 Chunngai Hui
Chunngai Hui Tao Ye
Tao Ye- Department of Applied Biology & Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Hong Kong
Lysine methyltransferase which catalyze methylation of histone and non-histone proteins, play a crucial role in diverse biological processes and has emerged as a promising target for the development of various human diseases, including cancer, inflammation, and psychiatric disorders. However, inhibiting lysine methyltransferases selectively has presented many challenges to medicinal chemists. During the past decade, lysine methyltransferase inhibitors covering many different structural classes have been designed and developed. In this review, we describe the development of selective, small-molecule inhibitors of lysine methyltransferases with an emphasis on their discovery and chemical synthesis. We highlight the current state of lysine methyltransferase inhibitors and discuss future directions and opportunities for lysine methyltransferase inhibitor discovery.
Introduction
Epigenetic aberrations often lead to cancer and other human diseases. Recent basic studies on histone methyltransferases indicated that deregulation of histone methylation plays a critical role in human carcinogenesis (Copeland et al., 2009; Spannhoff et al., 2009; Chi et al., 2010; Arrowsmith et al., 2012; Helin and Dhanak, 2013). In fact, protein lysine methylation has attracted considerable attention since the discovery of the first histone lysine methyltransferase, Suv39H1, in 2000 (Rea et al., 2000). After the discovery of Suv39H1 with lysine methyltransferases activities, additional proteins with methyltransferase activity were reported, such as G9a/GLP (Tachibana et al., 2002, 2005), EZH2 (Cao et al., 2002), MLLs (Milne et al., 2002), SET2 (Strahl et al., 2002), SET7/9 (Wang et al., 2001), DOT1 (Feng et al., 2002; Van Leeuwen et al., 2002), and SETD8 (Nishioka et al., 2002). These methyltransferases are classified as either SET-domain containing or non-SET-domain containing enzymes for methylation of lysine residue on histone (Spannhoff et al., 2009). The function of these enzymes includes serving as catalysts for the transfer of methyl group(s) from the co-factor S-adenosyl-L-methionine (SAM) to the lysine residues of histone (Yao et al., 2011) (Figure 1A). Specific lysine methyltransferase catalyzed the methylation of lysine residue in a site-dependent manner and catalyze the formation of distinct methylation state (Wagner et al., 2014) (Figure 1B). New findings of somatic mutations and mis-expression of genes coding for histone methyltransferase enzymes provided a strong impetus for therapeutic intervention via pharmacological modulation. Epigenetic drug development, especially the discovery of small molecule inhibitors for histone methyltransferases, has quickly gained widespread interest (Pachaiyappan and Woster, 2014). Substantial review effort was made on bioactivities and biological mode of actions of the recently developed lysine methyltransferase inhibitor (Copeland et al., 2009; He et al., 2012; Helin and Dhanak, 2013; Itoh et al., 2013; Knapp and Weinmann, 2013; Tian et al., 2013; Wang and Patel, 2013; Bojang and Ramos, 2014; Dhanak and Jackson, 2014). So far, no review work on chemical synthesis of lysine methyltransferase inhibitors has been reported. The aim of this review is to provide a concise summary of the research published in the recent years, with an emphasis on the chemical syntheses of lysine methyltransferase inhibitors. Special attention will be paid to inhibitors with prominent biological activities targeting protein lysine methyltransferases such as G9a/GLP, Suv391H1, DOT1L, EZH2, and SETD8.
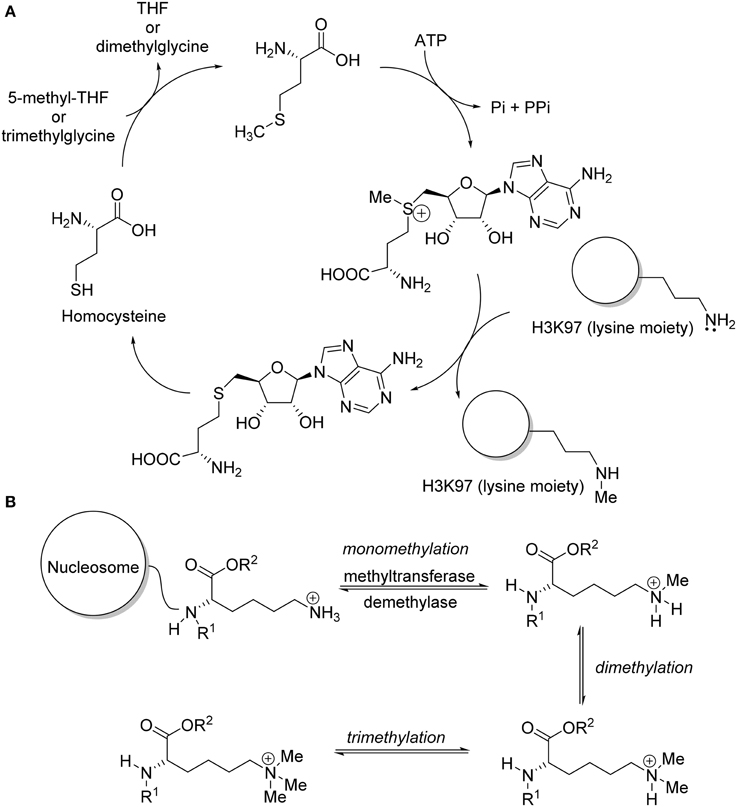
Figure 1. (A) Mechanism of DOT1L in methylation of lysine moiety of protein H3K79. (B) Specific lysine methyl transferase methylated lysine residues in histone lysine tail.
Suv39H1
Jenuwein and co-workers discovered that human Suv39H1 protein possesses histone H3K9 methyltransferases activities. Analysis of the Suv39H1 protein revealed that it carried a conserved motif of 130 amino acids which is corresponding to the SET domain (Jenuwein et al., 1998). Later, it was found that the SET domain was responsible for the histone lysine methylation function (Rea et al., 2000). Greiner and co-worker discovered that the fungal metabolite chaetocin 1 (Hauser et al., 1970), exhibited selectivity toward Su(var)3-9 methyltransferase (IC50 = 0.6 μM) (Greiner et al., 2005) is competitive with reactive methyl donor S-adenosyl-methionine (SAM) to facilitate enzymatic inhibition upon kinetic study (Figure 1). More recent work revealed that Chaetocin 1 was a non-specific inhibitor because it can inhibit many other unrelated enzymes (Cherblanc et al., 2013a). It also showed inhibitory effect toward G9a methyltransferase (Iwasa et al., 2010; Cherblanc et al., 2013b).
Structurally, Chaetocin 1 is classified as a complex epidithiodiketopiperazine (ETP) alkaloid (Iwasa et al., 2011b). Its total synthesis was accomplished by Sodeoka (Iwasa et al., 2010) and Movassaghi (Kim and Movassaghi, 2010), respectively in 2010. Sodeoka and coworkers also reported the synthesis of ent-chaetocin A 3 and the corresponding sulfur-deficient analogs (Iwasa et al., 2010, 2011a; Sodeoka et al., 2012; Fujishiro et al., 2013). Biological studies revealed that chaetocin A 1 and its ent-isomer 3 (ent-chaetocin A) inhibited G9a methyltransferase in similar extent (IC50 = 2.4 and 1.7 μM, respectively) (Iwasa et al., 2010). However, sulfur-deficient analog of chaetocin A 2 and its ent-isomer 4 were inactive toward G9a. (IC50 > 50 μM) (Figure 2).
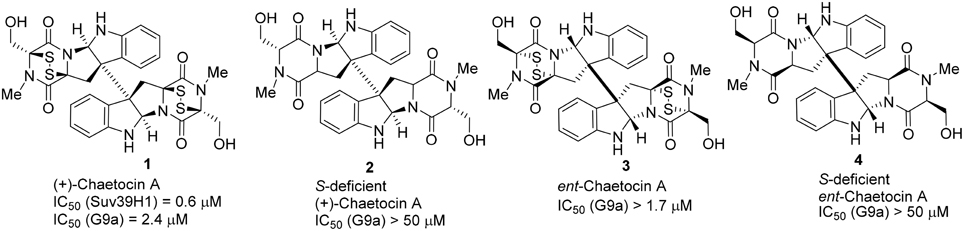
Figure 2. Example of inhibitors targeting protein Suv39H1.
Sodeoka and co-workers reported the first total synthesis of chaetocin A 1 in 2010 (Iwasa et al., 2010) (Scheme 1A). Diketopiperazine 7 was synthesized in five steps from Cbz protected N-methyl-D-serine 5 (Aurelio et al., 2003) and commercially available D-tryptophan methyl ester 6. Bromocyclization of 7 was promoted by NBS to afford 8 in 88% yield without formation of other diastereomers (Movassaghi et al., 2008; Kim et al., 2009). Stereoselective bromination of 8 was carried out using 2,2′-azobis-(2,4-dimethyl-4-methoxyvaleronitrile) (V-70) (Kita et al., 1997) as a radical initiator to afford tri-brominated intermediate 10, which was then treated with phosphate buffer to give rise to 11 in 47% yield. Reductive coupling of the freshly prepared diol 11 under Movassaghi Co(I)-catalyzed radical dimerization protocol (Movassaghi and Schmidt, 2007) furnished 12 in 55% yield. Construction of the disulfide bridge in 1 was accomplished by treating of dimmer 12 with H2S and BF3-Et2O, followed by I2 to afford chaetocin A (1) in 44% yield. Sodeoka also disclosed the method for the preparation of sulfur-deficient chaetocin A 2. Direct reductive coupling of 8 using Cobalt(I) as a catalyst produced dimer 9 in 47% yield and successive desilylation afforded sulfur-deficient chaetocin A 2 in 82% yield. In addition, the enantiomer of chaetocin A (3) (ent-chaetocin A) and its sulfur-deficient analog 4 were also prepared by the use of the same procedure (Iwasa et al., 2010, 2011a).
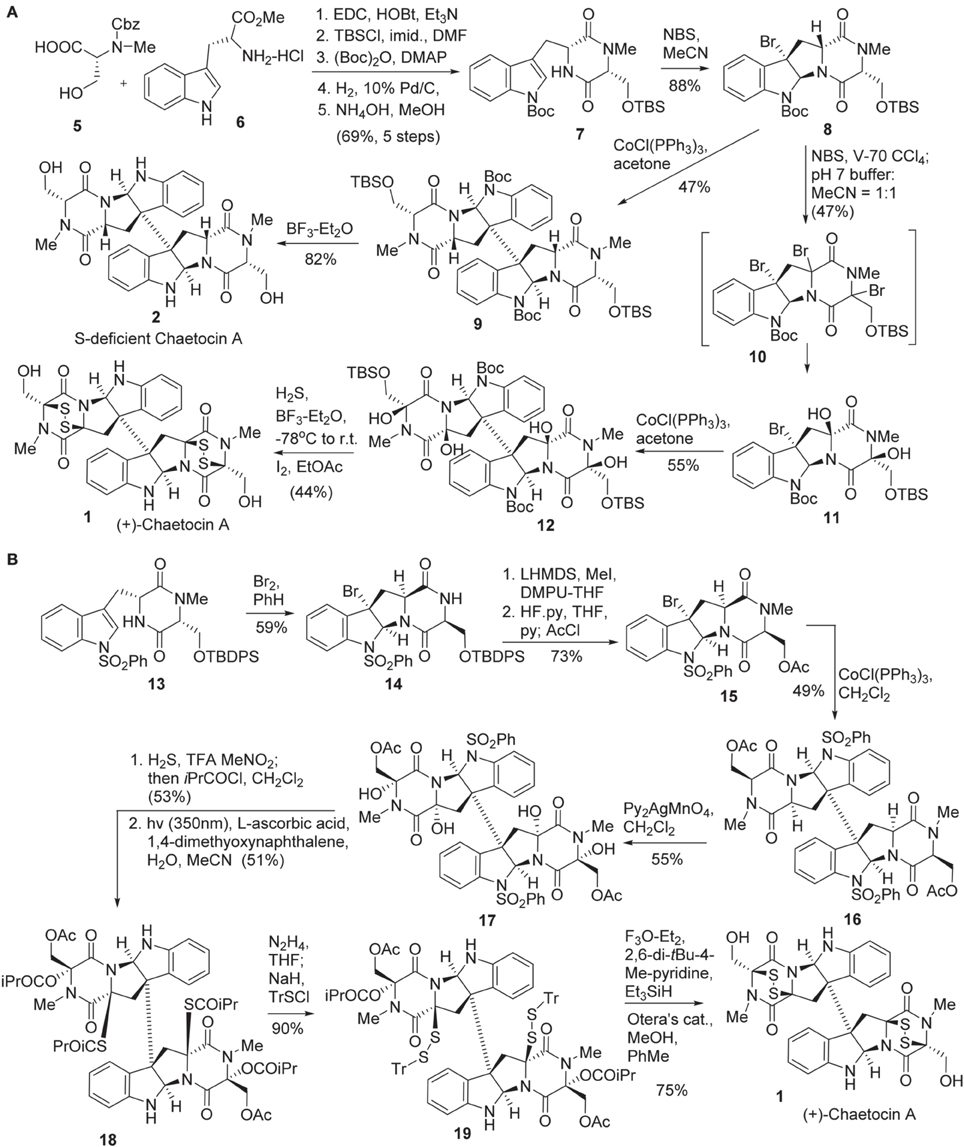
Scheme 1. (A) Total synthesis of (+)-Chaetocin A and its S-deficient analog by Sodeoka et al. (B) Total synthesis of (+)-Chaetocin A by Movassaghi and co-workers.
