 Lin Cheng1,2
Lin Cheng1,2 Xiaoyu Xie
Xiaoyu Xie Haibo Ma
Haibo Ma- 1College of Electrical and Electronic Engineering, Huazhong University of Science and Technology, Wuhan, China
- 2State Grid Electric Power Research Institute, Wuhan, China
- 3School of Electrical Engineering, Wuhan University, Wuhan, China
- 4School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, China
Octafluorocyclobutane, c-C4F8, and its derivatives are regarded as promising replacements of insulation gaseous SF6, which are currently widely used in electric equipment but suffer greatly from its greenhouse effect. Based on the recent finding that the dielectric and thermodynamics properties of insulating gases are greatly dependent on the molecule's microscopic electronic and vibrational parameters, in this work, we use density functional theory (DFT) to study the molecular structures, electron affinities, and IR-active vibrational frequencies as well as thermodynamic properties for c-C4F8 and a series of mono-, di-substituted c-C4F8 compounds. It is shown that DFT calculation of perfluoro-compounds is sensitive to the chosen functional. Although all chloro-substituted c-C4F8 molecules are found to have much larger electron affinities, only part of them have less IR intensity in the atmospheric IR “window” than c-C4F8. Such a study provides useful guideline for the pre-screening search for new insulation gases via electronic structure calculations.
Introduction
Because of its excellent electrical insulationc performance and high stability, Sulfur hexafluoride (SF6) is nowadays the most widely used insulating gas in electric equipment [e.g., gas insulated switchgear (GIS)] in the world (Okubo and Beroual, 2011). However, the current usage of SF6 suffers a great limitation because it is a greenhouse gas (GHG) regulated under the Kyoto Protocol with a global warming potential (GWP) of 23,900 over 100-year time horizon (Fang et al., 2013). As a consequence, nowadays investigations for new replacement gases for SF6 becomes highly necessary.
Due to its strong dielectric and relative low global warming potential (GWP = 8700 = 36% GWPSF6) (Christophorou and Olthoff, 2001), Octafluorocyclobutane, c-C4F8, and its derivatives as well as mixtures are now regarded as promising insulation gaseous substitutes of SF6 (Itoh et al., 1991; Yamamoto et al., 2001; Yamaji and Nakamura, 2003; Yamaji et al., 2004; Wu et al., 2006; de Urquijo and Juárez, 2009; Li et al., 2014; Zhao et al., 2016). c-C4F8 has been known to have more superior dielectric properties than SF6. The sparkover voltage of c-C4F8 is about 1.3 times that of SF6 at atmospheric pressure for an ac waveform, and the lightning impulse voltage is 1.3–1.4 times that of SF6 (Yamamoto et al., 2001). The critical reduced electric field strength (E/N)cr, where E and N represent the electric field and the particle number density respectively, was determined to be 359~434 Td (Naidu et al., 1972; de Urquijo and Basurto, 2001; Liu et al., 2007), higher than that of SF6 (362 Td). At the same time, c-C4F8 suffers from a severe shortcoming that the liquefaction temperature of c-C4F8 (−8°C at 0.1 MPa) is much higher than that of SF6(−64°C at 0.1 MPa), which greatly limits its application in cold regions and high gas pressure GIS devices (Li et al., 2014).
Nowadays it is also well-known that the dielectric and thermodynamics properties of insulating gases are greatly dependent on the molecule's microscopic electronic and vibrational parameters. (Rabie et al., 2013; Zhang et al., 2016) The recent multiple regression analysis by us Zhang et al. (2016) and Rabie et al. (2013) have indicated that the gas's relative dielectric strength is proportional to the polarizability and electron affinity of the molecule and the liquefaction temperature is proportional to its polarizability and dipole moment. Therefore, for designing ideal insulation gas with high dielectric strength and low liquefaction temperature which can be widely used in electric equipment, molecules with small dipole moments, large electron affinities and balanced polarizability are expected to be selected through effective screening via electronic structure studies among a large number of candidate molecules. Of course the gas's greenhouse effect should be also examined by infrared spectroscopy investigations. In the past several years, there have been intensive studies on the structural, vibrational characteristics and the electron binding energies of c-C4F8 and its anion through Raman, infrared, photoelectron spectrums or electron-spin resonance (ESR) experiments as well as quantum chemical calculations. (Lemaire and Livingston, 1952; Bauman and Bulkin, 1966; Chang et al., 1971; Miller and Capwbll, 1971; Beagley et al., 1987; Mao et al., 1988; Purchase et al., 1997; Gallup, 2004; ElSohly et al., 2005; Bopp et al., 2007; Choi et al., 2013) However, very few research works on the electronic structures for c-C4F8 derivatives or their corresponding anions has been done, hindering the further rational design of new SF6 replacement gases.
