 Bárbara Gionco1
Bárbara Gionco1 Eliandro R. Tavares2
Eliandro R. Tavares2 Admilton G. de Oliveira1
Admilton G. de Oliveira1 Sueli F. Yamada-Ogatta2Anderson O. do Carmo3
Sueli F. Yamada-Ogatta2Anderson O. do Carmo3 Ulisses de Pádua Pereira4
Ulisses de Pádua Pereira4 Roberta T. Chideroli4
Roberta T. Chideroli4 Ane S. Simionato1
Ane S. Simionato1 Miguel O. P. Navarro1Andreas L. Chryssafidis5
Miguel O. P. Navarro1Andreas L. Chryssafidis5 Galdino Andrade1*
Galdino Andrade1*- 1Microbial Ecology Laboratory, Department of Microbiology, Universidade Estadual de Londrina, Londrina, Brazil
- 2Molecular Biology Laboratory, Department of Microbiology, Universidade Estadual de Londrina, Londrina, Brazil
- 3Department of General Biology, Institute of Biologic Sciences, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
- 4Laboratory of Fish Bacteriology, Department of Preventive Veterinary Medicine, Universidade Estadual de Londrina, Londrina, Brazil
- 5Laboratory of Veterinary Toxicology, Department of Preventive Veterinary Medicine, Universidade Estadual de Londrina, Londrina, Brazil
The bacterial resistance for antibiotics is one of the most important problems in public health and only a small number of new products are in development. Antagonistic microorganisms from soil are a promising source of new candidate molecules. Products of secondary metabolism confer adaptive advantages for their producer, in the competition for nutrients in the microbial community. The biosynthesis process of compounds with antibiotic activity is the key to optimize their production and the transcriptomic study of microorganisms is of great benefit for the discovery of these metabolic pathways. Pseudomonas aeruginosa LV strain growing in the presence of copper chloride produces a bioactive organometallic compound, which has a potent antimicrobial activity against various microorganisms. The objective of this study was to verify overexpressed genes and evaluate their relation to the organometallic biosynthesis in this microorganism. P. aeruginosa LV strain was cultured in presence and absence of copper chloride. Two methods were used for transcriptomic analysis, genome reference-guided assembly and de novo assembly. The genome referenced analysis identified nine upregulated genes when bacteria were exposed to copper chloride, while the De Novo Assembly identified 12 upregulated genes. Nineteen genes can be related to an increased microbial metabolism for the extrusion process of exceeding intracellular copper. Two important genes are related to the biosynthesis of phenazine and tetrapyrroles compounds, which can be involved in the bioremediation of intracellular copper and we suggesting that may involve in the biosynthesis of the organometallic compound. Additional studies are being carried out to further prove the function of the described genes and relate them to the biosynthetic pathway of the organometallic compound.
Introduction
The increasing incidence of bacterial resistance against antibiotic compounds is a global problem in patients within a hospital environment. The multi-drug resistant (MDR) microorganisms have a significant impact in the mortality and in the hospitalization time of patients, increasing the cost of treatments associated with bacterial infection control (Rundramurthy et al., 2016). Currently, the high level of MDR bacteria dissemination is a worldwide major public health problem and the challenge is to find new antibiotics to control infections caused by these microorganisms (Bassetti et al., 2013). In the 90's, the pharmaceutical industry did not invest in new antibiotic products for many reasons, such as the time and cost for the development of new antibiotics. Due to a number of factors, antimicrobial resistance may emerge shortly after drug utilization and the future is uncertain, as some agencies estimate that none of the antibiotics currently in use will be effective against MDR bacteria in less than 10 years (Carlet et al., 2012).
The microbial community of soil was the source of many antimicrobials (Newman and Cragg, 2007). It is largely known that competing microorganisms produce some compounds to inhibit other microbial growth, using this mechanism as a survival strategy (Andrade, 2004). Therefore, to understand the interactions between microbial populations in the microcosm where they live and compete will certainly help to identify antagonism, and the bioactive compounds involved in these relations may be employed against bacterial infections (Fernandes, 2015).
Bacteria can produce thousands of compounds with bacteriostatic or bactericide activity, with potential use in the control of MDR bacteria (Butler et al., 2013). These compounds are usually products of secondary metabolism and would be involved in bacteria survival (Bérdy, 2005). Multiple studies described substances with bactericide or antifungal activity produced in the secondary metabolism of microorganisms, which could be applied in the management of human, animal and plant diseases (Stierle and Stierle, 2015; Depoorter et al., 2016; Thi et al., 2016).
