 Wei Ding
Wei Ding Xinjian Ji
Xinjian Ji Yongzhen Li
Yongzhen Li Qi Zhang
Qi Zhang- Department of Chemistry, Fudan University, Shanghai, China
Catalytic promiscuity plays a key role in enzyme evolution and the acquisition of novel biological functions. Because of the high reactivity of radical species, in our view enzymes involving radical-mediated mechanisms could intrinsically be more prone to catalytic promiscuity. This mini-review summarizes the recent advances in the study of NosL, a radical S-adenosyl-L-methionine (SAM)-dependent L-tryptophan (L-Trp) lyase. We demonstrate here the interesting chemistry and remarkable catalytic promiscuity of NosL, and attempt to highlight the high evolvability of radical SAM enzymes and the potential to engineer these enzymes for novel and improved activities.
It has long been assumed that enzymes have evolved to carry out specific functions with specific substrate recognition. While this assumption remains true in most cases, the past 2 decades have seen a dramatically growing number of examples where multiple functions are associated with single biomolecular entities (O'Brien and Herschlag, 1999; Copley, 2003, 2015; James and Tawfik, 2003; Bornscheuer and Kazlauskas, 2004; Khersonsky et al., 2006; Nobeli et al., 2009; Humble and Berglund, 2011; Pandya et al., 2014; Pabis and Kamerlin, 2016). The term “promiscuity” has been commonly used in biochemistry to describe enzymes that catalyze more than one reaction. The ability of enzymes to catalyze the same type of reactions on a series of substrates is referred to as substrate promiscuity or substrate ambiguity, whereas catalytic promiscuity means an enzyme catalyzes different chemical transformations via different reaction mechanisms (O'Brien and Herschlag, 1999; Copley, 2003; James and Tawfik, 2003; Bornscheuer and Kazlauskas, 2004; Khersonsky et al., 2006; Nobeli et al., 2009; Humble and Berglund, 2011; Pandya et al., 2014; Copley, 2015; Pabis and Kamerlin, 2016). Although the promiscuous activities usually are not of physiological relevance and were found by accident in biochemical analyses, they provide an important impetus for enzyme evolution, such as conferring selective advantages to members of a population where the promiscuous activities are beneficial for organismal fitness.
Radicals are commonly present in biochemistry and participate in myriads of cellular signaling and metabolic processes (Stubbe and van Der Donk, 1998; Frey et al., 2006; Buckel and Golding, 2012). Featuring the unpaired valence electrons, radicals are typically highly reactive, playing a pivotal role in many chemically challenging reactions by overcoming high kinetic and/or thermodynamic barriers. The high reactivity of radical species, on the other hand, can potentially lead to various side reactions, which, from a biochemistry point of view, are promiscuous reactions. Arguably, enzymes involving radical-mediated mechanisms are intrinsically more prone to promiscuity.
Among the most prominent radical enzyme families is the radical S-adenosyl-L-methionine (SAM) superfamily, which is a large and rapidly growing enzyme superfamily currently containing over 100,000 predicted members (Sofia et al., 2001; Frey et al., 2008; Booker and Grove, 2010; Vey and Drennan, 2011; Broderick et al., 2014; Wang et al., 2014). Radical SAM enzymes are found in all domains of life and believed to be among the earliest biological catalysts on earth (Frey et al., 2008; Broderick et al., 2014). These enzymes share a common mechanism for radical generation, utilizing a [4Fe–4S] cluster to bind SAM and reductively cleave its carbon-sulfur bond to produce a 5′-deoxyadenosyl (dAdo) radical. This primary carbon alkyl radical is highly reactive and initiates a remarkably diverse variety of reactions relevant to DNA repair, RNA and protein modification, and the biosynthesis of vitamins, coenzymes, and natural products (Bandarian, 2012; Zhang et al., 2012; Fluhe and Marahiel, 2013; Byer et al., 2015; Ding et al., 2015; Jarrett, 2015; Lanz and Booker, 2015; Mehta et al., 2015; Stojkovic and Fujimori, 2015; Yang and Li, 2015; Benjdia and Berteau, 2016; Hu and Ribbe, 2016; Landgraf et al., 2016). To achieve a specific catalytic outcome and avoid unwanted side reactions, the radical intermediates are presumably controlled by delicate van der Waals interactions in the enzyme active site, as exemplified by a prototypic radical SAM enzyme lysine 2,3-aminomutase (Lees et al., 2006; Horitani et al., 2015). Because of the intrinsic high reactivity of the radical intermediates, it may not be surprising that permutation of the van der Waals interactions in the enzyme active site by using unnatural substrates can result in catalytic promiscuity. An extensively-studied example is the radical SAM enzyme DesII involved in the biosynthesis of TDP-D-desosamine (Szu et al., 2009; Ruszczycky et al., 2012). DesII is a deaminase with regard to its native substrate TDP-4-amino-4,6-didoexy-D-glucose, but it acts as a dehydrogenase on an unnatural substrate TDP-D-quinovose, in which the C4 amino group is replaced with a hydroxyl group (Szu et al., 2009; Ruszczycky et al., 2012). In addition, DesII acts both as a dehydratase and a C3 epimerase on an unnatural substrate TDP-D-fucose (Ko et al., 2015), demonstrating its remarkable catalytic promiscuity.