Motivated by the above facts, in this work we presented a systematic study for the geometries, electron affinities, and vibrational frequencies as well as thermodynamic properties for the mono-, di-chloro-substituted c-C4F8 based on first principles density functional theory DFT (Hohenberg and Kohn, 1964; Kohn and Sham, 1965).
The rest of the paper is organized as follows. Computational details are reported in Section Computational Methods. In Section Results and Discussion, the ground state geometries, bonding energies, vibrational spectra, electron affinities, dipole moments, as well as thermodynamic properties are presented and analyzed. In Section Conclusions, the main points of this work are summarized and perspectives for future research are outlined.
Computational Methods
In this work, c-C4F8 (1) and its mono- and di-chloro-substituted derivatives (one mono-substitute c-C4F7Cl and five di-substitutes c-C4F6Cl2, see Figure 1) molecules (2-7) are selected and these molecules are studied using the first-principles DFT. DFT calculations are applied to optimize the molecule's geometry and describe its electronic structure of the selected molecules and corresponding negative ions, and frequency analysis is also performed. In our study, all the first-principle DFT calculations are performed using Gaussian09 D.01 software package (Frisch et al., 2009).
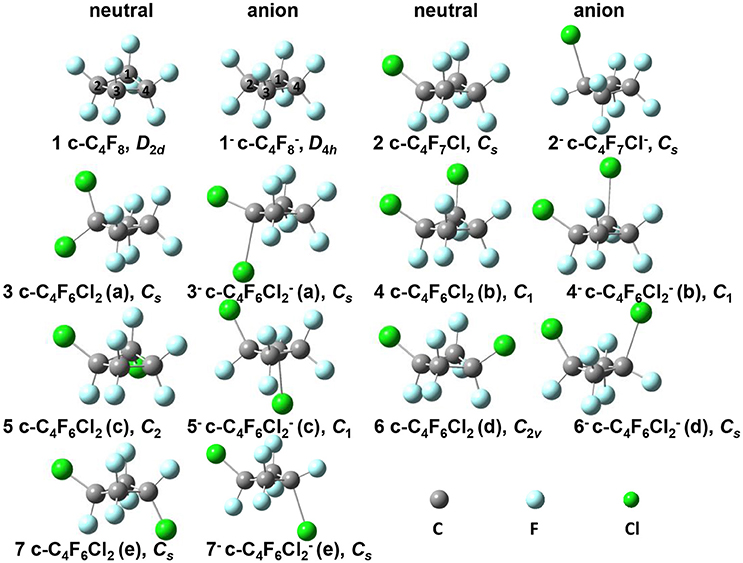
Figure 1. Optimized geometries for neutral c-C4F8−nCln (n = 0, 1, 2) series and their anions in their ground states. 1: c-C4F8, D2d symmetry; 1−: c-C4F, D4h symmetry; 2: c-C4F7Cl, Cs symmetry; 2−: c-C4F7Cl−, Cs symmetry; 3: c-C4F6Cl2(a), Cs symmetry; 3−: c-C4F6Cl(a), Cs symmetry; 4: c-C4F6Cl2(b), C1 symmetry; 4−: c-C4F6Cl(b), C1 symmetry; 5: c-C4F6Cl2(c), C2 symmetry; 5−: c-C4F6Cl(c), C1 symmetry; 6: c-C4F6Cl2(d), C2v symmetry; 6−: c-C4F6Cl(d), Cs symmetry; 7: c-C4F6Cl2(e), Cs symmetry; 7−: c-C4F6Cl(e), Cs symmetry.
The electron affinities are evaluated as the difference of the molecule's energy in the following manner: the adiabatic electron affinity is determined by, EAad = E(optimized neutral) − E(optimized anion), the vertical electron affinity by, EAvert = E(optimized neutral) − E(anion at optimized neutral geometry), and the vertical detachment energy of the anion by, VDE = E(neutral at optimized anion geometry) − E(optimized anion).
To benchmark the computational accuracy of our calculations for molecules with strong electronegative atoms like fluorine, seven DFT exchange-correlation functionals and four basis sets are tested, including GGA (Generalized gradient approximations) functional PBE (Perdew et al., 1996), hybrid functional B3LYP (Becke, 1993; Stephens et al., 1994) and M06 (Zhao and Truhlar, 2008), long-range corrected functional CAM-B3LYP (Yanai et al., 2004), ωB97-XD (Chai and Head-Gordon, 2008), and LC-ωPBE (Vydrov and Scuseria, 2006; Vydrov et al., 2006, 2007), as well as the newly developed meta-hybrid M06-2X (Zhao and Truhlar, 2008). The four tested basis sets include Pople basis sets (Krishnan et al., 1980; McLean and Chandler, 1980) 6–311+g(d) and 6–311+g(3df), and correlation-consistent basis sets aug-cc-pVTZ (Kendall et al., 1992) and aug-cc-pVQZ (Woon and Dunning, 1993).