Movassaghi and co-workers reported the second total synthesis of chaetocin 1 in 2010 (Kim and Movassaghi, 2010) (Scheme 1B). The synthesis began with bromocyclolization of diketopiperazine 13 to afford 14 in 59% yield with high diastereoselectivity (Movassaghi et al., 2008). Successive N-methylation using MeI/LiHMDS, desilylation, and acetoxylation afforded 15, which was then subjected to Cobalt(I)-catalyzed reductive coupling (Movassaghi and Schmidt, 2007; Movassaghi et al., 2008) to afford dimeric product 16 in 49% yield. Stereoselective tetra-hydroxylation of 16 was carried out by the action of Py2AgMnO4 to give rise to 17 in 55% yield (Kim et al., 2009). The high chemo- and stereo-selectivity of the above transformations enabled the advanced intermediates to be prepared in gram scale. Exposure of tetraol 17 to TFA and H2S allowed the formation of a bisthioaminal intermediate with high stereoselectivity in which both the diol and dithiol were immediately protected as the corresponding ester or thioester. Subsequent N1-desulphonylation under irradiation with a Black-light phosphor-coated lamp, 1,4-dimethyoxynaphthalene as photosensitizer and ascorbic acid as terminal reductant furnished the desired product 18 in 51% yield (Hamada et al., 1986). Chemoselective hydrazinolysis of thioester 18 afforded disulfide 19 in 90% yield. Sequential ionization of triphenylemthyl (Tr) group and cyclization with the loss of triphenylcarbocation afforded the disulfide-bridge-carrying chaetocin A precursor in 82% yield. Subsequent hydrolysis of acetate using Otera's catalyst afforded chaetocin A 1.
G9a/GLP
G9a and GLP are the primary enzymes for mono and dimethylation at Lys 9 of histone H3 (H3K9me1 and H3K9me2), and exist predominantly as a G9a–GLP heteromeric complex that appears to be a functional H3K9 methyltransferase in vivo (Shinkai and Tachibana, 2011). G9a-GLP complex regulates a wide range of biological activities including germ cell development, meiosis, DNA replication, cell proliferation, and cancer cell formation. Overexpression of protein G9a is observed in many cancers, including prostate, lung, colon cancer, and lymphocytic leukemia (Chen et al., 2010; Shinkai and Tachibana, 2011). It was reported that overexpression of G9a led to increasing metathesis and invasion in lung cancer (Chen et al., 2010). Since G9a is overexpressed in many different types of cancers, and has been shown to be responsible for various aspects of tumorigenesis, including cellular differentiation, proliferation, and epithelial to mesenchymal, indicates that G9a could be a feasible target for cancer therapy.
BIX 01294 20, the first G9a/GLP inhibitor, was discovered by Kubicek et al. (2007) by High-Throughput Screening of Compound Library against G9a (Kubicek et al., 2007) (Figure 3). The crystal structure of the catalytic SET domain of GLP in complex with BIX-01294 and S-adenosyl-L-homocysteine revealed that, the inhibitor is bound in the substrate peptide groove at the location where the histone H3 residues N-terminal to the target lysine lie in the previously solved structure of the complex with histone peptide (Chang et al., 2009). BIX-01294 (20) exhibited cellular toxicity at high concentration, which limited its further development. Synthesis of BIX-01294 was reported by Liu et al. (2009). In order to obtain more potent and selective inhibitors toward G9a/GLP, modification based on the structure of BIX01294 (20) has been carried out by Jin and co-workers since 2009 (Liu et al., 2010, 2011, 2013; Vedadi et al., 2011; Konze et al., 2014) (Scheme 2). Initial investigation on G9a-BIX01294 complex and SAR studies led to the discovery of the first selective G9a inhibitor, UNC0224 (21). The corresponding X-ray crystal structure of the G9a-21 complex, was also obtained by the same research group (Liu et al., 2009). Further crystal-structure-based optimization process resulted in the discovery of UNC0321 (22), which demonstrated higher cellular potency (Morrison Ki = 63 pM) (Liu et al., 2010). Successive development based on structural design and chemical synthesis gave rise to a potent and selective inhibitor UNC0638 (23), which also served as an effective chemical probe for G9a/GLP (Liu et al., 2011; Vedadi et al., 2011). Unfortunately, the poor pharmacokinetic properties of UNC0638 limited the further animal studies of this lead compound. In order to improve the in vivo pharmacokinetic properties of UNC0638, further optimization led to the development of a more promising lead compound UNC0642 (24) (Liu et al., 2013). This inhibitor displayed IC50 < 2.5 nM and excellent selectivity toward G9a protein over other methyl transferases. In addition, it also showed an improved in vivo pharmacokinetic properties. In 2014, a biotinylated tag of UNC0965 (25) was developed by the same research group for “chemiprecipitation” of G9a protein from whole cell lysates (Konze et al., 2014).
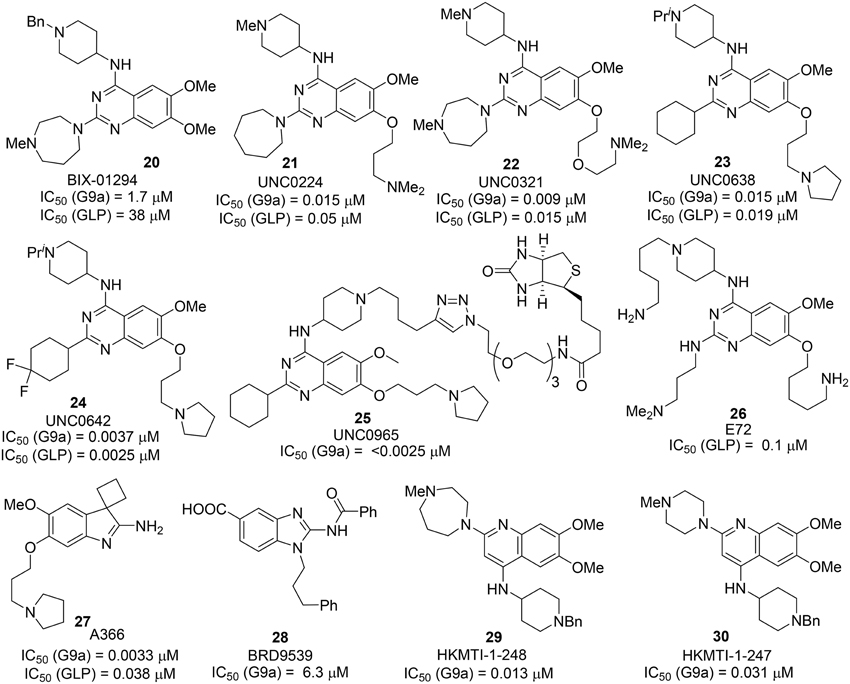
Figure 3. Example of inhibitors targeting protein G9a-GLP.
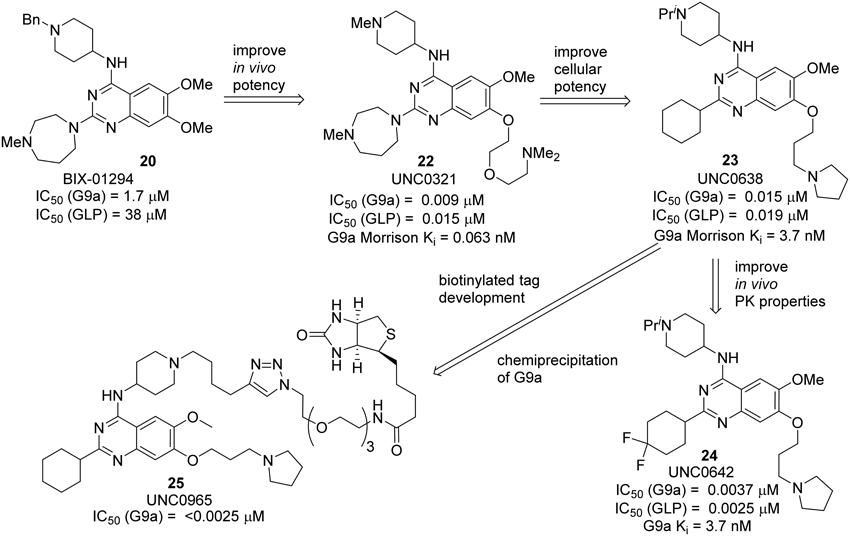
Scheme 2. Development of G9a-GLP inhibitors by Jin and co-workers.
In 2010, Chang et al. reported a potent and less toxic G9a inhibitor, E72 (26) (Chang et al., 2010) (Figure 2). However, its cellular potency is lower than that of BIX-01294 (20). Moreover, Yuan et al. and Sweis et al. independently reported BRD9539 (27) (Yuan et al., 2012) and A366 (28) (Sweis et al., 2014) as potent and selective G9a inhibitors in 2012 and 2014, respectively. In 2014, Srimongkolpithak et al. disclosed structures and the corresponding synthesis of potent G9a inhibitors Sirmongkolithak cmp 41 (29) and Sirmongkolithak cmp 41 (30) (Srimongkolpithak et al., 2014).
Synthesis of BIX-01294 (20) was reported by Liu et al. (2009) (Scheme 3A). Commercially available quinazoline derivative 31 was exposed to amine 32 in the presence of Hünig's base in DMF afforded the corresponding condensation intermediate, which was then subsequently reacted with amine 33 under acidic condition in microwave to furnish BIX01294 20 as the desired product.
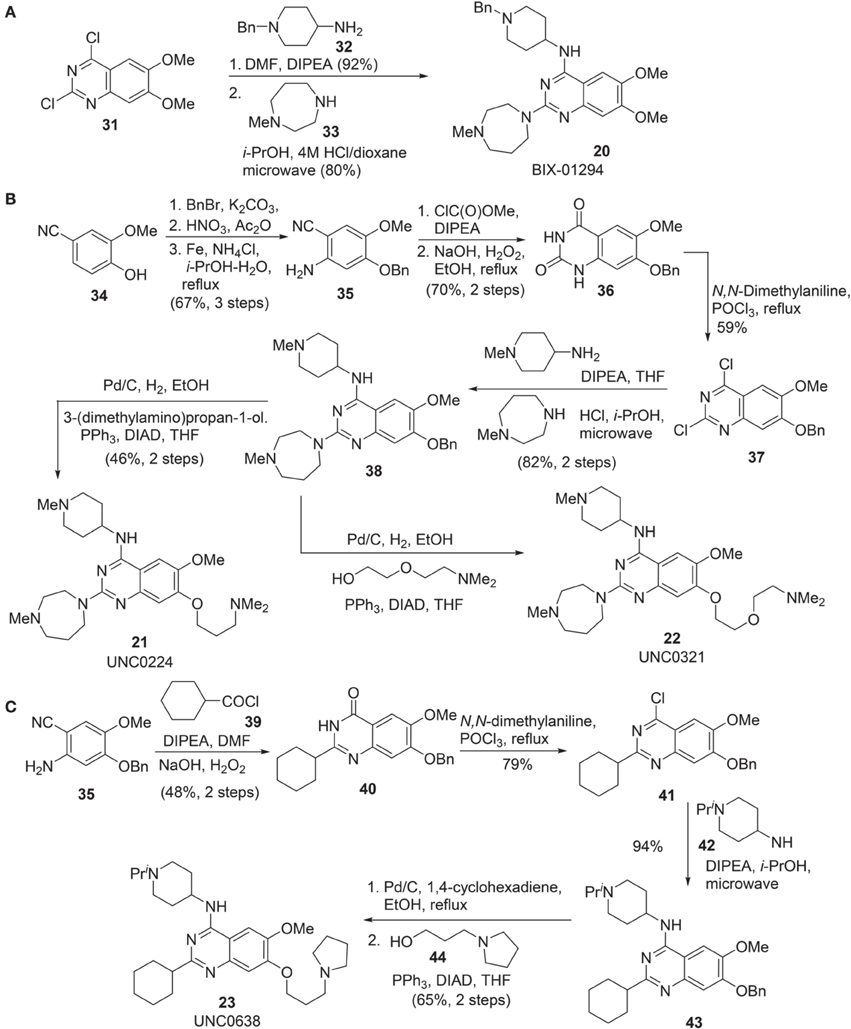
Scheme 3. (A) Synthesis of BIX-01294 by Liu et al. (B) Synthesis of UNC0224 and UNC0321 by Liu et al. (C) Synthesis of UNC0638 by Vedadi et al.
