate the H2 molecule. Although the Pt4 cluster is much smaller than a real catalyst particle, it has been demonstrated that the H2 dissociation energy and the hydrogen desorption energy are independent of the platinum particle size (Zhou et al., 2007).
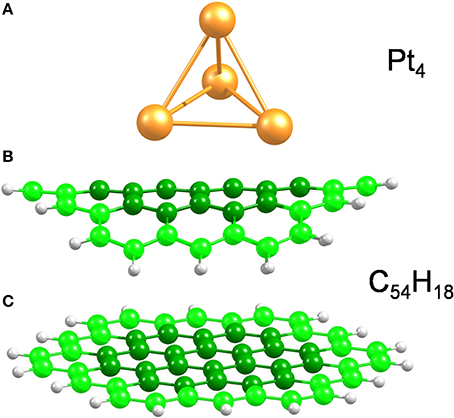
Figure 1. Model structures used in the present work. (A) Pt4 cluster, (B,C) bent and planar C54H18 structures to model small and large diameter SWCNTs, respectively. The bent carbon structure corresponds to the curvature of the (10,10) CNT. The carbon atoms at the edges are highlighted to stress that those, along with the H atoms, were kept fixed during structural optimizations.
Note that from the electronic structure point of view, our circumcoronene model is a semiconducting material, while the (10,10) CNT is actually a metallic system. However, the electronics should not significantly influence the binding energies, as shown earlier by Yildirim et al. (2001).
According to our spin-dependent calculations, we found that the distorted tetrahedral Pt4 cluster in the triplet spin state is by 0.47 eV and 0.13 eV more stable than the singlet and quintet spin configurations, respectively. Therefore, all calculations were performed using the triplet state, in agreement with the work of Sebetci (2006). We have tested four different DFT functionals [see Table S1 in Supporting Information (SI)] in terms of the electronic structure of Pt clusters and its geometry. We have selected the PBE (Perdew et al., 1996) density functional as it is one of the least expensive functionals, it is often used in similar studies, and, most importantly, it correctly predicts the triplet configuration of the Pt system in its tetrahedral geometry.
All calculations have been carried out using density-functional theory (DFT) (Hohenberg and Kohn, 1964; Kohn and Sham, 1965), with the PBE (Perdew et al., 1996) exchange-correlation functional in combination with the def2-TZVP basis set as implemented in the Turbomole 6.5 software (Ahlrichs et al., 1989; Eichkorn et al., 1997). The employed basis set accounts for relativistic effects by using an effective core potential (ECP) for the 60 core electrons for Pt. The geometry optimization criteria for the forces and energy threshold were set to be smaller than 10−4 hartree per bohr and 10−6 hartree, respectively. The convergence criterion for the energy calculation during the self-consistent-field procedure was set to 10−7 hartree. We have used the m4 grid in all simulations.
The coordinate-driven interpolation method of finding an estimate for the barrier, which we have used here, can be considered equivalent to the nudged elastic band technique (all other degrees of freedom are fully relaxed at each step) where the coordinate of interest would be the C-H bond length.
To analyze the whole spillover process, we divided the study into several steps. Firstly, we calculated the energy barriers for loading and unloading the carbon substrate with H2 in absence of any catalyst, for our small cluster models. Activation (E‡) and reaction (ΔE) energies (also called here adsorption or chemisorption energies) were calculated for two H atoms disposed at the center of the coronene structure in ortho, meta and para configurations. To calculate E‡ the energy was scanned over the C-H distance (dC−H) starting from the optimized structures (dC−H ~1.1 Å) up to the point at which the H2 molecule was already formed (dC−H ~3.6 Å) (see Movie 00 in SI). ΔE was calculated with reference to H2 at infinite distance from the C54H18 substrate. In these optimizations the carbon and hydrogen atoms at the edges (i.e., the 30 outer C atoms and the 18 H atoms) in C54H18 were kept fixed, while the rest of the structure was relaxed (see Figure 1).
In the second step, we have analyzed the dissociation of H2 on the Pt4 cluster omitting the influence of the carbon substrate. The calculations were performed by scanning over the Pt4–H2 distances starting from the optimized Pt4 and H2 molecules, separated by 2.5 Å and going down to 1.5 Å, where the hydrogen molecule was dissociated and H atoms were attached to the platinum cluster (See Figure S1 in SI). The only constraint imposed in this simulation was the Pt-H distance.
Next, we have studied the migration of hydrogen atoms to and from the hydrogenated catalyst (Pt4H2x with x = 1 − 6) to the carbon substrates (for an example, see Movie 01 in the SI). As before, we imposed constraints on the edges of the carbon models (see Figure 1) while the 24 carbon atoms at the center of the circumcoronene structure and the Pt4H2x cluster were allowed to move.