Results and Discussion
Benchmark Tests for DFT Functional and Basis Set
It is well-known that electronic structure properties of the molecules, especially the electron affinity energy, are very sensitive to the DFT functionals and basis sets. In Figure 2, we plotted DFT calculated EA results with different functional and basis sets for c-C4F8 together with comparisons to experimental reference (0.63 ± 0.05 eV) (Chang et al., 1971). It is clearly shown that with basis set including f -type functions, long-range corrected or meta-hybrid functionals can generally give results much closer to experimental references than local or standard hybrid functions. Among them, M06-2X and ωB97-XD are shown to give smallest deviations (about −25 to 25% error). It is because in perfluoro-compounds, the large number of F atoms is associated with intense non-bonding interactions. Therefore, functional with long-range corrected or meta-hybrid functionals are required for the accurate calculation of the electronic structures of perfluoro-compounds.
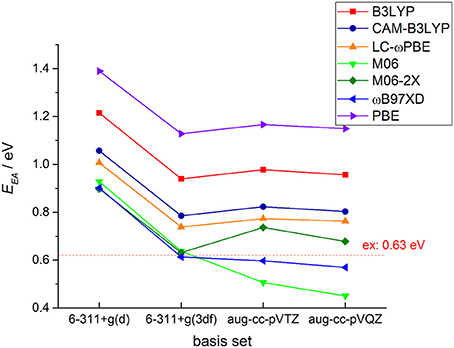
Figure 2. Exchange-correlation functional and basis set test for DFT calculations of the adiabatic electron affinity energy (EAad) for c-C4F8.
As for the basis set, from Figure 2 one may also clearly notice that not only the diffused function but also f -type polarized function are necessary for an accurate theoretical prediction of EA for possible insulating gas molecules. It is shown that 6–311+g(d) basis set gives results with large deviations from experimental references for all of DFT functionals we adopted, while 6–311+g(3df), aug-cc-pVTZ and aug-cc-pVQZ basis sets can give more reasonable results. From the data shown in the Supporting Information, one can further notice that the basis set difference only causes slight distinctions in the optimized geometries and the EA difference by different basis sets mainly comes from the single point energy difference. Considering that the further incorporation of g-type (aug-cc-pVQZ) polarized function do not yield an obvious improvement of the calculated results, we finally decided to perform our further systematic DFT calculations with 6–311+g(3df) basis set to achieve a good compromise between accuracy and computational efficiency.
Optimized Geometries
The equilibrium structures of the optimized molecules (1–7) in their both neutral and anion forms obtained from geometry optimization by DFT calculation at M06-2X/6–311g+(3df) level were shown in Figure 1, and the key structural parameters are listed in Table 1. It is shown that the molecule symmetry is decreased from D2d to C2v or Cs, C2 or even C1 with the increasing n for neutral forms of c-C4F8−nCln (n = 0, 1, 2), due to the symmetry break by chlorine substitution. Interestingly we notice that while the geometry of neutral c-C4F8 (1) is bent with D2d symmetry (∠C2C1C3C4 around 164.4°), that of its anion form (1−) becomes planar with D4h symmetry (∠C2C1C3C4 being 180.0°). This finding is consistent with earlier ESR experiments (ElSohly et al., 2005) and theoretical calculations (Choi et al., 2013). The previous computational results have shown that the added electron to c-C4F8 is delocalized in a “pi-like” orbital extending over the entire molecule, strengthening the four C-C bonds via π-bonding interactions, and weakening the eight C-F bonds via σ*-antibonding interactions. However, such a bent-to-planar transition was not observed for c-C4F8 derivatives upon attachment of an electron, due to the decrease of the symmetry by mono- and di-chloro-substitutions.
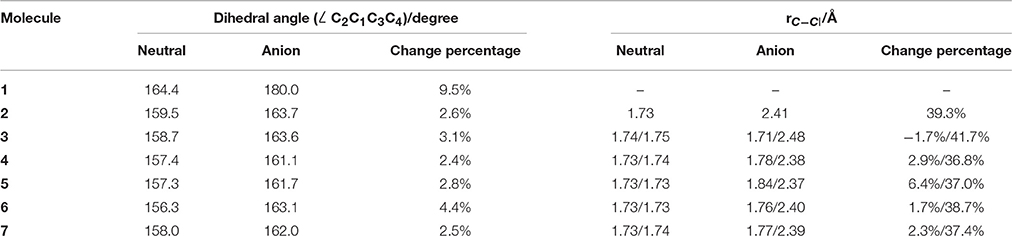
Table 1. Structural information of optimized molecules by DFT calculation at M06-2X/6–311g+(3df) level.
