 Kuan Zheng
Kuan Zheng Ran Hong
Ran Hong- CAS Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, China
Diverse array of biopolymers and second metabolites (particularly polyketide natural products) has been manufactured in nature through an enzymatic iterative assembly of simple building blocks. Inspired by this strategy, molecules with inherent modularity can be efficiently synthesized by repeated succession of similar reaction sequences. This privileged strategy has been widely adopted in synthetic supramolecular chemistry. Its value also has been reorganized in natural product synthesis. A brief overview of this approach is given with a particular emphasis on the total synthesis of polyol-embedded polyketides, a class of vastly diverse structures and biologically significant natural products. This viewpoint also illustrates the limits of known individual modules in terms of diastereoselectivity and enantioselectivity. More efficient and practical iterative strategies are anticipated to emerge in the future development.
Polyketides are a class of secondary metabolites being impressive not only with structurally intriguing carbon skeletons but also with strong pharmacological relevance. A wealth of biologically important activities including antitumor, antibiotic, cytostatic, antiparasitic and immunosuppressive properties, and many of them or their derivatives have become therapeutics for clinical use. Over the past five decades tremendous progress has been advanced in the field of chemical synthesis of polyketides including macrolides, polyphenols, polyethers, polyenes, and enediynes. Numerous innovative strategies and tactics have been pursued and illustrated in many elegant total syntheses. However, polyketide with a moderate–level complexity still posts a formidable challenge to cumulate enough quantity for clinical evaluation. The success of anticancer drug Halaven® (Eribulin) from Eisai and the Kishi's lab is considered as a triumph in the field of polyketide synthetic chemistry. Nevertheless, how many can polyketide drug leads be eventually evolved to clinical use from the synthetic laboratory? Sophisticated approaches to rapidly and flexibly access the stereoarrays are still highly demanded from the synthesis prospect.
It is informative when we closely analyze how Nature forms biomolecules in an iterative strategy by enzymatic assembly of simple building blocks. Inspired by this powerful strategy, chemists have developed similar assembly–line processes to synthesize biopolymers (such as polypeptides, oligonucleotides, and oligosaccharides) (Caruthers, 1985; Merrifield, 1985; Seeberger and Haase, 2000) and other supramolecular systems such as dendrimers, cyclacenes, oligophenylenes, and polyspiranes. This methodology becomes privileged in synthetic supramolecular chemistry (Feuerbacher and Vögtle, 1998). However, it is still undervalued in natural product synthesis, particularly in the field of polyketides. In this Review, we highlight a number of iterative total syntheses of some important classes of polyketide such as polyenes, skipped polyols and polypropionates. These examples are not meant to be comprehensive since many elegant syntheses are not included.
It is now wildly accepted that the polyketides synthases (PKS), which are similar to fatty acid synthases (FAS), are responsible for the biogenicity of polyketides (Katz, 1997; Staunton and Weissman, 2001). The key carbon–carbon bond formation in chain propagation is realized by repetitive decarboxylative Claisen condensation of thioester powered by PKS. Acid derivatives such as acetyl–CoA, malonyl-CoA, and methylmalonyl-CoA are employed as simple building blocks in chain elongation. A series of functional units or modules are ordered in sequence in the PKS, and each of the module contains several domains with different functions (Figure 1A). The modules are arranged in an ordered way that polyketides can be assembled iteratively and efficiently (Dutta et al., 2014; Whicher et al., 2014). Generally, at least 3 domains [ketosynthase (KS), acyltransferase (AT), and acyl carrier protein (ACP)], are required for one iterative cycle of the chain extension. Other domains include ketoreductase (KR), dehydratase (DH) and enoyl reductase (ER).
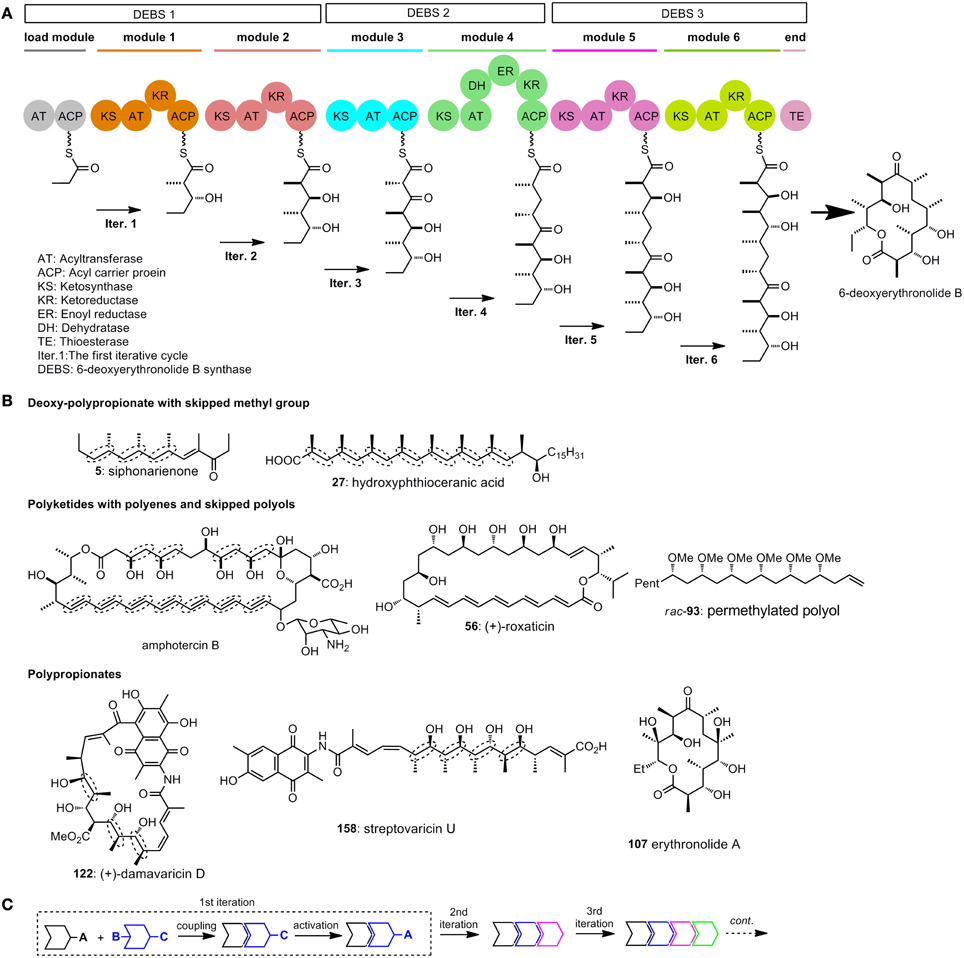
Figure 1. (A) Model for the domain organization of 6-deoxyerythronolide B synthase (Katz, 1997); (B) Selected polyketides with repetitive structure units. Elegant iterative synthesis of the deoxy-polypropionate, polyenes, skipped polys, and polypionates will be introduced in the following test; (C) A general strategy for iterative synthesis.
To illustrate the function of each module, we draw a model in which stepwise synthesis of 6-deoxyerythronolide B are showed (Staunton and Weissman, 2001) (Figure 1A). The PKS for biosynthesis of 6-deoxyerythronolide B (DEBS) are multienzyme polypeptides which consist of three synthases DEBS 1, 2, and 3. The load module in DEBS 1 have two domains, AT and ACP. These two domains indicate the beginning of polyketide assembly. The KR domain, as shown in the first iterative cycle (iter. 1), has the function of reducing ketones to alcohols. Module 3 does not contain the KR domain, so ketone group survives in iter. 3. DH has the function of forming double bonds, while ER will reduce the double bonds to saturated ones. The addition of DH and ER domains to a basic extending module (KR + AT + ACP) will give a saturate extension moiety, as shown in iter. 4. By altering the domains between AT and ACP in each module, different functional groups such as ketones, alcohols and alkenes can be arranged in the polyketide in an organized way.
The repetitive module 1 in a PKS will give the polypropionates, the repetitive module 4 in a PKS is expected to generate deoxy-polypropionates, while repetitive module 4 without ER domain in a PKS will provide polyenes. Polyketides are endowed with intrinsic repeatability through the enzyme catalyzed iterative synthesis (Figure 1B). As a imitation to nature, iterative sequences have already been used by many chemists to synthesize polyketides.
In a general illustration of monodirectional iterative sequence (Figure 1C), two different building blocks can either be utilized in the key linking reaction, between two of which the protected sites (such as B and C in Figure 1C) are activated. After a few iterations large arrays of targets with defined structures can be obtained in a straightforward manner.
Compared with other customized strategies, the whole synthetic sequence becomes much simpler and more flexible, thus holding the potential for automated production by non–specialists. In the context of polyketide total synthesis, the past decades have witnessed the development of several catalytic and non-catalytic processes where such streamlined construction of stereo-defined polyketide arrays can be realized. Two elegant reviews on iterative strategies focusing on deoxypropionates with skipped methyl groups have appeared (Hanessian et al., 2006; Horst et al., 2010).
