 Salvatore F. Altavilla1
Salvatore F. Altavilla1 Javier Segarra-Martí1*
Javier Segarra-Martí1* Artur Nenov1
Artur Nenov1 Irene Conti1
Irene Conti1 Ivan Rivalta2
Ivan Rivalta2 Marco Garavelli1,2*
Marco Garavelli1,2*- 1Dipartimento di Chimica “G. Ciamician,” Università di Bologna, Bologna, Italy
- 2École Normale Supérieure de Lyon, Centre National de la Recherche Scientifique, UMR 5182, Université de Lyon, Lyon, France
The photophysics and photochemistry of water-solvated guanine monophosphate (GMP) are here characterized by means of a multireference quantum-chemical/molecular mechanics theoretical approach (CASPT2//CASSCF/AMBER) in order to elucidate the main photo-processes occurring upon UV-light irradiation. The effect of the solvent and of the phosphate group on the energetics and structural features of this system are evaluated for the first time employing high-level ab initio methods and thoroughly compared to those in vacuo previously reported in the literature and to the experimental evidence to assess to which extent they influence the photoinduced mechanisms. Solvated electronic excitation energies of solvated GMP at the Franck-Condon (FC) region show a red shift for the ππ* La and Lb states, whereas the energy of the oxygen lone-pair nπ* state is blue-shifted. The main photoinduced decay route is promoted through a ring-puckering motion along the bright lowest-lying La state toward a conical intersection (CI) with the ground state, involving a very shallow stationary point along the minimum energy pathway in contrast to the barrierless profile found in gas-phase, the point being placed at the end of the minimum energy path (MEP) thus endorsing its ultrafast deactivation in accordance with time-resolved transient and photoelectron spectroscopy experiments. The role of the nπ* state in the solvated system is severely diminished as the crossings with the initially populated La state and also with the Lb state are placed too high energetically to partake prominently in the deactivation photo-process. The proposed mechanism present in solvated and in vacuo DNA/RNA chromophores validates the intrinsic photostability mechanism through CI-mediated non-radiative processes accompanying the bright excited-state population toward the ground state and subsequent relaxation back to the FC region.
Introduction
The genomic material and the mechanisms that process the disposal of the excess energy attained upon UV-light irradiation are of paramount importance from both biomedical and biotechnological standpoints. The initially populated excited states and their fate along the distinct deactivation routes present in the DNA/RNA double helix chains relate simultaneously to the intrinsic photostability of the genomic material (Callis, 1983; Crespo-Hernandez et al., 2004; Kohler, 2010), as well as to the damaging photo-reactions that ultimately yield mutations and single- and double-strand breaks that have been associated to increasingly featured diseases like skin cancer (Cadet et al., 2012; Noonan et al., 2012; Giussani et al., 2013b; Brash, 2015). Besides the biological relevance, the intricate photophysical properties of nucleobases can be also exploited to design optical photo-responsive nanomachines among a long list of prospective applications (Kamiya and Asanuma, 2014), yet it is their biological relevance that focuses our attention.
From a bio-physical and chemical point of view, fundamental knowledge on the deactivation processes, including the associated underlying molecular motions, are essential to rationalize the intrinsic photostability of the genomic material and to characterize the aforementioned routes leading to deleterious photochemical reactions (Markovitsi et al., 2010; Cadet et al., 2012). These different pathways increase in number and complexity as the size of the DNA/RNA single- or double-stranded chain enlarges, as the different de-excitation routes interact with one another yielding a ramification of complex photoinduced pathways that become extremely hard to discern, disentangle, and comprehend. This increasing degree of complexity arises partially due to the intermolecular interactions occurring from inter- and intra-strand conformations between the nucleobases compressing the DNA/RNA chains, related to their hydrogen-bonding motifs and π-stacking interactions, respectively. Extensive experimental work has been carried out recently in order to identify and separate the different contributions arising from these interactions, providing indicative fingerprints in the appearance of a long-lived spectroscopic signal originating from a π-stacking conformation within intra-strand sequences (Takaya et al., 2008; Su et al., 2012; Vayá et al., 2012; Chen et al., 2013, 2014; Chen and Kohler, 2014; Plasser et al., 2015) and to an ultrafast proton/hydrogen transfer stemming from a charge-transfer event in the hydrogen-bonding motifs responsible for the inter-strand interactions (Schwalb and Temps, 2007, 2008; Saurí et al., 2012). Given the abstruse nature of the photo-processes under study and their intrinsic difficulty, new development in methods and techniques are expected to play a crucial role on disentangling all the different deactivation channels (West et al., 2011, 2012; Krause et al., 2012; West and Moran, 2012; Fingerhut et al., 2013, 2014; Rivalta et al., 2014; Nenov et al., 2014a,b), even though the photophysical and photochemical studies on single nucleobases remain at the cornerstone of the field.
A proper characterization of the monomeric chromophores is therefore essential to understand the photoinduced events occurring in DNA/RNA from a bottom-up approach that can yield definitive answers regarding their role in photostability and photo-damage (Serrano-Andrés and Merchán, 2009). This avenue of research embodies the main efforts carried out by the research community over the last two decades (Kleinermanns et al., 2013; Giussani et al., 2013a; Barbatti et al., 2014; Chen et al., 2014; Mai et al., 2014), and even though plenty of advances have been made there is still no definitive consensus regarding the main deactivation routes present in the nucleobases (Crespo-Hernandez et al., 2004; Hudock et al., 2007; Barbatti et al., 2010; McFarland et al., 2014). Several decay paths have been identified both in vacuo (Giussani et al., 2013a) and in solution (Improta and Barone, 2014) and assigned to a number of processes, ranging from a barrierless deactivation through a ring-puckering motion of the bright ππ* state (Merchán and Serrano-Andrés, 2003; Gustavsson et al., 2006; Merchán et al., 2006; Serrano-Andrés et al., 2006, 2008; Conti et al., 2009; Improta and Barone, 2014) to a long-lived signal arising due to a possible crossing with a dark nπ* state (Hare et al., 2007), as well as an even longer-lived pathway widely attributed to the role of triplet states, specially prominent in non-canonical nucleobases with heavy-atom substitutions (Merchán et al., 2005; González-Luque et al., 2010; Martinez-Fernandez et al., 2012, 2014; Pollum et al., 2014).
In this study we turn out attention to water-solvated guanine monophosphate (GMP), one of the canonical DNA/RNA nucleobases. GMP has also been recently proposed to be an interesting compound for nanotechnological applications due to its outstanding capacity for self-assembly (Gupta et al., 2014), specially remarkable in its quadruplex form, which has even been located in DNA/RNA chains (Lam et al., 2013; Murat and Balasubramanian, 2014). Whereas, guanine in vacuo has been extensively studied at a high-level multireference ab initio level (Serrano-Andrés et al., 2008; Barbatti et al., 2010, 2011) as well as experimentally (Miannay et al., 2010; Chatterley et al., 2014), a lesser degree of scrutiny, mainly at a density functional theory level (Karunakaran et al., 2009; Parac et al., 2010; Santoro et al., 2010; Improta, 2014; Improta and Barone, 2014), has been considered on the effect of the phosphate group and the the solvent on the photoinduced processes occurring in this compound (Crespo-Hernandez et al., 2004). In the present work we propose a theoretical assessment of the deactivation routes embodying the main photophysical and photochemical features of GMP by employing high-level ab initio multireference perturbation theory methods coupled with a quantum-mechanical/molecular mechanics (QM/MM) approach, in order to ascertain the role of the environmental perturbations in these type of systems as they remain relatively unknown (Conti et al., 2010; Nachtigallová et al., 2010; Barbatti, 2014). Further knowledge on the environmental effects affecting the photo-processes occurring in the DNA/RNA chromophores upon UV-light irradiation will provide essential information that can be properly translated to water-solvated DNA/RNA systems such as those found in cells.
Computational Details
Sections MM Dynamics and Sampling and QM/MM Calculations describe the computational strategies and methodological details considered in the computations of the MM sampling dynamics and electronic structure QM/MM calculations carried out.