Figure 3 gives the highest occupied molecular orbitals (HOMO) and the lowest unoccupied molecular orbitals (LUMO) of the selected molecules 1, 2, and 6. For c-C4F8 (1), the LUMO is similar to a delocalized π-orbitals of the four carbon atoms, therefore, the attachment of an additional electron would make the carbon cycle more planar. On the contrary, for mono- and di-substituted molecules, the p-orbitals of chlorine atoms have much greater contributions to the LUMO, indicating a more localized feature at the Cl atom. As a consequence, the electron attached to the chloro-substitutes of c-C4F8 (2–7) will locate at the Cl atom to a great extent with an elongated C-Cl bond length and will not remarkably alter the planarity. This can be verified from the structure in Figure 1 and the data in Table 1, in which the ∠C2C1C3C4 change is less than 7° and the bond length difference between two C-Cl bonds are as large as 0.5–0.8 Å for the chloro-substitutes of c-C4F8 (2–7) upon electron attachment.
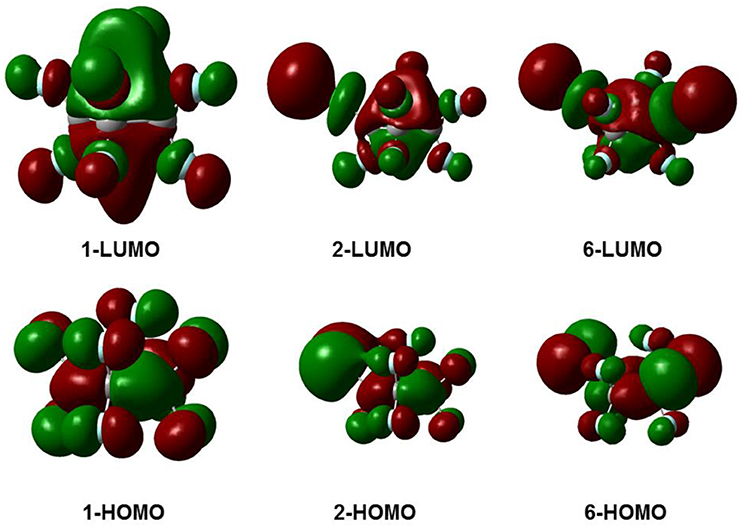
Figure 3. The frontier molecular orbitals for selected typical molecules (1, 2, 6).
The results for binding energies and dipole moments for c-C4F8−nCln (n = 0, 1, 2) are listed in Table 2. The c-C4F8 (1) molecule is found to have the highest binding energy as compared to its mono- and di-chloro-substituted derivatives (2–7), and a zero dipole moment due to its relatively high symmetry of D2d. At the same time, we notice that the binding energy is not sensitive to the different conformations for the isomers of c-C4F6Cl2 (3–7), but the dipole moment is found to be highly dependent on them. Because recent multiple regression analysis (Rabie et al., 2013; Zhang et al., 2016) have shown that the magnitudes of dipole moment are correlated to the liquefaction temperature of the gases, this implies that searching for new insulating gases which can work in extremely areas should carefully examine the molecule's possible different dipole moments caused by various isomerized conformations.
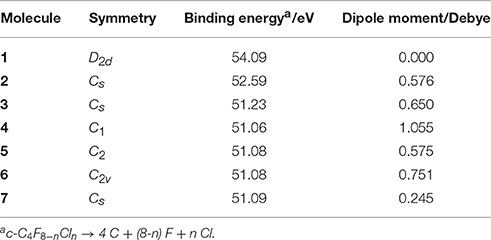
Table 2. Binding energies and dipole moments of optimized molecules by DFT calculations at M06-2X/6–311g+(3df) level.
Electronic Affinities
As the electron affinity feature has been revealed to be highly correlated with the insulating properties of the potential new insulating gases (Rabie et al., 2013; Zhang et al., 2016), in this work we also performed calculations for various electron affinity parameters of c-C4F8−nCln (n = 0, 1, 2). The calculated EAad, EAvert, and VDE by both ωB97-XD and M06-2X functionals are here listed in Table 3. As we have shown in Section Benchmark Tests for DFT Functional and Basis Set, by using suitable basis set (e.g., 6–311g+(3df)), DFT exchange-correlation functionals of ωB97-XD and M06-2X can yield very similar numerical results which are close to available experimental value. It is also shown that the electron affinity and VDA values are remarkably increased when the c-C4F8 (1) molecule is chloro-substituted. Considering that a larger EA value is beneficial to the improvement of relative electric strength (Rabie et al., 2013; Zhang et al., 2016), it would be useful to investigate the various halogen substitutions of currently used insulating gases of perfluoro-compounds for future improvement or replacement.