Recent studies on the radical SAM enzyme NosL indicated that the catalytic promiscuity of radical SAM enzymes could be even more intriguing. NosL catalyzes the carbon-chain rearrangement of L-Trp (1) to produce 3-methyl-2-indolic acid (MIA, 4) (Figure 1A), which is a key intermediate in the biosynthesis of a clinically interesting thiopeptide antibiotic nosiheptide (Yu et al., 2009; Zhang et al., 2011; Zhang and Kelly, 2012; Zhang and Liu, 2013; Just-Baringo et al., 2014). This enzyme shares sequence similarities with the L-tyrosine (L-Tyr) lyase family enzymes, including the hydrogenase-maturating enzyme HydG (Kuchenreuther et al., 2013; Duffus et al., 2014; Shepard et al., 2014; Dinis et al., 2015), thiamine biosynthesis protein ThiH (Martinez-Gomez et al., 2004; Kriek et al., 2007; Challand et al., 2010), and the F420 biosynthesis protein CofG (Decamps et al., 2012; Philmus et al., 2015); all of these enzymes are radical SAM-dependent and cleave the Cα-Cβ bond of L-Tyr. Similar to these L-Tyr lyases, in vitro assays showed that NosL cleaved the Cα-Cβ bond of L-Trp and produced significantly amount of 3-methylindole (7) (Zhang et al., 2011; Bhandari et al., 2015; Ji et al., 2015). These results lead to a hypothesis that MIA biosynthesis starts from scission of the L-Trp Cα-Cβ bond and then proceeds via an unusual recombination process that installs the carboxyl group on the C2 of the indole ring (Figure 1A; Zhang et al., 2011; Bhandari et al., 2015).
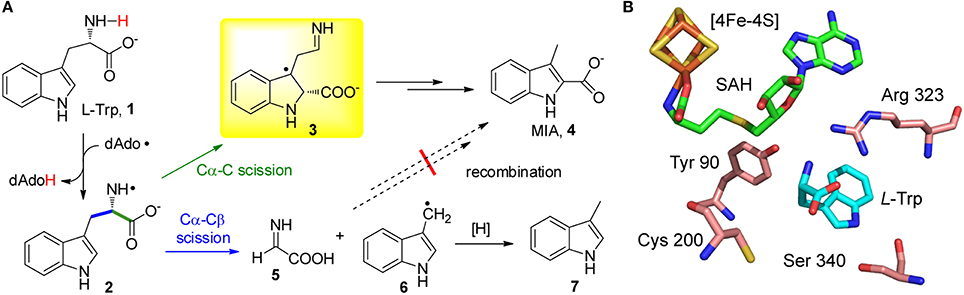
Figure 1. NosL-catalyzed MIA biosynthesis. (A) Proposed mechanism for NosL catalysis. The two fragmentation patterns of L-Trp are shown in green and blue, respectively. The key radical intermediate identified by Nicolet et al. (Sicoli et al., 2016) is shown in a yellow box. (B) The active site architecture of NosL (PDB ID: 4R33).
However, a recent study by Nicolet, Gambarelli, and coworkers demonstrated the presence of an unusual radical intermediate (3) in the NosL-catalyzed reaction, which contains a carboxyl group attached to the C2 of the indole ring (Figure 1A; Bridwell-Rabb and Drennan, 2016; Sicoli et al., 2016). Identification of this radical intermediate suggested that, in contrast to the previously proposed fragmentation-recombination mechanism, MIA biosynthesis likely proceeds via cleavage of the L-Trp Cα-C bond and subsequent migration of the carboxyl-fragment radical to the indole C2. This finding not only revealed the unusual radical chemistry in MIA biosynthesis but also highlighted the remarkable catalytic promiscuity of NosL, which diverts L-Trp into two parallel reaction pathways—one produces MIA (4) whereas the second produces glyoxylate (5) and 3-methylindole (7) (Figure 1A). Catalytic promiscuity is always associated with mutant enzymes and/or unnatural substrates, and the fact that a wild type enzyme exhibits two different types of activities with its genuine substrate is, to the best of our knowledge, unprecedented in enzymology.