MM Dynamics and Sampling
MM simulations were performed for GMP in water to obtain a representative starting geometry to be employed for all subsequent computations and analyses. The MM dynamics calculations were carried out using the Amber-11 suite of programs making use of the Parm99 force field (Case et al., 2005, 2011). Initially, a cubic solvent box comprising 700 water molecules of explicit TIP3P (Jorgensen et al., 1983) with one Na+ counterion were considered. The entire system was then heated from 0 to 300 K for 1 ns at constant volume and constant pressure (1 atm), and then finally performing a production run for 100 ns recording the snapshots every 200 fs. To select the initial geometry we performed a cluster analysis based on a Root Mean Square (RMS) coordinate deviation analysis on the guanine moiety over all snapshots recorded along the MM dynamics run within a 2.0 Å difference using the MMTSB toolbox. We obtained three different clusters, denoted a, b, and c in Figure 1, with populations relating to the 93, 6, and 1% of the total number of structures obtained along the dynamics run, respectively. The selected initial structure was therefore chosen as the snapshot with the closest geometrical parameters to the centroid of the average structures obtained in cluster A, being the most representative.
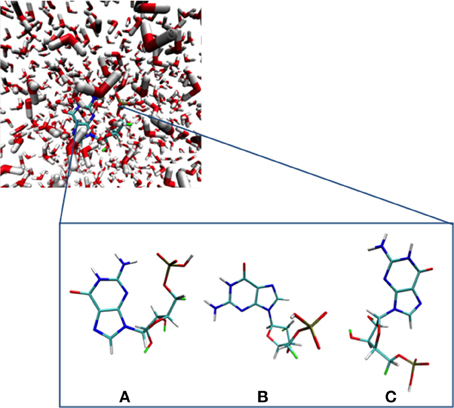
Figure 1. GMP and its different main conformations along the MM dynamics run obtained through a RMS deviation cluster analysis. (A–C) depict the most important conformations extracted from the molecular dynamics simulation (see text).
QM/MM Calculations
QM/MM calculations were performed using the COBRAMM interface developed in our group (Altoé et al., 2007a,b). The cut between the QM and MM regions has been done so that it includes all guanine atoms in the QM region, placing the link atom between the N9 of guanine and the carbon of the sugar ring directly attached to the nucleobase (see Figure 2). The choice of the cut between the QM and MM regions is justified in terms of charge redistribution on the nucleobase and its π-system in order to better reflect the covalent link between guanine and the monophosphate group. A three-layer approach (high, medium, and low) was used throughout (Altoé et al., 2007a): guanine was included in the QM region (high layer, ball, and stick representation in Figure 2), whereas the medium layer comprises the movable MM atoms within a 10 Å radius surrounding the GMP moiety, the remainder of the MM system being kept frozen during all optimization procedures in the low layer. Equilibrium geometries and photoreaction paths (Garavelli, 2006) were determined by using fully unconstrained optimizations and minimum energy path (MEP) computations on the relevant potential energy hypersurfaces by employing the intrinsic reaction coordinate (IRC) and optimization algorithms as implemented in the Gaussian 09 program package (Frisch et al., 2009) interfaced with COBRAMM. CI optimizations were performed with the gradient projection algorithm of Bearpark et al. (1994) as implemented in COBRAMM at the QM/MM level. Further details can be found in Conti et al. (2015).
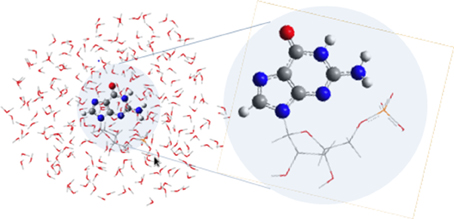
Figure 2. GMP and the three different QM/MM partitions defined in the present study. The high layer treating the guanine moiety at the QM level (ball and stick representation), the medium layer encompassing the movable MM water molecules and the phosphate group enclosed within a 10 Å radius distance from GMP, and the low layer containing the remainder of the MM region that is kept frozen throughout the calculations (see text).
Energies and gradients in the QM region were computed making use of the complete active space self-consistent field (CASSCF) and complete active space second-order perturbation theory (CASPT2) methods (Andersson et al., 1992; Roca-Sanjuán et al., 2012), as implemented in the MOLPRO-2010 (Werner et al., 2012a,b) and MOLCAS-7 (Aquilante et al., 2010, 2013) suite of programs, respectively. All gradient and non-adiabatic coupling calculations have been performed with the CASSCF implementation of the MOLPRO-2010 code. Subsequent CASPT2 calculations on the key structures obtained along the optimizations and MEPs have been carried out as implemented in the MOLCAS package in order to correct the energy values due to the lack of dynamic correlation present in the CASSCF method. This procedure is referred to as the CASSCF/CASPT2 protocol, and has been successfully employed over the years to tackle a variety of photoinduced phenomena from a theoretical standpoint (Garavelli, 2006; González-Ramírez et al., 2012; Segarra-Martí et al., 2012; Segarra-Martí and Coto, 2014). The active space selected comprised the full π space with the exception of the lowest occupied π orbital plus the three lone-pair orbitals to provide a proper description of the nπ* states, thus making 18 electrons in 13 orbitals. The removal of the lowest occupied π orbital is carried out due to its occupation number being very close to two and given that its removal does not affect the energy values obtained while speeding up the computations. An imaginary level shift of 0.2 a.u. was employed in the perturbation step to avoid intruder states. Two different basis sets were employed throughout: 6-31G* was used in order to map the hypersurfaces at the CASSCF level whereas atomic natural orbital (ANO) type (Pierloot et al., 1995) basis set with the large (ANO-L) primitive set C,N,O(14s9p4d3f)/H(8s4p3d) contracted to O[3s2p1d]/C[3s2p1d]/N[3s2p1d]/H[2s1p] (ANO-L 321/21 hereafter) was employed in order to refine the single-point CASPT2 energies.
Results and Discussion
The results are divided in three different sections: First, the main geometrical parameters of the ground-state Franck-Condon minimum of 9H-guanine both in vacuo and in solvated GMP are presented, together with its corresponding vertical spectra, drawing some conclusions on the influence the solvent and phosphate group have on the relative position of the excited states. Next, the different minima and conical intersections describing the photo-process are presented. Finally, a rationale of the photoinduced mechanisms in GMP is drawn in conjunction with previous results and experimental data acquired from the literature, yielding concluding notes on the photophysical and photochemical decay channels featuring in GMP and leading to its intrinsic photostability.
UV Absorption at the Franck-Condon Region
The optimized FC structure of GMP in solution (Figure 3) shows a strong resemblance with the in vacuo 9H-Guanine CASSCF/6-31G* structure previously reported in the literature (Serrano-Andrés et al., 2008). Table 1 presents the main geometrical parameters of both structures. As can be seen, bond lengths and dihedral angles are analogous giving rise to a planar structure. Small solvation effects are noticed in the GS structure mainly due to its relatively small dipole moment, providing slight shortenings in the C6-N1 bond distance in the presence of the solvent. A very shallow stationary point (La)sp, not present in the gas phase, has been located at the end of the La MEP featuring a pronounced elongation of the C2-N3 and a shortening of the N3-C4 distances, similar to those featured by the conical intersections between the La and GS states both in vacuo and in solution. The two CIs located between the La and GS states do show pronounced differences due to the solvent, yielding large elongations in the C2-N3 bond compared to its in vacuo counterparts. This is mainly due to the large dipole moment featured by the La state, which makes it a more influenced state upon solvation. A similar behavior can be seen in the CIs between the La and Lb states, prompting large deviations in the C4-C5, C5-C6 and specially pronounced in the C6-O distance, where water-solvated GMP suffers an elongation of 0.15 Å. The nOπ* state minimum does not show significant differences highlighting the scarce role polar solvation has on these types of states. The CIs between nOπ* and the polar ππ* La and Lb states are profoundly influenced by the solvent comparing them to their in vacuo counterparts, featuring large differences in the C4-C5 bond distance. These differences are mainly attributed to the polar character of the ππ* La/Lb states than to the effects on the nOπ* state. Overall, it can be concluded that solvation has an important effect on the polar ππ* La/Lb states, while being negligible for the nOπ* state. These states feature excitations that are prominently placed on the six-member ring of guanine featuring noticeable changes in the structure upon solvation, whereas the five-member ring remains relatively unchanged. A possible cause for this effect could lie on the presence of the phosphate group, which is tied to the five-ring member and could be shielding that molecular moiety from the surrounding water molecules thus mitigating its exposure as compared to the six-member ring and justifying why the latter suffers such pronounced changes upon solvation.