Finally, we have investigated the mobility of the Pt4 cluster on the carbon substrates. We have taken into account the influence of the Pt4 cluster loading with hydrogen where we have compared bare and fully saturated clusters as limiting cases, e.i C54H18–Pt4 and C54H18–Pt4H12, respectively. The tetrahedral Pt4 and Pt4H12 clusters were pre-optimized on top of C54H18 substrate using different starting points. During these optimizations the edges of the C54H18 structure were kept fixed, while the Pt4 and Pt4H12 clusters and the central carbon atoms were allowed to move. All the different starting points studied here resulted in only three different configurations; bare Pt4 cluster with tip down (end on), tip up (face on) and saturated Pt4H12 cluster with edge on. Afterwards, for the resulting structures, the platinum clusters were shifted along the carbon substrate with a step size of 0.5 Å (see Movie 02 in the SI) and rotated parallel to the substrate in steps of 2° (see Movie 03 in the SI). We have kept the C-Pt distance fixed during the scanning to estimate the influence of the substrate's curvature on the mobility of the catalyst.
Results
Hydrogen Chemisorption and Recombination in Absence of Catalyst
The energy required to hydrogenate the carbon substrate in the absence of any catalyst was calculated for the bent and planar models according to the reaction H2 + C54H18 → H2@C54H18(see Table 1 and Figure 2). The results show that the process is rather endothermic, with ΔE of 2.0, 3.4, and 1.9 eV for two H atoms attached to the large diameter CNT model in ortho, meta, and para positions, respectively. The corresponding activation energies to chemisorb (E‡,s) the two H atoms are 4.0 (ortho), 3.8 (meta) and 3.5 eV (para). For the curved (10,10) CNT, the endothermicity is reduced by ~0.6 eV to 1.4, 2.9 and 1.3 eV for the two H atoms attached in ortho, meta and para positions, respectively. This is in accordance with previous studies on the influence of curvature on the adsorption energy in CNTs, which showed strong reduction with increased curvature (Gao et al., 2011). The E‡,s is oppositely affected by the tube curvature, increasing by 0.2–0.7 eV. According to our calculations, ortho and para are the preferred adsorption positions for hydrogen, which is in agreement with earlier works (Andree et al., 2006; Hornekær et al., 2006b). Becher et al. (2003) concluded that reversible hydrogen storage at ambient temperature is unlikely based on the endothermic adsorption energies, which range from 2–3 eV. It should be noted, however, that the adsorption energy per hydrogen atom of nearly fully hydrogenated structures is different compared to the initial step (initial hydrogenation of the substrate); depending on the curvature, these values can get much smaller (Gao et al., 2011).
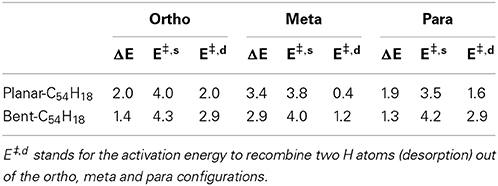
Table 1. Reaction (ΔE) and activation (E‡,s) energies (eV) for the sorption of two H atoms on the planar and bent C54H18 substrates disposed at ortho, meta, and para positions.
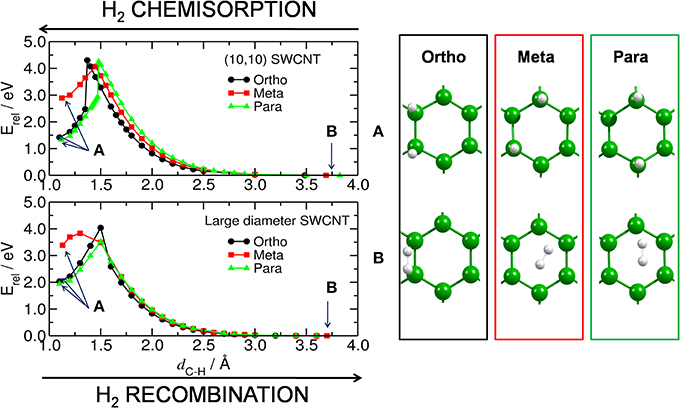
Figure 2. H2 chemisorption/recombination on carbon substrate. (Left) energy barriers for the chemisorption/recombination (E‡,s/E‡,d) of two H atoms on the bent (upper) and planar (lower) C54H18 substrates, corresponding to the (10,10) and large diameter SWCNT models, respectively. Blue arrows inside the plot point to the equilibrium energy for (A) the structure with two H atoms attached to the substrate (i.e., H2@C54H18) and, (B) for the H2 detached from C54H18 and located at a distance of ~3.6 Å from the substrate. (Right) corresponding central part of the equilibrated structures for H2@C54H18 and H2 + C54H18.
The discontinuities in the computed potential energy curves are not due to an inappropriate reaction coordinate, but rather due to hysteresis effects in the applied DFT technique when treating a curve crossing. Forming the two C-H bonds from closed-shell H2 and the coronene requires a spin-uncoupling of both systems to reach the bonding singlet state formed from triplet H2 and triplet coronene (Triguero et al., 1998). This internal rehybridization of both systems simultaneously may be difficult to achieve and the computed curves erroneously follow the repulsive state beyond the crossing point before collapsing to the correct lower state. However, since even the energy barrier estimated from the lower state is too high for the reaction to be realistic we take this lower value as estimate of the barrier.