Previous studies at the Microbial Ecology Laboratory showed that Pseudomonas aeruginosa LV strain produced an organometallic compound with antimicrobial activity against MDR bacteria, including Klebsiella pneumoniae carbapenemase producer KPC (K. pneumoniae carbapenemase) (Kerbauy et al., 2016), and Staphylococcus aureus methicillin resistant (Cardozo et al., 2013). Additionally, the same strain produced a Phenazine-1-Carboxylic Acid (even if in low quantity) which shown antifungal potential against Botrytis cinerea (Simionato et al., 2017). Apparently, the copper chloride induces the production of this compounds, since no antimicrobial activity can be detected in the supernatant of P. aeruginosa strain LV cultured in its absence (De Oliveira et al., 2016).
In this context, the objective of this work was to find the expressed genes related to the production of the bioactive organometallic compound in the presence of copper and related to the biosynthesis pathway in the P. aeruginosa strain LV using the transcriptomic approach.
Materials and Methods
Culture of Bacterial Strain
P. aeruginosa LV strain was cultured in nutrient broth at 28°C for 24 h in the presence or absence of copper chloride (0.1 g L−1), according to patent (PI0803350-1 – INPI 12/09/20092008; http://www.inpi.gov.br). The cells were recovered by centrifugation in a Jouan CR3 equipment (9,000 rpm/20 min, 4°C), resuspended in TRIZOL (INVITROGEN) and kept at −80°C. The confirmation of the antibiotic activity of the supernatants was confirmed as described elsewhere (De Oliveira et al., 2016). Both cultures were performed in biological triplicate (three samples for each condition, corresponding to three experiments separated spatially and temporally).
Total RNA Extraction and Ribosomal RNA Depletion
The total RNA was isolated using the illustra RNAspin kit™ (Ge Healthcare, UK) and ribosomal RNA depletion was performed using the RiboMinus™ transcriptome Kits (TermoFisher Scientific), followed the manufacturer's recommendation. The integrity of RNA was checked in agarose gel (1.5%) in a denaturant condition and the amount of RNA was determined using a spectrophotometer (Figures S7, S8) (BioTek Sinergy HT).
cDNA Library Construction and Sequencing
The construction of library was carried at Biotechnology and Molecular Markers Laboratory in the Universidade Federal de Minas Gerais (Belo Horizonte, MG, Brazil). The rRNA-depleted RNA (1 μg) was used to build cDNA library using the kit TruSeq® RNA Sample Preparation v.2 (Illumina, USA). The libraries were quantified with KAPA Library Quantification Illumina® (KAPA Biosystems) by quantitative PCR. The libraries were normalized to the concentration of 4 nM and sequencing was performed with MiSeq sequencer (Illumina) using MiSeq Reagent v3 (600 cycles) paired-end 2 × 300.
Detection of Differentially Transcribed Genes
Reference-Guided Genome Analysis
Due the absence of complete genome of P. aeruginosa LV strain, a more close genome for used for this analysis. For that, ten complete genomes of P. aeruginosa was randomly choiced for read mapping step. For the reason of more percentage of read mapped, the reference genome of P. aeruginosa deposited at the NCBI accession number NC_002516.2 was picked for Reference-guided genome analysis. The quality of the reads was analyzed in the platform of CLC Genomics Workbench version 8.5.1 (CLC bio, Denmark). The analysis was carried out using FastQC, which showed a high quality of reads and the use of filter was discarded, in order to guarantee the whole information about the sequences. The data was analyzed by CLC Genomics Workbench platform to quantify the expression by RPKM values (Reads per Kilobase per Million of Mapped Reads). Empirical analysis of DGE (Robinson and Smyth, 2008) hosted in the CLC platform was used to determine the differentially transcribed genes between copper-growth conditions. The genes were normalized and used for the dispersion analysis. The differential transcripts in presence of copper were identified and filtered applying the corrector Fold Change > than 1.5 and p < 0.001.