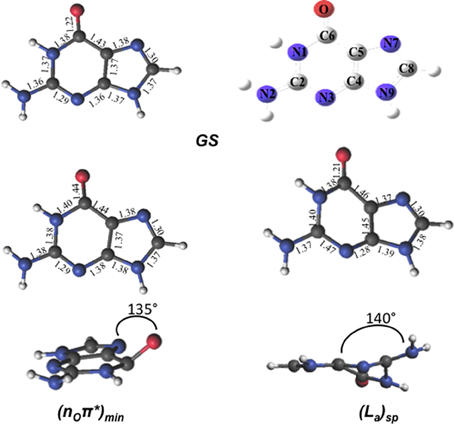
Figure 3. Geometries and main geometrical parameters of the Franck-Condon structure and excited-state stationary points characterized in GMP for its lowest-lying excited states computed at the CASSCF/6-31G* level of theory.
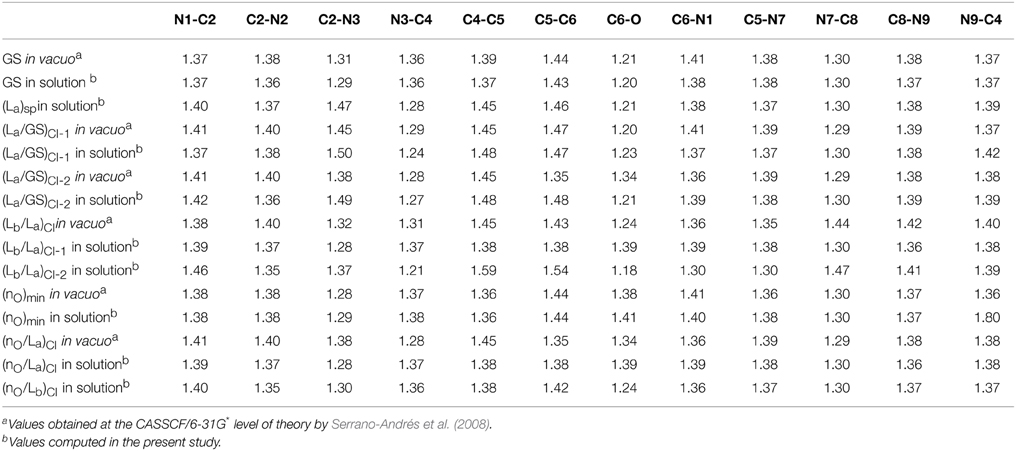
Table 1. Bond distances (in Å) characterizing the key structures involved in the photoinduced events of GMP upon UV-light irradiation.
Table 2 contains the vertical excitation energies at the CASPT2 level computed in the present study, together with several other computations and experimental values reported in the literature. We have used Platt's nomenclature (Platt, 1949), where La represents the ππ* excited state characterized by the largest contribution of the configuration HOMO (H) → LUMO (L) to the CASSCF wave function, whereas Lb depicts the ππ* excited state with a predominance of H → L+1 and H-1 → L configurations. By inspection of Table 2, it can be readily seen that there is a qualitative difference in the relative order of the lowest-lying excited states in vacuo and in solution: both cases feature the π π* La state as the energetically lowest-lying excited state, at 4.93 and 4.77 eV respectively, but while in vacuo S2 has nOπ* nature (5.54 eV, involving the oxygen lone pair of the purinic base), in solution S2 isthe ππ* Lb state, placed at 5.44 eV. This effect has been widely reported in the literature being related to a bathochromic effect (Nakayama et al., 2013), with the nOπ* featuring a dipole moment of 5.69 D (3.51 D in vacuo) being less stabilized than the ππ* Lb state (7.96 D, 4.92 D in vacuo) as it can be also noticed by looking at their change in dipole moment. The solvent therefore affects the absorption spectra prominently at the FC region by blue-shifting the nOπ* state by 0.23 and red-shifting the ππ* Lb by 0.33 eV with respect to the gas-phase, a change comparable to those previously reported in other QM/MM studies in solution (Parac et al., 2010; Nakayama et al., 2013). It is worth noting that the oscillator strengths associated to the transitions to the La and Lb states are also affected by the effect of the solvent. Whereas, La features as the brightest lowest-lying excited state in the gas phase and the Lb state presents a slightly smaller oscillator strength (0.158 and 0.145 respectively), in solvent La and Lb present an inverse order in the values for the oscillator strength associated to their transitions (0.09 and 0.17 respectively), being in agreement with the experimental data reported by femtosecond broad-band transient spectroscopy (Karunakaran et al., 2009).
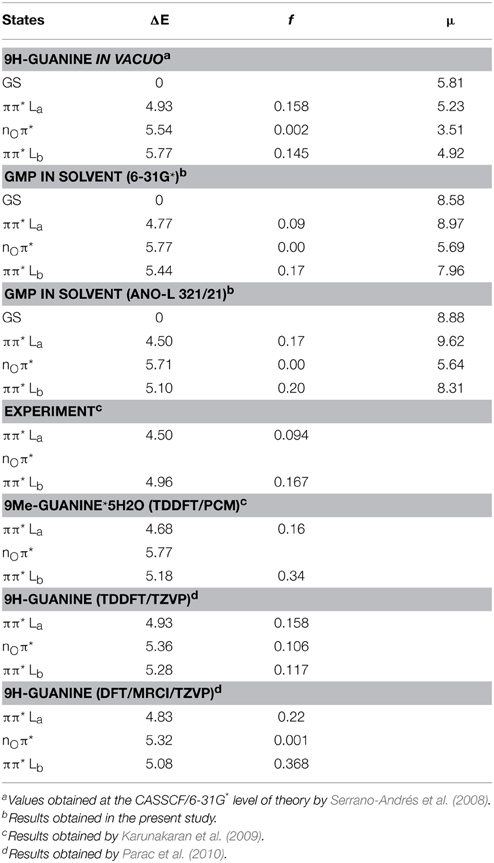
Table 2. Vertical absorption energies (ΔE, in eV), oscillator strengths (f) and dipole moments (μ, in Debye) computed at the FC region, together with previous theoretical and experimental data.
The effect of the basis set on the excitation energies has also been studied by employing both 6-31G* and ANO-L 321/21 basis sets. The 6-31G* CASPT2 computations report values of 4.77, 5.44, and 5.77 eV for the La, Lb, and nOπ* transitions, respectively, whereas the ANO-L yields absorption energy values of 4.50, 5.10, and 5.71 eV for the La, Lb, and nOπ* states respectively, consistent with those reported experimentally (Karunakaran et al., 2009), which place the absorption maximum of La at 4.50 eV. Table 2 also reports several theoretical approaches found in the literature employing different methods to simulate solvated GMP. As can be seen, more sophisticated DFT/MRCI methods agree qualitatively with the values here reported at the CASPT2 level of theory, yielding values within 0.33 and 0.02 for the La and Lb states respectively with respect to the CASPT2/ANO-L values here obtained, and a slight energetic deviation is also found when comparing to the values computed at a TDDFT level. This small difference present in TDDFT results employing a polarization continuum model (PCM) (see Table 2) could also be due to the fact that (Karunakaran et al., 2009) used a methylated guanine to model the effect of the phosphate group whereas here the phosphate group is explicitly included even if just at the MM level. On overall we can conclude that the absorption values reported here at the CASPT2/ANO-L level are consistent with the experimental data and with the highest-level theoretical estimates present in the literature, thus highlighting the capabilities of the CASSCF/CASPT2 protocol to treat excited states in a balanced manner (Roos et al., 1996) and its usage for mapping photochemical reaction paths (Garavelli, 2006).
Excited-State Evolution
The excited-state evolution is here tracked by means of static approaches through excited-state optimizations and the characterization of the conical intersections representing the crossings among the energetically lowest-lying excited states, ultimate protagonists in the deactivation photo-process. Additionally, the MEPs connecting the initially accessed states and subsequent photoinduced events have been mapped making use of the IRC technique.