Deoxy-polypropionate with Skipped Methyl Groups
When methylmalonate units are incorporated into the polyketide chain and the resulting β-keto functionalities undergo fully enzymatic reductions and dehydrations, the skipped methyl carbon chain is formed. Pedant methyl groups in naturally derived deoxypropionate have a bias for all-syn orientation in order to alleviate the number of destabilizing interactions during the chain-elongation process (Hoffmann, 2000). Abiko and Masamune (1996) have employed a chiral auxiliary-based asymmetric alkylation in the first synthesis of (+)-siphonarienone (5, Figure 2). The potassium enolate of benzopyrano-isoxazolidine 2 reacted with the triflate of β-branched alcohol 1 (obtained via resolution) with good diastereoselectivity. The reductive removal of the chiral auxiliary is needed to set the next iterative alkylation. Another noteworthy approach toward 5 was reported via the Negishi Zr-catalyzed asymmetric carboalumination (ZACA) (Magnin-Lachaux et al., 2004). Staring from homoallylic alcohol 6, the ZACA protocol introduced an ω-n-propyl group in the presence of chiral zirconocene derivative 7. Three iterative carboaluminations furnished an all syn-product 4 with high diastereoselectivity while those minor diastereomers were readily depleted by column chromatography. The total synthesis of the potent antitumor agent (−)-doliculide (18) by Ghosh and Liu (2001) disclosed a repetitive cyclopropanation-fragmentation (Figure 3). The synthesis of its polyketide unit 17 commenced with the Charette asymmetric cyclopropanation of allylic alcodol 12 by dioxaborolane 13. Subsequent iodination and lithium-holagen exchange-initiated ring-opening delivered alkene 15. A four-step sequence is further required to deliver a homologated allylic alcohol 16 for next iteration.
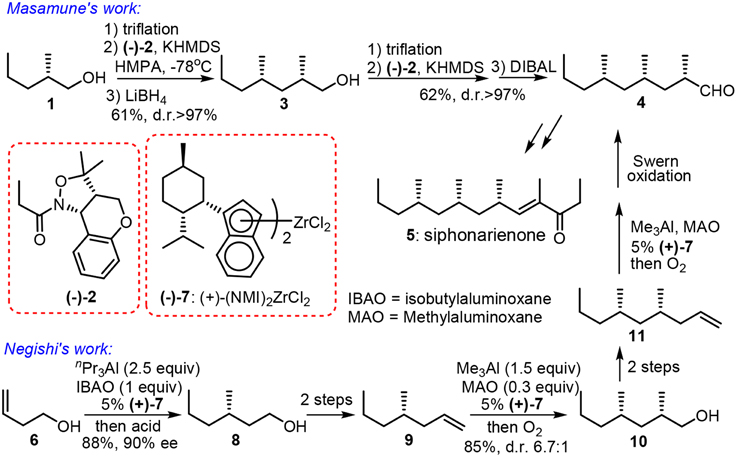
Figure 2. Synthesis of siphonarienone (Abiko and Masamune, 1996; Magnin-Lachaux et al., 2004).
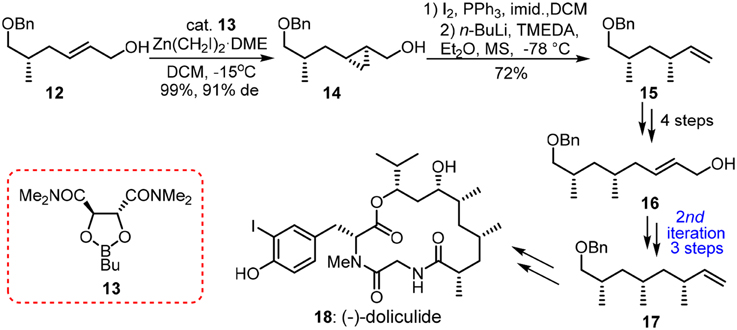
Figure 3. Synthesis of (−)-doliculide (Ghosh and Liu, 2001).
Hydroxyphthioceranic acid (27, Figure 4) is a constituent of the cell-wall lipid of pathogenic germ Mycobacterium tuberculosis. Pioneering work in iterative construction of its all-syn skipped methyl deoxypropionate array was dictated by Minnaard and Feringa group via catalytic iterative 1,4-addition (Geerdink et al., 2013). Aggarwal and co-workers have developed a traceless iterative strategy based on the Matteson rearrangement (Matteson, 2013; Rasappan and Aggarwal, 2014). Bifunctional building blocks 20 and 21 can be synthesized in several steps from the commercially available alcohol 19. Homologated boronic ester 22 was obtained via a stereospecific 1,2-migration reaction in TS-1. Treatment of α-lithiated 22 with 4-tert-butylcatechol/Mn(OAc)3·2H2O and subsequent carbamoylation furnished the protodeboronated product 23. Next round of lithiation-borylation-protodeboronation incorporating 21 was found to be challenging and MeLi should be used instead of nBuLi to form a more stereo–demanding boronate complex. Undoubtedly, a more reliable protodeboronation protocol which is especially suitable for bulky alkyl pinacol boronic esters is still in request. More resonates with Hendrickson's ideal synthesis, the same group has revised an assembly-line strategy toward 27 (Balieu et al., 2015), where 16 staggered homologations are sequentially required. The required methylene units were inserted via boronic ester homologations with α-chloromethyllithium 30. Other homologations were dominated by chiral lithiated building blocks 29 and 32. Aggarwal's lithiation-borylation method can be also used in the stereoselective synthesis of other challenging targets (Leonori and Aggarwal, 2014), such as alkyl chain with adjacent acyclic quaternary-tertiary motifs (Blair et al., 2014) or alkyl chain with ten contiguous methyl groups (Burns et al., 2014).
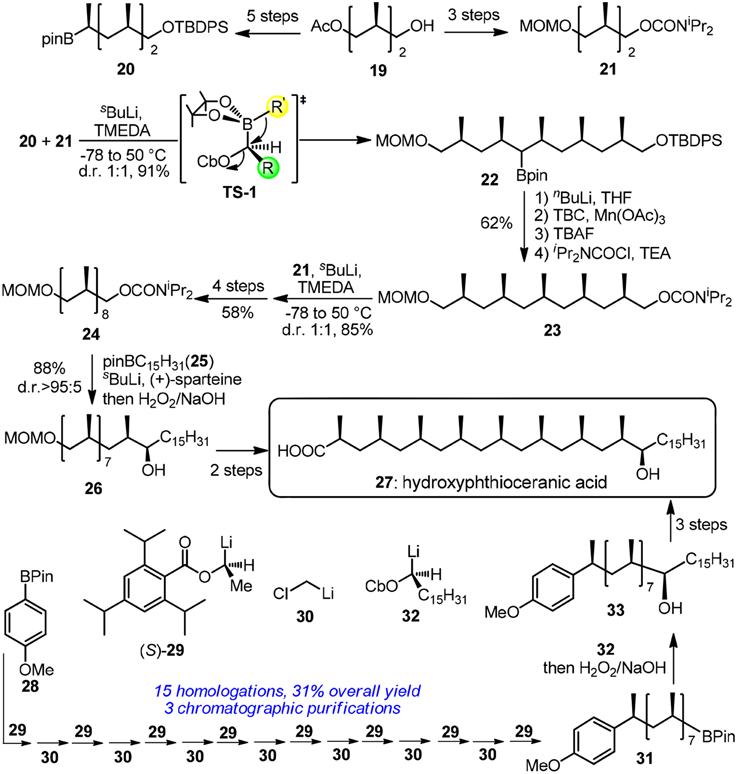
Figure 4. Synthesis of hydroxyphthioceranic acid (Rasappan and Aggarwal, 2014; and Balieu et al., 2015).
Polyene-type Polyketides
In biosynthetic machinery leads to polyene-type polyketides, enoyl reductase (ER) takes no active part after each β-keto reduction and dehydration in several consecutive chain elongation processes, thus providing natural products with considerable structural unsaturation. Synthetic approaches to such conjugated double bond frameworks are made challenging by their sensitivities to oxygen, light and acidic conditions, as well as the issues concerning good stereocontrol. Controlled iterative cross coupling reactions play an important role in the construction of conjugated organic compounds (Wang and Glorius, 2008). Very recently, Burke and co-workers have devised a versatile iterative bifunctional building block-based cross-coupling strategy for accessing polyene natural products such as fatty acids, polyketides, hydrid peptide/polyketides and polyterpines (Gillis and Burke, 2007, 2009). The instability of alkenyl boronic acid has limited its usage in the synthesis. In Burke's N-methyliminodiacetic acid (MIDA) protected boronic acid, the hybrid orbital of boron changes from sp2 to sp3. This has greatly increased the stability of MIDA borates and attenuated the reactivity of boronic acid. Oligomerization is inhibited when bifunctional MIDA borates are used in iterative cross coupling. Meanwhile, the MIDA protecting group can be easily removed under basic aqueous conditions. Currently, over 170 MIDA building blocks are commercially available from Sigma-Aldrich (Woerly et al., 2014).
Taking their modular synthesis of the heptaene core of vacidin A as an illustration (41, Figure 5). In the structure of which a problematic cis-diene is embedded (Lee et al., 2010), the target 41 as well as other polyene macrolides such as amphotericin B and nystatin, are found to display an unique mode of action by forming ion channels when dealing with systematic fungal infections. Bornic aicd 34 couples with vinyl iodide moiety of bifunctional building block 35 to form 36 successfully under the palladium catalyzed conditions without oligomerization 35. The MIDA protecting group in 36 can be easily unmasked with NaOH aqueous solution and subsequent coupling reaction of the protecting group removed boronic acid with 37 will generate the challenging cis-diene moiety in 38. The last iterative Suzuki-Miyaura coupling reaction of 38 and building block 39 will form the polyene moiety of vacidin A. By uniting a collection of MIDA building blocks, a vast number of complex polyene motifs can be made with excellent stereocontrolled iterative cross-coupling reaction. Recently, great achievements have been made by Burke group (Li et al., 2015) in the field automated process. The authors have synthesized 14 distinct classes of small molecules using this automated process. Interestingly, the challenging purification problem is solved by using MIDA borates' unusual binary affinity for silica gel with certain pairs of eluents. The MIDA borates show minimal mobility on thin-layer silica gel chromatography when eluting with MeOH/Et2O, while they are rapidly eluted when THF is used.