De novo Assembly of Transcripts
The analysis of raw RNA-seq data was performed using the Rockhopper 2 software platform (Tjaden, 2015). The FASTQ files were uploaded directly to the platform using a paired-end library approach. De novo assembly of bacterial transcriptomes was done using the software default settings. Rockhopper 2 normalized each RNA-seq data set using upper quartile normalization and employed a modified RPKM value and foldchange to quantify transcript abundance levels. The difference in expression was evaluated based on the results of transcript foldchange. The negative binomial distribution was used as the statistical model in order to detect significance levels that indicated the probability of observing difference in a transcript expression level under presence and absence of copper chloride. Resulting p-values were corrected and q-values were employed to control false positives, using the Benjamini-Hochberg procedure (Tjaden, 2015). In summary, Rockhopper 2 estimated the variance of a transcript expression, using local regression to obtain a smooth estimate of the variance, and performed a statistical test to determine whether a transcript presented different level of expression under the presence or absence of copper.
Identification of Functional Genes Differentially Expressed
The transcripts differentially expressed in presence of copper were analyzed using BLAST (Basic Local Alignment Search Tool), INTERPRO (Protein Sequence Analysis and Classification), UNIPROT (Universal Protein Resource) e PFAM (Protein Domain) databases, separately and manually classified according to the similarity of sequences description and function.
Results
The gene expression analysis of P. aeruginosa LV strain was realized in two different growth conditions, in the presence or absence of copper, using two bioinformatics strategies, genome reference-based and de novo assembly, with the identification of differently expressed genes and their putative function. The results suggest that some genes are involved in the biosynthesis of the organometallic compound with antimicrobial activity and other genes are related with the detoxification, or influx reduction, of copper inside the bacterial cells.
Reads Obtention
The generated RNA-Seq library was composed by 30 million reads, obtained by RNA extraction and depletion and synthesis and sequencing of resulting cDNA (Figure 1). The reads were separated in two groups, corresponding to the P. aeruginosa strain LV culture conditions (presence or absence of copper chloride). The first group (absence of copper) was composed of 14,783,618 reads, and the second group (presence of copper) contained 15,091,252 read (Table 1). The triplicates of each group were analyzed together and separately, generating similar results.
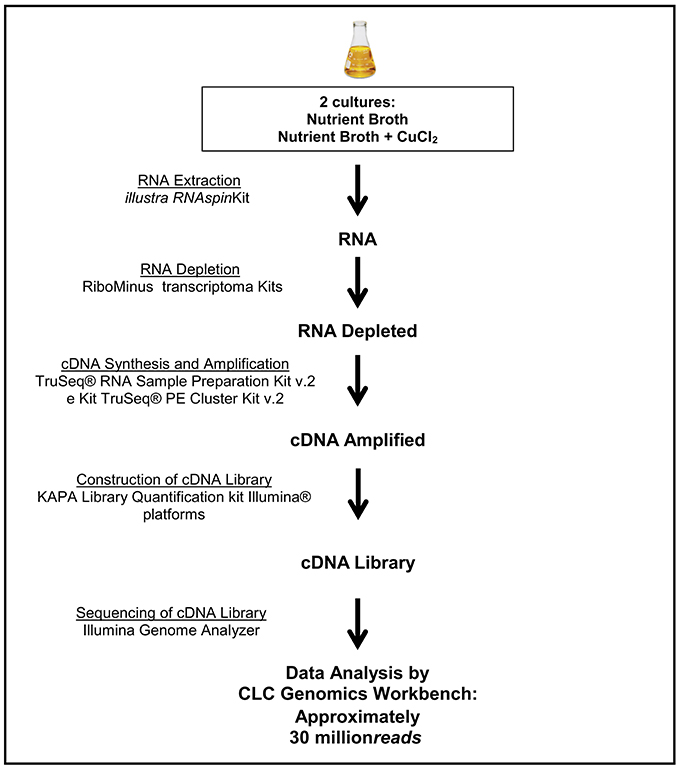
Figure 1. Workflow of RNA extraction steps, RNA depletion, cDNA synthesis, construction, and sequencing of genomic libraries.