The catalytic promiscuity of NosL was firstly revealed by an effort aiming to locate the hydrogen abstraction site in NosL catalysis (Ji et al., 2015), which was initially believed to be the indole nitrogen (Zhang et al., 2011) but later was suggested to be the amino group of L-Trp according to the NosL crystal structure (Figure 1B; Nicolet et al., 2014). An unnatural substrate 2-amino-3-(benzofuran-3-yl)propanoic acid (ABPA, 8) was transformed to 3-methyl-2-benzofuranic acid (MBA, 9) (Figure 2A), a benzofuran analog of MIA, thereby excluding the possibility that the dAdo radical-mediated hydrogen abstraction is from the indole nitrogen (Ji et al., 2015). Similar to this analysis, Begley et al. reached the same conclusion by using a thiophenyl substrate analog (12) and by using 1-methyl-L-Trp (13) in combination with mutagenesis (Figures 2B,C; Bhandari et al., 2015). Interestingly, when ABPA (8) was used in the reaction, the major product is neither MBA (9) nor 3-methylbenzofuran (10) but 2-(benzofuran-3-yl)ethanamine (BEA, 11) a decarboxylated product of ABPA (Figure 2A), suggesting that NosL mainly serves as a non-oxidative decarboxylase on ABPA (Ji et al., 2015).
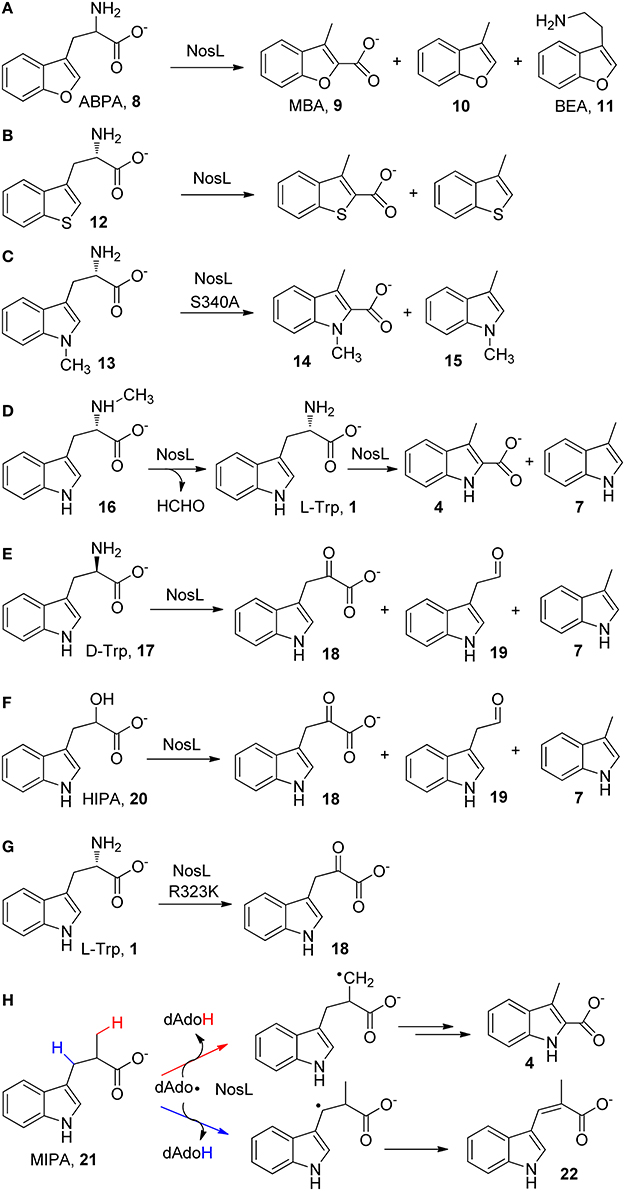
Figure 2. Catalytic promiscuity of NosL. (A–H) shows the in vitro reactions of various L-Trp analogs with NosL wild type or mutant enzymes. Simultaneous cleavage of both the Cα-Cβ and Cα-COO(H) bonds have been observed in most cases.