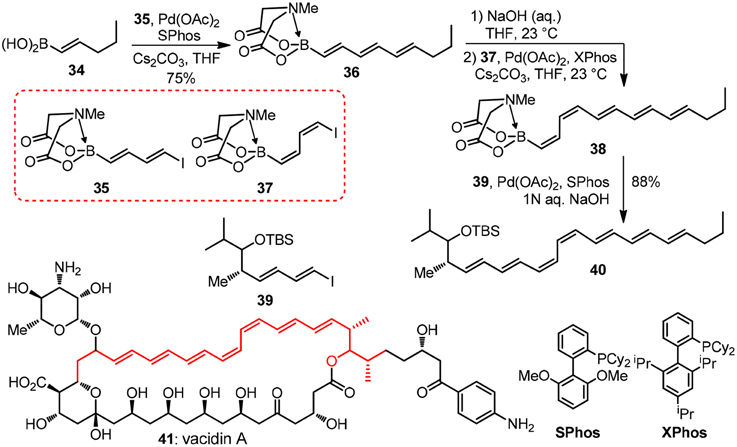
Figure 5. Synthesis of the heptaene fragment of vacidin A (Lee et al., 2010).
Polyketides with Skipped Polyols
As one of the key structural elements, polyacetate-derived 1,3-polyol unit is featured in vast collection of polyketide natural products. During the past decades, numerous synthetic methodologies have been advanced to stereoselectively introduce 1,3-diols or higher order 1,3-polyol arrays (Bode et al., 2006). Methods based on allylation of carbonyl groups are frequently encountered in literature. The repetitive allylmetallation of aldehydes is of great interest since typically only three steps including allylation, protection and oxidative aldehyde regeneration are required in a single iteration. An illustrative example of Brown's asymmetric allylation is showed in Marco's synthesis of α-pyrone natural product passifloricin A (48, Figure 6A) (Garcia-Fortanet et al., 2003). After the first iteration, the resulting β-silyoxy aldehyde 46 was subjected to second allylation to yield homoallylic alcohol 45.
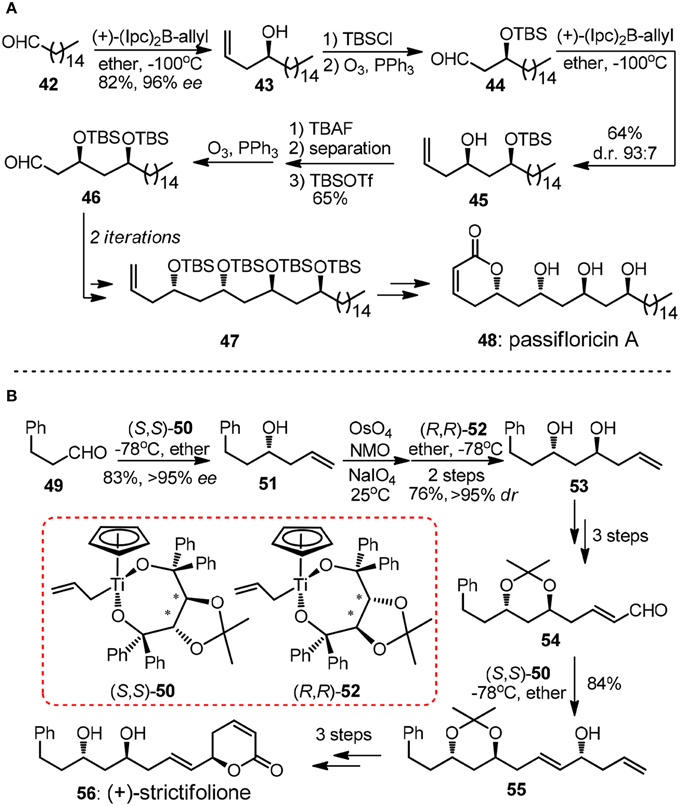
Figure 6. (A) Synthesis of published structure of passifloricin A (Garcia-Fortanet et al., 2003); (B) Synthesis of (+)-strictifolione (BouzBouz and Cossy, 2003).
The undesired diastereomer of 45 could only be removed as the corresponding diol after desilylation by column chromatography, thus addition steps were required in order to afford the product in a high optical purity. Apart from chiral allylborane, Duthaler-Hafner reagent 50 and 52 were nicely implemented in repetitive synthetic operations during the concise synthesis of antifungal α-pyrone (+)-strictifolione (56, Figure 6B) (BouzBouz and Cossy, 2003). It is notable that 1,3-anti diol 53 can be produced via direct allylation of the intermediacy β-hydroxyl aldehyde in excellent diastereoselectivity.
Evidently, it is of immense potential to conduct allylation in a catalytic version so as to preclude the usage of stoichiometric chiral reagent. An intriguing chiral sulfonamide-catalyzed allylchromation protocol was described by Kishi group (Zhang et al., 2008). In the same year, Krische and co-workers showcased a breakthrough Ir-catalyzed transfer hydrogenation allylation reaction (Kim et al., 2008). Homoallylic alcohols of high optical purity can be obtained using a modified chiral iridium C,O-benzoate complex 58. Oxo-pentaene macrolide (+)-roxaticin (62, Figure 7) constitute an appealing target due to its strict polyacetate origin. An impressing short synthesis of 62 by Krische group relies principally on an iterative two-dimensional assembly of 1,3-diol 57, which can be considered as unstable malondialdehyde equivalent (Han et al., 2010). The first allylation can be conducted on a large scale using a less costly (R)-BINAP analog to yield C2-symmetric diol 59 with same excellent levels of stereocontrol, albeit in moderate isolating yield. It is quite remarkable that in each dimensional allylation, the stereochemical bias of the in situ generated iridium catalyst overrides the intrinsic facial selectivity of the transient chiral β-branched aldehydes. The acetonide-protected polyacetate fragment 61 processing six chiral centers was prepared in an elegant nine-step sequence.
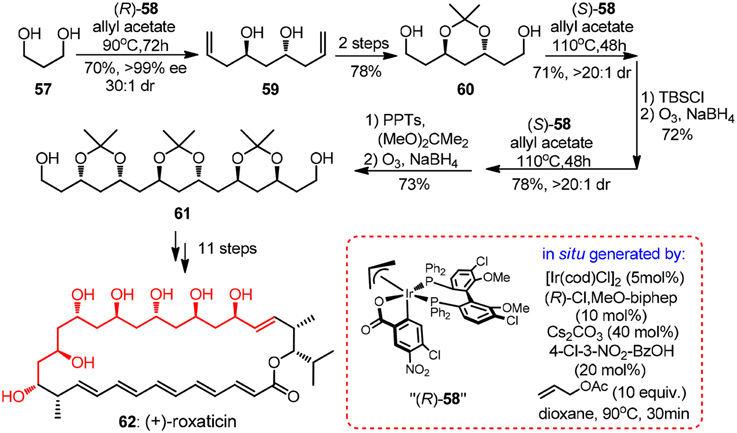
Figure 7. Synthesis of (+)-roxaticin (Han et al., 2010).
Synthetic pursuit of the C19-C28 polyacetate unit of macrolide RK-397 (68, Figure 8) was initiated by Evans and co-workers to demonstrate the convenience of their newly developed bismuth-mediated protocol for diastereoselective formation syn-1,3-dioxanes (Evans et al., 2012). Treatment of chiral δ-trialkylsilyloxy α,β-unsaturated aldehyde 64 or δ-hydroxy α,β-unsaturated ketone 66 with excessive amount of acetaldehyde and bismuth(III) nitrate as a substoichiometric mediator furnished ethylidene acetal-protected syn-1,3-dioxane 65 or 67 in good yield. The reaction proceeds via a thermodynamically controlled hemiacetal/oxa-conjugate addition process, and gives rise to product bearing electron-withdrawing group which can be directly utilized in consecutive Keck-Maruoka allylation.
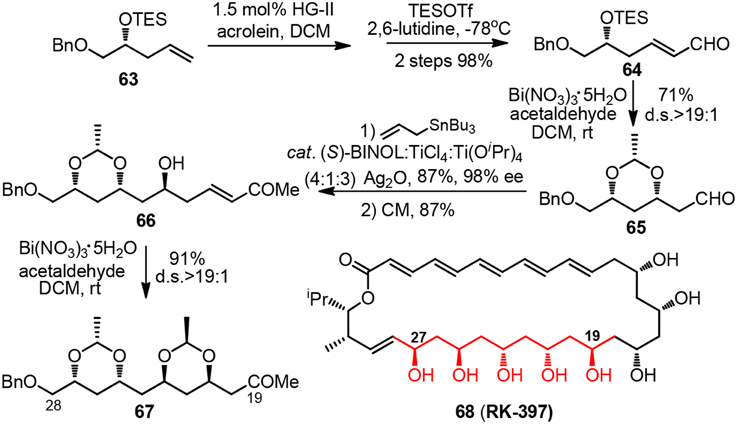
Figure 8. Synthesis of the C18-C28 polyol fragment of RK-397 (Evans et al., 2012).
In analogy to Krische's two-directional modular synthesis of (+)-roxaticin, the rapid growth of alternating polyol segment can also be achieved by iterative stereoselctive alkylation of cyanohydrin acetonides. As was exemplified by Rychnovsky's early convergent synthesis of isotactic permethylated polyol (77, Figure 9) (Rychnovsky and Griesgraber, 1992), the alkylation of 69 with its electrophilic-activated derivative 70 gave a single isomer 71 owing to a kinetic anometric effect. A secondary alkylation followed by electrophilic activation delivered 73, which was subjected to the final alkylation with cyanohydrin acetonide 74 to yield pentanitrile 75. The featured reductive removal of the five axial cyano groups was realized by dissolving metal reduction with complete retention of configuration.