Furthermore, Hornekær et al. (2006a) reported, using scanning tunneling microscopy and DFT calculations, that recombination of two H atoms on graphite occurs only from the para position with a barrier of 1.4 eV. This is in close agreement with our E‡,d from the para position of 1.6 eV for the planar model of a large diameter CNT. We obtained an E‡,d of 2.9 eV for the (10,10) CNT. The E‡,d from the ortho position was found to be 2.0 and 2.9 eV for the planar and bent models, respectively. These results indicate that, without catalyst, the chemisorption and recombination of hydrogen on the carbon materials would require large energies to overcome the barriers. Considering finally the least favorable adsorption site, the meta position, we find low values of 1.2 and 0.4 eV for the recombination on the curved and planar substrates, respectively. This is not surprising since the meta position is the least favored one.
Catalyzed Hydrogen Chemisorption and Recombination on Carbon Substrates
Platinum nanoparticles are commonly used as transition-metal catalysts to hydrate surfaces via the spillover process (Somorjai, 1994; Lueking and Yang, 2004). In agreement with previous reports (Zhou et al., 2007; Chen et al., 2009; Gomez et al., 2011) we found that the dissociation of H2 on Pt4 occurs without any energy barrier and up to 6 H2 molecules can be dissociated and accommodated on this catalyst. Figure 3 shows the optimized structures of the hydrogenated Pt4 cluster attached to the bent and planar carbon substrates. It is important to mention that the fully hydrogenated Pt4 cluster retains its tetrahedral shape, but an increase of the Pt–Pt distance of maximum 0.3–0.5 Å is generated due to saturation. Chen et al. have reported an increase of the Pt–Pt bond lengths of 0.01–0.3 Å for a Pt6 cluster at high H coverage (Chen et al., 2007b).
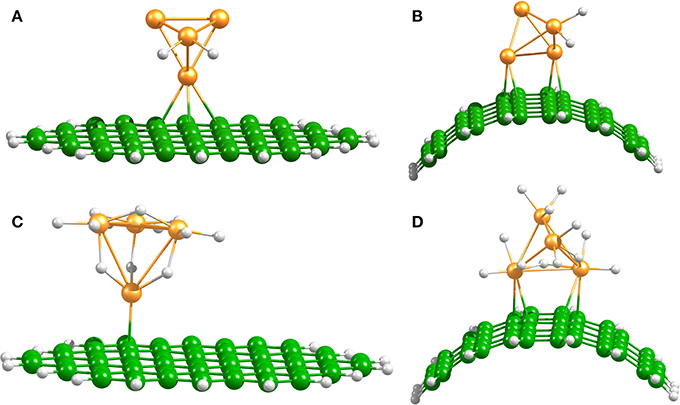
Figure 3. Spillover models. Optimized Pt4–2H and Pt4–12H structures on planar (A,C), and bent (B,D) C54H18 structures.
Independent of the number of H atoms attached to the catalyst, the Pt4 cluster binds to the planar substrate with one Pt atom close to the C atoms and with the H atoms oriented nearly parallel to the carbon surface. The binding energies for the bare and fully saturated Pt4 cluster to the planar substrate are strongly exothermic with values of −2.25 and −1.50 eV, respectively. Surprisingly, we find that the presence of the Pt4 cluster on the substrate does not affect the structure of the substrate. On the bent substrate we find a Pt4 structure with two Pt atoms bonded to the CNT. The binding energies for the bare and fully saturated Pt4 with the bent C54H18 are in this case −2.81 and −2.04 eV, respectively. Moreover, on the bent substrate the H atoms are oriented nearly perpendicular to the carbon surface. For the subsequent migration of two H atoms to the carbon substrate, it is required to rotate the H atoms to face the carbon substrate in a parallel manner (Figure 4). The rotation barrier of around 0.14 eV was calculated using the same constraints as shown in Figure 1, but additionally a dihedral angle was fixed to avoid the rotation of the H atoms in each point of the energy scan.
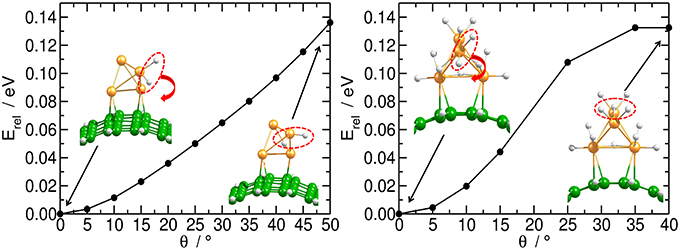
Figure 4. Rotation of H atoms on Pt4 catalyst. Energy profile for the rotation of two H atoms on bi-hydrogenated (left) and dodeca-hydrogenated (right) clusters on bent C54H18 structure. The energy is related to the equilibrium geometry.