The lowest-lying excited state is the ππ* La state, which is expected to be the main spectroscopic state due to its lowest-lying position and relatively large oscillator strength. This state is generally assumed to present an easily accessible CI along its relaxation pathway toward the ground-state characterized by a ring-puckering motion (see Figure 4) widely featured in the DNA/RNA nucleobases (Giussani et al., 2013a) in vacuo. This profile is slightly altered in the presence of polar environments as it has been previously reported for other purine nucleobases (Conti et al., 2010), where a shallow stationary point (La)sp arises along the MEP close to the CI with the ground state being placed at 1.15 eV vertically and adiabatically at 3.30 eV from the ground state (see Figure 5). Two different CIs have been characterized in the vicinity of this stationary point, one optimized directly in solvent corresponding to the minimum energy conical intersection (MECI) also reported by Serrano-Andrés et al. (2008), and another relating to the CI found in the same study along the MEP in vacuo, which we have tentatively named (La/GS)CI-1 and (La/GS)CI-2 in the present study, respectively. Both CIs as well as the (La)sp present ring-puckering structures in the A6 cycle yielding slight bond-length alterations (C2-N3, N1-C2, N3-C4, and C4-C5) compared to the FC structure and featuring prominent N1-N-C2-N3 dihedral angle distortions at 133°, 132°, and 140° for the (La/GS)CI-1, (La/GS)CI-2, and (La)sp structures, respectively. These distortions are quantitatively different to those reported in vacuo, stressing out the importance of the solvent where the (La/GS)CI-1 presents a ~143° dihedral angle as compared to its 133° solvated counterpart, together with slightly pronounced bond-length differences as can be seen in Table 1. Both CIs here characterized present very similar structures (see Figure 4) and charge distributions very close to those found in the gas phase, presenting an inversion in their energetic order in solution being (La/GS)CI-1 the energetically lowest-lying adiabatically at 3.31 eV, (La/GS)CI-2 being placed 0.3 eV higher in energy and 3.60 eV adiabatically from the FC region.
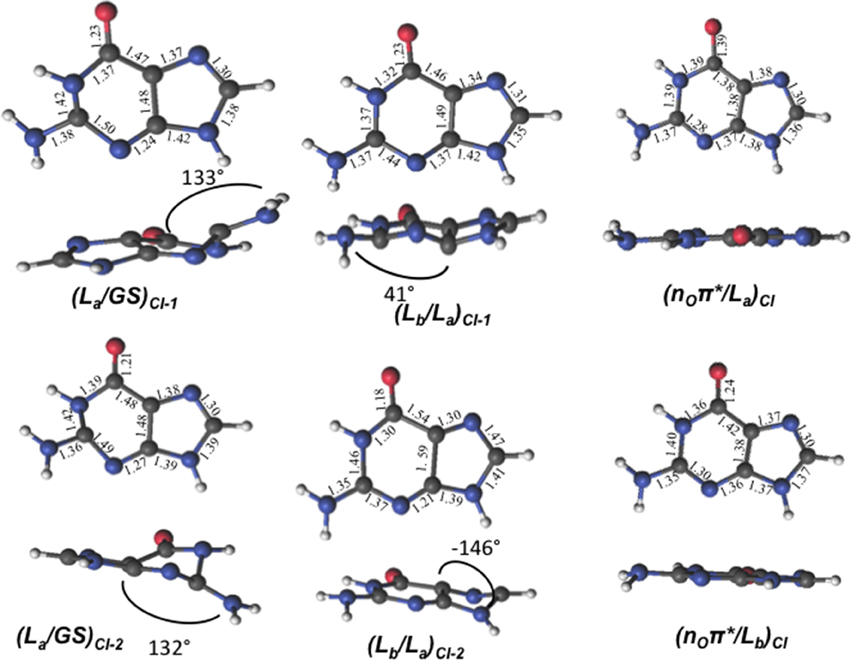
Figure 4. GMP geometries and main geometrical parameters of the characterized conical intersections between the lowest-lying excited states computed at the CASSCF/6-31G* level of theory.
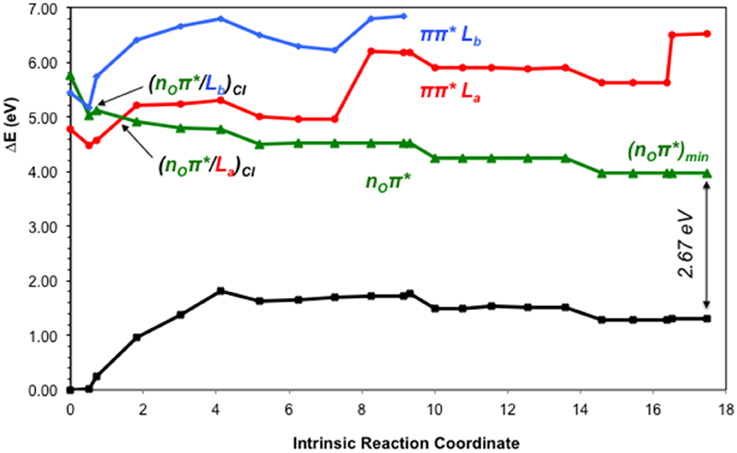
Figure 5. CASPT2 energies of the ground (GS) and lowest-lying singlet excited states (ππ* La, ππ* Lb and nOπ*) of the GMP from the FC geometry and along the ππ* La CASSCF MEP.
Two different CIs connecting the ππ* La and ππ* Lb states have also been located. Direct CI optimization leads to (Lb/La)CI-2, its geometry presenting a deformation near the N9-C8-N7 angle with respect to the ground state. The bond lengths suffer large distortions, observing a shortening of the N1-C6, C5-N7, C4-N3, N3-C2, C6-O, and C2-N bonds, and an increase in the length of the remaining bonds with respect to the FC region (see Figure 4). This CI is placed at 7.62 eV vertically from the FC region, which is higher in energy than both ππ* La and ππ* Lb vertical excitation energies, placed at 4.77 and 5.44 respectively, thus hinting toward a sloped CI profile preventing their non-adiabatic interaction. A second CI, (Lb/La)CI-1, obtained along the Lb MEP computed within a 4in4 active space to avoid excessive wave function mixing, has been located being placed ~5 eV adiabatically from the FC region. This CI presents geometrical similarities with its in vacuo counterpart and provide an accessible channel to funnel the initially populated Lbstate down to La. Further attempts to optimize the Lb state toward a possible minimum have been fruitless due to the large wave function mixing and root flipping problems experienced along the optimization procedure, preventing us to obtain further information on this specific state.
As previously stated, nOπ* represents the third energetically wise excited state for the GMP in solvent. The optimized nOπ* minimum found in solvent is prominently characterized by an elongation of the C6-O bond distance with respect to the ground state. Table 1 shows the main differences in bond lengths, where the C6-O bond is elongated in this minimum from 1.22 Å at the FC region to 1.41 Å. The minimum of this excited state in solvent is very similar to the one reported in vacuo (Serrano-Andrés et al., 2008), as would be expected given the small effects provided by the solvent on nπ* excited states. Energetically it is placed 3.97 eV adiabatically and 2.67 eV vertically with respect to the ground state (see Figure 6), which constitutes a stabilization of 1.8 eV from its initial value at the FC region.
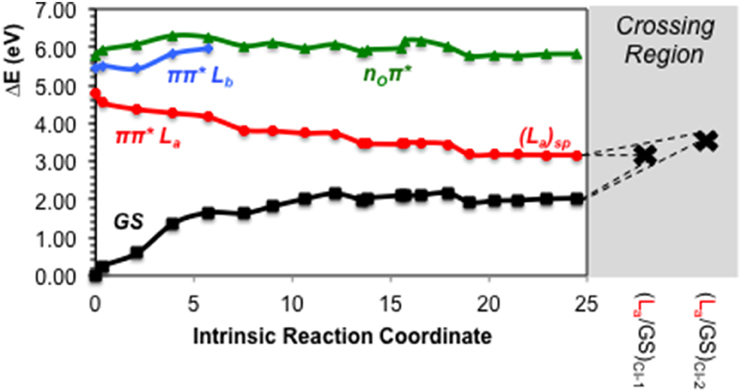
Figure 6. CASPT2 energies of the ground (GS) and lowest-lying singlet excited states (ππ* La, π π* Lb and nOπ*) of the GMP from the FC geometry and along the nOπ* CASSCF MEP.
We have also located the CI between the states nOπ* and π π* La, (nO/La)CI, its geometry presenting a planar structure like the ground state, but showing an elongation of the C6-O bond and a shortening of the C6-C5 bond. We observe similarities between this geometry and that of the CI optimized in vacuo, as they both present the same kind of geometrical distortions given the small solvent effects in the nOπ* state, yielding a relative energy of 4.90 eV with respect to the FC region. The CI connecting the nOπ* and π π* Lb states, (nO/Lb)CI, has also been located, featuring a planar structure and differences in the bond lengths in the six-member ring, in particular in the differences in the N1-C2, N1-C6 bonds.