The catalytic promiscuity of NosL was further demonstrated by Begley and coworkers by using a series of substrate analogs (Bhandari et al., 2015). When Nα-methyl-L-Trp (16) was used in the reaction, the enzyme acted as a demethylase and produced L-Trp (1), which was further transformed to MIA (4) and 3-methylindole (7) (Figure 2D). When D-Trp (17), the enantiomer of L-Trp, was used in the reaction, three different products corresponding to three possible β-scission reactions of the tryptophanyl radical (2) (i.e., deprotonation, decarboxylation, and Cα-Cβ bond cleavage) were observed in the reaction (Figure 2E). Similar β-scission reactions were also observed when 2-hydroxy-3-(indol-3-yl)propanoic acid (HIPA, 20), an alcohol analog of Trp was used as a substrate (Figure 2F; Bhandari et al., 2015; Ji et al., 2015). Notably, while the decarboxylation reactions observed for several substrate analogs (Figures 2A,E,F) appear to be consistent with the mechanistic proposal by Nicolet et al. that involves Cα-C bond cleavage (Figure 1A), no decarboxylated product was observed in the reaction with L-Trp (Bhandari et al., 2015; Ji et al., 2015). The catalytic promiscuity of NosL was also demonstrated by a mutagenesis study, showing that change of an arginine residue (Arg 323) (Figure 2B) to lysine transformed NosL into an oxidative deaminase, which produced 3-(indol-3-yl)-2-oxopropanoic acid (18) from L-Trp and did not produce MIA and 3-methylindole anymore (Figure 2G; Bhandari et al., 2015).
More intriguingly, a recent study using 2-methyl-3-(indol-3-yl)propanoic acid (MIPA, 21), a Trp analog in which a methyl group replaces the amino group, showed that the dAdo radical produced in NosL catalysis abstracted hydrogen atoms from both the methyl group and the Cβ of MIPA (21), thereby diverting MIPA into two different reactions that produce MIA (4) and a desaturated product (22), respectively (Figure 2H; Ji et al., 2016). These results demonstrated the conformational diversity of the enzyme active site.
In contrast to its remarkable catalytic promiscuity, NosL only exhibits moderate substrate ambiguity. This enzyme was shown to transform 5-fluoro-Trp and 6-fluoro-Trp to the corresponding fluorinated MIAs, and the latter compounds can be further incorporated into nosiheptide framework by downstream enzymes to produce fluorinated antibiotics (Zhang et al., 2011). In addition, as mentioned above, NosL converted the benzofuranyl and thiophenyl Trp analogs into the corresponding MIA analogs (Figures 2A,B; Bhandari et al., 2015; Ji et al., 2015). However, for many tested Trp analogs (5-hydroxy-L-Trp, 5-bromo-Trp, 6-methyl-Trp, to name a few), NosL failed to produce detectable amounts of the corresponding MIA analogs (Zhang et al., 2011). A NosL mutant carrying a Ser-to-Ala mutation (S340A) was shown to transform 1-methyl-L-Trp (13) to 1,3-dimethyl-2-indolic acid (14) and 1,3-dimethylindole (15) (Figure 2C; Bhandari et al., 2015).
An important question regarding NosL catalysis is how the same amino-centered tryptophanyl radical (2) is partitioned roughly equally into two parallel reaction pathways in the active site (Figure 1A). Like other members of the radical SAM superfamily, NosL adopts a triose phosphate isomerase (TIM) barrel fold (Nicolet et al., 2014), which is renowned for high evolvability of diverse enzyme functions (Zhang and DeLisi, 2001). The TIM barrel folds of the radical SAM superfamily enzymes have evolved to accommodate highly diverse substrates ranging from small molecules (e.g., L-Trp for NosL) to large biomolecules such as proteins and nucleic acids (Vey and Drennan, 2011). A key arginine residue (Arg 323) that directly interacts with L-Trp via a salt bridge resides in a loop region (Figure 1B; Nicolet et al., 2014), reflecting the plasticity of the enzyme active site that accommodates the substrate. When in silico scanning the conformation of the radical intermediate (2) (Figure 1A), Nicolet et al. found two thermally accessible stable conformations whose spin densities are consistent with scission of the Cα-Cβ and Cα-C bonds, respectively (Sicoli et al., 2016), suggesting the final catalytic outcome could be fine-tuned by subtle conformational change of the radical intermediates. Nicolet et al. also showed the key radical intermediate (3) mainly adopted two conformations that exhibited different spin relaxation properties (Sicoli et al., 2016), again highlighting the conformational diversity of enzyme active site and the flexible interactions between enzyme and radical intermediates, both of which account for the observed promiscuity of NosL.