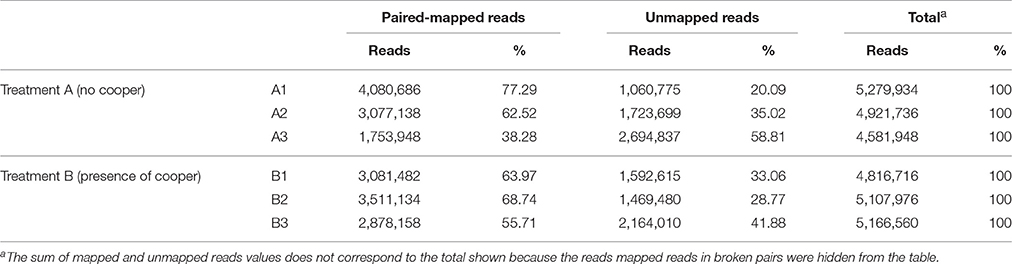
Table 1. The table shows the number and percentage of mapped and unmapped reads the triplicates of each treatment.
Measure of Expression Levels and Differential Expression Analysis in Guided Genome
The mean covering of each treatment was greater than 100× on the reference genome. Approximately 63% of reads from group one and 65% of group two were mapped (Table 1). The quantification of gene expression used the values of RPKM. The mapping data obtained between the different growth conditions were compared for determining the levels and differences in the gene expression. Nine genes were significantly upregulated (Figure 2), suggesting their relation with the biosynthesis of the bioactive organometallic compound, which only occurs in the presence of copper. On the other hand, four genes related to copper efflux were downregulated in cells cultured in the presence of copper.
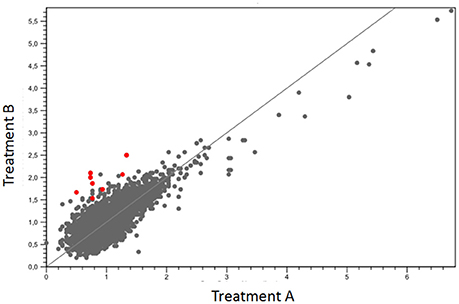
Figure 2. Dispersion of the expressed genes between the treatments when compared to the reference genome PA01. Red dots indicate hyperexpressed genes with lower levels of significance.
Functional Analysis
Upregulated genes were filtered and classified in functional categories, based on the similarity of sequence description for reference-guided genome analysis (Table 2) and de novo assembly (Table 3). Downregulated genes were also identified (Table 4).
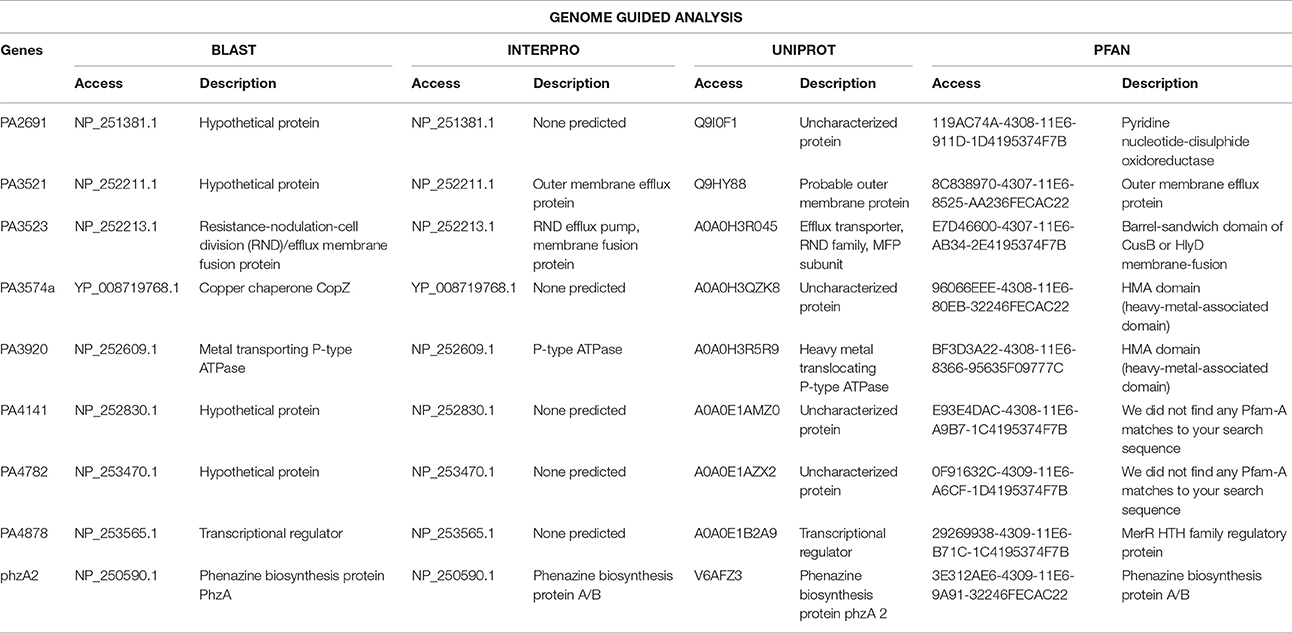
Table 2. Functional categories of overexpressed genes with statistical significance for the four analyzed databases and their access in Genome Guided Analysis.