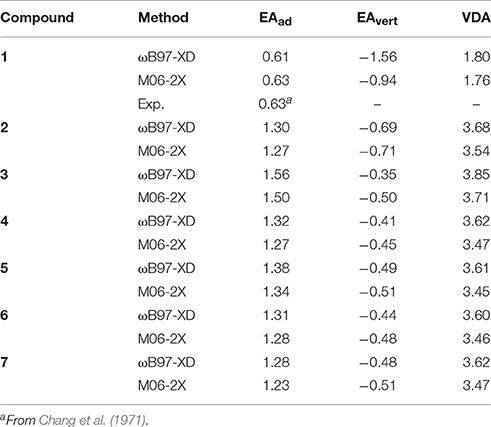
Table 3. Adiabatic and vertical electron affinities of the neutral c-C4F8 and mono-, di-chloro-substituted c-C4F8 compounds as well as vertical detachment energies of their anions in units of eV.
Vibrational Frequencies and IR Intensities
For the purpose of studying the greenhouse effect of the c-C4F8 and its derivatives, here we list the total infrared (IR) intensities and the cumulative IR intensities in the atmospheric IR “window” (800–1200 cm−1) in Table 4, the latter of which is widely used as a descriptor for qualitatively measuring the gas's greenhouse effect (Sturges et al., 2000). All these values can be calculated by frequency analysis using Gaussian09 D.01 package. It is clear that both the total IR intensity and cumulative IR intensity in the atmospheric IR “window” are increased upon the attachment of an electron for c-C4F8−nCln (n = 0, 1, 2). Meanwhile, most chloro-substituted c-C4F8 compounds (4, 5, 7) show higher IR intensity in the atmospheric IR “window” than c-C4F8, implying a worse greenhouse effect. However, there are also some chloro-substituted c-C4F8 compounds (2, 3, 6) showing lower IR intensity in the atmospheric IR “window” than the c-C4F8 (1) molecule, implying a decreased greenhouse effect. Especially, a di-chloro-substituted c-C4F8 (6) has much less IR intensity (222.8 km × mol−1) in the atmospheric IR “window” than that of the c-C4F8 (1) molecule (411.4 km × mol−1). Therefore, new c-C4F8 derivatives like 6 can be expected to be possibly used as new insulating gases in electric equipment with much suppressed environmental problems.
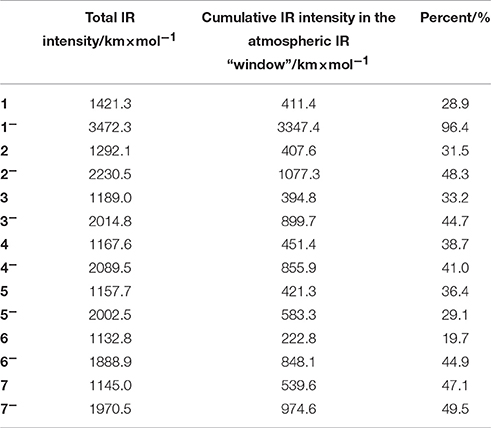
Table 4. IR intensities by DFT calculations at M06-2X/6–311g+(3df) level.
Thermodynamic Properties
Basic thermodynamic properties such as zero-point vibrational energy (ZPVE) and entropy as well as heat capacity have been also calculated for c-C4F8−nCln (n = 0, 1, 2), and the results are presented in Table 5. It is shown that while all the translational, rotational and vibrational movements contribute considerably to the total entropy, only vibrational ones make the dominant contributions to the total heat capacity. It can be also noticed that the ZPVE decreases upon chloro-substitution, but the entropy and the heat capacity increase upon chloro-substitution.
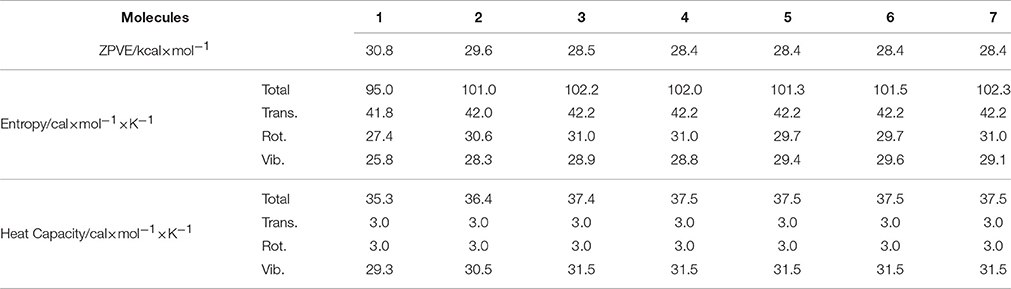
Table 5. Zero-point vibrational energy (ZPVE) and entropy as well as heat capacity of c-C4F8 and its mono-, di-chloro-substituted derivatives.