The activation barrier for one H atom to migrate from the Pt4 cluster to carbon has been calculated by Wu et al. (2011) and by Psofogiannakis and Froudakis (2009) using DFT. The authors found that the estimated barrier for migration of one H atom from the fully saturated Pt4 cluster to the carbon substrate is 2.7 eV (Wu et al., 2011) and 2.6 eV, (Psofogiannakis and Froudakis, 2009) respectively. Our results show that the barrier for the simultaneous migration of two H atoms from the dodeca-hydrogenated Pt4 cluster to the bent and planar surface is 2.0 eV and 2.3 eV, respectively (see Figure 5 and the Movie 01 in SI). On the other hand, we have observed that when bending the saturated Pt4 cluster toward the substrate, the carbon structure is puckered down. This additionally supports the fact that the hydrogen migration from the catalyst to the carbon is not a straightforward process and even a rotational motion of the catalyst cluster does not lower the energy barrier (see Figure S2 in SI).
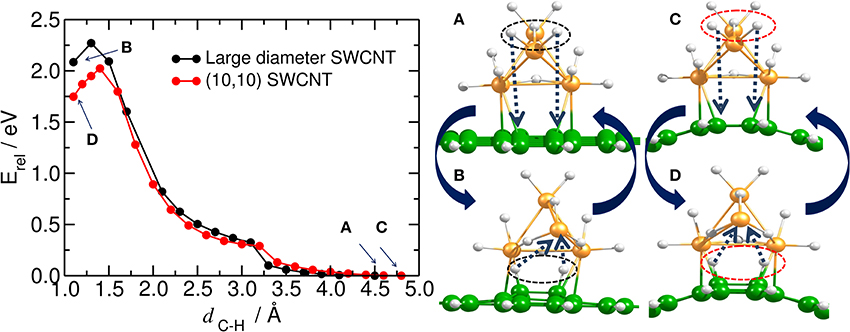
Figure 5. Hydrogen spillover on carbon substrate. (Left) Energy profile for the simultaneous migration of the first two H atoms from the dodeca-hydrogenated Pt4 cluster to the bare carbon substrate (i.e., initial hydrogenation). Equilibrium energy for the structures in which all H atoms are initially attached to the platinum cluster and the carbon substrate is bare are pointed out by blue arrows as (A,C), and shown in the right upper panel. (B, D) inside the plot (and in the right lower panel) stand for the equilibrium energy (and structures) in which the first two H atoms are chemisorbed by the carbon substrate. This plot can also be read as the migration of the H atoms from the carbon substrate to the deca-hydrogenated Pt4 cluster.
Wu et al. (2011) have shown that for pure graphene, in which an H atom was manually put on a C atom, the hydrogen migrates back to the Pt4 cluster after structural relaxation. In our calculations, we observed that this effect depends on the degree of saturation of the Pt4 cluster and the substrate itself. From simple optimization calculations, we observed that the Pt4 cluster with eight hydrogen atoms attached (Pt4H8) spontaneously collects two out of four H atoms that were located manually on the substrate in one hexagon ring, but it does not collect any when only one pair of H atoms is attached to the substrate in ortho position (see Movie 04 and Movie 05 in SI). Similarly, we observed that the bare Pt4 cluster spontaneously collects H atoms from the graphitic surface, in which two hydrogen atoms were manually attached (see Movie 06 in SI).
To investigate the role of the substrate and catalyst saturation, we calculated the energy profile for subsequent hydrogen migrations; a second pair of H atoms migrating from the dodeca-hydrogenated (Pt4H12) cluster to the (10,10) CNT surface modified such that there were two H atoms initially attached to the substrate. These two hydrogens were disposed into three different positions of the same hexagonal ring (Figure S3 in SI). From these calculations we found that the energy barrier for the subsequent migration remains essentially unchanged (~2.0 eV), while the adsorption energy for the subsequent hydrogenations decreases by around 0.5 and 0.1 eV depending on the relative arrangement of the first two hydrogens and the second pair (see Figure S3 in SI). This means that the adsorption of H atoms becomes less endothermic.
To investigate the effect of the cluster saturation we also performed similar calculations for the migration of the first two hydrogens from a deca-hydrogenated (Pt4H10) cluster to the bare C54H18 surface. However, we did not observe any significant change in the energetics. In conclusion, the energy barrier for the initial hydrogenation of the substrate is around 2 eV and is essentially unaffected by the level of pre-hydrogenation of the substrate. Nevertheless, the presence of the H pair on the substrate lowers the hydrogen adsorption energy up to 0.5 eV, lowering the level of the endothermic process.
Platinum Cluster Mobility on Carbon Nanotubes
Figure 6 shows the relative energy for the migration of the platinum catalyst along and around the (10,10) CNT. All energies are related to the optimized C54H18–Pt4 and C54H18–Pt4H12 structures. We have found that the Pt4 cluster moves along the CNT axis more easily than around the tube. The largest barriers for the Pt4 mobility along the CNT axis were calculated for the case when the cluster binds to the substrate with the lowest coordination number (i.e., that is tip-down), resulting in an activation barrier of about 0.30 eV. This barrier is further reduced for the Pt4 cluster with tip-up or the saturated cluster for which we find barriers of 0.22 eV and 0.16 eV, respectively (see Figure 6A). Thus, platinum clusters move more easily on the carbon substrate at the full saturation limit.