Syntheses of UNC0224 21 (Liu et al., 2009) and UNC0321 22 (Liu et al., 2010) were reported by Liu et al. (2009, 2010), respectively. (Scheme 3B) Successive benzylation, nitration, and reduction of commercially available 34 afforded aniline 35 in good yield. Treatment of aniline 35 with methyl chloroformate followed by hydrolysis of the nitrile group and simultaneous ring closure produced quinazolinedione 36 in 70% yield over two steps. Dichloroquinazoline 37, obtained by the treatment of 36 with POCl3, was subjected to two sequential chlorine displacement reactions with two different amines to afford 38 in 82% yield over two steps. With the key intermediate 38 in hand, the synthesis of UNC0224 21 and UNC0321 22 was completed by reductive debenzylation followed by elongation of the resultant phenol under Mitsunobu condition.
Synthesis of UNC0638 (23) was reported by Vedadi et al. (2011) (Scheme 3C). Intermediate 35, which has been employed in the synthesis of UNC0224 (21) (Liu et al., 2009), can be selected as the starting material for the construction of UNC0638 (23). Acylation of 35 with cyclohexanoyl chloride 39 followed by an oxidative cyclization afforded 40 in 48% yield over two steps. Chloroquinazoline 41, obtained by the treatment of 40 with POCl3, was then condensed with amine 42 to give rise to 43. Successive debenzylation followed by elongation of the resultant phenol under Mitsunobu condition produced UNC638 (23) in 65% yield.
Synthesis of UNC0642 (24) was reported by Liu et al. (2013) (Scheme 4A). Commercially available benzoate 45 was subjected to nucleophilic substitution with 1-chloro-3-iodopropane 46 followed by nitration gave rise to 47 in 75% yield over two steps. Substitution of chlorine of 47 with pyrrolidine and reduction of nitrate with Fe dust afforded 48. Dichloro-quinazoline 49 was prepared from this intermediate in 50% yield via a three-step sequence including urea formation, cyclization followed by chlorination. The consecutive displacement of two chlorine atoms with two different amines produced UNC0642 (24) in 75% yield.
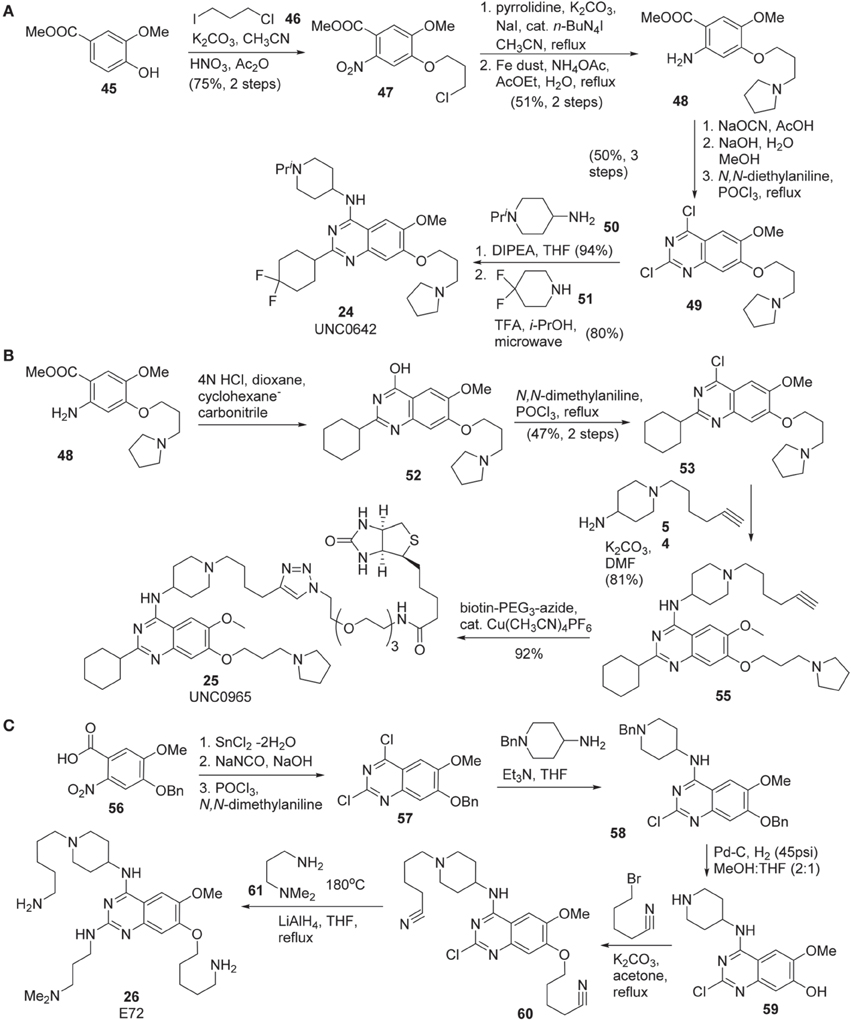
Scheme 4. (A) Synthesis of UNC0642 by Liu et al. (B) Synthesis of UNC0965 by Konze et al. (C) Synthesis of E72 by Chang et al.
Synthesis of UNC0965 25 was reported by Konze et al. (2014) (Scheme 4B). Cyclization of the known intermediate 48 afforded 52 and subsequent chlorination furnished 53 in 47% yield over two steps. Treatment of this intermediate with amine 54 afforded 55, which was then subjected to a Copper-catalyzed click reaction with commercially available biotin-PEG3-azide to afford UNC0965 (25) in 92% yield.
The preparation of E72 (26) was disclosed by Chang et al. (2010) (Scheme 4C). This synthesis is straight-forward, however, no yield for each step of the synthesis was reported. The starting material 56, prepared according to the previously described procedure (Thurston et al., 1990), was reduced by the action of SnCl2 (Hu et al., 2001) and then converted into dichloroquinazoline 57 via a two step sequence including treatment of the acid with sodium isocyanate followed by chlorination by the use of POCl3(Andrus et al., 2002; Smits et al., 2008). Displacement of the more reactive chlorine atom with 4-amino-1-benzylpiperidine afforded 58, which was subjected to catalytic hydrogenolysis to furnish 59. Treatment of 59 with 5-bromopentanenitrile in the presence of K2CO3 in refluxing acetone to produce 60. Displacement of chlorine atom with N, N, -dimethylaminopropylamine at high temperature followed by the reduction of nitriles to primary amines using lithium aluminum hydride to furnish E72 (26) as the desired product.
Synthesis of A366 (27) was reported by Sweis et al. (2014) (Scheme 5A). Treatment of 62 with 1,3-dibromopropane under basic condition afforded the corresponding cyclobutane-containing derivative 63, which was then subjected to nitration to give rise to 64. Successive debenzylation and nucleophilic substitution with 1-(3-bromopropyl)pyrrolidine produced 65. Intramolecular cyclization was accomplished under reductive conditions to furnish A366 (27) in 28% yield.
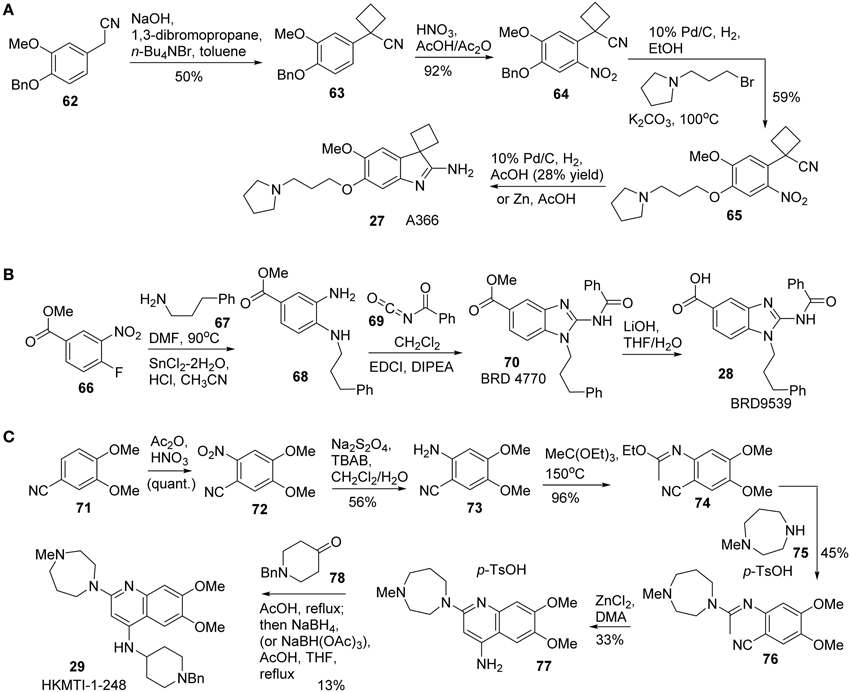
Scheme 5. (A) Synthesis of A366 by Sweis et al. (B) Synthesis of BRD9539 by Yuen et al. (C) Synthesis of HKMTI-1-248 by Srimongkolpithak et al.
Synthesis of BRD9539 (28) was disclosed by Yuan et al. (2012). The detailed synthesis is shown in Scheme 5B, however, no yield was reported for the synthesis. Nucleophilic aromatic substitution of fluorobenzoate derivative 66 with phenylpropylamine 67 followed by a reduction of nitro group with SnCl2 afforded 68. Treatment of 68 with benzoyl isocyanate 69 afforded the corresponding benzoyl urea, which was then undergoing a EDCI-mediated cyclization to give rise to BRD4770 (70). Saponification of 70 produced BRD9539 (27).
Quinoline derivative HKMTI-1-248 was reported by Srimongkolpithak et al. (2014). The synthesis commenced from the nitration of commercially available substrate 71 afforded 72. A sequential reduction of 72 to the corresponding amine 73, followed by condensation of the resulting amine with triethyl orthoacetate at high temperature to afford N-arylimidic ester 74. Treatment of 74 with 1-methylhomopiperazine 75 in the presence of p-toluenesulfonic acid produced 76, which was then subjected to a ZnCl2-promoted ring closure process to afford 77 (Moore and Kornreich, 1963). Condensation of aniline 77 with ketone 78 followed by a reductive amination to furnish HKMTI-1-248 (Scheme 5C).
EZH2
EZH2 is one of the first histone lysine methyltransferases found to be related to human cancers (Varambally et al., 2002; Kleer et al., 2003). EZH2 over expression has been linked to breast, prostate, and bladder cancers. EZH2/EZH1 is one of the core components within the PRC2 complex while EZH2 is the enzymatic subunit responsible for histone H3K27 methylation (Czermin et al., 2002; Kuzmichev et al., 2002; Muller et al., 2002; Margueron and Reinberg, 2011). It has been reported that ablation of EZH2 in tumor cells using RNA interference technology suggested that the enzyme is a promising drug target for cancer treatment (Varambally et al., 2002).
Three selective and potent EZH2 inhibitors namely EPZ005687 (79) (Knutson et al., 2012), GSK126 (80) (Mccabe et al., 2012), and Novartis EI1 (81) (Qi et al., 2012) were reported in 2012 (Figure 4). These inhibitors exhibited high selectivity over other histone methyltransferase. Verma et al. disclosed a high potent EZH2 inhibitor GSK343 (84) (Verma et al., 2012), while Konze et al. reported a selective EZH2 inhibitor UNC1999 (85) which possesses structural characteristics similar to those of EPZ005687 (79) and GSK 126 (80) with a pyridine-amide scaffold (Konze et al., 2013). UNC1999 (85) has better physicochemical properties and oral bioavailability when compared with previously reported inhibitors. Further optimization led to the development of the latest version of EZH2 inhibitor EPZ-6438 (82), which exhibited excellent potency, selectivity, and pharmacokinetic properties over EPZ005687 (79) (Knutson et al., 2013, 2014), and which has recently progressed into Phase-I trial for treatment of lymphoma.