It is generally believed that enzymes have evolved to satisfy cellular metabolism and their catalytic efficiencies stop improving unless the selection pressure remains. Apparently, NosL is not an efficient enzyme and far from being well evolved, a fact that is consistent with the role of NosL in biosynthesizing a secondary metabolite—although NosL is not an efficient MIA synthase, its activity is likely enough to support nosiheptide biosynthesis by the producer. From an evolutionary point of view, the ability of NosL to perform various promiscuous activities (i.e., a generalist) (Khersonsky et al., 2006; Khersonsky and Tawfik, 2010) could possibly render the organism selective advantages under certain circumstances, whereas being a highly efficient and specific enzyme (i.e., a specialisit) (Khersonsky et al., 2006; Khersonsky and Tawfik, 2010) is not necessary for the cell in this case. Given the intrinsically high reactivity of radical species and the fact that many radical SAM enzymes are involved in secondary metabolism, it is likely that the catalytic promiscuity of the radical SAM superfamily enzymes remains largely underestimated. The catalytic promiscuity of these enzymes could provide new opportunities to engineer novel biocatalysts with improved properties, which could be of enormous academic and industrial value.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants from the State Key Laboratory of Microbial Technology (M2015-1 to WD) and from the Chinese National Natural Science Foundation (31500028 to QZ). QZ would also like to thank the Thousand Talents Program for support.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fchem.2016.00027
References
Bandarian, V. (2012). Radical SAM enzymes involved in the biosynthesis of purine-based natural products. Biochim. Biophys. Acta 1824, 1245–1253. doi: 10.1016/j.bbapap.2012.07.014
Benjdia, A., and Berteau, O. (2016). Sulfatases and radical SAM enzymes: emerging themes in glycosaminoglycan metabolism and the human microbiota. Biochem. Soc. Trans. 44, 109–115. doi: 10.1042/BST20150191
Bhandari, D. M., Xu, H., Nicolet, Y., Fontecilla-Camps, J. C., and Begley, T. P. (2015). Tryptophan lyase (NosL): mechanistic insights from substrate analogues and mutagenesis. Biochemistry 54, 4767–4769. doi: 10.1021/acs.biochem.5b00764
Booker, S. J., and Grove, T. L. (2010). Mechanistic and functional versatility of radical SAM enzymes. F1000 Biol. Rep. 2, 52. doi: 10.3410/b2-52
Bornscheuer, U. T., and Kazlauskas, R. J. (2004). Catalytic promiscuity in biocatalysis: using old enzymes to form new bonds and follow new pathways. Angew. Chem. Int. Ed. Engl. 43, 6032–6040. doi: 10.1002/anie.200460416
Bridwell-Rabb, J., and Drennan, C. L. (2016). Biochemistry. A radically unexpected mechanism. Science 351, 1266–1267. doi: 10.1126/science.aaf4942
Broderick, J. B., Duffus, B. R., Duschene, K. S., and Shepard, E. M. (2014). Radical S-adenosylmethionine enzymes. Chem. Rev. 114, 4229–4317. doi: 10.1021/cr4004709
Buckel, W., and Golding, B. T. (2012). “Radical enzymes,” in Encyclopedia of Radicals in Chemistry, Biology and Materials, eds C. Chatgilialoglu and A. Studer (Chichester, UK: John Wiley & Sons, Ltd.), 1501–1546.