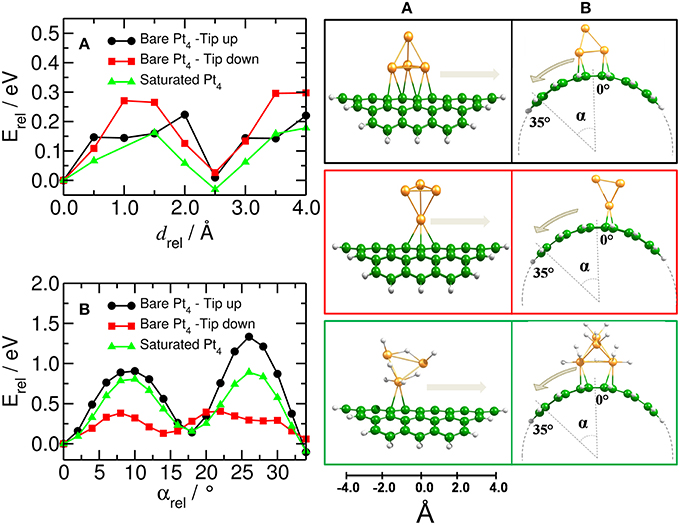
Figure 6. Mobility of catalyst on bent carbon substrate. (Left) Energy profile for moving the Pt4 cluster (A) along and (B) around the curved C54H18 structure. (Right) Corresponding equilibrium positions of the Pt4 clusters with tip up (black) and down (red) and for the fully saturated Pt4 cluster (green). Energy is related to the respective initial optimized structures.
In order to move the saturated Pt4 cluster around the tube, it is required to overcome a barrier five times higher than when moving it along the tube (0.89 eV against 0.16 eV, cf. green lines in Figures 6A,B), suggesting that the curvature of the surface has an important effect on the Pt4 mobility. This was also suggested by Chen et al. (2007a).
We have analyzed the mobility of the Pt4 cluster also on the flat C54H18 structure, to model the situation of large diameter carbon nanotubes (Figure 7). We found that the energy barrier does not depend on the direction of the cluster motion (zigzag or armchair), but it does depend on the C-Pt coordination number. Thus, the highest barrier for the Pt4 mobility (of about 1.0 eV) was found for the situation where the cluster binds to more than three carbon atoms at an average C-Pt distance of 2.28 Å and a binding energy of −2.25 eV. Similar to the bent model, the saturated cluster with binding energy of −1.50 eV moves easily along the carbon surface with barrier of only 0.2 eV. These results show that the migration of the catalyst is preferable for the saturated catalyst and it is strongly influenced by the curvature of the tube.
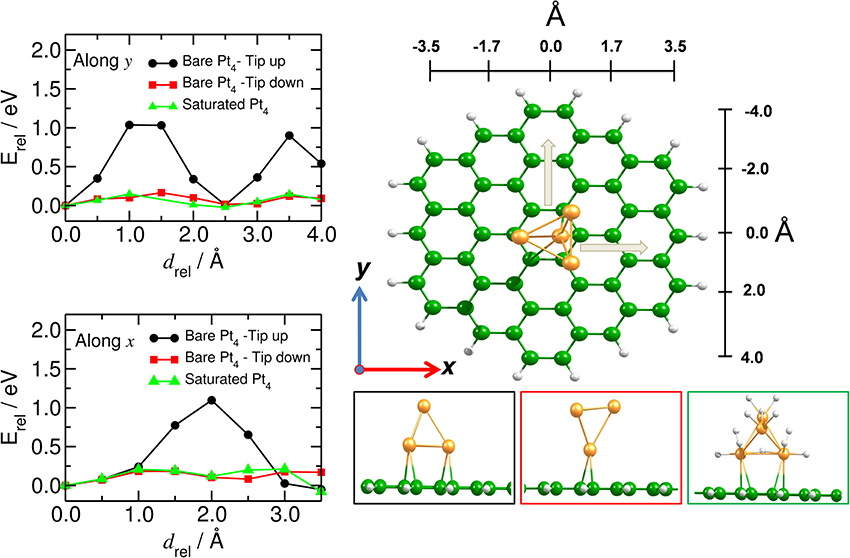
Figure 7. Mobility of catalyst on planar carbon substrate. (Left) Energy profile of the mobility of the Pt4 cluster (along, y, and across, x) on the flat C54H18 structure. (Right) Corresponding equilibrium positions of the Pt4 cluster with tip up (black), tip down (red) and for the fully saturated Pt4 cluster (green). x and y indicate the directions of the Pt4 motion.
Conclusions
We have investigated the individual steps of hydrogen storage by the spillover mechanism and complemented our calculations with data from the literature. First, on carbon nanostructures, such as small-diameter SWCNT, large-diameter SWCNT and graphene, hydrogen can chemisorb and desorb only by overcoming a significant energy barrier. This suggests that those structures can store hydrogen for a long time at very high gravimetric and volumetric capacities. Hydrogen is covalently bound, and the endothermic adsorption energies are between 1.3 and 2.9 eV for the small diameter SWCNT and between 1.9 and 3.4 eV for the large diameter SWCNT. For smaller SWCNTs, even lower energy values are reported in the literature (Gao et al., 2011). The work of Chen et al. (2007a) reports a very low hydrogen mobility on the carbon surface with a barrier of 1.42, 1.09 and 0.78 eV associated to its motion on (5,5), (9,9) CNTs and graphene, respectively. Thus, a traditional spillover mechanism where hydrogen atoms migrate quickly along the surface is not possible for graphitic structures.