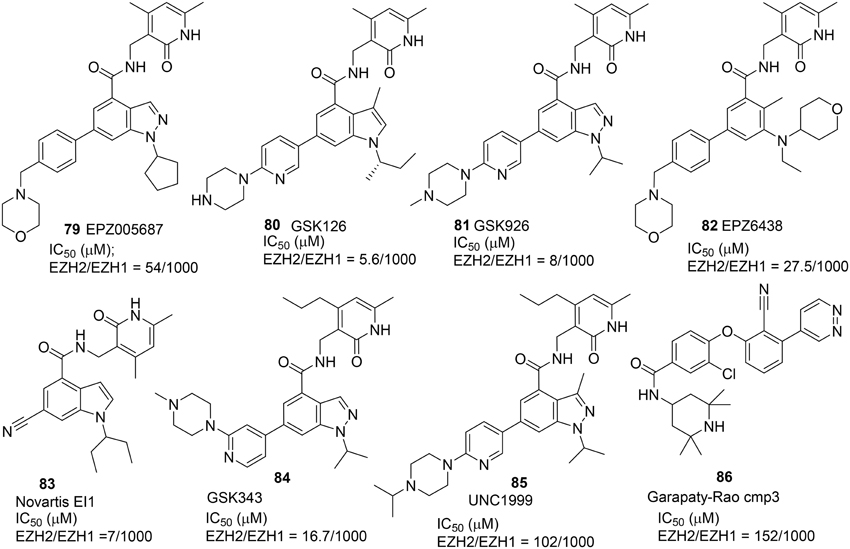
Figure 4. Example of inhibitors targeting protein EZH.
All these EZH2 inhibitors (Figure 4) are SAM-competitive. So far, no synthesis for Novartis EI1 (83) has been reported. The synthesis of EPZ-6438 (82) was described in a patent (WO2012142504). Synthesis of EPZ005687 (79) was reported by Knutson et al. (2012) (Scheme 6A). meta-Selective bromination of benzoic acid derivative 87 with 1,3-dibromo-5,5-dimethyl-2,4-imidzaolidinedione (DBDMH) 88 afforded 89 in 98% yield (Eguchi et al., 1994). Methylation of benzoic acid group of 89 using sodium carbonate and iodomethane afforded ester 90, which was then converted into aniline 91 by reduction of the nitro group. Acetylation of aniline 91 followed by nitrozation with tert-butyl nitrite under phase-transfer condition afforded indazole derivative 92, which was subjected to deacetylation to afford 93 in 72% yield. Alkylation of 93 with bromocyclopentane was facilitated by cesium carbonate to give rise to 94 in 29% yield. Saponification of 94 afforded the free carboxylic acid, which was condensed with amine 95 by the action of PyBop to produce 96 in 56% yield. Suzuki coupling between aryl bromide 96 and boronate 97 furnished EPZ005687 (79) in 68% yield.
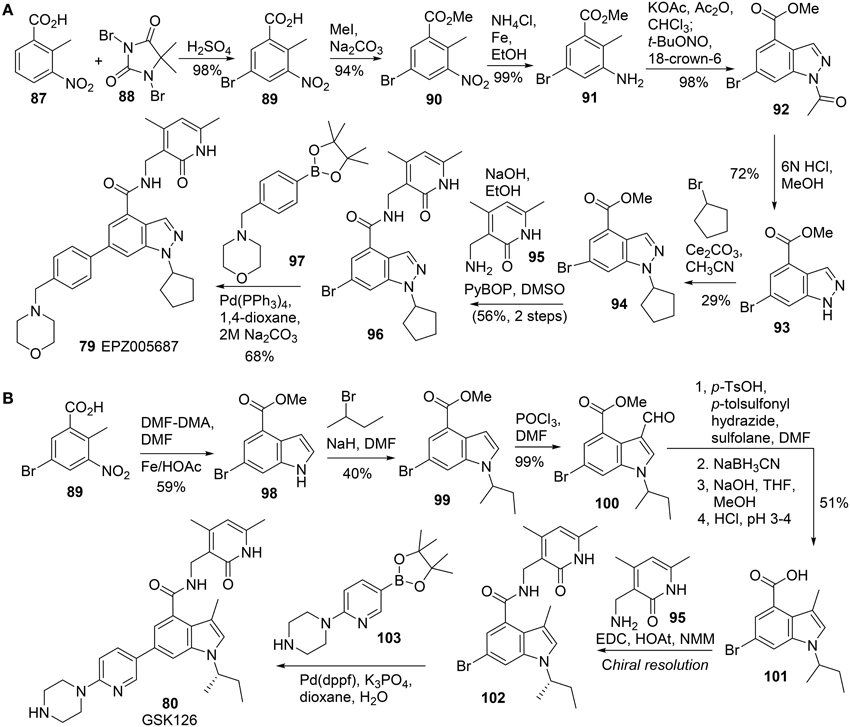
Scheme 6. (A) Synthesis of EPZ005687 by Kuntson et al. (B) Synthesis of GSK126 by McCabe et al.
GSK126 (80) was prepared by Mccabe et al. (2012) (Scheme 6B), according to an approach which is similar to the one reported by Knutson (Knutson et al., 2012). Following Leimgruber-Batcho indole synthesis, methyl 5-bromo-2-methyl-3-nitrobenzoate 89 was readily converted into methyl 6-bromo-1H-indole-4-carboxylate 98 in 59% yield. (Batcho and Leimgruber, 1985) Alkylation of indole nitrogen of 98 with 2-bromobutane followed by formylation of 99 at C-3 position delivered 100 in 40% yield over two steps. Deoxygenative reduction of aldehyde 100 was mediated by N-tosylhydrazine with sodium cyanoborohydride as the reductant to afford the corresponding reduction product, which was then saponified to the corresponding acid 101. Condensation of acid 101 with amine 95 was mediated by EDC-HOAt to furnish the corresponding amide which was then subjected to chiral resolution to afford the desired (S)-isomer (102). Suzuki cross-coupling of aryl bromide A 102 and boronate 103 completed the synthesis of GSK126 (80).
Synthesis of GSK926 (81) and GSK 343 (84) was reported by Verma et al. (2012) (Scheme 7A). Alkylation of 93 with 2-bromoisopropane afforded 104 in 43% yield. Saponification of 104 followed by coupling of the resultant acid 105 with amine 95 furnished amide 106, which was then subjected to Suzuki coupling with boronic ester 107 to give rise to GSK926 (81) in 56% yield. GSK343 (84) was prepared following the sequences described for the synthesis of GSK926 (81). (Scheme 7A) Thus, condensation of acid 105 and amine 108 produced the corresponding amide 109, which underwent a highly efficient Suzuki coupling reaction with 110 to complete the synthesis.
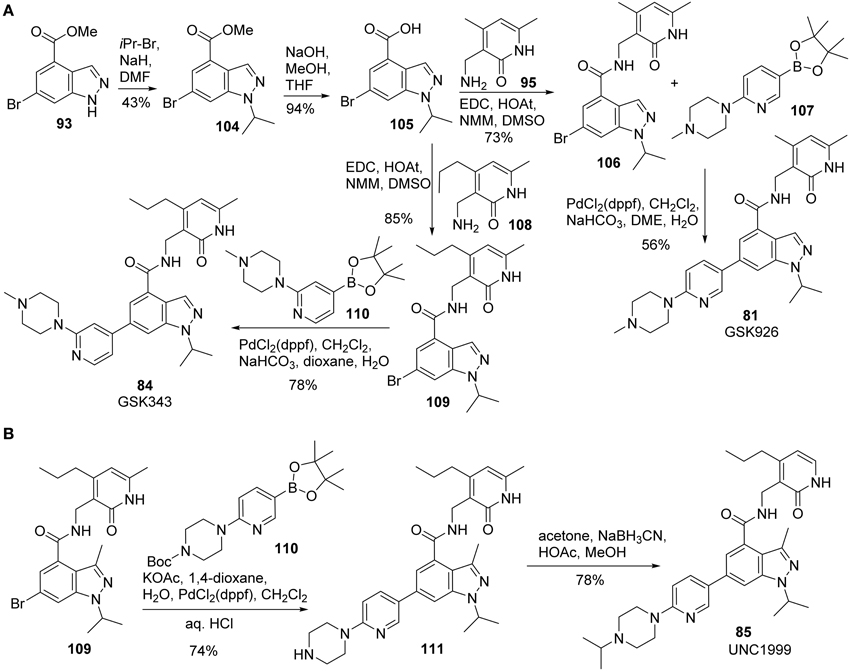
Scheme 7. (A) Synthesis of GSK926 and GSK343 by Verma et al. (B) Synthesis of UNC1999 by Konze et al.
Synthesis of UNC1999 (85) was reported by Konze et al. (2013) (Scheme 7B). The synthetic strategy for UNC1999 (85) is similar to that employed for the preparation of GSK926 (81) and GSK343 (84) (Verma et al., 2012). With the key intermediate 109 in hand, Suzuki coupling of 109 with boronate 110 followed by deprotection of Boc group afforded 111 in 74% yield. Reductive amination of the second amine with acetone by sodium cyanoborohydride then furnished UNC1999 (85) in 85% yield.
Synthesis of Garapaty-Rao cmp3 (86) (Garapaty-Rao et al., 2013) was accomplished by Nasveschuk et al. and disclosed in 2014 (Nasveschuk et al., 2014) (Scheme 8). Treatment of phenol 112 with fluoroarene 113 underwent a SNAr reaction to afford biaryl ether 114 in 81% yield (Zhu, 1997). Condensation of acid 114 with amine 115 was facilitated by the action of EDCI to furnish amide 116. Stille coupling of freshly prepared 116 with tributyltin derivative 117 under standard condition afforded cmp3 (86) in 60% yield.
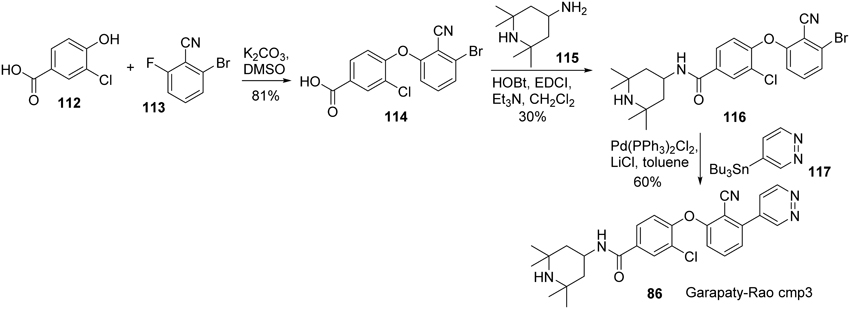
Scheme 8. Synthesis of Garapaty-Rao cmp3 by Nasveschuk et al.
DOT1L
DOT1 and DOT1L (DOT1-Like protein) are the only known enzymes which targeted H3K79 on the nucleosome core (Feng et al., 2002; Lacoste et al., 2002; Ng et al., 2002; Van Leeuwen et al., 2002), and it methylates on the lysine residue (Frederiks et al., 2008). DOT1L was later found to be important for telomeric silencing. DOT1L-mediated H3K79 methylation was found to be related to transcriptional activation, which involves regulation of a wide range of biological processes, including transcriptional regulation, DNA repair, cell cycle regulation and myocardial genesis (Nguyen and Zhang, 2011). Unlike other histone methyltransfer, Dot1 does not contain a SET domain, and it specifically methylates nucleosomal histone H3 (Min et al., 2003). Moreover, DOT1L also interacts with interact with MLL (mixed lineage leukemia) fusion proteins (Ayton and Cleary, 2001; Okada et al., 2005; Nguyen and Zhang, 2011), which would result in the misregulation of hox gene expression, and lead to lukemogensis. Significant advances in the development of DOT1 inhibitors for acute leukemias bearing MLL rearrangements have led to the first reported histone methyltransferase inhibitor to enter human clinical trials (Daigle et al., 2013).