Conclusions
In this work, we use DFT to investigate the molecular structures, electron affinities, and IR-active vibrational frequencies as well as thermodynamic properties for c-C4F8 and a series of mono-, di-substituted c-C4F8 compounds. It is shown that functional with long-range corrected or meta-hybrid functionals and f-component containing basis set are obligatory for DFT calculations of the electronic structures of perfluoro-compounds to reproduce the experimental results.
We found that the bending geometry (D2d symmetry) of neutral c-C4F8 becomes planar (D4h symmetry) upon the attachment of an electron as its LUMO delocalized in a “p-like” orbital extending over the entire molecule. On the contrary, the chloro-substituted c-C4F8 molecules remains non-planar upon electron attachment because their LUMOs have great contributions from the Cl atoms and become asymmetric.
Although all chloro-substituted c-C4F8 molecules are found to have much larger electron affinities, only part of them have less IR intensity in the atmospheric IR “window” than c-C4F8. This implies that new insulation gas with improved dielectric and environmental properties can be fabricated from some chloro-substituted c-C4F8 molecules after careful selection.
Author Contributions
KZ, XX, and HM designed the project. LC, ZQ, CZ, and HS did the calculations and analyzed the results. KZ, XX, and HM wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by State Grid Corporation of China grant 2014-1192, National Natural Science Foundation of China (Grant No. 21373109) and Special Program for Applied Research on Super Computation of the NSFC-Guangdong Joint Fund (the second phase).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fchem.2016.00047/full#supplementary-material
References
Bauman, R. P., and Bulkin, B. J. (1966). Spectra and structure of perfluoro-cyclobutane. J. Chem. Phys. 45, 496–498. doi: 10.1063/1.1727596
Beagley, B., Calladine, R., Pritchard, R., and Taylor, S. (1987). The molecular structure of perfluoro-thiirane in the presence of perfluorocyclobutane: a gas-phase electron diffraction study. J. Mol. Struct. 158, 309–314. doi: 10.1016/0022-2860(87)80027-3
Becke, A. D. (1993). Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. doi: 10.1063/1.464913
Bopp, J. C., Roscioli, J. R., Johnson, M. A., Miller, T. M., Viggiano, A., Villano, S. M., et al. (2007). Spectroscopic characterization of the isolated SF6-and C4F8-anions: observation of very long harmonic in progressions symmetric deformation modes upon photo detachment. J. Phys. Chem. A 111, 1214–1221. doi: 10.1021/jp0665372
Chai, J.-D., and Head-Gordon, M. (2008). Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620. doi: 10.1039/b810189b
Chang, C., Porter, R., and Bauer, S. (1971). The molecular structures of perfluorocyclobutane and perfluorocyclobutane determined by electron diffraction. J. Mol. Struct. 7, 89. doi: 10.1016/0022-2860(71)90010-X
Choi, H., Park, Y. C., Lee, Y. S., and Baeck, K. K. (2013). Density functional theory calculations for simple prototypes of perfluorocarbons: neutral and anionic c-C4F8 and 2-C4F8. J. Fluo. Chem. 146, 46–52. doi: 10.1016/j.jfluchem.2013.01.002
Christophorou, L. G., and Olthoff, J. K. (2001). Electron interactions with c-C4F8. J. Phys. Chem. Ref. Data 30, 449–473. doi: 10.1063/1.1372164
de Urquijo, J., and Basurto, E. (2001). Electron attachment, ionization and drift in c-C4F8. J. Phys. D Appl. Phys. 34, 1352. doi: 10.1088/0022-3727/34/9/311
de Urquijo, J., and Juárez, A. M. (2009). The mobility of negative ions in CF3I, CF3I-N2, CF3I-Ar, CF3I-Xe, C2F6, and CH3F, and of positive ions in C2F4 and c-C4F8. IEEE Trans. Plasma Sci. 37, 1665–1669. doi: 10.1109/TPS.2009.2022362
ElSohly, A. M., Tschumper, G. S., Crocombe, R. A., Wang, J. T., and Williams, F. (2005). Computational and esr studies of electron attachment to decafluorocyclopentane, octafluorocyclobutane, and hexafluoro-cyclopropane: electron affinities of the molecules and the structures of their stable negative ions as determined from 13c and 19f hyperfine coupling constants. J. Am. Chem. Soc. 127, 10573–10583. doi: 10.1021/ja0505898
Fang, X., Hu, X., Janssens-Maenhout, G., Wu, J., Han, J., Su, S., et al. (2013). Sulfur hexafluoride (SF6) emission estimates for China: an inventory for 1990–2010 and a projection to 2020. Environ. Sci. Technol. 47, 3848–3855. doi: 10.1021/es304348x
Frisch, M., Trucks, H. G., Schlegel, Scuseria, G., Robb, M., Cheese man, J., et al. (2009). Gaussian 09, Revision D.01. Wallingford, CT: Gaussian, Inc.