Loading and unloading these structures with hydrogen without a catalyst would require overcoming barriers of more than 3.5 eV and 1.5 eV to chemisorb and to remove, respectively, a pair of H atoms from ortho and para positions. However, the barrier for loading the carbon substrate can be significantly reduced by using a catalyst. Moreover, the sorption energy becomes less endothermic for smaller diameter SWCNTs.
The most promising catalysts for hydrogen activation are platinum nanoparticles. These particles spontaneously dissociate H2 from the gas phase and can take up a large number of hydrogen molecules. For example, our Pt4 model cluster is saturated after adsorbing six H2 molecules. Bare and hydrogenated Pt clusters bind to the sp2 carbon surface with binding energies of at least −2.25 eV and −1.50 eV, respectively. The catalyst lowers the barriers for the initial surface hydrogenation by about 50%. In addition, we found that the presence of a pair of H atoms on the substrate does not affect the activation barrier for subsequent hydrogenation, but it reduces the endothermic adsorption energy for the next H pair by about 0.5 eV.
We have further established that Pt nanoparticles are very mobile on the sp2 carbon substrates and can relocate nearly barrier-free. Thus, instead of the traditional spillover mechanism from a static catalyst particle followed by hydrogen diffusion along the support, it is more likely that the Pt nanoparticles move over the substrate surface and thus load/unload H2 locally. The high mobility of the Pt nanoparticles has, however, a crucial drawback: due to their high mobility we expect the particles to cluster to larger Pt aggregates, which ultimately terminates the efficiency of the catalyst and the working principle of a hydrogen storage device based on this concept.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the EU Indo-European network HYPOMAP FP7-NMP-2008-EU-India-2 (Grant agreement no.: 233482). We thank Dr. Binit Lukose for his support and Deutsche Forschungsgemeinschaft (HE 3543/17-1).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fchem.2015.00002/abstract
References
Ahlrichs, R., Bär, M., Häser, M., Horn, H., and Kölmel, C. (1989). Electronic structure calculations on workstation computers: the program system turbomole. Chem. Phys. Lett. 162, 165–169. doi: 10.1016/0009-2614(89)85118-8
Andree, A., Lay, M. L., Zecho, T., and Küpper, J. (2006). Pair formation and clustering of D on the basal plane of graphite. Chem. Phys. Lett. 425, 99–104. doi: 10.1016/j.cplett.2006.05.015
Becher, M., Haluska, M., Hirscher, M., Quintel, A., Skakalova, V., Dettlaff-Weglikovska, U., et al. (2003). Hydrogen storage in carbon nanotubes. C. R. Phys. 4, 1055–1062. doi: 10.1016/S1631-0705(03)00107-5
Bhowmick, R., Rajasekaran, S., Friebel, D., Beasley, C., Jiao, L., Ogasawara, H., Dai, H., et al. (2011). Hydrogen spillover in pt-single-walled carbon nanotube composites: formation of stable C-H bonds. J. Am. Chem. Soc. 133, 5580–5586. doi: 10.1021/ja200403m
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bonfanti, M., Martinazzo, R., Tantardini, G. F., and Ponti, A. (2007). Physisorption and Diffusion of Hydrogen atoms on graphite from correlated calculations on the H-Coronene model system. J. Phys. Chem. C 111, 5825–5829. doi: 10.1021/jp070616b
Chen, C.-H., and Huang, C.-C. (2007). Hydrogen storage by KOH-modified multi-walled carbon nanotubes. Int. J. Hydrogen Energy 32, 237–246. doi: 10.1016/j.ijhydene.2006.03.010
Chen, C.-H., and Huang, C.-C. (2008). Enhancement of hydrogen spillover onto carbon nanotubes with defect feature. Microporous Mesoporous Mater 109, 549–559. doi: 10.1016/j.micromeso.2007.06.003
Chen, H., and Yang, R. T. (2010). Catalytic effects of TiF3 on hydrogen spillover on Pt/Carbon for hydrogen storage. Langmuir 26, 15394–15398. doi: 10.1021/la100172b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, L., Cooper, A. C., Pez, G. P., and Cheng, H. (2007a). Mechanistic study on hydrogen spillover onto graphitic carbon materials. J. Phys. Chem. C 111, 18995–19000. doi: 10.1021/jp074920g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, L., Cooper, A. C., Pez, G. P., and Cheng, H. (2007b). Density functional study of sequential H2 dissociative chemisorption on a Pt6 cluster. J. Phys. Chem. C 111, 5514–5519. doi: 10.1021/jp070181s
Chen, L., Zhou, C.-G., Wu, J.-P., and Cheng, H.-S. (2009). Hydrogen adsorption and desorption on the Pt and Pd subnano clusters—a review. Front. Phys. China 4, 356–366. doi: 10.1007/s11467-009-0050-6
Cheng, H., Chen, L., Cooper, A. C., Sha, X., and Pez, G. P. (2008). Hydrogen spillover in the context of hydrogen storage using solid-state materials. Energy Environ. Sci. 1, 338–354. doi: 10.1039/B807618A
Conner, W. C., and Falconer, J. L. (1995). Spillover in heterogeneous catalysis. Chem. Rev. 95, 759–788. doi: 10.1021/cr00035a014
Eichkorn, K., Weigend, F., Treutler, O., and Ahlrichs, R. (1997). Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor. Chem. Acc. 97, 119–124.