A small molecule SAM-mimic compound called EPZ004777 (118) was identified as a potent DOT1L inhibitor by Daigle et al. (2011) (Figure 5). It selectively inhibited H3K27 methylation with high potency (IC50 = 0.4 nM) and suppress lukemogenesis by selectively killing leukemic cells that bear MLL translocation genes (Daigle et al., 2011). Later, the same research group reported the DOT1L inhibitor EPZ003696 (119) is 43 times less potent than EPZ004777 (118) on DOT1L inhibition (Basavapathruni et al., 2012). Meanwhile, Yu et al. reported the brominated analog of EPZ004777, SGC946 (120), exhibited more significant potency in cell than that of EPZ004777 (118) (Yu et al., 2012). In 2013, Daigle et al. identified the optimized SAM-mimic, EPZ5676 (124), is a potent DOT1L inhibitor that causes tumor regressions in a rat xenograft model of MLL-rearranged leukemia. This inhibitor was more than 37 000-fold selective for DOT1L over 16 other protein methyltransferases and has entered a Phase I clinical trial (Daigle et al., 2013). Studies on pharmokinetics and metabolism of EPZ5676 (124) were also reported (Basavapathruni et al., 2014). EPZ5676 (124) was developed by Epizyme and the detailed synthesis was reported in a patent (Olhava et al., 2012). Besides, Yao et al. and Yu et al. revealed two highly potent and selective DOT1L inhibitors, Yao cmp4 (122) (Yao et al., 2011, 2012) and BrSAH (123) (Yu et al., 2013b), in 2011 and 2013, respectively. Yao cmp4 (122) inhibits DOT1L as a SAM analog at the SAM binding site. Discovery of BrSAH (123) by Yu et al. demonstrated that the addition of a single halogen at the critical position of cofactor SAH (by-product of SAM DOT1L-catalyzed histone methylation) could result in a 8-fold increase in potency against DOT1L. All these DOT1L inhibitors (Figure 5) shown are SAM-competitive.
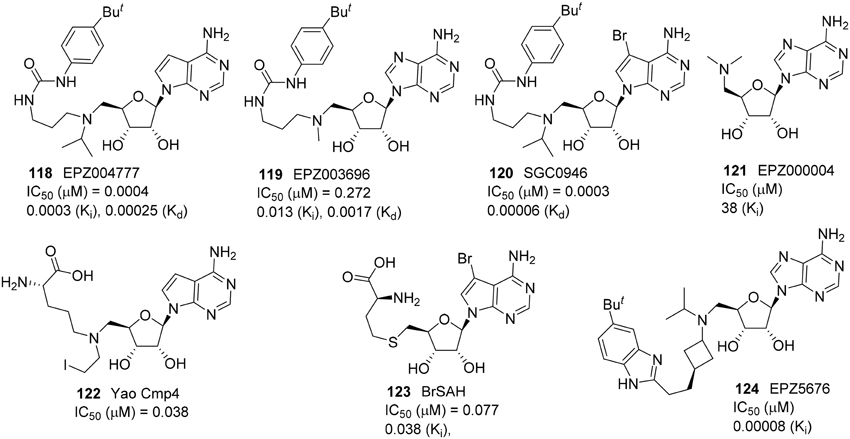
Figure 5. Example of inhibitors targeting protein DOL1L.
Synthesis of EPZ004777 (118) was reported by Basavapathruni et al. (2012) (Scheme 9). Treatment of 7-chloro tubercidin (125) with 1-(2,4-dimethyoxyphenyl) methanamine at 110°C afforded 126 in 90% yield. The cis-diol of 126 was protected as its acetonide 127 and the remaining primary alcohol was then converted into its corresponding azide 128 under Mitsunobu condition. The azide moiety of 128 was reduced to amine 129 by the action of PMe3, followed by a reductive amination to give rise to secondary amine 130. N-alkylation of amine 130 with N-(3-Bromopropyl)phthalimide furnished 131 in 72% yield. Cleavage of the N-phthaloyl group of 131, followed by coupling of the resultant primary amine with 1-tert-butyl-4-isocyanotobenzene to afford the desired urea derivative 132 in 73% yield over two steps. Finally, deprotection of both acetonide and DMP protecting groups using TFA produced EPZ004777 (118) in 46% yield.

Scheme 9. Synthesis of EPZ004777 by Basavapathruni et al.
Yu and co-workers reported the synthesis of EPZ004777 (118) using a different route in which both the primary amine and pyrimidin-4-amine were installed via the corresponding azide substitution and subsequent reduction (Yu et al., 2012, 2013a). In addition, a brominated analog of EPZ004777 (118), named SGC946 (120), was also synthesized by the same research group (Yu et al., 2012, 2013a) (Scheme 10A). Thus, treatment of bis-protected D-ribose (133) with 134 in the form of sodium salt afforded 136 in 70% yield. Similar reaction with 135 afforded 137 in 44% yield. 137 could also be obtained by bromination of 136 in essentially quantitative yield. Amine 144 could be constructed from the key intermediate 136 by sequential transformations including cleavage of the TBS protecting group, conversion of the resultant primary alcohol into the corresponding iodide, azide displacement followed by catalytic hydrogenation of the resultant azide derivative. Amine 144 was subjected to two sequential reductive amination processes to afford urea derivative 147 in 80% yield over two steps. Both acetone and oxazinanol 146 were employed as reactants during the respective reductive amination process (Scheme 10A). Acid hydrolysis of the acetonide protecting group furnished EPZ004777 (118) in 92% yield.
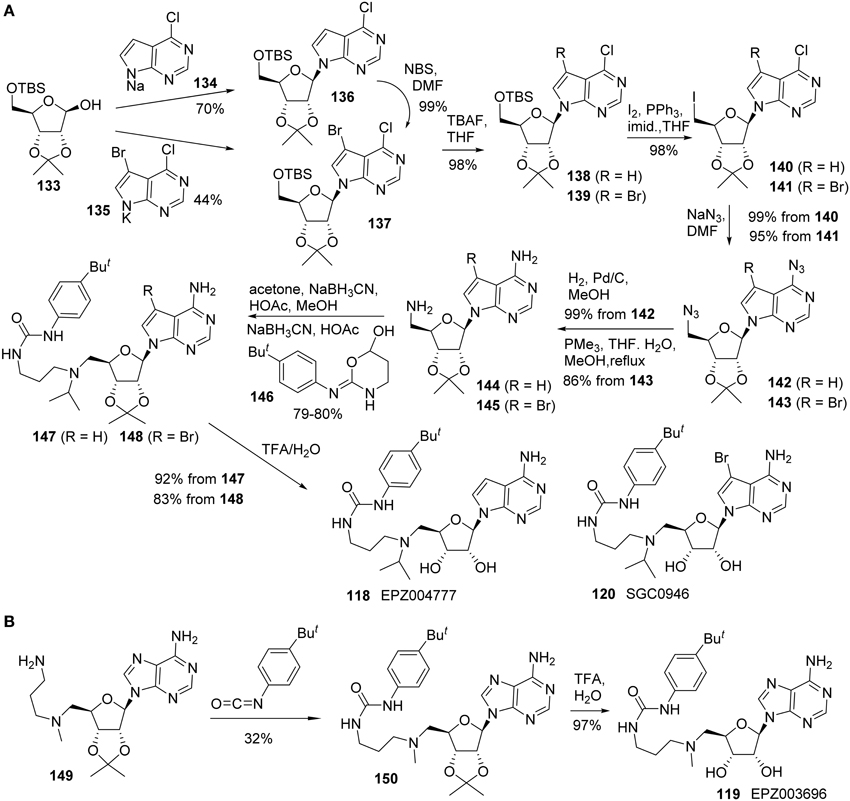
Scheme 10. (A) Synthesis of EPZ004777 and SGC0946 by Yu et al. (B) Synthesis of EPZ003696 by Basavapathruni et al.
Amine 145 was converted into SGC0946 (120) using the same sequence of reactions and Staudinger reduction was employed for the conversion of azide 143 to the corresponding amine 145.
Synthesis of EPZ003696 (119) was reported by Basavapathruni et al. (2012) (Scheme 10B). The synthesis began with the known amine 149, which was prepared as previously described (Mccloskey et al., 2009). Treatment of amine 149 with 1-tert-butyl-4-isocyanotobenzene afforded the corresponding urea derivative 150 in 32% yield. TFA-mediated removal of acetonide afforded EPZ003696 119 in 97% yield.
Synthesis of Yao cmp4 (122) is illustrated in Scheme 11A (Yao et al., 2011). The cis-diol of adenosine 151 was protected as the corresponding acetonide. Direct N-alkylation of a phthalimide with the primary alcohol under Mitsunobu conditions afforded the corresponding N-phthaloyl protected amine, which was cleaved with hydrazine to give rise to amine 152. This amine (152) was alkylated with ethyl bromoacetate followed by reduction of the ethyl ester with LiAlH4 to afford 153 in 75% yield over five steps. Reductive amination of 153 with aldehyde 154 furnished 155 in 55% yield. Conversion of the hydroxy group to the corresponding iodide was mediated by the action of PPh3, iodine and imidazole. Global deprotection of the resultant iodide furnished Yao Cmp4 (122) in 75% yield over two steps.
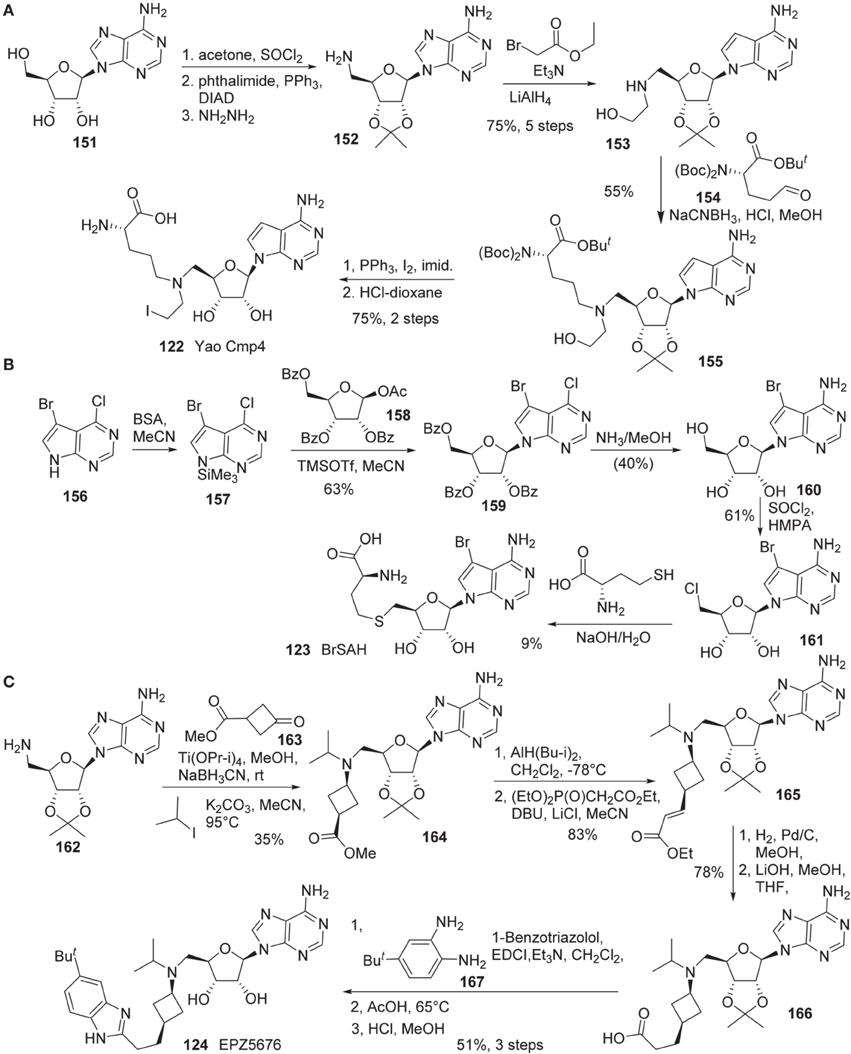
Scheme 11. (A) Synthesis of Yao cmp4 by Yao by Yu et al. (B) Synthesis of BrSHA by Yu et al. (C) Synthesis of EPZ5676 by Olhava et al.
Synthesis of BrSAH (123) was reported by Yu et al. (2013b) (Scheme 11B). Treatment of 5-bromo-4-chloro-7H-pyrrolo[2,3-d]pyrimidine 160 with N,O-Bis(trimethylsilyl)-acetamide (BSA) in acetonitrile furnished silylated intermediate 157, which was subjected to glycosylation with 158 to afford 159 in 63% yield (Seela and Ming, 2007). Simultaneous displacement of chlorine atom with ammonia and hydrolysis of benzoyl groups furnished 160 in 40% yield. The primary hydroxy group of 160 was converted into the corresponding chloride with SOCl2to give rise to 161 in 61% yield. Nucleophilic substitution of 161 with L-homocysteine under alkaline conditions afforded the desired product BrSAH 123 in 9% yield.