Byer, A. S., Shepard, E. M., Peters, J. W., and Broderick, J. B. (2015). Radical S-adenosyl-L-methionine chemistry in the synthesis of hydrogenase and nitrogenase metal cofactors. J. Biol. Chem. 290, 3987–3994. doi: 10.1074/jbc.R114.578161
Challand, M. R., Martins, F. T., and Roach, P. L. (2010). Catalytic activity of the anaerobic tyrosine lyase required for thiamine biosynthesis in Escherichia coli. J. Biol. Chem. 285, 5240–5248. doi: 10.1074/jbc.M109.056606
Copley, S. D. (2003). Enzymes with extra talents: moonlighting functions and catalytic promiscuity. Curr. Opin. Chem. Biol. 7, 265–272. doi: 10.1016/S1367-5931(03)00032-2
Copley, S. D. (2015). An evolutionary biochemist's perspective on promiscuity. Trends Biochem. Sci. 40, 72–78. doi: 10.1016/j.tibs.2014.12.004
Decamps, L., Philmus, B., Benjdia, A., White, R., Begley, T. P., and Berteau, O. (2012). Biosynthesis of F0, precursor of the F420 cofactor, requires a unique two radical-SAM domain enzyme and tyrosine as substrate. J. Am. Chem. Soc. 134, 18173–18176. doi: 10.1021/ja307762b
Ding, W., Li, Y. Z., and Zhang, Q. (2015). Substrate-controlled stereochemistry in natural product biosynthesis. ACS Chem. Biol. 10, 1590–1598. doi: 10.1021/acschembio.5b00104
Dinis, P., Suess, D. L., Fox, S. J., Harmer, J. E., Driesener, R. C., De La Paz, L., et al. (2015). X-ray crystallographic and EPR spectroscopic analysis of HydG, a maturase in [FeFe]-hydrogenase H-cluster assembly. Proc. Natl. Acad. Sci. U.S.A. 112, 1362–1367. doi: 10.1073/pnas.1417252112
Duffus, B. R., Ghose, S., Peters, J. W., and Broderick, J. B. (2014). Reversible H atom abstraction catalyzed by the radical S-adenosylmethionine enzyme HydG. J. Am. Chem. Soc. 136, 13086–13089. doi: 10.1021/ja504618y
Fluhe, L., and Marahiel, M. A. (2013). Radical S-adenosylmethionine enzyme catalyzed thioether bond formation in sactipeptide biosynthesis. Curr. Opin. Chem. Biol. 17, 605–612. doi: 10.1016/j.cbpa.2013.06.031
Frey, P. A., Hegeman, A. D., and Reed, G. H. (2006). Free radical mechanisms in enzymology. Chem. Rev. 106, 3302–3316. doi: 10.1021/cr050292s
Frey, P. A., Hegeman, A. D., and Ruzicka, F. J. (2008). The radical SAM superfamily. Crit. Rev. Biochem. Mol. Biol. 43, 63–88. doi: 10.1080/10409230701829169
Horitani, M., Byer, A. S., Shisler, K. A., Chandra, T., Broderick, J. B., and Hoffrnan, B. M. (2015). Why nature uses radical SAM enzymes so widely: electron nuclear double resonance studies of lysine 2,3-aminomutase show the 5′-dAdo center dot “Free Radical” is never free. J. Am. Chem. Soc. 137, 7111–7121. doi: 10.1021/jacs.5b00498
Hu, Y., and Ribbe, M. W. (2016). Maturation of nitrogenase cofactor-the role of a class E radical SAM methyltransferase NifB. Curr. Opin. Chem. Biol. 31, 188–194. doi: 10.1016/j.cbpa.2016.02.016
Humble, M. S., and Berglund, P. (2011). Biocatalytic promiscuity. Eur. J. Org. Chem. 2011, 3391–3401. doi: 10.1002/ejoc.201001664
James, L. C., and Tawfik, D. S. (2003). Conformational diversity and protein evolution–a 60-year-old hypothesis revisited. Trends Biochem. Sci. 28, 361–368. doi: 10.1016/S0968-0004(03)00135-X
Jarrett, J. T. (2015). The biosynthesis of thiol- and thioether-containing cofactors and secondary metabolites catalyzed by radical S-adenosylmethionine enzymes. J. Biol. Chem. 290, 3972–3979. doi: 10.1074/jbc.R114.599308
Ji, X., Li, Y., Ding, W., and Zhang, Q. (2015). Substrate-tuned catalysis of the radical S-Adenosyl-L-methionine enzyme NosL involved in nosiheptide biosynthesis. Angew. Chem. Int. Ed Engl. 54, 9021–9024. doi: 10.1002/anie.201503976