Gallup, G. A. (2004). The structures of c-C4F8 and the adiabatic electron affinity of c-C4F8. Chem. Phys. Lett. 399, 206–209. doi: 10.1016/j.cplett.2004.09.147
Hohenberg, P., and Kohn, W. (1964). Inhomogeneous electron gas. Phys. Rev. 136:B864. doi: 10.1103/PhysRev.136.B864
Itoh, H., Miyachi, T., Kawaguchi, M., Nakao, Y., and Tagashira, H. (1991). Electron transport coefficients in SF6 and c-C4F8 mixtures. J. Phys. D Appl. Phys. 24, 277. doi: 10.1088/0022-3727/24/3/008
Kendall, R. A., Dunning, T. H. Jr., and Harrison, R. J. (1992). Electron affinities of the first-row atoms revisited systematic basis sets and wave functions. J. Chem. Phys. 96, 6796–6806. doi: 10.1063/1.462569
Kohn, W., and Sham, L. J. (1965). Self-consistent equations including exchange and correlation effects. Phys. Rev. 140:A1133. doi: 10.1103/PhysRev.140.A1133
Krishnan, R., Binkley, J. S., Seeger, R., and Pople, J. A. (1980). Self-consistent molecular orbital methods XX: a basis set for correlated wave functions. J. Chem. Phys. 72, 650–654. doi: 10.1063/1.438955
Lemaire, H. P., and Livingston, R. (1952). The molecular structures of octafluorocyclobutane and of methylcy-clobutane1. J. Am. Chem. Soc. 74, 5732–5736. doi: 10.1021/ja01142a048
Li, X., Zhao, H., Jia, S., and Murphy, A. B. (2014). Prediction of the dielectric strength for c-C4F8 mixtures with CF4, CO2, N2, O2 and air by Boltzmann equation analysis. J. Phys. D Appl. Phys. 47, 425204. doi: 10.1088/0022-3727/47/42/425204
Liu, X., Wang, J., Wang, Y., Zhang, Z., and Xiao, D. (2007). Analysis of the insulation characteristics of c-C4F8/CO2 gas mixtures by the Monte Carlo method. J. Phys. D Appl. Phys. 41, 015206. doi: 10.1088/0022-3727/41/1/015206
Mao, C., Nie, C.-S., and Zhu, Z.-Y. (1988). Normal coordinate analyses of C4-C6 perfluorocycloparaffins. Spectrochim. Acta A Mol. Spectros. 44, 1093–1098. doi: 10.1016/0584-8539(88)80077-1
McLean, A., and Chandler, G. (1980). Contracted Gaussian basis sets for molecular calculations I: second row atoms, z = 11-18. J. Chem. Phys. 72, 5639–5648. doi: 10.1063/1.438980
Miller, F. A., and Capwbll, R. J. (1971). The infrared and Raman spectra of octachloro-, octafluoro-, and octahydroxycycfobutane. Spectrochim. Acta A Mol. Spectros. 27, 1113–1131. doi: 10.1016/0584-8539(71)80194-0
Naidu, M., Prasad, A., and Craggs, J. (1972). Electron transport, attachment and ionization in c-C4F8 and iso-C4F8. J. Phys. D Appl. Phys. 5, 741. doi: 10.1088/0022-3727/5/4/313
Okubo, H., and Beroual, A. (2011). Recent trend and future perspectives in electrical insulation techniques in relation to sulfur hexafluoride (SF6) substitutes for high voltage electric power equipment. IEEE Electr. Insul. Mag. 27, 34–42. doi: 10.1109/MEI.2011.5739421
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865. doi: 10.1103/PhysRevLett.77.3865
Purchase, R., Smith, D., and Fischer, R. (1997). The ring-puckering motion in perfluorocyclobutane. J. Mol. Struct. 405, 159. doi: 10.1016/S0022-2860(96)09590-7
Rabie, M., Dahl, D. A., Donald, S. M., Reiher, M., and Franck, C. M. (2013). Predictors for gases of high electrical strength. IEEE Trans. Dielectr. Electr. Insul. 20, 856–863. doi: 10.1109/TDEI.2013.6518955
Stephens, P., Devlin, F., Chabalowski, C., and Frisch, M. J. (1994). Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627. doi: 10.1021/j100096a001