The detailed synthesis of EPZ5676 (124) was described by Epizyme in a patent (Olhava et al., 2012) (Scheme 11C). Thus, 5′-Amino-5′-deoxy-2′,3′-O-(1-methylethylidene)-adenosine (162) was transformed into the corresponding bis-alkylated product (164) in a two step sequence involving reductive amination with ketone 163, and alkylation with isopropyl iodide. Dibal reduction of 164 in dichloromethane at -78°C provided the corresponding aldehyde, which underwent Wadsworth-Emmons olefination to afford 165 in 83% yield in two steps. Catalytic hydrogenation of α,β-unsaturated ester, followed by saponification of the ethyl ester by treatment with aqueous lithium hydroxide in methanl/tetrahydrofuran afforded acid 166 in 78% yield. Acid 166 was converted into EPZ5676 (124) in 51% yield by a three-step sequence involving EDCI-promoted coupling of acid 166 with diamine 167, acid-mediated benzimidazole cyclization and cleavage of the acetonide moiety.
SET8
SET8 (also known as PR-Set7/9, SETD8, KMT5A), a member of the SET domain-containing methyltransferase family is solely responsible for the catalysis of monomethylation of histone protein H4K20 (Fang et al., 2002; Nishioka et al., 2002; Beck et al., 2012). Since H4K20 methylation is widespread in the genome and appears to be regulated during the cell cycle, SET8 plays important roles in various of biological processes, which involved in regulation of a range of biological processes, namely gene transcription (Congdon et al., 2010; Li et al., 2011), cell-cycle progression and development (Jorgensen et al., 2007; Abbas et al., 2010; Centore et al., 2010; Wu et al., 2010), genome integrity (Houston et al., 2008; Oda et al., 2009), DNA replication, DNA double-strand breaks and DNA damage response (Dulev et al., 2014). Moreover, SET8 demonstrated its ability on methylation of non-histone proteins including p53 tumor suppressor protein (Shi et al., 2007). This methylation process resulted in the suppression of p53 mediated transcription activation of highly responsive target genes. In addition, SET8 catalyzes monomethylation of proliferating cell nuclear antigen (PCNA) that results stabilization of PCNA protein and consequentially promotes cancer cell proliferation (Takawa et al., 2012). SET8 also played a novel role in tumor invasion and metastasis and provided a molecular mechanism underlying TWIST-promoted EMT (Yang et al., 2012).
Selective small-molecule inhibitors of SET8 are important tools for investigating the biology of this emerging target. However, only a limited number of selective inhibitors of SET8 have been reported (Williams et al., 2013; Blum et al., 2014; Ma et al., 2014a,b) (Figure 6). Marine natural product nahuoic acid A (162), the first known selective inhibitor of SET8, is competitive with the cofactor SAM and non-competitive with the peptide substrate (Williams et al., 2013). Very recently, Ma et. al. disclosed the structure of UNC0379 (163), which is the first synthetic small molecule inhibitor of SET8 (Ma et al., 2014a,b). Later, Blum et al. reported the validation of three irreversible SET8 methyltransferase inhibitors with different mode of inhibition. (Blum et al., 2014). Biological studies demonstrated that the inhibition of SET8 by SPS8I1 (164) and SPS8I3 (166) are substrate-dependent while SPS8I2 (165) facilitates substrate-independent inhibition of SET8 (Blum et al., 2014). Nahouic acid A is SAM-competitive inhibitor of SET8 (Williams et al., 2013). However, no total synthesis has been reported up to date. UNC0379 is a substrate-competitive inhibitor (Ma et al., 2014a), and the corresponding synthesis is shown in Scheme 12. Treatment of commercially available 2,4-dichloro-6,7-dimethoxyquinazoline (167) with 5-(pyrrolidin-1-yl)pentan-1-amine (168) and N,N-diisopropylethylamine at room temperature afforded 169, which was then reacted with pyrrolidine in the present of N,N-diisopropylethylamine and under microwave irradiation to give rise to UNC0379 (163) in 33% yield over two steps. (Ma et al., 2014a).
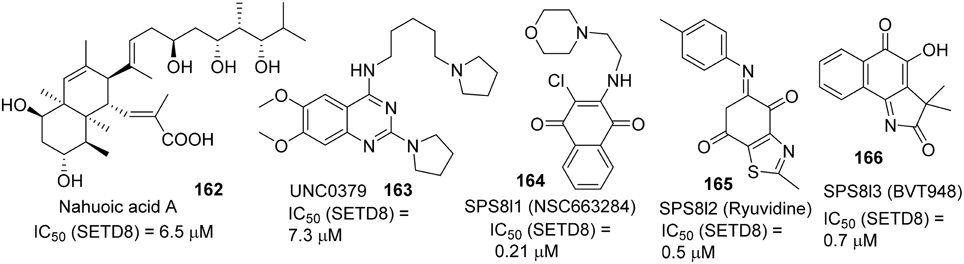
Figure 6. Example of inhibitors targeting protein SETD8.
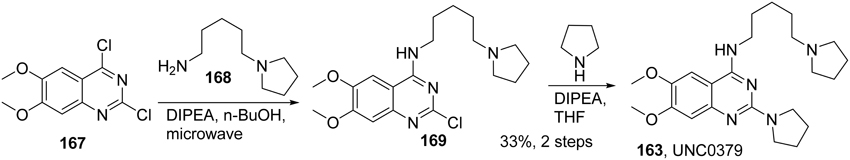
Scheme 12. Synthesis of UNC0379 by Ma et al.
Conclusions
Lysine methyltransferases continue to be an attractive drug design target for the treatment of various disease ranging from cancer, inflammation, psychiatric disorders to rheumatoid arthritis. This review outlines the development and chemical synthesis of a variety of structural chemotypes of lysine methyltransferase inhibitors that have been reported during the recent decade. The determination of the X-ray structure of a number of mechanism-based inhibitors and lysine methyltransferase complexes have provided important information for the structure-based design of potent lysine methyltransferase inhibitors. Many of these inhibitors exhibited impressive intrinsic potency. Subsequent lead optimization provided a broad range of lysine methyltransferase inhibitors with promising pharmacological properties. In recent years, a few of these new classes of lysine methyltransferase inhibitors have been advanced to pre-clinical development, and selective inhibitors of DOT1L and EZH2, have entered phase I clinical studies. The major issue that needs to be addressed is the selectivity among the different histone methyltransferases. By applying a non-competitive approach that preserves the physiological nature of endogenousl ligand-methyltransferase signaling, it may be possible to produce therapeutic agents that are safer than conventional competitive drugs. We predict that efforts through structure-based design would lead to the development of novel allosteric lysine methyltransferase inhibitors with superior selectivity and improving the overall side-effect profile.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge financial support from Hong Kong Research Grants Council (Projects: PolyU 5037/11P, 5020/12P; 5030/13P, 153035/14P); Fong Shu Fook Tong Foundation and Joyce M. Kuok Foundation.
Abbreviations
2,2-DMP, 2,2-dimethoxypropane; BSA, N,O-Bis(trimethylsilyl)acetoamide; DIAD, Diisopropyl azodicarboxylate; DIPEA, Diisopropylethylamine; DMAPA, dimethylaminopropylamine; DMPU, 1,3-Dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone; HMPA, Hexamethylphosphoramide; HMTs, histone methyltransferases; MLL, mixed lineage leukemia; NMM, N-Methylmorpholine; PKMTs, protein lysine methyltransferases; SAH, S-Adenosyl-L-homocysteine; SAM, S-Adenosyl-L-methionine; SAR, structure-activity relationship; V-70, 2,2′-azobis-(2,4-dimethyl-4-methoxyvaleronitrile).
References
Abbas, T., Shibata, E., Park, J., Jha, S., Karnani, N., and Dutta, A. (2010). CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol. Cell 40, 9–21. doi: 10.1016/j.molcel.2010.09.014
Andrus, M. B., Mettath, S. N., and Song, C. (2002). A modified synthesis of Iodoazidoaryl Prazosin. J. Org. Chem. 67, 8284–8286. doi: 10.1021/jo026217o
Arrowsmith, C. H., Bountra, C., Fish, P. V., Lee, K., and Schapira, M. (2012). Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discov. 11, 384–400. doi: 10.1038/nrd3674
Aurelio, L., Box, J. S., Brownlee, R. T. C., Hughes, A. B., and Sleebs, M. M. (2003). An efficient synthesis of N-methyl amino acids by way of intermediate 5-oxazolidinones. J. Org. Chem. 68, 2652–2667. doi: 10.1021/jo026722l
Ayton, P. M., and Cleary, M. L. (2001). Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene 20, 5695–5707. doi: 10.1038/sj.onc.1204639
Basavapathruni, A., Jin, L., Daigle, S. R., Majer, C. R. A., Therkelsen, C. A., Wigle, T. J., et al. (2012). Conformational adaptation drives potent, selective and durable inhibition of the human protein Methyltransferase DOT1L. Chem. Biol. Drug Des. 80, 971–980. doi: 10.1111/cbdd.12050
Basavapathruni, A., Olhava, E. J., Daigle, S. R., Therkelsen, C. A., Jin, L., Boriack-Sjodin, P. A., et al. (2014). Nonclinical pharmacokinetics and metabolism of EPZ-5676, a novel DOT1L histone methyltransferase inhibitor. Biopharm. Drug Dispos. 35, 237–252. doi: 10.1002/bdd.1889
Batcho, A. D., and Leimgruber, W. (1985). Indoles from 2-methylnitrobenzenes by condensation with formamide acetals followed by reduction: 4-benzyloxyindole. Org. Synth. 63, 214–220. doi: 10.15227/orgsyn.063.0214
Beck, D. B., Oda, H., Shen, S. S., and Reinberg, D. (2012). PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Gene. Dep. 26, 325–337. doi: 10.1101/gad.177444.111
Blum, G., Ibáñez, G., Rao, X., Shum, D., Radu, C., Djaballah, H., et al. (2014). Small-Molecule Inhibitors of SETD8 with Cellular Activity. ACS Chem. Biol. 9, 2471–2478. doi: 10.1021/cb500515r
Bojang, P. Jr., and Ramos, K. S. (2014). The promise and failures of epigenetic therapies for cancer treatment. Cancer. Treat. Rev. 40, 153–169. doi: 10.1016/j.ctrv.2013.05.009
Cao, R., Wang, L. J., Wang, H. B., Xia, L., Erdjument-Bromage, H., Tempst, P., et al. (2002). Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 298, 1039–1043. doi: 10.1126/science.1076997
Centore, R. C., Havens, C. G., Manning, A. L., Li, J. M., Flynn, R. L., Tse, A., et al. (2010). CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell 40, 22–33. doi: 10.1016/j.molcel.2010.09.015
Chang, Y., Ganesh, T., Horton, J. R., Spannhoff, A., Liu, J., Sun, A., et al. (2010). Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J. Mol. Biol. 400, 1–7. doi: 10.1016/j.jmb.2010.04.048
Chang, Y., Zhang, X., Horton, J. R., Upadhyay, A. K., Spannhoff, A., Liu, J., et al. (2009). Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat. Struct. Mol. Biol. 16, 312–317. doi: 10.1038/nsmb.1560
Chen, M.-W., Hua, K.-T., Kao, H.-J., Chi, C.-C., Wei, L.-H., Johansson, G., et al. (2010). H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 70, 7830–7840. doi: 10.1158/0008-5472.CAN-10-0833
Cherblanc, F. L., Chapman, K. L., Brown, R., and Fuchter, M. J. (2013a). Chaetocin is a nonspecific inhibitor of histone lysine methyltransferases. Nat. Chem. Biol. 9, 136–137. doi: 10.1038/nchembio.1187
Cherblanc, F. L., Chapman, K. L., Reid, J., Borg, A. J., Sundriyal, S., Alcazar-Fuoli, L., et al. (2013b). On the histone lysine methyltransferase activity of fungal metabolite chaetocin. J. Med. Chem. 56, 8616–8625. doi: 10.1021/jm401063r
Chi, P., Allis, C. D., and Wang, G. G. (2010). Covalent histone modifications - miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 10, 457–469. doi: 10.1038/nrc2876
Congdon, L. M., Houston, S. I., Veerappan, C. S., Spektor, T. M., and Rice, J. C. (2010). PR-Set7-mediated monomethylation of histone H4 lysine 20 at specific genomic regions induces transcriptional repression. J. Cell. Biochem. 110, 609–619. doi: 10.1002/jcb.22570
Copeland, R. A., Solomon, M. E., and Richon, V. M. (2009). Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discov. 8, 724–732. doi: 10.1038/nrd2974
Czermin, B., Melfi, R., Mccabe, D., Seitz, V., Imhof, A., and Pirrotta, V. (2002). Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111, 185–196. doi: 10.1016/S0092-8674(02)00975-3
Daigle, S. R., Olhava, E. J., Therkelsen, C. A., Basavapathruni, A., Jin, L., Boriack-Sjodin, P. A., et al. (2013). Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 122, 1017–1025. doi: 10.1182/blood-2013-04-497644
Daigle, S. R., Olhava, E. J., Therkelsen, C. A., Majer, C. R., Sneeringer, C. J., Song, J., et al. (2011). Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65. doi: 10.1016/j.ccr.2011.06.009
Dhanak, D., and Jackson, P. (2014). Development and classes of epigenetic drugs for cancer. Biochem. Biophys. Res. Commun. 455, 58–69. doi: 10.1016/j.bbrc.2014.07.006
Dulev, S., Tkach, J., Lin, S., and Batada, N. N. (2014). SET8 methyltransferase activity during the DNA double-strand break response is required for recruitment of 53BP1. EMBO Rep. 15, 1163–1174. doi: 10.15252/embr.201439434