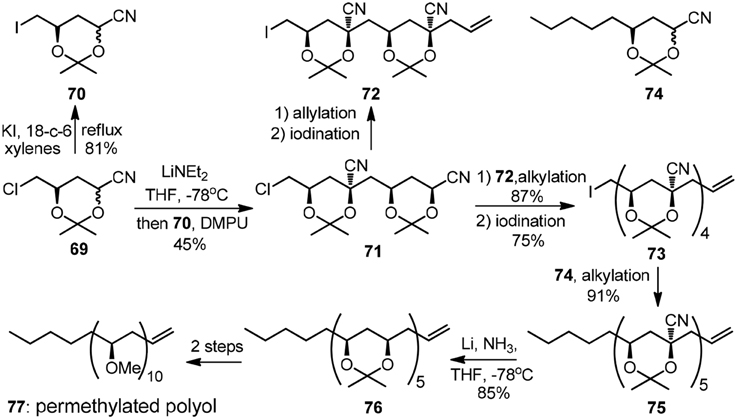
Figure 9. Synthesis of the permethylated polyol (Rychnovsky and Griesgraber, 1992).
The pioneering studies on iterative 1,3,5-triols synthesis based on asymmetric epoxidation followed by reductive epoxide ring-opening was reported by the group of Sharpless (Katsuki et al., 1982). The chain extension protocol was modified by Nicolaou et al. (1987) and further applied in their total synthesis of amphotericin B. The Shibasaki group, on the other hand, established a new catalyst-controlled epoxidation promoted by complex 79 (Tosaki et al., 2004). All possible isomers of 1,3,5,7-tetraol array were readily accessible with equally high efficiency. Synthetic endeavor on cryptocaryolone diacetate (85, Figure 10) was presented to showcase its versatility. α,β-Epoxy morpholinyl amide 80 was subjected to Claisen-type condensation to form β-keto ester 81, the later of which underwent subsequent 6 steps to complete one iteration. Yadav et al. (2007) also demonstrated a chiral pool strategy toward 85 by means of consecutive use of Prins cyclisation followed by reductive tetrahydropyran ring opening.
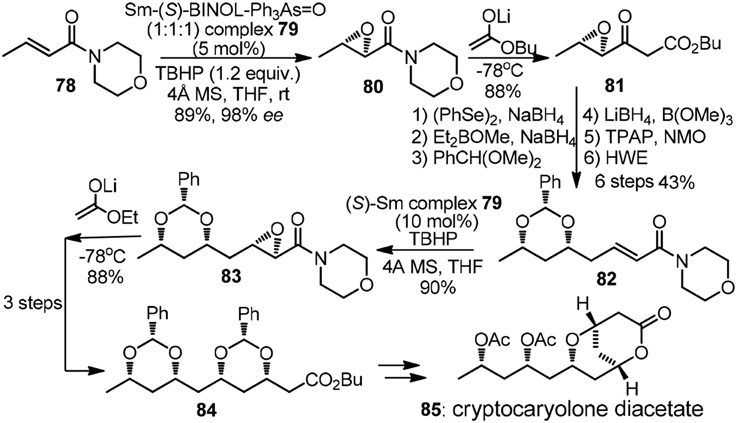
Figure 10. Synthesis of cryptocaryolone diacetate (Tosaki et al., 2004).
An iterative organocatalytic approach was recently displayed by Kumar et al. (2012) in their total synthesis of revised structure of passifloricin A (93, Figure 11). In typical iteration, palmitaldehyde 86, upon sequential treatment of nitrosobenzene and D-proline followed by Horner-Wadsworth-Emmons (HWE) olefination was transformed into enantioenriched O-amino allylic alcohol which was hydrogenated to provide δ-hydroxy ester. C2-homologated aldehyde 87 was afforded after silylation and DIBAL-H reduction. The stereochemical outcome of α-aminoxylation can be rationalized via TS-2.
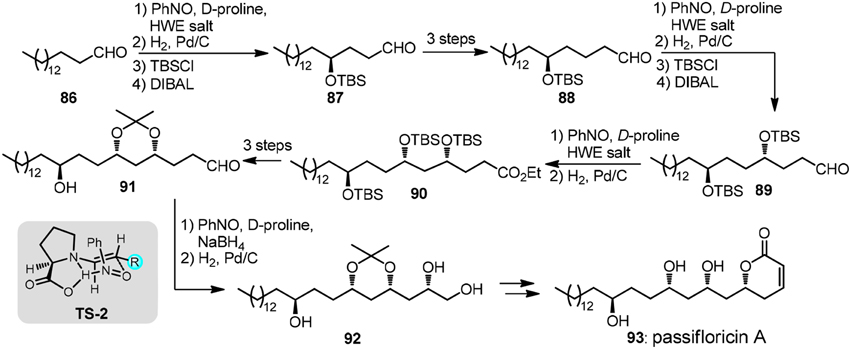
Figure 11. Synthesis of passifloricin A (Kumar et al., 2012).
The strategic use of aldol chemistry plays a fundamental role in efficient assembly of variant polyoxygenated targets, particularly those of polyketide origin (Mahrwald, 2013). When it comes to the construction of polyacetate unit by iterative acetate aldol reactions, very few examples have been advanced to date. Although the above mentioned tactic may be in principle more closely related to biosynthetic origin, great challenges have been posed on stereochemical controlling issues. One-pot polyaldol cascade reactions elegantly developed by Yamamoto group allowed for the rapid access to natural products such as anticancer compound EBC-23 (96) and polymethoxy-1-alkene (99, Figure 12) (Albert and Yamamoto, 2010, 2011). In each case, aldol adducts 95 and 98 were formed as already protected tris(trimethylsilyl)silyl ether with high 1,3-stereoselection. The rational of the high syn–selectivity as is depicted in TS-3, which is arising from the extreme bulkiness of the supersilyl group. An asymmetric version remains a formidable challenge so far.
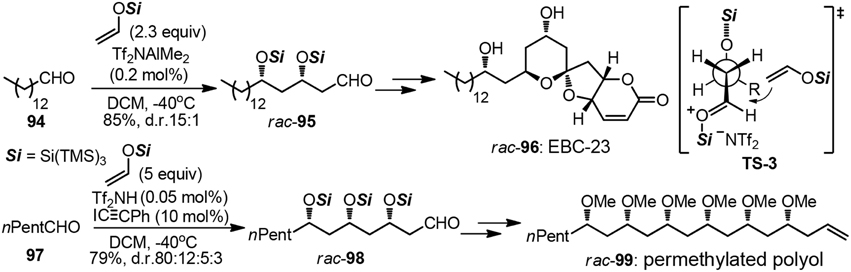
Figure 12. Synthesis of rac-EBC-23 and rac-permethylated polyol (Albert and Yamamoto, 2011).
Other noteworthy iterative methods for the construction of 1,3-polyols have been used in the total synthesis of polyrhacitides B. Polyrhacitides B was envisioned by Kirsch group to be assembled by alternate Pd-catalyzed asymmetric allylic esterification and chain extension (Menz and Kirsch, 2009). Mohapatra and coworkers completed the synthesis of polyrhacitides B based on the iterative usage of Maruoaka allylation and iodocyclization (Mohapatra et al., 2014).
Polypropionates
In a prototypical polypropionate chain, vicinal Me-OH carrying chiral centers are densely packed. The challenge of making 2-methyl-1,3-polyol array assemblage in laboratory arises from rapid access to enantiomerically pure products of enormous stereochemical diversity. Before aldol and related carbonyl addition reactions were privileged, stereocontrol in loose open chain systems have been achieved via rigid ring templates. In Woodward's masterful synthesis of erythromycin (Woodward et al., 1981), the repeated tactical use of cis fused dithiadecalin building block enabled diastereoselective formation of eight chiral centers found on the macrocylic aglycon. After Woodward's landmark work, great achievement has also been made in using thiopyran as template to access polypropionates in the past decades (Ward, 2011). Different from Woodward's strategy, Stork and Rychnovsky (1987) made use of an iterative butenolide construction of polypropionate chains to synthesize (+)-(9S)-dihydroerythronolide A.
Chiral pyranoid moieties may also serve as potential templates in acylic stereochontrol. Danishefsky and co-workers devised a synthesis of (±)-6-deoxyerythronolide (107, Figure 13A) featured by reiterative application of Lewis acid catalyzed cycloaddition of diene and aldehyde to stereoselectively generate trans-disubstituted dihydropyrone (Myles and Danishefsky, 1990). Danishefsky diene 101 and other dimethylated diene analogs equipped with exploitable functionality can be viewed as versatile dipropionyl building blocks. Following the same logic, polypropionate units in (+)-zincophorin and rifamycin S (120) have been conquered (Danishefsky and Selnick, 1987). Apart form those cycloaddition methods, an unique iterative sigmatropic strategy was used by Cywin and Kallmerten (1993) in their synthesis of C1-C11 subunit of zincophorin.