The differences observed in the geometries of the excited state minima and of the different CIs characterized both in vacuo and in solvent highlight the importance of the solvent that is considered explicit in our calculations, together with the explicit presence of the ribose and phosphate groups on GMP, providing more accurate estimates to relate to the photoinduced events in the cellular system.
Photophysics and Photochemistry of GMP: Elucidating Its Intrinsic Photostability
Figure 7 features a scheme with the tentative photo-processes occurring upon UV irradiation in GMP based on our present computations and the theoretical and experimental data available in the literature. Experimentally, time-resolved transient absorption in solution by Karunakaran et al. (2009) suggest either bi- or triexponential decay depending on the spectral region measured, providing τ1 = 0.25 ps, τ2 = 1.0 ps, and τ3 = 2.5 ps for the 270–400 nm region in the non-protonated GMP at neutral pH. (Karunakaran et al., 2009) justify these ultrafast signals in terms of TDDFT calculations due to ring-puckering CIs between the π π* La and GS states, in accordance to what was previously proposed in vacuo by Serrano-Andrés et al. (2008) on the basis of CASSCF/CASPT2 computations. Molecular dynamics simulations by Serrano-Andrés et al. (2008) proposed that the ultrafast decay from the π π* La state could occur already within the first 100 fs, an assumption that has been challenged by Barbatti et al. (2011), pushing the timescale toward the 500 fs mark. (Lan et al., 2009), on the other hand, support a biexponential decay based on their MD simulations where they obtain a faster ~190 fs and a slower ~400 fs decays through two different CIs between the π π* La and GS states, thus highlighting the complex process under study and the difficulty to simulate it coherently (Mai et al., 2014). The most recent experimental data to our knowledge is based on the photoelectron spectroscopy of GMP both in vacuo and in solution (Chatterley et al., 2014), yielding a biexponential decay of τ1 = 50 fs and τ2 = 600 fs in vacuo and τ1 = 120 fs and τ2 = 680 fs in solution, which reveals a striking similarity among the two different set of values as they are within experimental error and therefore provide evidence of the negligible role played by the solvent in the photoinduced decay paths present in this system. This biexponential decay is obtained through a 4.66 eV pump, which also suggests the possibility of only probing those kinetic processes undergone after direct π π* La population, whereas the time-resolved transient data by Karunakaran et al. (2009) yields a third exponent related to a slower timescale possibly arisen through an initial population of the π π* Lb state.
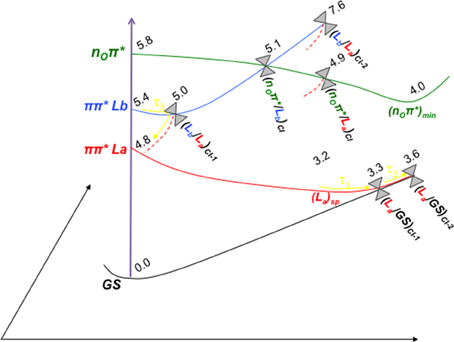
Figure 7. Scheme of the photoinduced processes occurring in solvated GMP upon UV-light irradiation. The energetic values (in eV) and the different crossing points and minima depicted refer to the CASPT2 computations carried out in the present study. The associated time constants marked in yellow have been taken tentatively from the ultrafast pump-probe transient absorption experiments reported by Karunakaran et al. (2009). Three different decay channels have been assigned to the experimental evidence: τ1 refers to the ultrafast decay path from an initially accessed π π* La excitation to the ring-puckering CI with the GS, τ2 to the initial population of the π π* Lb state and subsequent non-adiabatic population transfer to the La state finalizing in the CI between La and GS, and τ3 that is tentatively assigned mainly to deactivation processes along the Lb state (See text for details).
Our present results point toward an ultrafast deactivation along the La MEP characterized by a ring-puckering motion centered in the C2 atom, which brings the excited-state population to interact non-adiabatically with the ground state, funneling down the population that is further relaxed to the FC region. This kind of process has been widely attributed to the DNA/RNA nucleobases and is permanently linked with their intrinsic photostability (Serrano-Andrés and Merchán, 2009) and therefore extends to the photostability of the genomic material. Our calculations in the presence of the solvent and the sugar and phosphate groups reveal a very shallow stationary point at the end of the MEP right before reaching the intersection seam (see Figure 5), which has also been previously documented in other nucleobases at a TDDFT level of theory to be related to a transition state with an imaginary reaction coordinate that drives the system toward the ring-puckering CI (Picconi et al., 2011). This stationary point is explained in terms of the planarity of the π π* potential energy hypersurfaces of all nucleobases (Giussani et al., 2013a; Improta and Barone, 2014), and is expected to be overcome in order to reach the CIs with the ground state in an ultrafast manner. In contrast, we have shown recently (Conti et al., 2010, 2015) that the corresponding internal conversion path in adenine is controlled by a larger barrier, which may explain its longer lifetimes. Two different CIs have been located between the π π* La and GS states, namely (La/GS)CI-1 and (La/GS)CI-2. These two CIs are analogous to those found in vacuo by Serrano-Andrés et al. (2008) but present an energetic inversion due to the embedding effect of the solvent. Both are expected to belong to the same intersection seam given their similar structures, and both are considered to be accessible from the La stationary point previously characterized. (La/GS)CI-1, being the lowest-lying energy wise is expected to embody the fastest decay route, relating to the τ1 = 0.25 ps recorded through time-resolved transient and τ1 = 120 fs in time-resolved photoelectron spectroscopy experiments. This assignment is analogous to that previously reported for 9H-Guanine in vacuo, and supports recent experiments reporting analogous excited-state decays for GMP in gas-phase and in solution (Chatterley et al., 2014). The second conical intersection with the ground state, (La/GS)CI-2, might be responsible for the second exponential measured at τ2 = 1.0 ps and τ2 = 680 fs in time-resolved transient and photoelectron spectroscopy, respectively, given its higher-lying energetic position and a relatively small barrier of ~0.3 eV to be overcome in order to access it once reaching the intersection seam. Nevertheless, the existence of another CI between the La and GS states not characterized in the present contribution could better explain that second lifetime, which will be related to the La surface as it has been recently reported experimentally (Chatterley et al., 2014). These assignments are in line with the MD studies by Lan et al. (2009), reporting two different decay routes with their associated timescales to two distinct CIs between the π π* La and the ground state, and are in line with other theoretical CASPT2 and TDDFT studies that point toward the La state as the responsible for both ultrafast decay timescales.
The π π* Lb state is predicted to be involved in the photoinduced events being related possibly only to the longest-lived τ3. This is explained in terms of an initial population of the Lb state, which presents a sizable oscillator strength, and subsequent funneling of the population toward the La state mediated by the (Lb/La)CI-1, placed at ~5 eV along the Lb relaxation pathway. The second CI characterized between Lb and La states, (Lb/La)CI-2, is expected to be irrelevant to the photo-process due to its high-lying energetic position preventing its accessibility. The lone-pair excited state, nOπ*, is blue-shifted in solution as has been already mentioned above. Its elevated vertical absorption energy prevents it to be one of the main spectroscopic states, yet its close-lying position to the absorbing π π* Lb state facilitates a possible population of this state and subsequent relaxation toward its minimum, (nOπ*)min. This minimum is placed at 3.97 eV adiabatically and 2.67 eV vertically from the GS, and could be partly responsible of the longest-lived signal reported experimentally at τ3 = 2.5 ps or at τ3 = 167 ps at low pH, given that such nπ*-mediated processes have been already characterized experimentally to be close to the ~100 ps timescale on pyrimidines both in vacuo and in solution (Hare et al., 2007).
The present study elucidates the photoinduced events in GMP in terms of an ultrafast decay along the main spectroscopic and initially accessed π π* La state characterized by a ring-puckering motion, which would cover the experimental timescales τ1 and τ2 through different CIs with the ground state, whereas the longest-lived component would be attributed to the decay routes mediated through the π π* Lb and, to a minor extent, to the nOπ* state.