Eguchi, H., Kawaguchi, H., Yoshinaga, S., Nishida, A., Nishiguchi, T., and Fujisaki, S. (1994). Halogenation using <i>N</i>-Halogenocompounds. II. Acid catalyzed bromination of aromatic compounds with 1,3-Dibromo-5,5-dimethylhydantoin. Bull. Chem. Soc. Jpn. 67, 1918–1921. doi: 10.1246/bcsj.67.1918
Fang, J., Feng, Q., Ketel, C. S., Wang, H. B., Cao, R., Xia, L., et al. (2002). Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr. Biol. 12, 1086–1099. doi: 10.1016/S0960-9822(02)00924-7
Feng, Q., Wang, H. B., Ng, H. H., Erdjument-Bromage, H., Tempst, P., Struhl, K., et al. (2002). Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 12, 1052–1058. doi: 10.1016/S0960-9822(02)00901-6
Frederiks, F., Tzouros, M., Oudgenoeg, G., Van Welsem, T., Fornerod, M., Krijgsveld, J., et al. (2008). Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat. Struct. Mol. Biol. 15, 550–557. doi: 10.1038/nsmb.1432
Fujishiro, S., Dodo, K., Iwasa, E., Teng, Y., Sohtome, Y., Hamashima, Y., et al. (2013). Epidithiodiketopiperazine as a pharmacophore for protein lysine methyltransferase G9a inhibitors: reducing cytotoxicity by structural simplification. Bioorg. Med. Chem. Lett. 23, 733–736. doi: 10.1016/j.bmcl.2012.11.087
Garapaty-Rao, S., Nasveschuk, C., Gagnon, A., Chan, E. Y., Sandy, P., Busby, J., et al. (2013). Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse Large B cell lymphoma cell growth. Chem. Biol. 20, 1329–1339. doi: 10.1016/j.chembiol.2013.09.013
Greiner, D., Bonaldi, T., Eskeland, R., Roemer, E., and Imhof, A. (2005). Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9. Nat. Chem. Biol. 1, 143–145. doi: 10.1038/nchembio721
Hamada, T., Nishida, A., and Yonemitsu, O. (1986). Selective removal of electron-accepting Para-toluenesulfonyl and naphthalenesulfonyl protecting groups for amino function via photoinduced donor-acceptor ion-pairs with electron-donating aromatics. J. Am. Chem. Soc. 108, 140–145. doi: 10.1021/ja00261a023
Hauser, D., Weber, H. P., and Sigg, H. P. (1970). Isolation and structure elucidation of chaetocin. Helv. Chim. Acta 53, 1061–1073. doi: 10.1002/hlca.19700530521
He, Y. L., Korboukh, I., Jin, J., and Huang, J. (2012). Targeting protein lysine methylation and demethylation in cancers. Acta Biochim. Biophys. Sin. 44, 70–79. doi: 10.1093/abbs/gmr109
Helin, K., and Dhanak, D. (2013). Chromatin proteins and modifications as drug targets. Nature 502, 480–488. doi: 10.1038/nature12751
Houston, S. I., McManus, K. J., Adams, M. M., Sims, J. K., Carpenter, P. B., Hendzel, M. J., et al. (2008). Catalytic function of the PR-Set7 histone H4 lysine 20 monomethyltransferase is essential for mitotic entry and genomic stability. J. Biol. Chem. 283, 19478–19488. doi: 10.1074/jbc.M710579200
Hu, W. P., Wang, J. J., Lin, F. L., Lin, Y. C., Lin, S. R., and Hsu, M. H. (2001). An efficient synthesis of pyrrolo [2,1-c] [1,4]benzodiazepine. Synthesis of the antibiotic DC-81. J. Org. Chem. 66, 2881–2883. doi: 10.1021/jo010043d
Itoh, Y., Suzuki, T., and Miyata, N. (2013). Small-molecular modulators of cancer-associated epigenetic mechanisms. Mol. Biosyst. 9, 873–896. doi: 10.1039/c3mb25410k
Iwasa, E., Hamashima, Y., Fujishiro, S., Hashizume, D., and Sodeoka, M. (2011a). Total syntheses of chaetocin and ent-chaetocin. Tetrahedron 67, 6587–6599. doi: 10.1016/j.tet.2011.05.081
Iwasa, E., Hamashima, Y., Fujishiro, S., Higuchi, E., Ito, A., Yoshida, M., et al. (2010). Total synthesis of (+)-Chaetocin and its analogues: their histone methyltransferase G9a inhibitory activity. J. Am. Chem. Soc. 132, 4078–4079. doi: 10.1021/ja101280p
Iwasa, E., Hamashima, Y., and Sodeoka, M. (2011b). Epipolythiodiketopiperazine alkaloids: total syntheses and biological activities. Isr. J. Chem. 51, 420–433. doi: 10.1002/ijch.201100012
Jenuwein, T., Laible, G., Dorn, R., and Reuter, G. (1998). SET domain proteins modulate chromatin domains in eu- and heterochromatin. Cell. Mol. Life Sci. 54, 80–93. doi: 10.1007/s000180050127
Jorgensen, S., Elvers, I., Trelle, M. B., Menzel, T., Eskildsen, M., Jensen, O. N., et al. (2007). The histone methyltransferase SET8 is required for S-phase progression. J. Cell. Biol. 179, 1337–1345. doi: 10.1083/jcb.200706150
Kim, J., Ashenhurst, J. A., and Movassaghi, M. (2009). Total synthesis of (+)-11,11 '-Dideoxyverticillin A. Science 324, 238–241. doi: 10.1126/science.1170777
Kim, J., and Movassaghi, M. (2010). General approach to epipolythiodiketopiperazine alkaloids: total synthesis of (+)-chaetocins A and C and (+)-12,12 '-dideoxychetracin A. J. Am. Chem. Soc. 132, 14376–14378. doi: 10.1021/ja106869s
Kita, Y., Sano, A., Yamaguchi, T., Oka, M., Gotanda, K., and Matsugi, M. (1997). Additions of malononitrile radicals to alkenes under mild conditions using 2,2′-azobis-(2,4-dimethyl-4-methoxyvaleronitrile) (V-70) as an initiator. Tetrahedron Lett. 38, 3549–3552. doi: 10.1016/S0040-4039(97)00700-4
Kleer, C. G., Cao, Q., Varambally, S., Shen, R. L., Ota, L., Tomlins, S. A., et al. (2003). EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 100, 11606–11611. doi: 10.1073/pnas.1933744100
Knapp, S., and Weinmann, H. (2013). Small-molecule modulators for epigenetics targets. ChemMedChem 8, 1885–1891. doi: 10.1002/cmdc.201300344
Knutson, S. K., Kawano, S., Minoshima, Y., Warholic, N. M., Huang, K.-C., Xiao, Y., et al. (2014). Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-hodgkin lymphoma. Mol. Cancer Ther. 13, 842–854. doi: 10.1158/1535-7163.MCT-13-0773
Knutson, S. K., Warholic, N. M., Wigle, T. J., Klaus, C. R., Allain, C. J., Raimondi, A., et al. (2013). Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc. Natl. Acad. Sci. U.S.A. 110, 7922–7927. doi: 10.1073/pnas.1303800110
Knutson, S. K., Wigle, T. J., Warholic, N. M., Sneeringer, C. J., Allain, C. J., Klaus, C. R., et al. (2012). A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 8, 890–896. doi: 10.1038/nchembio.1084
Konze, K. D., Ma, A., Li, F., Barsyte-Lovejoy, D., Parton, T., Macnevin, C. J., et al. (2013). An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem. Biol. 8, 1324–1334. doi: 10.1021/cb400133j
Konze, K. D., Pattenden, S. G., Liu, F., Barsyte-Lovejoy, D., Li, F. L., Simon, J. M., et al. (2014). A chemical tool for in vitro and in vivo precipitation of lysine methyltransferase G9a. ChemMedChem 9, 549–553. doi: 10.1002/cmdc.201300450
Kubicek, S., O'sullivan, R. J., August, E. M., Hickey, E. R., Zhan, Q., Teodoro, M. L., et al. (2007). Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 25, 473–481. doi: 10.1016/j.molcel.2007.01.017
Kuzmichev, A., Nishioka, K., Erdjument-Bromage, H., Tempst, P., and Reinberg, D. (2002). Histone methyltransferase activity associated with a human multiprotein complex containing the enhancer of Zeste protein. Gene. Dev. 16, 2893–2905. doi: 10.1101/gad.1035902
Lacoste, N., Utley, R. T., Hunter, J. M., Poirier, G. G., and Cote, J. (2002). Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J. Biol. Chem. 277, 30421–30424. doi: 10.1074/jbc.C200366200
Li, Z., Nie, F., Wang, S., and Li, L. (2011). Histone H4 Lys 20 monomethylation by histone methylase SET8 mediates Wnt target gene activation. Proc. Natl. Acad. Sci. U.S.A. 108, 3116–3123. doi: 10.1073/pnas.1009353108
Liu, F., Barsyte-Lovejoy, D., Allali-Hassani, A., He, Y., Herold, J. M., Chen, X., et al. (2011). Optimization of cellular activity of G9a inhibitors 7-Aminoalkoxy-quinazolines. J. Med. Chem. 54, 6139–6150. doi: 10.1021/jm200903z
Liu, F., Barsyte-Lovejoy, D., Li, F., Xiong, Y., Korboukh, V., Huang, X.-P., et al. (2013). Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J. Med. Chem. 56, 8931–8942. doi: 10.1021/jm401480r
Liu, F., Chen, X., Allali-Hassani, A., Quinn, A. M., Wasney, G. A., Dong, A., et al. (2009). Discovery of a 2,4-Diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J. Med. Chem. 52, 7950–7953. doi: 10.1021/jm901543m
Liu, F., Chen, X., Allali-Hassani, A., Quinn, A. M., Wigle, T. J., Wasney, G. A., et al. (2010). Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-Diamino-7-aminoalkoxy-quinazolines. J. Med. Chem. 53, 5844–5857. doi: 10.1021/jm100478y
Ma, A., Yu, W., Li, F., Bleich, R. M., Herold, J. M., Butler, K. V., et al. (2014a). Discovery of a selective, substrate-competitive inhibitor of the lysine methyltransferase SETD8. J. Med. Chem. 57, 6822–6833. doi: 10.1021/jm500871s
Ma, A., Yu, W., Xiong, Y., Butler, K., Brown, P., and Jin, J. (2014b). Structure-activity relationship studies of SETD8 inhibitors. Medchemcomm 5, 1892–1898. doi: 10.1039/C4MD00317A
Margueron, R., and Reinberg, D. (2011). The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349. doi: 10.1038/nature09784
Mccabe, M. T., Ott, H. M., Ganji, G., Korenchuk, S., Thompson, C., Van Aller, G. S., et al. (2012). EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112. doi: 10.1038/nature11606
Mccloskey, D. E., Bale, S., Secrist, J. A., Tiwari, A., Moss, T. H., Valiyaveettil, J., et al. (2009). New insights into the design of inhibitors of human S-adenosylmethionine decarboxylase: studies of adenine C8 substitution in structural analogues of S-adenosylmethionine†. J. Med. Chem. 52, 1388–1407. doi: 10.1021/jm801126a
Milne, T. A., Briggs, S. D., Brock, H. W., Martin, M. E., Gibbs, D., Allis, C. D., et al. (2002). MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. Cell 10, 1107–1117. doi: 10.1016/S1097-2765(02)00741-4
Min, J. R., Feng, Q., Li, Z. Z., Zhang, Y., and Xu, R. M. (2003). Structure of the catalytic domain of human DOT1L, a Non-SET domain nucleosomal histone methyltransferase. Cell 112, 711–723. doi: 10.1016/S0092-8674(03)00114-4
Moore, J. A., and Kornreich, L. D. (1963). A direct synthesis of 4-aminoquinolines. Tetrahedron Lett. 4, 1277–1281. doi: 10.1016/S0040-4039(01)90818-4
Movassaghi, M., and Schmidt, M. A. (2007). Concise total synthesis of (-)-calycanthine, (+)-chimonanthine, and (+)-folicanthine. Angew. Chem. Int. Ed. 46, 3725–3728. doi: 10.1002/anie.200700705
Movassaghi, M., Schmidt, M. A., and Ashenhurst, J. A. (2008). Concise Total Synthesis of (+)-WIN 64821 and (-)-ditryptophenaline. Angew. Chem. Int. Ed. 47, 1485–1487. doi: 10.1002/anie.200704960
Muller, J., Hart, C. M., Francis, N. J., Vargas, M. L., Sengupta, A., Wild, B., et al. (2002). Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111, 197–208. doi: 10.1016/S0092-8674(02)00976-5
Nasveschuk, C. G., Gagnon, A., Garapaty-Rao, S., Balasubramanian, S., Campbell, R., Lee, C., et al. (2014). Discovery and optimization of tetramethylpiperidinyl benzamides as inhibitors of EZH2. ACS Med. Chem. Lett. 5, 378–383. doi: 10.1021/ml400494b
Ng, H. H., Feng, Q., Wang, H., Erdjument-Bromage, H., Tempst, P., Zhang, Y., et al. (2002). Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Gene. Dev. 16, 1518–1527. doi: 10.1101/gad.1001502
Nguyen, A. T., and Zhang, Y. (2011). The diverse functions of Dot1 and H3K79 methylation. Gene. Dep. 25, 1345–1358. doi: 10.1101/gad.2057811
Nishioka, K., Rice, J. C., Sarma, K., Erdjument-Bromage, H., Werner, J., Wang, Y. M., et al. (2002). PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell 9, 1201–1213. doi: 10.1016/S1097-2765(02)00548-8
Oda, H., Okamoto, I., Murphy, N., Chu, J., Price, S. M., Shen, M. M., et al. (2009). Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol. Cell. Biol. 29, 2278–2295. doi: 10.1128/MCB.01768-08
Okada, Y., Feng, Q., Lin, Y. H., Jiang, Q., Li, Y. Q., Coffield, V. M., et al. (2005). hDOT1L links histone methylation to leukemogenesis. Cell 121, 167–178. doi: 10.1016/j.cell.2005.02.020
Olhava, E. J., Chesworth, R., Kuntz, K. W., Richon, V. M., Pollock, R. M., and Daigle, S. R. (2012). Preparation of substituted purine and 7-deazapurine compounds as modulators of epigenetic enzymes. PCT Int. Appl. WO 2012075381.