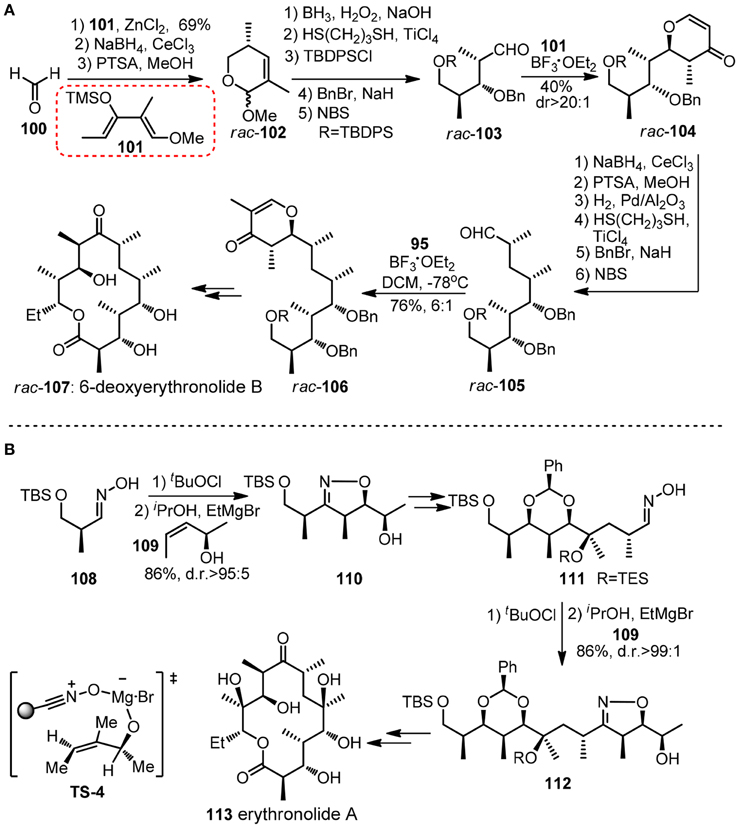
Figure 13. (A) Synthesis of rac-6-deoxyerythronolide B (Myles and Danishefsky, 1990); (B) Synthesis of erythronolide A (Bode et al., 2001).
Besides Hetero Diels-Alder reaction, another iterative cycloaddition-based approach in polypropionate construction was disclosed by Carreira group (Bode et al., 2001). Their erythronolide A (113, Figure 13B) synthesis commenced with Kanemasa's Mg-mediated cycloaddtion between 108-derived nitrile oxide and chiral allylic alcohol 109 to give alcohol 110, whose isoxazoline unit served as a template to ensure a high diastereo-bias in subsequent tertiary hydroxy group formation. A repeated succession of 109 provided 111 as a masked β-hydroxyketone.
The crotylmetal-aldehyde addition reaction has proven to be an universal approach for the modular preparation of polypropionate-derived natural products. Homoallylic alcohols of different stereodyads can be accessed and homologated aldehyde can be unveiled from its latent alkene functionality. The power of substrate-controlled crotylation was illustrated by Kishi's synthesis of aliphatic segment of ansamycin-type antibiotic rifamysin S (120, Figure 14A) (Nagaoka and Kishi, 1981). The Nozaki-Hiyama crotylchromium reagent, in situ prepared from either trans- or cis-crotyl iodide, smoothly reacted with α-methyl chiral aldehyde 114 to yield Felkin-adduct 115. Although the substrate-directed approach may be attractive in terms of exceptionally low cost of crotylating reagent, its drawback is also apparent since its inability to realize diversity-oriented synthesis. Recently, progress has been made in the synthesis of 1,3,5-triols with iterative Cr-mediated catalytic asymmetric allylation reactions (Zhang et al., 2008).
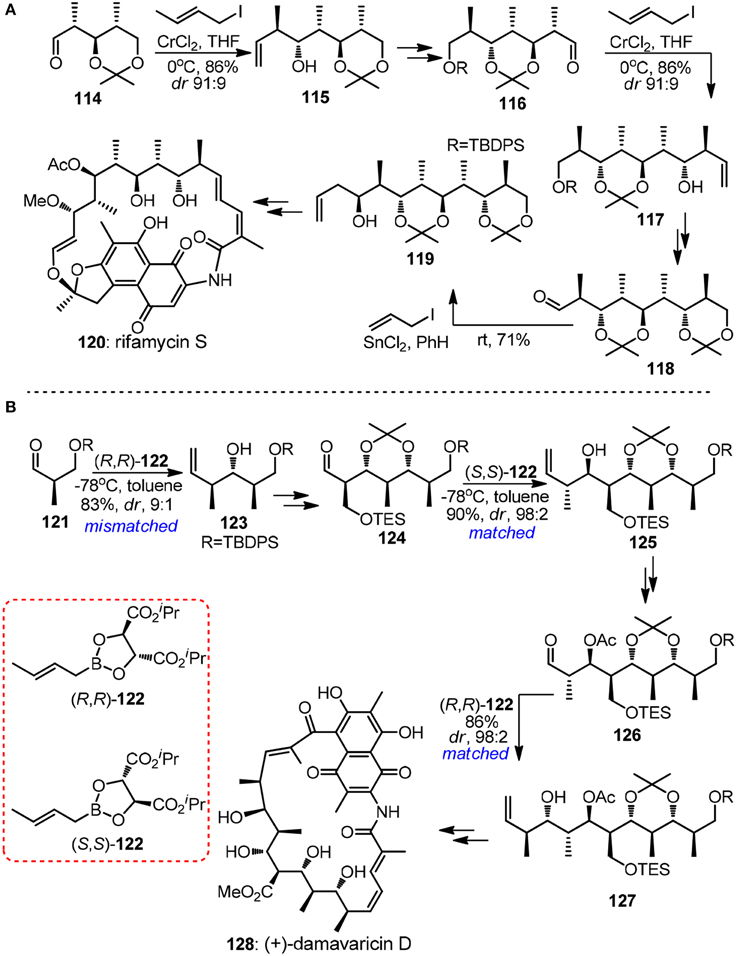
Figure 14. (A) Synthesis of rifamycin S (Nagaoka and Kishi, 1981); (B) Synthesis of (+)-damavaricin D (Roush et al., 1997).
The (+)-damavaricin D (128, Figure 14B) campaign in Roush's laboratory was marked with three highly reagent-controlled crotylations (Roush et al., 1997). Mismatched double asymmetric reaction between 123 and boronate (R,R)-122, which generated an desired anti, anti-stereotriad, was strategically designed to conduct at the very beginning of the whole synthetic sequence in order to obtain useful levels of diastereoselection. For advanced aldehyde intermediates 124 and 126 where overwhelming diastereofacial bias were expected as a result of the existence of more sterically demanding α-substituents, matched crotylation proceeded with exhibited excellent selectivity. Grée and co-workers (Ruiz et al., 2015) had demonstrated a new iterative tandem isomerization-aldolization sequence, where α-hydroxyallylsilanes can be viewed as propionaldehyde enolates equivalents. Aldehydes were generated from corresponding protected β-hydroxyacylsilanes via either photolytic cleavage or hydrogenolysis. Stereotetrads with syn-anti derivatives were obtained with only moderate stereoselectivity, which originates from preferentially anti-Felkin addition to chiral aldehyde.
The past three decades have witnessed the marvelous potentialities of aldol chemistry in assembling of complex polyoxygenated natural products as well as non-natural analogs. However, one of the most popular strategies involves a late-stage coupling of several advanced appropriately matched non-racemic fragments. As a laboratory emulation of nature's genius in polyketide biosynthesis, the iterative, monodirectional logic paves the way for rapid streamlined access of structural diversity through fine-tuning of the stereochemical nature of each aldolisations. Pioneering explorations in developing mono- and dipropionate synthones of various kinds had been led by Paterson and Scott (1999), Evans, etc. (Mahrwald, 2013).
Crimmins and Slade (2006) reported a formal synthesis of 6-deoxyerythronolide 107 validating the power of N-acylthiazolidinethiones as building blocks in consecutive aldol reactions. After that, a short enantioselective synthesis of (−)-pironetin (136, Figure 15A) was showcased (Crimmins and Dechert, 2009). The strictly linear sequence consisted of three highly selective auxiliary-based aldolisations including Urpi anti acetal aldol of chiral dimethylacetal 129, acetal aldol using chlorotitanium enolate derived from mesityl-substituted 132 and a final syn–aldol. Note in each chain elongation cycle the reductive removal of the chiral thiazolidinethione is high yielding and straightforward, thus rendering the whole iteration particularly attractive.
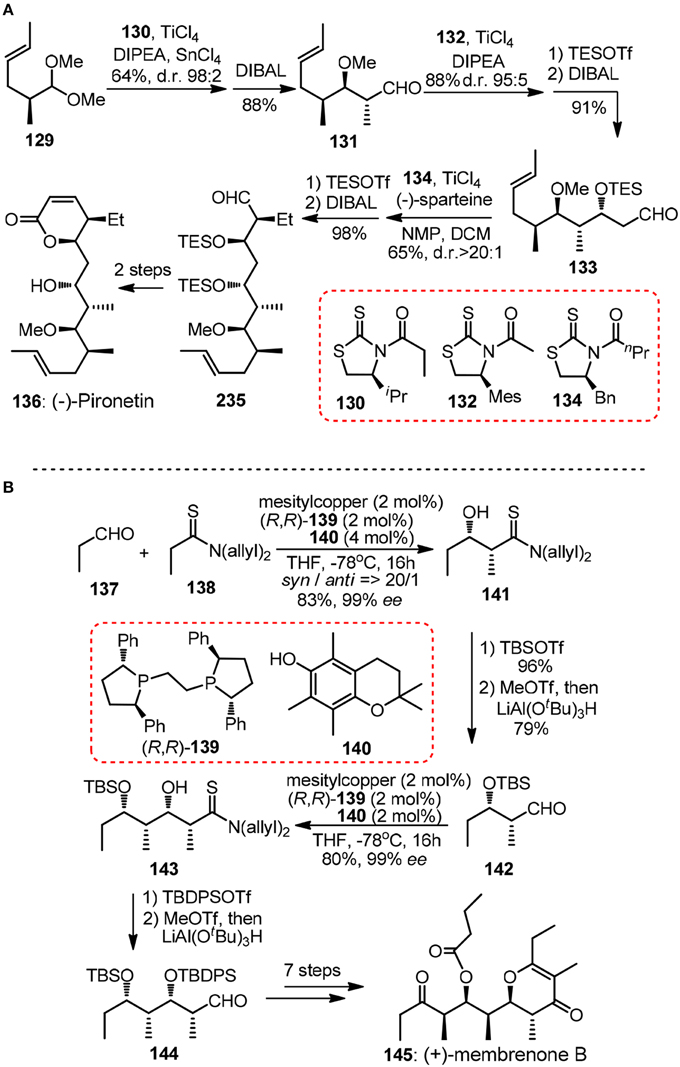
Figure 15. (A) Synthesis of (−)-pironetin (Crimmins and Dechert, 2009); (B) Synthesis of (+)-membrenone B (Alagiri et al., 2014).