Ji, X., Li, Y., Jia, Y., Ding, W., and Zhang, Q. (2016). Mechanistic Insights into the Radical S-adenosyl-l-methionine Enzyme NosL From a Substrate Analogue and the Shunt Products. Angew. Chem. Int. Ed Engl. 55, 3337. doi: 10.1002/anie.201509900
Just-Baringo, X., Albericio, F., and Alvarez, M. (2014). Thiopeptide antibiotics: retrospective and recent advances. Mar. Drugs 12, 317–351. doi: 10.3390/md12010317
Khersonsky, O., Roodveldt, C., and Tawfik, D. S. (2006). Enzyme promiscuity: evolutionary and mechanistic aspects. Curr. Opin. Chem. Biol. 10, 498–508. doi: 10.1016/j.cbpa.2006.08.011
Khersonsky, O., and Tawfik, D. S. (2010). Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu. Rev. Biochem. 79, 471–505. doi: 10.1146/annurev-biochem-030409-143718
Ko, Y., Ruszczycky, M. W., Choi, S. H., and Liu, H. W. (2015). Mechanistic studies of the radical S-adenosylmethionine enzyme DesII with TDP-D-fucose. Angew. Chem. Int. Ed Engl. 54, 860–863. doi: 10.1002/anie.201409540
Kriek, M., Martins, F., Leonardi, R., Fairhurst, S. A., Lowe, D. J., and Roach, P. L. (2007). Thiazole synthase from Escherichia coli: an investigation of the substrates and purified proteins required for activity in vitro. J. Biol. Chem. 282, 17413–17423. doi: 10.1074/jbc.M700782200
Kuchenreuther, J. M., Myers, W. K., Stich, T. A., George, S. J., Nejatyjahromy, Y., Swartz, J. R., et al. (2013). A radical intermediate in tyrosine scission to the CO and CN- ligands of FeFe hydrogenase. Science 342, 472–475. doi: 10.1126/science.1241859
Landgraf, B. J., McCarthy, E. L., and Booker, S. J. (2016). Radical S-adenosylmethionine enzymes in human health and disease. Annu. Rev. Biochem. 85, 485–514. doi: 10.1146/annurev-biochem-060713-035504
Lanz, N. D., and Booker, S. J. (2015). Auxiliary iron-sulfur cofactors in radical SAM enzymes. Biochim. Biophys. Acta. 1853, 1316–1334. doi: 10.1016/j.bbamcr.2015.01.002
Lees, N. S., Chen, D. W., Walsby, C. J., Behshad, E., Frey, P. A., and Hoffman, B. M. (2006). How an enzyme tames reactive intermediates: positioning of the active-site components of lysine 2,3-aminomutase during enzymatic turnover as determined by ENDOR spectroscopy. J. Am. Chem. Soc. 128, 10145–10154. doi: 10.1021/ja061282r
Martinez-Gomez, N. C., Robers, M., and Downs, D. M. (2004). Mutational analysis of ThiH, a member of the radical S-adenosylmethionine (AdoMet) protein superfamily. J. Biol. Chem. 279, 40505–40510. doi: 10.1074/jbc.M403985200
Mehta, A. P., Abdelwahed, S. H., Mahanta, N., Fedoseyenko, D., Philmus, B., Cooper, L. E., et al. (2015). Radical S-adenosylmethionine (SAM) enzymes in cofactor biosynthesis: a treasure trove of complex organic radical rearrangement reactions. J. Biol. Chem. 290, 3980–3986. doi: 10.1074/jbc.R114.623793
Nicolet, Y., Zeppieri, L., Amara, P., and Fontecilla-Camps, J. C. (2014). Crystal structure of tryptophan lyase (NosL): evidence for radical formation at the amino group of tryptophan. Angew. Chem. Int. Ed Engl. 53, 11840–11844. doi: 10.1002/anie.201407320
Nobeli, I., Favia, A. D., and Thornton, J. M. (2009). Protein promiscuity and its implications for biotechnology. Nat. Biotechnol. 27, 157–167. doi: 10.1038/nbt1519
O'Brien, P. J., and Herschlag, D. (1999). Catalytic promiscuity and the evolution of new enzymatic activities. Chem. Biol. 6, R91–R105. doi: 10.1016/s1074-5521(99)80033-7
Pabis, A., and Kamerlin, S. C. (2016). Promiscuity and electrostatic flexibility in the alkaline phosphatase superfamily. Curr. Opin. Struct. Biol. 37, 14–21. doi: 10.1016/j.sbi.2015.11.008
Pandya, C., Farelli, J. D., Dunaway-Mariano, D., and Allen, K. N. (2014). Enzyme promiscuity: engine of evolutionary innovation. J. Biol. Chem. 289, 30229–30236. doi: 10.1074/jbc.R114.572990