Pachaiyappan, B., and Woster, P. M. (2014). Design of small molecule epigenetic modulators. Bioorg. Med. Chem. Lett. 24, 21–32. doi: 10.1016/j.bmcl.2013.11.001
Qi, W., Chan, H., Teng, L., Li, L., Chuai, S., Zhang, R., et al. (2012). Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. U.S.A. 109, 21360–21365. doi: 10.1073/pnas.1210371110
Rea, S., Eisenhaber, F., O'carroll, N., Strahl, B. D., Sun, Z. W., Schmid, M., et al. (2000). Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599. doi: 10.1038/35020506
Seela, F., and Ming, X. (2007). 7-Functionalized 7-deazapurine β-d and β-l-ribonucleosides related to tubercidin and 7-deazainosine: glycosylation of pyrrolo[2,3-d]pyrimidines with 1-O-acetyl-2,3,5-tri-O-benzoyl-β-d or β-l-ribofuranose. Tetrahedron 63, 9850–9861. doi: 10.1016/j.tet.2007.06.107
Shi, X., Kachirskaia, L., Yamaguchi, H., West, L. E., Wen, H., Wang, E. W., et al. (2007). Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell 27, 636–646. doi: 10.1016/j.molcel.2007.07.012
Shinkai, Y., and Tachibana, M. (2011). H3K9 methyltransferase G9a and the related molecule GLP. Gene. Dep. 25, 781–788. doi: 10.1101/gad.2027411
Smits, R. A., De Esch, I. J. P., Zuiderveld, O. P., Broeker, J., Sansuk, K., Guaita, E., et al. (2008). Discovery of quinazolines as histamine H4 receptor inverse agonists using a scaffold hopping approach. J. Med. Chem. 51, 7855–7865. doi: 10.1021/jm800876b
Sodeoka, M., Dodo, K., Teng, Y. O., Iuchi, K., Hamashima, Y., Iwasa, E., et al. (2012). Synthesis and biological activities of chaetocin and its derivatives. Pure Appl. Chem. 84, 1369–1378. doi: 10.1351/PAC-CON-11-10-31
Spannhoff, A., Hauser, A. T., Heinke, R., Sippl, W., and Jung, M. (2009). The emerging therapeutic potential of histone methyltransferase and demethylase inhibitors. ChemMedChem 4, 1568–1582. doi: 10.1002/cmdc.200900301
Srimongkolpithak, N., Sundriyal, S., Li, F., Vedadi, M., and Fuchter, M. J. (2014). Identification of 2, 4-diamino-6, 7-dimethoxyquinoline derivatives as G9a inhibitors. Medchemcomm 5, 1821–1828. doi: 10.1039/C4MD00274A
Strahl, B. D., Grant, P. A., Briggs, S. D., Sun, Z. W., Bone, J. R., Caldwell, J. A., et al. (2002). Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell. Biol. 22, 1298–1306. doi: 10.1128/MCB.22.5.1298-1306.2002
Sweis, R. F., Pliushchev, M., Brown, P. J., Guo, J., Li, F. L., Maag, D., et al. (2014). Discovery and development of potent and selective inhibitors of histone methyltransferase G9a. ACS Med. Chem. Lett. 5, 205–209. doi: 10.1021/ml400496h
Tachibana, M., Sugimoto, K., Nozaki, M., Ueda, J., Ohta, T., Ohki, M., et al. (2002). G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Gene. Dep. 16, 1779–1791. doi: 10.1101/gad.989402
Tachibana, M., Ueda, J., Fukuda, M., Takeda, N., Ohta, T., Iwanari, H., et al. (2005). Histone methyltransferases G9a and GLP form heteromeric complexes and are both crucial for methylation of euchromatin at H3-K9. Gene. Dep. 19, 815–826. doi: 10.1101/gad.1284005
Takawa, M., Cho, H.-S., Hayami, S., Toyokawa, G., Kogure, M., Yamane, Y., et al. (2012). Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res. 72, 3217–3227. doi: 10.1158/0008-5472.CAN-11-3701
Thurston, D. E., Murty, V. S., Langley, D. R., and Jones, G. B. (1990). O-Debenzylation of a pyrrolo[2,1-c][1,4]benzodiazepine in the presence of a carbinolamine functionality: synthesis of DC-81. Synthesis 1, 81–84. doi: 10.1055/s-1990-26795
Tian, X., Zhang, S., Liu, H. M., Zhang, Y. B., Blair, C. A., Mercola, D., et al. (2013). Histone lysine-specific methyltransferases and demethylases in carcinogenesis: new targets for cancer therapy and prevention. Curr. Cancer Drug Targets 13, 558–579. doi: 10.2174/1568009611313050007
Van Leeuwen, F., Gafken, P. R., and Gottschling, D. E. (2002). Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109, 745–756. doi: 10.1016/S0092-8674(02)00759-6
Varambally, S., Dhanasekaran, S. M., Zhou, M., Barrette, T. R., Kumar-Sinha, C., Sanda, M. G., et al. (2002). The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629. doi: 10.1038/nature01075
Vedadi, M., Barsyte-Lovejoy, D., Liu, F., Rival-Gervier, S., Allali-Hassani, A., Labrie, V., et al. (2011). A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 7, 566–574. doi: 10.1038/nchembio.599
Verma, S. K., Tian, X., Lafrance, L. V., Duquenne, C., Suarez, D. P., Newlander, K. A., et al. (2012). Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med. Chem. Lett. 3, 1091–1096. doi: 10.1021/ml3003346
Wagner, T., Robaa, D., Sippl, W., and Jung, M. (2014). Mind the methyl: methyllysine binding proteins in epigenetic regulation. ChemMedChem 9, 466–483. doi: 10.1002/cmdc.201300422
Wang, H. B., Cao, R., Xia, L., Erdjument-Bromage, H., Borchers, C., Tempst, P., et al. (2001). Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol. Cell 8, 1207–1217. doi: 10.1016/S1097-2765(01)00405-1
Wang, Z., and Patel, D. J. (2013). Small molecule epigenetic inhibitors targeted to histone lysine methyltransferases and demethylases. Q. Rev. Biophys. 46, 349–373. doi: 10.1017/S0033583513000085
Williams, D. E., Dalisay, D. S., Li, F. L., Amphlett, J., Maneerat, W., Chavez, M., et al. (2013). Nahuoic acid a produced by a streptomyces sp isolated from a marine sediment is a selective SAM-competitive inhibitor of the histone methyltransferase SETD8. Org. Lett. 5, 414–417. doi: 10.1021/ol303416k
Wu, S., Wang, W., Kong, X., Congdon, L. M., Yokomori, K., Kirschner, M. W., et al. (2010). Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Gene. Dev. 24, 2531–2542. doi: 10.1101/gad.1984210
Yang, F., Sun, L., Li, Q., Han, X., Lei, L., Zhang, H., et al. (2012). SET8 promotes epithelial–mesenchymal transition and confers TWIST dual transcriptional activities. EMBO J. 31, 110–123. doi: 10.1038/emboj.2011.364
Yao, Y., Chen, P. H., Diao, J. S., Cheng, G., Deng, L. S., Anglin, J. L., et al. (2012). Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies. J. Am. Chem. Soc. 134, 17834–17834. doi: 10.1021/ja309785w
Yao, Y., Chen, P., Diao, J., Cheng, G., Deng, L., Anglin, J. L., et al. (2011). Selective inhibitors of histone methyltransferase dot1l: design, synthesis, and crystallographic studies. J. Am. Chem. Soc. 133, 16746–16749. doi: 10.1021/ja206312b
Yu, W., Chory, E. J., Wernimont, A. K., Tempel, W., Scopton, A., Federation, A., et al. (2012). Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 3:1288. doi: 10.1038/ncomms2304
Yu, W., Chory, E. J., Wernimont, A. K., Tempel, W., Scopton, A., Federation, A., et al. (2013a). Corrigendum: catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors (vol 3, 1288, 2012). Nat. Commun. 4:1893. doi: 10.1038/Ncomms2845
Yu, W., Smil, D., Li, F. L., Tempel, W., Fedorov, O., Nguyen, K. T., et al. (2013b). Bromo-deaza-SAH: a potent and selective DOT1L inhibitor. Bioorg. Med. Chem. 21, 1787–1794. doi: 10.1016/j.bmc.2013.01.049
Yuan, Y., Wang, Q., Paulk, J., Kubicek, S., Kemp, M. M., Adams, D. J., et al. (2012). A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem. Biol. 7, 1152–1157. doi: 10.1021/cb300139y
Keywords: epigenetics, lysine methyltransferase, inhibitors, synthesis, S-adenosyl-l-methionine
Citation: Hui C and Ye T (2015) Synthesis of lysine methyltransferase inhibitors. Front. Chem. 3:44. doi: 10.3389/fchem.2015.00044
Received: 24 May 2015; Accepted: 06 July 2015;
Published: 23 July 2015.
Edited by:
Xuechen LI, The University of Hong Kong, Hong KongReviewed by:
George Kokotos, University of Athens, GreeceHongyan Sun, City University of Hong Kong, Hong Kong
Copyright © 2015 Hui and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Ye, Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Room Y846 of Lee Shau Kee Building, Hung Hom, Hong Kong,dGFvX3llMzVAaG90bWFpbC5jb20=