Iterative application of catalytic asymmetric reactions in polypropionate construction has also been evolved by Shibasaki group in their synthesis of marine natural product (+)-membrenone B (145, Figure 15B) (Alagiri et al., 2014). Chemoselective enolization of thiopropionamide 138 proceeded catalytically under a soft Lewis acid/hard Brønsted base cooperative system. Aldehydes 142 and 144 can be delivered by facile reduction of more electrophilic iminium thioesters derived from silylated thioamide adducts.
Other noteworthy synthetic effort toward 136 came from Nelson and co-workers (Shen et al., 2006). Their featured asymmetric acyl halide-aldehyde cyclocondensations can be catalyzed by either cinchona alkaloid derived 147 and 150 or in situ generated aluminum complex 153 (Figure 16). A similar strategy for 1,3-polyols, in which iterative [2 + 2] asymmetric cycloaddition reaction of ketenes to O-protected α-hydroxy imines was involved, was developed by Palomo et al. (1993).
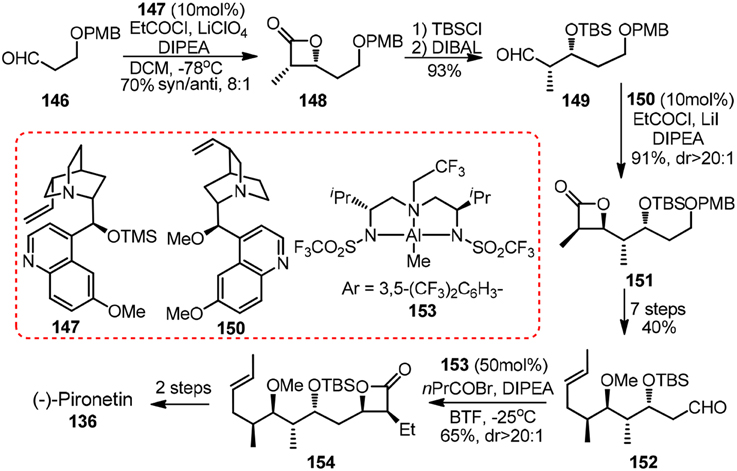
Figure 16. Synthesis of (−)-pironetin (Shen et al., 2006).
Apart from the above mentioned carbonyl chemistry-based approach, a stereoselective chiral epoxide ring-opening strategy has also been elaborated by several synthetic groups, albeit to a lesser extent. Excellent attempts in this area were conbtributed by Kishi (Corey and Hase, 1979; Nagaoka et al., 1980; Bartlett et al., 1982; Lipshutz and Kozlowski, 1984; Miyashita et al., 1991) and Shimizu (Oshima et al., 1989). An impressive example of alkenyloxirane methylation was disclosed by Miyashita's lab (1991) in their reiterative entry to the polypropionate ansa chain of antibiotic streptovaricin U (164, Figure 17). The key methylation reactions of γ,δ-epoxy acrylates 156, 158, 160, and 162 occurred regioselectively at γ-position stereoselectively with net inversion of configuration. Chiral epoxy groups during each intervening homologation were introduced by Katsuki-Sharpless asymmetric epoxidation or substrate-controlled epoxidation. Water was found to be crucial to achieve the desired transformation. The major drawback, however, was that tedious protection group manipulations (not shown in Figure 17) were required. Triethylsilyl protection was necessary in the chain extension, but methylation of the corresponding polysilyloxy substrates turned out to be extremely sluggish. Additionally, chiral α,γ-dimethyl-γ,δ-epoxy acrylates were also developed to serve as propionate building blocks by Shimizu and co-workers (Oshima et al., 1989). The key epoxide ring-opening was realized by a stereoselective and regioselective Pd-catalyzed intramolecular hydride delivery. This methodology was further explored by Pujari and Kaliappan (2012) in their recent synthetic endeavor targeting all syn stereopentad framework of callystatin A.
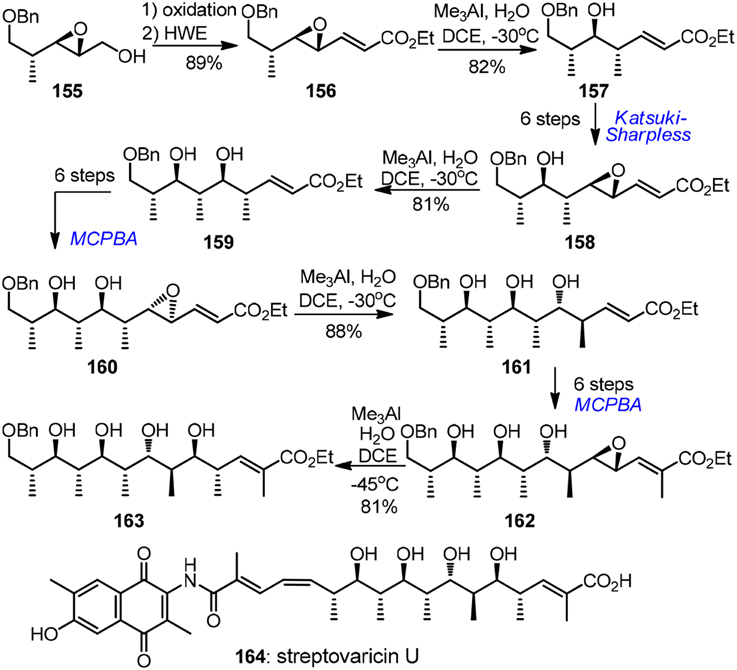
Figure 17. Synthesis of streptovaricin U (Miyashita et al., 1991).
The iterative strategies have also been successfully used in the total synthesis of polycyclic ethers (Zakarian et al., 2003; Kadota and Yamamoto, 2005; Clark, 2006). Though there are still many problems for an ideal automation synthesis, purification constitutes one of the tough ones. Recently, flow chemistry (Newton et al., 2014) and solid-phase synthesis (Umarye et al., 2006; Lee et al., 2007) were applied to the iterative synthesis of natural products and their derivatives.
Future Directions
In Reinhard W. Hoffmann's seminar review (Hoffmann, 1987) on the stereoselective syntheses of stereotrids as important precursors of polyketide natural products, he declared “at first surprising, preferences for linear synthesis strategies is, in principle, the possibility of using an iterative procedure, which is amenable to automatization… However, reliable procedures for the specific construction of the stereotriads A to D are first required.” It is clear that the intrinsic modular nature of diversified architectures found in complex polyketide natural products offers an ideal forum to demonstrate the salient utility of iterative strategies. Contrast to traditional laboratory synthesis where personal preference for designing synthetic plan to access different modules are uniquely designed, iterative synthesis facilitates synthetic effort by using similar reactants and well-developed conventional reaction conditions, thus holds considerable promise for rapid streamlined assembly of the entire spectrum of functionality and stereochemical permutations as will. Several elegant examples have been illustrated in the synthesis of catechin oligomers by Suzuki and co-workers (Ohmori et al., 2011) and McDonald's interative synthesis of poly-deoxysugers based on the alkynol cycloisomerization (McDonald and Zhu, 1998). Despite the fact that the emergence of iterative design in polyketide synthesis over the last few decades has been rather dramatic, a number of gaps have remained. Later comprise in terms of the stoichiometric use of chiral controlling reagents, tediously lengthy protocols in building block preparation as well as intervening re-functionalization operations, repeated use of protecting groups to suppress uncontrolled polymerization as well as other side reactions, the elusive and intractable stereo-controlling nature of the crucial reactions that used in a reiterative fashion. Nevertheless, these problems seem to cast light on the challenges the synthetic communities have to cope with in developing an ideal biomimetic iteration strategy. The robustness of the strategy will enabled more spectacular synthetic accomplishments in the coming future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial supports from the National Natural Science Foundation of China (No. 21290184), and the MOST–973 (No. 2011CB710800) are highly appreciated.