Gao, X., Wang, Y., Liu, X., Chan, T.-L., Irle, S., Zhao, Y., et al. (2011). Regioselectivity control of graphene functionalization by ripples. Phys. Chem. Chem. Phys. 13, 19449–19453. doi: 10.1039/C1CP22491C
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ghorbani-Asl, M., Zibouche, N., Wahiduzzaman, M., Oliveira, A. F., Kuc, A., and Heine, T. (2013). Electromechanics in MoS2 and WS2: nanotubes vs. monolayers. Sci. Rep. 3, 1–5. doi: 10.1038/srep02961
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gomez, T., Florez, E., Rodriguez, J. A., and Illas, F. (2011). Reactivity of transition metals (Pd, Pt, Cu, Ag, Au) toward molecular hydrogen dissociation: extended surfaces versus particles supported on TiC(001) or small is not always better and large is not always bad. J. Phys. Chem. C 115, 11666–11672. doi: 10.1021/jp2024445
Han, S. S., Jung, H., Jung, D. H., Choi, S.-H., and Park, N. (2012). Stability of hydrogenation states of graphene and conditions for hydrogen spillover. Phys. Rev. B 85:155408. doi: 10.1103/PhysRevB.85.155408
Hohenberg, P., and Kohn, W. (1964). Inhomogeneous electron gas. Phys. Rev. 136, B864–B871. doi: 10.1103/PhysRev.136.B864
Hornekær, L., Rauls, E., Xu, W., Šljivanèanin, Ž., Otero, R., Stensgaard, I., et al. (2006b). Clustering of chemisorbed H(D) atoms on the graphite (0001) surface due to preferential sticking. Phys. Rev. Lett. 97: 186102. doi: 10.1103/PhysRevLett.97.186102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hornekær, L., Sljivancanin, Z., Xu, W., Otero, R., Rauls, E., Stensgaard, I., et al. (2006a). Metastable structures and recombination pathways for atomic hydrogen on the graphite (0001) surface. Phys. Rev. Lett. 96:156104. doi: 10.1103/PhysRevLett.96.156104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jeloaica, L., and Sidis, V. (1999). {DFT} investigation of the adsorption of atomic hydrogen on a cluster-model graphite surface. Chem. Phys. Lett. 300, 157–162. doi: 10.1016/S0009-2614(98)01337-2
Kohn, W., and Sham, L. J. (1965). Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138. doi: 10.1103/PhysRev,0.140.A.1133
Lachawiec, A. J., Qi, G., and Yang, R. T. (2005). Hydrogen storage in nanostructured carbons by spillover: bridge-building enhancement. Langmuir 21, 11418–11424. doi: 10.1021/la051659r
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Y., and Yang, R. T. (2006a). Hydrogen storage in metal-organic frameworks by bridged hydrogen spillover. J. Am. Chem. Soc. 128, 8136–8137. doi: 10.1021/ja061681m
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Y., and Yang, R. T. (2006b). Significantly enhanced hydrogen storage in metal-organic frameworks via spillover. J. Am. Chem. Soc. 128, 726–727. doi: 10.1021/ja056831s
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, Y.-Y., Zeng, J.-L., Zhang, J., Xu, F., and Sun, L.-X. (2007). Improved hydrogen storage in the modified metal-organic frameworks by hydrogen spillover effect. Int. J. Hydrogen Energy 32, 4005–4010. doi: 10.1016/j.ijhydene.2007.04.029
Lueking, A. D., and Yang, R. T. (2004). Hydrogen spillover to enhance hydrogen storage—study of the effect of carbon physicochemical properties. Appl. Catal. A 265, 259–268. doi: 10.1016/j.apcata.2004.01.019
Lueking, A., and Yang, R. T. (2003). Hydrogen storage in carbon nanotubes: residual metal content and pretreatment temperature. AIChE J. 49, 1556–1568. doi: 10.1002/aic.690490619
Marella, M., and Tomaselli, M. (2006). Synthesis of carbon nanofibers and measurements of hydrogen storage. Carbon 44, 1404–1413. doi: 10.1016/j.carbon.2005.11.020
Meregalli, V., and Parrinello, M. (2001). Review of theoretical calculations of hydrogen storage in carbon-based materials. Appl. Phys. A 72, 143–146. doi: 10.1007/s003390100789
Mitchell, P. C. H., Ramirez-Cuesta, A. J., Parker, S. F., and Tomkinson, J. (2003a). Inelastic neutron scattering in spectroscopic studies of hydrogen on carbon-supported catalysts-experimental spectra and computed spectra of model systems. J. Mol. Struct. 651–653, 781–785. doi: 10.1016/S0022-2860(03)00124-8