Conclusions
The present study encompasses a photophysical and photochemical appraisal of water-solvated GMP by means of theoretical multireference perturbation theory QM/MM techniques. An initial MD simulation has been carried out in order to characterize the geometrical parameters of the FC region. The vertical excitation energies have been computed and compared to recent data found in the literature and to the results obtained in vacuo, highlighting the importance of the environment yielding qualitative differences for the π π* La and π π* Lb states being red-shifted and for the nOπ* state being blue-shifted as compared to their gas-phase counterpart. The π π* La state is predicted to be the main spectroscopic state driving the ultrafast deactivation processes occurring in GMP upon UV-light irradiation based on a ring-puckering motion that enhances its non-adiabatic interaction with the ground state in a radiationless manner. A shallow stationary point toward the end of the π π* La MEP has been characterized, together with two different CIs with the ground state that help rationalize the two fastest decay times measured experimentally. Upon initial Lb absorption, two CIs between the π π* Lb and La states have also been located, one of them along the Lb decay path pointing toward a population funneling down to the La state and another being too high in energy to contribute prominently in the photo-process. The CIs connecting the nOπ* state and the π π* Lb and La states have also been characterized along its relaxation route, yielding a minimum in the nOπ* state expected to emit vertically at ~2.7 eV. Both π π* Lb and nOπ* are suggested to contribute to the longest-lived experimental timescale, the latter providing a lesser contribution given the relatively fast kinetic timescale and the long-lived timescales expected in nπ* fluorescent states. Upcoming QM/MM dynamics simulations are expected to shed some more light on the photoinduced events occurring in water-solvated GMP and its specific decay timescales in order to provide a more specific molecular counterpart to the experiment and better explain the photochemical and photophysical processes resulting in the intrinsic stability of the genomic material.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MG acknowledges support by the European Research Council Advanced Grant STRATUS (ERC-2011-AdG No. 291198). We acknowledge the use of HPC resources of the “Pôle Scientifique de Modélisation Numérique” at the ENS-Lyon, France.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fchem.2015.00029/abstract
References
Altoé, P., Stenta, M., Bottoni, A., and Garavelli, M. (2007a). A tunable QM/MM approach to chemical reactivity, structure and physico-chemical properties prediction. Theor. Chem. Acc. 118, 219–240. doi: 10.1007/s00214-007-0275-9
Altoé, P., Stenta, M., Bottoni, A., and Garavelli, M. (2007b). COBRAMM: a tunable QM/MM approach to complex molecular architectures. Modelling the excited and ground state properties of sized molecular systems. AIP Conf. Proc. 963, 491–505. doi: 10.1063/1.2827033
Andersson, K., Malmqvist, P.-Å., and Roos, B. O. (1992). 2nd-order perturbation-theory with a complete active space self-consistent field reference function. J. Chem. Phys. 96, 1218–1226. doi: 10.1063/1.462209
Aquilante, F., De Vico, L., Ferre, N., Ghigo, G., Malmqvist, P.-A., Neogrady, P., et al. (2010). Software news and update MOLCAS 7: the next generation. J. Comput. Chem. 31, 224–247. doi: 10.1002/jcc.21318
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aquilante, F., Pedersen, T. B., Veryazov, V., and Lindh, R. (2013). MOLCAS—a software for multiconfigurational quantum chemistry calculations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 3, 143–149. doi: 10.1002/wcms.1117
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barbatti, M., Aquino, A. J. A., Szymczak, J. J., Nachtigallova, D., Hobza, P., and Lischka, H. (2010). Relaxation mechanisms of UV-photoexcited DNA and RNA nucleobases. Proc. Natl. Acad. Sci. U.S.A. 107, 21453–21458. doi: 10.1073/pnas.1014982107
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barbatti, M., Borin, A., and Ullrich, S. (2014). Photoinduced Processes in Nucleic Acids. Berlin Heidelberg: Springer.
Barbatti, M., Szymczak, J. J., Aquino, A. J., Nachtigallová, D., and Lischka, H. (2011). The decay mechanism of photoexcited guanine - a nonadiabatic dynamics study. J. Chem. Phys. 134:014304. doi: 10.1063/1.3521498
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Barbatti, M. (2014). Photorelaxation induced by water–chromophore electron transfer. J. Am. Chem. Soc. 136, 10246–10249. doi: 10.1021/ja505387c
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Bearpark, M. J., Robb, M. A., and Bernhard Schlegel, H. (1994). A direct method for the location of the lowest energy point on a potential surface crossing. Chem. Phys. Lett. 223, 269–274. doi: 10.1016/0009-2614(94)00433-1
Brash, D. E. (2015). UV signature mutations. Photochem. Photobiol. 91, 15–26. doi: 10.1111/php.12377
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Cadet, J., Mouret, S., Ravanat, J.-L., and Douki, T. (2012). Photoinduced damage to cellular DNA: direct and photosensitized reactions†. Photochem. Photobiol. 88, 1048–1065. doi: 10.1111/j.1751-1097.2012.01200.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Callis, P. R. (1983). Electronic states and luminiscence of nucleic-acid systems. Annu. Rev. Phys. Chem. 34, 329–357. doi: 10.1146/annurev.pc.34.100183.001553
Case, D., Darden, T. A., Cheatham, T. E., Simmerling, C., Wang, J., Duke, R., et al. (2011). Amber 11. San Francisco, CA: University of California.
Case, D. A., Cheatham, T. E., Darden, T., Gohlke, H., Luo, R., Merz, K. M., et al. (2005). The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688. doi: 10.1002/jcc.20290
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chatterley, A. S., West, C. W., Stavros, V. G., and Verlet, J. R. R. (2014). Time-resolved photoelectron imaging of the isolated deprotonated nucleotides. Chem. Sci. 5, 3963–3975. doi: 10.1039/C4SC01493F
Chen, J., and Kohler, B. (2014). Base stacking in adenosine dimers revealed by femtosecond transient absorption spectroscopy. J. Am. Chem. Soc. 136, 6362–6372. doi: 10.1021/ja501342b
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, J., Thazhathveetil, A. K., Lewis, F. D., and Kohler, B. (2013). Ultrafast excited-state dynamics in hexaethyleneglycol-linked DNA homoduplexes made of A·T base pairs. J. Am. Chem. Soc. 135, 10290–10293. doi: 10.1021/ja4049459
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Chen, J., Zhang, Y., and Kohler, B. (2014). Excited States in DNA Strands Investigated by Ultrafast Laser Spectroscopy. Berlin; Heidelberg: Springer.
Conti, I., Altoe, P., Stenta, M., Garavelli, M., and Orlandi, G. (2010). Adenine deactivation in DNA resolved at the CASPT2//CASSCF/AMBER level. Phys. Chem. Chem. Phys. 12, 5016–5023. doi: 10.1039/b926608a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Conti, I., Garavelli, M., and Orlandi, G. (2009). Deciphering low energy deactivation channels in adenine. J. Am. Chem. Soc. 131, 16108–16118. doi: 10.1021/ja902311y
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Conti, I., Nenov, A., Höfinger, S., Flavio Altavilla, S., Rivalta, I., Dumont, E., et al. (2015). Excited state evolution of DNA stacked adenines resolved at the CASPT2//CASSCF/Amber Level: from the bright to the excimer state and back. Phys. Chem. Chem. Phys. 17, 7291–7302. doi: 10.1039/C4CP05546B
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Crespo-Hernandez, C. E., Cohen, B., Hare, P. M., and Kohler, B. (2004). Ultrafast excited-state dynamics in nucleic acids. Chem. Rev. 104, 1977–2019. doi: 10.1021/cr0206770
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fingerhut, B. P., Dorfman, K. E., and Mukamel, S. (2013). Monitoring nonadiabatic dynamics of the RNA base uracil by UV pump–IR probe spectroscopy. J. Phys. Chem. Lett. 4, 1933–1942. doi: 10.1021/jz400776r
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Fingerhut, B. P., Dorfman, K. E., and Mukamel, S. (2014). Probing the conical intersection dynamics of the RNA base uracil by UV-pump stimulated-Raman-probe signals; ab initio simulations. J. Chem. Theory Comput. 10, 1172–1188. doi: 10.1021/ct401012u
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09. Wallingford, CT: Gaussian, Inc.