References
Abiko, A., and Masamune, S. (1996). Synthesis of (+)-siphonarienone: asymmetric alkylation using a chiral benzopyrano-isoxazolidine auxiliary. Tetrahedron Lett. 37, 1081–1083. doi: 10.1016/0040-4039(95)02353-4
Alagiri, K., Lin, S.-Q., Kumagai, N., and Shibasaki, M. (2014). Iterative direct aldol strategy for polypropionates: enantioselective total synthesis of (−)-membrenone A and B. Org. Lett. 16, 5301–5303. doi: 10.1021/ol5024932
Albert, B. J., and Yamamoto, H. (2010). A triple-aldol cascade reaction for the rapid assembly of polyketides. Angew. Chem. Int. Ed. 49, 2747–2749. doi: 10.1002/anie.200907076
Albert, B. J., and Yamamoto, H. (2011). Rapid total syntheses utilizing “supersilyl” chemistry. Angew. Chem. Int. Ed. 50, 2610–2612. doi: 10.1002/anie.201007210
Balieu, S., Hallett, G. E., Burns, M., Bootwicha, T., Studley, J., and Aggarwal, V. K. (2015). Toward ideality: the synthesis of (+)-kalkitoxin and (+)-hydroxyphthioceranic acid by assembly-line synthesis. J. Am. Chem. Soc. 137, 4398–4403. doi: 10.1021/ja512875g
Bartlett, P. A., Meadows, J. D., Brown, E. G., Morimoto, A., and Junstedt, K. K. (1982). Carbonate extension. A versatile procedure for functionalization of acyclic homoallylic alcohols with moderate stereocontrol. J. Org. Chem. 47, 4013–4018. doi: 10.1021/jo00142a002
Blair, D. J., Fletcher, C. J., Wheelhouse, K., Atherine, M. P., and Aggarwal, V. K. (2014). Stereocontrolled synthesis of adjacent acyclic quaternary-tertiary motifs: application to a concise total synthesis of (−)-filiformin. Angew. Chem. Int. Ed. 53, 5552–5555. doi: 10.1002/anie.201400944
Bode, J. W., Fraefel, N., Muri, D., and Carreira, E. M. (2001). A general solution to the modular synthesis of polyketide building blocks by Kanemasa hydroxy-directed nitrile oxide cycloadditions. Angew. Chem. Int. Ed. 40, 2082–2085. doi: 10.1002/1521-3773(20010601)40:11<2082::AID-ANIE2082>3.0.CO;2-1
Bode, S. E., Wolberg, M., and Müller, M. (2006). Stereoselective synthesis of 1,3-diols. Synthesis 557–588. doi: 10.1055/s-2006-926315
BouzBouz, S., and Cossy, J. (2003). Total synthesis of (+)-strictifolione. Org. Lett. 5, 1995–1997. doi: 10.1021/ol034619s
Burns, M., Essafi, S., Bame, J. R., Bull, S. P., Matthew, P., Balieu, S., et al. (2014). Assembly-line synthesis of organic molecules with tailored shapes. Nature 513, 183–188. doi: 10.1038/nature13711
Caruthers, M. H. (1985). Gene synthesis machines: DNA chemistry and its uses. Science 230, 281–285. doi: 10.1126/science.3863253
Clark, J. S. (2006). Construction of fused polycyclic ethers by strategies involving ring-closing metathesis. Chem. Commun. 3571–3581. doi: 10.1039/b601839d
Corey, E. J., and Hase, T. (1979). Studies on the total synthesis of rifamycin. Highly stereoselective synthesis of intermediates for construction of the C(15) to C(29) chain. Tetrahedron Lett. 20, 335–338. doi: 10.1016/S0040-4039(01)85964-5
Crimmins, M. T., and Dechert, A.-M. R. (2009). Enantioselective total synthesis of (−)-pironetin: iterative aldol reactions of thiazolidinethiones. Org. Lett. 11, 1635–1638. doi: 10.1021/ol9003228
Crimmins, M. T., and Slade, D. J. (2006). Formal synthesis of 6-deoxyerythronolide B. Org. Lett. 8, 2191–2194. doi: 10.1021/ol0607241
Cywin, C. L., and Kallmerten, J. (1993). Synthetic studies of the ionophore antibiotic zincophorin. 2. Iterative sigmatropic construction of the C1-C11 subunit. Tetrahedron Lett. 34, 1103–1106. doi: 10.1016/S0040-4039(00)77501-0
Danishefsky, S. J., and Selnick, H. G. (1987). Total synthesis of zincophorin. J. Am. Chem. Soc. 109, 1572–1574. doi: 10.1021/ja00239a049
Dutta, S., Whicher, J. R., Hansen, D. A., Hale, W. A., Chemler, J. A., Congdon, G. R., et al. (2014). Structure of a modular polyketide synthase. Nature 510, 512–517. doi: 10.1038/nature13423
Evans, P. A., Grisin, A., and Lawler, M. J. (2012). Diastereoselective construction of syn-1,3-dioxanes via a Bismuth-mediated two-component hemiacetal/oxa-conjugate addition reaction. J. Am. Chem. Soc. 132, 15559–15561. doi: 10.1021/ja208668u
Feuerbacher, N., and Vögtle, F. (1998). “Iterative,” in Dendrimers, ed F. Vögtle (Berlin; Heidelberg: Springer), 1–18.
Garcia-Fortanet, J., Murga, J., Carda, M., and Alberto, J. (2003). On the structure of passifloricin A: asymmetric synthesis of the α-lactones of (2Z, 5S, 7R, 9S, 11S)- and (2Z, 5R, 7R, 9S, 11S)-tetrahydroxyhexacos-2-enoic Acid. Org. Lett. 5, 1447–1449. doi: 10.1021/ol034182o
Geerdink, D., Horst, T. B., Lepore, M., Mori, L., Puzo, G., Hirsch, A. K., et al. (2013). Total synthesis, stereochemical elucidation and biological evaluation of Ac2SGL; a 1,3-methyl branched sulfoglycolipid from Mycobacterium tuberculosis. Chem. Sci. 4, 709–716. doi: 10.1039/c2sc21620e
Ghosh, A. K., and Liu, C. (2001). Total synthesis of antitumor depsipeptide (−)-doliculide. Org. Lett. 3, 635–638. doi: 10.1021/ol0100069
Gillis, E. P., and Burke, M. D. (2007). A simple and modular strategy for small molecule synthesis: iterative Suzuki-Miyaura coupling of B-protected haloboronic acid building blocks. J. Am. Chem. Soc. 129, 6716–6717. doi: 10.1021/ja0716204
Gillis, E. P., and Burke, M. D. (2009). Iterative cross-couplng with MIDA boronates: towards a general platform for small molecule synthesis. Aldrichimica Acta. 42, 17–27.
Han, S. B., Hassan, A., Kim, I. S., and Krische, M. J. (2010). Total synthesis of (+)-roxaticin via C-C bond forming transfer hydrogenation: a departure from stoichiometric chiral reagents, auxiliaries, and premetalated nucleophiles in polyketide construction. J. Am. Chem. Soc. 132, 15559–15561. doi: 10.1021/ja1082798
Hanessian, S., Giroux, S., and Mascitti, V. (2006). The iterative synthesis of acyclic deoxypropionate units and their implication in polyketide-derived natural products. Synthesis 1057–1076. doi: 10.1055/s-2006-926376
Hoffmann, R. W. (1987). Stereoselective syntheses of building blocks with three consecutive stereogenic centers: important precursors of polyketide natural products. Angew. Chem. Int. Ed. 26, 489–503. doi: 10.1002/anie.198704893
Hoffmann, R. W. (2000). Conformation design of open chain compounds. Angew. Chem. Int. Ed. 39, 2054–2070. doi: 10.1002/1521-3773(20000616)39:12<2054::AID-ANIE2054>3.0.CO;2-Z
Horst, B. T., Feringa, B. L., and Minnaard, A. J. (2010). Iterative strategies for the synthesis of deoxypropionates. Chem. Commun. 46, 2535–2547. doi: 10.1039/B926265B
Kadota, I., and Yamamoto, Y. (2005). Synthetic strategies of marine polycyclic ethers via intramolecular allylations: linear and convergent approaches. Acc. Chem. Res. 38, 423–432. doi: 10.1021/ar040118a
Katsuki, T., Lee, A. W., Ma, M. P., Martin, V. S., Masamune, S., Sharpless, K. B., et al. (1982). Synthesis of saccharides and related polyhydroxylated natural products. 1. Simple Alditols. J. Org. Chem. 47, 1373–1378. doi: 10.1021/jo00346a051
Katz, L. (1997). Manipulation of modular polyketide synthases. Chem. Rev. 97, 2557–2575. doi: 10.1021/cr960025+
Kim, I. S., Ngai, M.-Y., and Krische, M. J. (2008). Enantioselective Iridium-catalyzed carbonyl allylation from the alcohol or aldehyde oxidation level using allyl acetate as an allyl metal surrogate. J. Am. Chem. Soc. 130, 6340–6341. doi: 10.1021/ja802001b
Kumar, P., Pandey, M., Guptaa, P., and Dhavale, D. D. (2012). Organocatalytic stereoselective synthesis of passifloricin A. Org. Biomol. Chem. 10, 1820–1825. doi: 10.1039/c2ob06711k
Lee, S. J., Anderson, T. M., and Burke, M. D. (2010). A simple and general platform for generating stereochemically complex polyene frameworks by iterative cross-coupling. Angew. Chem. Int. Ed. 49, 8860–8863. doi: 10.1002/anie.201004911
Lee, S., Lee, T., Lee, Y., Kim, D., and Kim, S. (2007). Solid-phase library synthesis of polyynes similar to naturalproducts. Angew. Chem. Int. Ed. 46, 8422–8425. doi: 10.1002/anie.200703208
Leonori, D., and Aggarwal, V. K. (2014). Lithiation-borylation methodology and its application in synthesis. Acc. Chem. Res. 47, 3174–3183. doi: 10.1021/ar5002473
Li, J., Ballmer, S. G., Gillis, E. P., Fujii, S., Schmidt, M. J., Palazzolo, A. M., et al. (2015). Synthesis of many different types of organic small molecules using one automated process. Science 347, 1221–1226. doi: 10.1126/science.aaa5414
Lipshutz, B. H., and Kozlowski, J. A. (1984). A reiterative route to chiral all-syn-1,3-polyols. J. Org. Chem. 49, 1147–1149. doi: 10.1021/jo00180a045
Magnin-Lachaux, M., Tan, Z., Liang, B., and Negishi, E. (2004). Efficient and selective synthesis of siphonarienolone and related reduced polypropionates via Zr-catalyzed asymmetric carboalumination. Org. Lett. 6, 1425–1427. doi: 10.1021/ol0497483
Mahrwald, R. (2013). Modern Methods in Stereoselective Aldol Reactions. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA.