Sturges, W. T., Wallington, T. J., Hurley, M. D., Shine, K. P., Sihra, K., Engel, A., et al. (2000). A potent greenhouse gas identified in the atmosphere: SF5CF3. Science 289, 611–613. doi: 10.1126/science.289.5479.611
Vydrov, O. A., Heyd, J., Krukau, A. V., and Scuseria, G. E. (2006). Importance of short-range versus long-range Hartree-Fock exchange for the performance of hybrid density functionals. J. Chem. Phys. 125, 074106. doi: 10.1063/1.2244560
Vydrov, O. A., and Scuseria, G. E. (2006). Assessment of a long-range corrected hybrid functional. J. Chem. Phys. 125, 234109. doi: 10.1063/1.2409292
Vydrov, O. A., Scuseria, G. E., and Perdew, J. P. (2007). Tests of functionals for systems with fractional electron number. J. Chem. Phys. 126, 154109. doi: 10.1063/1.2723119
Woon, D. E., and Dunning, T. H. Jr. (1993). Gaussian basis sets for use in correlated molecular calculations III the atoms aluminum through argon. J. Chem. Phys. 98, 1358–1371. doi: 10.1063/1.464303
Wu, B.-T., Xiao, D.-M., Liu, Z.-S., Zhang, L.-C., and Liu, X.-L. (2006). Analysis of insulation characteristics of c-C4F8 and N2 gas mixtures by the Monte Carlo method. J. Phys. D Appl. Phys. 39, 4204. doi: 10.1088/0022-3727/39/19/012
Yamaji, M., and Nakamura, Y. (2003). Measurements of electron transport in the coefficients 0.468% and 4.910% c-C4F8/air mixtures and pure c-C4F8. J. Phys. D Appl. Phys. 36, 640. doi: 10.1088/0022-3727/36/6/305
Yamaji, M., Nakamura, Y., and Morokuma, Y. (2004). Measurements of ionization and attachment coefficients in 0.468% and 4.910% c-C4F8/air mixtures and pure c-C4F8. J. Phys. D Appl. Phys. 37, 432. doi: 10.1088/0022-3727/37/3/021
Yamamoto, O., Takuma, T., Hamada, S., Yamakawa, Y., and Yashima, M. (2001). Applying a gas mixture containing c-C4F8 as an insulation medium. IEEE Trans. Dielectr. Electr. Insul. 8, 1075–1081. doi: 10.1109/94.971467
Yanai, T., Tew, D. P., and Handy, N. C. (2004). A new hybrid exchange-correlation functional using the coulomb-attenuating method (cam-b3lyp). Chem. Phys. Lett. 393, 51–57. doi: 10.1016/j.cplett.2004.06.011
Zhang, C., Shi, H., Cheng, L., et al. (2016). First principles based computational scheme for designing new SF6 replacements. IEEE Trans. Dielectr. Electr. Insul. 23, 2572–2257. doi: 10.1109/TDEI.2016.7736814
Zhao, X., Jiao, J., Li, B., and Xiao, D. (2016). The electronegativity analysis of c-C4F8 as a potential insulation substitute of SF6 supported by national natural science foundation of China (no. 51337006). Plasma Sci. Technol. 18, 292. doi: 10.1088/1009-0630/18/3/13
Zhao, Y., and Truhlar, D. G. (2008). The m06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four m06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241. doi: 10.1007/s00214-007-0310-x
Keywords: insulating gas, density functional theory, octafluorocyclobutane, electron affinity, vibrational frequency
Citation: Cheng L, Qin Z, Zhang C, Shi H, Zhao K, Xie X and Ma H (2016) Theoretical Investigation of Mono- and Di-Chloro-Substitient Effects on the Insulation and Greenhouse Properties of Octafluorocyclobutane. Front. Chem. 4:47. doi: 10.3389/fchem.2016.00047
Received: 11 November 2016; Accepted: 30 November 2016;
Published: 15 December 2016.
Edited by:
Yong Wang, Lanzhou Institute of Chemical Physics (CAS), ChinaCopyright © 2016 Cheng, Qin, Zhang, Shi, Zhao, Xie and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyu Xie, bmp1eHh5MjAxM0BnbWFpbC5jb20=
Haibo Ma, aGFpYm9Abmp1LmVkdS5jbg==