Garavelli, M. (2006). Computational organic photochemistry: strategy, achievements and perspectives. Theor. Chem. Acc. 116, 87–105. doi: 10.1007/s00214-005-0030-z
Giussani, A., Segarra-Martí, J., Roca-Sanjuán, D., and Merchán, M. (2013a). Excitation of nucleobases from a computational perspective I: reaction paths. Top. Curr. Chem. 355, 57–97. doi: 10.1007/128_2013_501
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Giussani, A., Serrano-Andrés, L., Merchán, M., Roca-Sanjuán, D., and Garavelli, M. (2013b). Photoinduced formation mechanism of the thymine–thymine (6–4) adduct. J. Phys. Chem. B 117, 1999–2004. doi: 10.1021/jp307200g
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
González-Luque, R., Climent, T., González-Ramírez, I., Merchán, M., and Serrano-Andrés, L. (2010). Singlet-triplet states interaction regions in DNA/RNA nucleobase hypersurfaces. J. Chem. Theory Comput. 6, 2103–2114. doi: 10.1021/ct100164m
González-Ramírez, I., Segarra-Martí, J., Serrano-Andrés, L., Merchán, M., Rubio, M., and Roca-Sanjuán, D. (2012). On the N1–H and N3–H Bond dissociation in uracil by low energy electrons: a CASSCF/CASPT2 study. J. Chem. Theory Comput. 8, 2769–2776. doi: 10.1021/ct300153f
Gupta, A. P., Taylor, W. J., McGown, L. B., and Kempf, J. G. (2014). NMR studies of the chiral selectivity of self-assembled guanosinemonophosphate. J. Phys. Chem. B. 118, 14243–14256. doi: 10.1021/jp5075016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Gustavsson, T., Banyasz, A., Lazzarotto, E., Markovitsi, D., Scalmani, G., Frisch, M. J., et al. (2006). Singlet excited-state behavior of uracil and thymine in aqueous solution: a combined experimental and computational study of 11 uracil derivatives. J. Am. Chem. Soc. 128, 607–619. doi: 10.1021/ja056181s
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hare, P. M., Crespo-Hernández, C. E., and Kohler, B. (2007). Internal conversion to the electronic ground state occurs via two distinct pathways for pyrimidine bases in aqueous solution. Proc. Natl. Acad. Sci. U.S.A. 104, 435–440. doi: 10.1073/pnas.0608055104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hudock, H. R., Levine, B. G., Thompson, A. L., Satzger, H., Townsend, D., Gador, N., et al. (2007). Ab initio molecular dynamics and time-resolved photoelectron spectroscopy of electronically excited uracil and thymine. J. Phys. Chem. A 111, 8500–8508. doi: 10.1021/jp0723665
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Improta, R. (2014). Quantum mechanical calculations unveil the structure and properties of the absorbing and emitting excited electronic states of guanine quadruplex. Chem. Eur. J. 20, 8106–8115. doi: 10.1002/chem.201400065
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Improta, R., and Barone, V. (2014). Excited States Behavior of Nucleobases in Solution: Insights from Computational Studies. Berlin Heidelberg: Springer.
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. doi: 10.1063/1.445869
Kamiya, Y., and Asanuma, H. (2014). Light-driven DNA nanomachine with a photoresponsive molecular engine. Acc. Chem. Res. 47, 1663–1672. doi: 10.1021/ar400308f
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Karunakaran, V., Kleinermanns, K., Improta, R., and Kovalenko, S. A. (2009). Photoinduced dynamics of guanosine monophosphate in water from broad-band transient absorption spectroscopy and quantum-chemical calculations. J. Am. Chem. Soc. 131, 5839–5850. doi: 10.1021/ja810092k
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Kleinermanns, K., Nachtigallova, D., and de Vries, M. S. (2013). Excited state dynamics of DNA bases. Int. Rev. Phys. Chem. 32, 308–342. doi: 10.1080/0144235X.2012.760884
Kohler, B. (2010). Nonradiative decay mechanisms in DNA model systems. J. Phys. Chem. Lett. 1, 2047–2053. doi: 10.1021/jz100491x
Krause, P., Matsika, S., Kotur, M., and Weinacht, T. (2012). The influence of excited state topology on wavepacket delocalization in the relaxation of photoexcited polyatomic molecules. J. Chem. Phys. 137:22A537. doi: 10.1063/1.4748580
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lam, E. Y., Beraldi, D., Tannahill, D., and Balasubramanian, S. (2013). G-quadruplex structures are stable and detectable in human genomic DNA. Nat. Commun. 4, 1796. doi: 10.1038/ncomms2792
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Lan, Z., Fabiano, E., and Thiel, W. (2009). Photoinduced nonadiabatic dynamics of 9H-guanine. Chemphyschem 10, 1225–1229. doi: 10.1002/cphc.200900030
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mai, S., Richter, M., Marquetand, P., and González, L. (2014). Excitation of Nucleobases from a Computational Perspective II: Dynamics. Berlin Heidelberg: Springer.
Markovitsi, D., Gustavsson, T., and Vayá, I. (2010). Fluorescence of DNA duplexes: from model helices to natural DNA. J. Phys. Chem. Lett. 1, 3271–3276. doi: 10.1021/jz101122t
Martínez-Fernández, L., Corral, I., Granucci, G., and Persico, M. (2014). Competing ultrafast intersystem crossing and internal conversion: a time resolved picture for the deactivation of 6-thioguanine. Chem. Sci. 5, 1336–1347. doi: 10.1039/c3sc52856a
Martinez-Fernandez, L., Gonzalez, L., and Corral, I. (2012). An ab initio mechanism for efficient population of triplet states in cytotoxic sulfur substituted DNA bases: the case of 6-thioguanine. Chem. Commun. 48, 2134–2136. doi: 10.1039/c2cc15775f
McFarland, B. K., Farrell, J. P., Miyabe, S., Tarantelli, F., Aguilar, A., Berrah, N., et al. (2014). Ultrafast X-ray Auger probing of photoexcited molecular dynamics. Nat. Commun. 5:4235. doi: 10.1038/ncomms5235
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Merchán, M., González-Luque, R., Climent, T., Serrano-Andrés, L., Rodriguez, E., Reguero, M., et al. (2006). Unified model for the ultrafast decay of pyrimidine nucleobases. J. Phys. Chem. B 110, 26471–26476. doi: 10.1021/jp066874a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Merchán, M., and Serrano-Andrés, L. (2003). Ultrafast internal conversion of excited cytosine via the lowest pi pi* electronic singlet state. J. Am. Chem. Soc. 125, 8108–8109. doi: 10.1021/ja0351600
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Merchán, M., Serrano-Andrés, L., Robb, M. A., and Blancafort, L. (2005). Triplet-state formation along the ultrafast decay of excited singlet cytosine. J. Am. Chem. Soc. 127, 1820–1825. doi: 10.1021/ja044371h
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Miannay, F.-A., Gustavsson, T., Banyasz, A., and Markovitsi, D. (2010). Excited-state dynamics of dGMP measured by steady-state and femtosecond fluorescence spectroscopy†. J. Phys. Chem. A 114, 3256–3263. doi: 10.1021/jp909410b
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Murat, P., and Balasubramanian, S. (2014). Existence and consequences of G-quadruplex structures in DNA. Curr. Opin. Genet. Dev. 25, 22–29. doi: 10.1016/j.gde.2013.10.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nachtigallová, D., Zelenı, T., Ruckenbauer, M., Müller, T., Barbatti, M., Hobza, P., et al. (2010). Does stacking restrain the photodynamics of individual nucleobases? J. Am. Chem. Soc. 132, 8261–8263. doi: 10.1021/ja1029705
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nakayama, A., Arai, G., Yamazaki, S., and Taketsugu, T. (2013). Solvent effects on the ultrafast nonradiative deactivation mechanisms of thymine in aqueous solution: excited-state QM/MM molecular dynamics simulations. J. Chem. Phys. 139:214304. doi: 10.1063/1.4833563
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nenov, A., Rivalta, I., Cerullo, G., Mukamel, S., and Garavelli, M. (2014a). Disentangling peptide configurations via two-dimensional electronic spectroscopy: ab initio simulations beyond the Frenkel Exciton Hamiltonian. J. Phys. Chem. Lett. 5, 767–771. doi: 10.1021/jz5002314
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Nenov, A., Segarra-Martí, J., Giussani, A., Conti, I., Rivalta, I., Dumont, E., et al. (2014b). FD 177: probing deactivation pathways of DNA nucleobases by two-dimensional electronic spectroscopy: first principles simulations. Faraday Discuss. doi: 10.1039/C4FD00175C. (in press).