Matteson, D. S. (2013). Boronic esters in asymmetric synthesis. J. Org. Chem. 78, 10009-10023. doi: 10.1021/jo4013942
McDonald, F. E., and Zhu, H. Y. H. (1998). Novel strategy for oligosaccharide synthesis featuring reiterative alkynol cycloisomerization. J. Am. Chem. Soc. 120, 4246–4247. doi: 10.1021/ja980196r
Menz, H., and Kirsch, S. F. (2009). Total synthesis of polyrhacitides A and B by use of an iterative strategy for the stereoselective synthesis of 1,3-polyol arrays. Org. Lett. 14, 5634–5637. doi: 10.1021/ol902135v
Merrifield, R. B. (1985). Solid phase synthesis (Nobel Lecture). Angew. Chem. Int. Ed. 24, 799–810. doi: 10.1002/anie.198507993
Miyashita, M., Hoshino, M., and Yoshikoshi, A. (1991). Stereospecific methylation of.gamma., delta.-epoxy acrylates by trimethylaluminum: a method for the iterative construction of polypropionate chains. J. Org. Chem. 56, 6483–6485. doi: 10.1021/jo00023a001
Mohapatra, D. K., Krishnarao, P. S., Bhimireddy, E., and Yadav, J. S. (2014). Iterative iodocyclization: total synthesis of polyrhacitide B. Synthesis 46, 1639–1647. doi: 10.1055/s-0033-1341154
Myles, D. C., and Danishefsky, S. J. (1990). Development of a fully synthetic stereoselective route to 6-deoxyerythronolide B by reiterative applications of the Lewis acid catalyzed diene aldehyde cyclocondensation reaction: a remarkable instance of diastereofacial selectivity. J. Org. Chem. 55, 1636–1648. doi: 10.1021/jo00292a045
Nagaoka, H., and Kishi, Y. (1981). Further synthetic studies on rifamycin S. Tetrahedron 37, 3873–3888.
Nagaoka, H., Rutsch, W., Schimid, G., Lio, H., Johnson, M. R., and Kishi, Y. (1980). Total synthesis of rifamycins. 1. stereocontrolled synthesis of the aliphatic building block. J. Am. Chem. Soc. 102, 7962–7965. doi: 10.1021/ja00547a037
Newton, S., Carter, C. F., Pearson, C. M., Alves, L., de, C., Lange, H., et al. (2014). Accelerating spirocyclic polyketide synthesis using flow chemistry. Angew. Chem. Int. Ed. 53, 4915–4920. doi: 10.1002/anie.201402056
Nicolaou, K. C., R., Daines, A., Chakraborty, T. K., and Ogawa, Y. (1987). Total synthesis of amphotericin B. J. Am. Chem. Soc. 109, 2821–2822. doi: 10.1021/ja00243a043
Ohmori, K., Shono, T., Hatakoshi, Y., Yano, T., and Suzuki, K. (2011). Integrated synthetic strategy for higher catechin oligomers. Angew. Chem. Int. Ed. 50, 4862–4867. doi: 10.1002/anie.201007473
Oshima, M., Yamazaki, H., Shimizu, I., Nisar, M., and Tsuji, J. (1989). Palladium-catalyzed selective hydrogenolysis of alkenyloxiranes with formic acid. Stereoselectivity and synthetic utility. J. Am. Chem. Soc. 111, 6280–6287. doi: 10.1021/ja00198a045
Palomo, C., Aizpurua, J. M., Urchegui, R., and Garcfa, J. M. (1993). A new entry to 1,3-Polyols, 2-amino 1,3-polyols, and beta-(l-hydroxyalkyl)isoserines using azetidinone frameworks as chiral templates via iterative asymmetric [2 + 2] cycloaddition reactions. J. Org. Chem. 58, 1646–1648. doi: 10.1021/jo00059a003
Paterson, I., and Scott, J. P. (1999). Laboratory emulation of polyketide biosynthesis: an iterative, aldol-based, synthetic entry to polyketide libraries using (R)- and (S)-1-(benzyloxy)-2-methylpentan-3-one, and conformational aspects of extended polypropionates. J. Chem. Soc. Perkin Trans. 1, 1003–1014. doi: 10.1039/A809818B
Pujari, S. A., and Kaliappan, K. P. (2012). An iterative Shimizu non-aldol approach for the stereoselective synthesis of C13-C22 fragment of callystatin A. Org. Biomol. Chem. 10, 1750–1753. doi: 10.1039/c2ob06838a
Rasappan, R., and Aggarwal, V. K. (2014). Synthesis of hydroxyphthioceranic acid using a traceless lithiation–borylation–protodeboronation strategy. Nat. Chem. 6, 810–814. doi: 10.1038/nchem.2010
Roush, W. R., Coffey, D. S., and Madar, D. J. (1997). Total synthesis of (+)-damavaricin D. J. Am. Chem. Soc. 119, 11331–11332. doi: 10.1021/ja971828x
Ruiz, J., Murthy, A. S., Roisnel, T., Chandrasekhar, S., and Grée, R. (2015). α-Hydroxyallylsilanes as propionaldehyde enolate equivalents and their use toward iterative aldol reactions. J. Org. Chem. 80, 2364–2375. doi: 10.1021/acs.joc.5b00031
Rychnovsky, S. D., and Griesgraber, G. (1992). An iterative and convergent synthesis of syn polyols. J. Org. Chem. 57, 1559–1563. doi: 10.1021/jo00031a041
Seeberger, P. H., and Haase, W.-C. (2000). Solid-phase oligosaccharide synthesis and combinatorial carbohydrate libraries. Chem. Rev. 100, 4349–4393. doi: 10.1021/cr9903104
Shen, X., Wasmuth, A. S., Zhao, J., Zhu, C., and Nelson, S. G. (2006). Catalytic asymmetric assembly of stereodefined propionate units: an enantioselective total synthesis of (−)-pironetin. J. Am. Chem. Soc. 128, 7438–7439. doi: 10.1021/ja061938g
Staunton, J., and Weissman, K. J. (2001). Polyketide biosynthesis: a millennium review. Nat. Prod. Rep. 18, 380–416. doi: 10.1039/a909079g
Stork, G., and Rychnovsky, S. D. (1987). Iterative butenolide construction of polypropionate chains. Application to an efficient synthesis of (+)(9S)-dihydroerythronolide A. Pure Appl. Chem. 59, 345–352. doi: 10.1351/pac198759030345
Tosaki, S.-Y., Horiuchi, Y., Nemoto, T., Ohshima, T., and Shibasaki, M. (2004). Strategy for enantio- and diastereoselective syntheses of all possible stereoisomers of 1,3-Polyol arrays based on a highly catalyst-controlled epoxidation of α,β-unsaturated morpholinyl amides: application to natural product synthesis. Chem. Eur. J. 10, 1527–1544. doi: 10.1002/chem.200305709
Umarye, J. D., Leßmann, T., García, A. B., Mamane, V., Sommer, S., and Waldmann, H. (2006). Biology-oriented synthesis of stereochemically diverse natural-product-derived compound collections by iterative allylations on a solid support. Chem. Eur. J. 13, 3305–3319. doi: 10.1002/chem.200601698
Wang, C., and Glorius, F. (2008). Controlled iterative cross-coupling: on the way to the automation of organic synthesis. Angew. Chem. Int. Ed. 48, 5240–4244. doi: 10.1002/anie.200901680
Ward, D. E. (2011). The thiopyran route to polypropionates. Chem. Commun. 47, 11375–11393. doi: 10.1039/c1cc13323c
Whicher, J. R., Dutta, S., Hansen, D. A., Hale, W. A., Chemler, J. A., Dosey, A. M., et al. (2014). Structural rearrangements of a polyketide synthase module during its catalytic cycle. Nature 510, 560–564. doi: 10.1038/nature13409
Woerly, E. M., Roy, J., and Burke, M. D. (2014). Synthesis of most polyene natural product motifs using just 12 building blocks and one coupling reaction. Nat. Chem. 6, 484–491. doi: 10.1038/NCHEM.1947
Woodward, R. B., Logusch, E., Nambiar, K. P., Sakan, K., Ward, D. E., Au-Yeung, B. W., et al. (1981). Asymmetric total synthesis of erythromycin. 2. Synthesis of an erythronolide a lactone system. J. Am. Chem. Soc. 103, 3210–3213. doi: 10.1021/ja00401a050
Yadav, J. S., Rao, P. P., Reddy, M. S., Rao, N. V., and Prasad, A. R. (2007). Stereoselective synthesis of (+)-cryptocarya diacetate by an iterative Prins cyclisation and reductive cleavage sequence. Tetrahedron Lett. 48, 1469–1471. doi: 10.1016/j.tetlet.2006.12.068
Zakarian, A., Batch, A., and Holton, R. A. (2003). A convergent total synthesis of hemibrevetoxin B. J. Am. Chem. Soc. 26, 7822–7824. doi: 10.1021/ja029225v
Keywords: polyketides, iterative synthesis, bioinspired synthesis, polyol, biosynthesis
Citation: Zheng K, Xie C and Hong R (2015) Bioinspired iterative synthesis of polyketides. Front. Chem. 3:32. doi: 10.3389/fchem.2015.00032
Received: 04 January 2015; Accepted: 27 April 2015;
Published: 21 May 2015.
Edited by:
Bastien Nay, Centre National de la Recherche Scientifique, FranceReviewed by:
Ahmed A. Al-Amiery, National University of Malaysia, MalaysiaLaurent Evanno, Université Paris-Sud, France
Copyright © 2015 Zheng, Xie and Hong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ran Hong, CAS Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China,cmhvbmdAc2lvYy5hYy5jbg==