Philmus, B., Decamps, L., Berteau, O., and Begley, T. P. (2015). Biosynthetic versatility and coordinated action of 5′-deoxyadenosyl radicals in deazaflavin biosynthesis. J. Am. Chem. Soc. 137, 5406–5413. doi: 10.1021/ja513287k
Ruszczycky, M. W., Ogasawara, Y., and Liu, H. W. (2012). Radical SAM enzymes in the biosynthesis of sugar-containing natural products. Biochim. Biophys. Acta 1824, 1231–1244. doi: 10.1016/j.bbapap.2011.11.006
Shepard, E. M., Mus, F., Betz, J. N., Byer, A. S., Duffus, B. R., Peters, J. W., et al. (2014). [FeFe]-hydrogenase maturation. Biochemistry 53, 4090–4104. doi: 10.1021/bi500210x
Sicoli, G., Mouesca, J. M., Zeppieri, L., Amara, P., Martin, L., Barra, A. L., et al. (2016). Fine-tuning of a radical-based reaction by radical S-adenosyl-L-methionine tryptophan lyase. Science 351, 1320–1323. doi: 10.1126/science.aad8995
Sofia, H. J., Chen, G., Hetzler, B. G., Reyes-Spindola, J. F., and Miller, N. E. (2001). Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 29, 1097–1106. doi: 10.1093/nar/29.5.1097
Stojkovic, V., and Fujimori, D. G. (2015). Radical SAM-mediated methylation of ribosomal RNA. Meth. Enzymol. 560, 355–376. doi: 10.1016/bs.mie.2015.03.002
Stubbe, J., and van Der Donk, W. A. (1998). Protein radicals in enzyme catalysis. Chem. Rev. 98, 705–762. doi: 10.1021/cr9400875
Szu, P. H., Ruszczycky, M. W., Choi, S. H., Yan, F., and Liu, H. W. (2009). Characterization and mechanistic studies of DesII: a radical S-adenosyl-L-methionine enzyme involved in the biosynthesis of TDP-D-desosamine. J. Am. Chem. Soc. 131, 14030–14042. doi: 10.1021/ja903354k
Vey, J. L., and Drennan, C. L. (2011). Structural insights into radical generation by the radical SAM superfamily. Chem. Rev. 111, 2487–2506. doi: 10.1021/cr9002616
Wang, J., Woldring, R. P., Roman-Melendez, G. D., McClain, A. M., Alzua, B. R., and Marsh, E. N. (2014). Recent advances in radical SAM enzymology: new structures and mechanisms. ACS Chem. Biol. 9, 1929–1938. doi: 10.1021/cb5004674
Yang, L., and Li, L. (2015). Spore photoproduct lyase: the known, the controversial, and the unknown. J. Biol. Chem. 290, 4003–4009. doi: 10.1074/jbc.R114.573675
Yu, Y., Duan, L., Zhang, Q., Liao, R., Ding, Y., Pan, H., et al. (2009). Nosiheptide biosynthesis featuring a unique indole side ring formation on the characteristic thiopeptide framework. ACS Chem. Biol. 4, 855–864. doi: 10.1021/cb900133x
Zhang, C., and DeLisi, C. (2001). Protein folds: molecular systematics in three dimensions. Cell. Mol. Life Sci. 58, 72–79. doi: 10.1007/PL00000779
Zhang, F., and Kelly, W. L. (2012). In vivo production of thiopeptide variants. Meth. Enzymol. 516, 3–24. doi: 10.1016/B978-0-12-394291-3.00022-8
Zhang, Q., and Liu, W. (2013). Biosynthesis of thiopeptide antibiotics and their pathway engineering. Nat. Prod. Rep. 30, 218–226. doi: 10.1039/C2NP20107K
Zhang, Q., Li, Y., Chen, D., Yu, Y., Duan, L., Shen, B., et al. (2011). Radical-mediated enzymatic carbon chain fragmentation-recombination. Nat. Chem. Biol. 7, 154–160. doi: 10.1038/nchembio.512
Keywords: promiscuity, evolution, metalloenzyme, enzyme engineering, biosynthesis
Citation: Ding W, Ji X, Li Y and Zhang Q (2016) Catalytic Promiscuity of the Radical S-adenosyl-L-methionine Enzyme NosL. Front. Chem. 4:27. doi: 10.3389/fchem.2016.00027
Received: 12 April 2016; Accepted: 09 June 2016;
Published: 22 June 2016.
Edited by:
Lei Li, Indiana University – Purdue University Indianapolis, USAReviewed by:
Minkui Luo, Memorial Sloan Kettering Cancer Center, USADali Liu, Loyola University Chicago, USA
Copyright © 2016 Ding, Ji, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qi Zhang, cWl6aGFuZ0BzaW9jLmFjLmNu