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Noonan, F. P., Zaidi, M. R., Wolnicka-Glubisz, A., Anver, M. R., Bahn, J., Wielgus, A., et al. (2012). Melanoma induction by ultraviolet A but not ultraviolet B radiation requires melanin pigment. Nat. Commun. 3, 884. doi: 10.1038/ncomms1893
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Parac, M., Doerr, M., Marian, C. M., and Thiel, W. (2010). QM/MM calculation of solvent effects on absorption spectra of guanine. J. Comput. Chem. 31, 90–106. doi: 10.1002/jcc.21233
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Picconi, D., Barone, V., Lami, A., Santoro, F., and Improta, R. (2011). The interplay between ππ*/nπ* excited states in gas-phase thymine: a quantum dynamical study. Chemphyschem 12, 1957–1968. doi: 10.1002/cphc.201001080
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pierloot, K., Dumez, B., Widmark, P. O., and Roos, B. O. (1995). Density matrix averaged atomic natural orbital (ANO) basis sets for correlated molecular wave functions. IV. Medium size basis sets for the atoms H-Kr. Theor. Chim. Acta 90, 87–114. doi: 10.1007/BF01113842
Plasser, F., Aquino, A. J. A., Lischka, H., and Nachtigallová, D. (2015). Electronic excitation processes in single-strand and double-strand DNA: a computational approach. Top. Curr. Chem. 356, 1–37. doi: 10.1007/128_2013_517
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Platt, J. R. (1949). Classification of spectra of cata−condensed hydrocarbons. J. Chem. Phys. 17, 484–495. doi: 10.1063/1.1747293
Pollum, M., Martínez-Fernández, L., and Crespo-Hernández, C. (2014). Photochemistry of Nucleic Acid Bases and Their Thio- and Aza-Analogues in Solution. Berlin Heidelberg: Springer.
Rivalta, I., Nenov, A., Cerullo, G., Mukamel, S., and Garavelli, M. (2014). Ab initio simulations of two-dimensional electronic spectra: the SOS//QM/MM approach. Int. J. Quantum Chem. 114, 85–93. doi: 10.1002/qua.24511
Roca-Sanjuán, D., Aquilante, F., and Lindh, R. (2012). Multiconfiguration second-order perturbation theory approach to strong electron correlation in chemistry and photochemistry. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 585–603. doi: 10.1002/wcms.97
Roos, B. O., Andersson, K., Fulscher, M. P., Malmqvist, P. A., Serrano-Andrés, L., Pierloot, K., et al. (1996). Multiconfigurational perturbation theory: applications in electronic spectroscopy. Adv. Chem. Phys. Vol. Xciii 93, 219–331. doi: 10.1002/9780470141526.ch5
Santoro, F., Barone, V., Lami, A., and Improta, R. (2010). The excited electronic states of adenine-guanine stacked dimers in aqueous solution: a PCM/TD-DFT study. Phys. Chem. Chem. Phys. 12, 4934–4948. doi: 10.1039/b925108a
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Saurí, V., Gobbo, J. P., Serrano-Pérez, J. J., Lundberg, M., Coto, P. B., Serrano-Andrés, L., et al. (2012). Proton/hydrogen transfer mechanisms in the guanine-cytosine base pair: photostability and tautomerism. J. Chem. Theory Comput. 9, 481–496. doi: 10.1021/ct3006166
Schwalb, N. K., and Temps, F. (2007). Ultrafast electronic relaxation in guanosine is promoted by hydrogen bonding with cytidine. J. Am. Chem. Soc. 129, 9272–9273. doi: 10.1021/ja073448
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Schwalb, N. K., and Temps, F. (2008). Base sequence and higher-order structure induce the complex excited-state dynamics in DNA. Science 322, 243–245. doi: 10.1126/science.1161651
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Segarra-Martí, J., and Coto, P. B. (2014). A theoretical study of the intramolecular charge transfer in 4-(dimethylamino)benzethyne. Phys. Chem. Chem. Phys. 16, 25642–25648. doi: 10.1039/C4CP03436H
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Segarra-Martí, J., Roca-Sanjuán, D., Merchán, M., and Lindh, R. (2012). On the photophysics and photochemistry of the water dimer. J. Chem. Phys. 137, 244309. doi: 10.1063/1.4772187
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Serrano-Andrés, L., and Merchán, M. (2009). Are the five natural DNA/RNA base monomers a good choice from natural selection? A photochemical perspective. J. Photochem. Photobiol. C Photochem. Rev. 10, 21–32. doi: 10.1016/j.jphotochemrev.2008.12.001
Serrano-Andrés, L., Merchán, M., and Borin, A. C. (2006). Adenine and 2-aminopurine: paradigms of modern theoretical photochemistry. Proc. Natl. Acad. Sci. U.S.A. 103, 8691–8696. doi: 10.1073/pnas.0602991103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Serrano-Andrés, L., Merchán, M., and Borin, A. C. (2008). A three-state model for the photophysics of guanine. J. Am. Chem. Soc. 130, 2473–2484. doi: 10.1021/ja0744450
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Su, C., Middleton, C. T., and Kohler, B. (2012). Base-stacking disorder and excited-state dynamics in single-stranded adenine homo-oligonucleotides. J. Phys. Chem. B 116, 10266–10274. doi: 10.1021/jp305350t
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Takaya, T., Su, C., Harpe, K. D. L., Crespo-Hernández, C. E., and Kohler, B. (2008). UV excitation of single DNA and RNA strands produces high yields of exciplex states between two stacked bases. Proc. Natl. Acad. Sci. U.S.A. 105, 10285–10290. doi: 10.1073/pnas.0802079105
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Vayá, I., Gustavsson, T., Douki, T., Berlin, Y., and Markovitsi, D. (2012). Electronic excitation energy transfer between nucleobases of natural DNA. J. Am. Chem. Soc. 134, 11366–11368. doi: 10.1021/ja304328g
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Werner, H.-J., Knowles, P. J., Knizia, G., Manby, F. R., and Schütz, M. (2012a). Molpro: a general-purpose quantum chemistry program package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 242–253. doi: 10.1002/wcms.82
Werner, H.-J., Knowles, P. J., Knizia, G., Manby, F. R., Schütz, M., Celani, P., et al. (2012b). MOLPRO, Version 2012.1. A Package of ab Initio Programs.
West, B. A., and Moran, A. M. (2012). Two-dimensional electronic spectroscopy in the ultraviolet wavelength range. J. Phys. Chem. Lett. 3, 2575–2581. doi: 10.1021/jz301048n
West, B. A., Womick, J. M., and Moran, A. M. (2011). Probing ultrafast dynamics in adenine with mid-UV four-wave mixing spectroscopies. J. Phys. Chem. A 115, 8630–8637. doi: 10.1021/jp204416m
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
West, B. A., Womick, J. M., and Moran, A. M. (2012). Interplay between vibrational energy transfer and excited state deactivation in DNA components. J. Phys. Chem. A 117, 5865–5874. doi: 10.1021/jp306799e
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: DNA, purine, CASSCF/CASPT2, photochemistry, QM/MM, photostability, GMP, guanine
Citation: Altavilla SF, Segarra-Martí J, Nenov A, Conti I, Rivalta I and Garavelli M (2015) Deciphering the photochemical mechanisms describing the UV-induced processes occurring in solvated guanine monophosphate. Front. Chem. 3:29. doi: 10.3389/fchem.2015.00029
Received: 22 January 2015; Accepted: 30 March 2015;
Published: 20 April 2015.
Edited by:
Antonio Monari, Université de Lorraine, FranceReviewed by:
Michael Bearpark, Imperial College London, UKChantal Daniel, Centre National de la Recherche Scientifique, France
Copyright © 2015 Altavilla, Segarra-Martí, Nenov, Conti, Rivalta and Garavelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Javier Segarra-Martí and Marco Garavelli, École Normale Supérieure de Lyon, Centre National de la Recherche Scientifique, UMR 5182, Université de Lyon, 46 Allée d'Italie, Lyon 69364, FranceamF2aWVyLnNlZ2FycmFAdW5pYm8uaXQ=;bWFyY28uZ2FyYXZlbGxpQHVuaWJvLml0