Mitchell, P. C. H., Ramirez-Cuesta, A. J., Parker, S. F., Tomkinson, J., and Thompsett, D. (2003b). Hydrogen spillover on carbon-supported metal catalysts studied by inelastic neutron scattering. Surface vibrational states and hydrogen riding modes. J. Phys. Chem. B 107(28), 6838–6845. doi: 10.1021/jp0277356
Nikitin, A., Li, X., Zhang, Z., Ogasawara, H., Dai, H., and Nilsson, A. (2008). Hydrogen storage in carbon nanotubes through the formation of stable C-H bonds. Nano Lett. 8, 162–167. doi: 10.1021/nl072325k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nikitin, A., Ogasawara, H., Mann, D., Denecke, R., Zhang, Z., Dai, H., Cho, K., et al. (2005). Hydrogenation of single-walled carbon nanotubes. Phys. Rev. Lett. 95, 225507. doi: 10.1103/PhysRevLett.95.225507
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi: 10.1103/PhysRevLett.77.3865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Psofogiannakis, G. M., and Froudakis, G. E. (2009). DFT study of the hydrogen spillover mechanism on Pt-Doped graphite. J. Phys. Chem. C 113, 14908–14915. doi: 10.1021/jp902987s
Saito, R., Dresselhaus, G., and Dresselhaus, M. S. (2000). Trigonal wraping effect of carbon nanotubes. Phys. Rev. B, 61, 2981–2985. doi: 10.1103/PhysRevB.61.2981
Sebetci, A. (2006). A density functional study of bare and hydrogenated platinum clusters. Chem. Phys. 331, 9–18. doi: 10.1016/j.chemphys.2006.09.037
Somorjai, G. A. (1994). Introduction to Surface Chemistry and Catalysis. New York, NY: Wiley-Interscience.
Tibbetts, G. G., Meisner, G. P., and Olk, C. H. (2001). Hydrogen storage capacity of carbon nanotubes, filaments, and vapor-grown fibers. Carbon 39, 2291–2301. doi: 10.1016/S0008-6223(01)00051-3
Triguero, L., Pettersson, L. G. M., Minaev, B., and Ågren, H. (1998). Spin Uncoupling in Surface Chemisorption of unsaturated hydrocarbons. J. Chem. Phys. 108, 1193–1205
White, C. T., and Todorov, T. N. (1998). Carbon nanotubes as long ballistic conductors. Nature 393, 240–242.
Wu, H.-Y., Fan, X., Kuo, J.-L., and Deng, W.-Q. (2011). DFT study of hydrogen storage by spillover on graphene with boron substitution. J. Phys. Chem. C 115, 9241–9249. doi: 10.1021/jp200038b
Yang, F. H., Lachawiec, A. J., and Yang, R. T. (2006). Adsorption of spillover hydrogen atoms on single-wall carbon nanotubes. J. Phys. Chem. B 110, 6236–6244. doi: 10.1021/jp056461u
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, R. T., and Wang, Y. (2009). Catalyzed hydrogen spillover for hydrogen storage. J. Am. Chem. Soc. 131, 4224–4226. doi: 10.1021/ja808864r
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yildirim, T., Gülseren, O., and Ciraci, S. (2001). Exohydrogenated single-wall carbon nanotubes. Phys. Rev. B 64:075404 doi: 10.1103/PhysRevB.64.075404
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zacharia, R., Rather, S., Hwang, S. W., and Nahm, K. S. (2007). Spillover of physisorbed hydrogen from sputter-deposited arrays of platinum nanoparticles to multi-walled carbon nanotubes. Chem. Phys. Lett. 434, 286–291. doi: 10.1016/j.cplett.2006.12.022
Zhou, C., Wu, J., Nie, A., Forrey, R. C., Tachibana, A., and Cheng, H. (2007). On the sequential hydrogen dissociative chemisorption on small platinum clusters: a density functional theory study. J. Phys. Chem. C 111, 12773–12778. doi: 10.1021/jp073597e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: carbon nanotube, hydrogen spillover, hydrogen storage, DFT, Pt catalyst, circumcoronene
Citation: Juarez-Mosqueda R, Mavrandonakis A, Kuc AB, Pettersson LGM and Heine T (2015) Theoretical analysis of hydrogen spillover mechanism on carbon nanotubes. Front. Chem. 3:2. doi: 10.3389/fchem.2015.00002
Received: 09 October 2014; Accepted: 09 January 2015;
Published online: 02 February 2015.
Edited by:
Alister Page, The University of Newcastle, AustraliaReviewed by:
Terry J. Frankcombe, The Australian National University, AustraliaMichael Hirscher, Max Planck Institute for Intelligent Systems, Germany
Copyright © 2015 Juarez-Mosqueda, Mavrandonakis, Kuc, Pettersson and Heine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Heine, School of Engineering and Science, Jacobs University Bremen, Campus Ring 1, 28759 Bremen, Germany e-mail:dC5oZWluZUBqYWNvYnMtdW5pdmVyc2l0eS5kZQ==