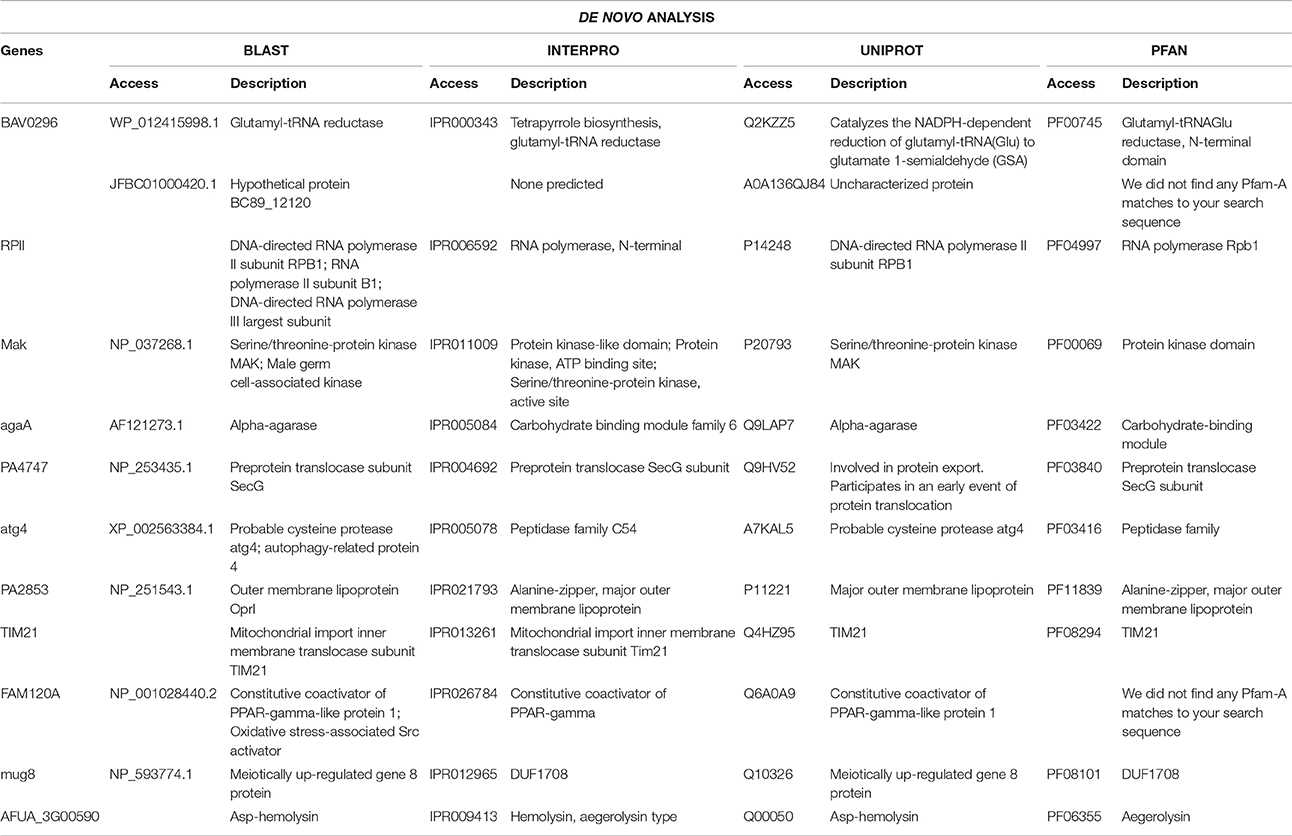
Table 3. Functional categories of overexpressed genes with statistical significance for the four analyzed databases and their access, in De novo analysis.
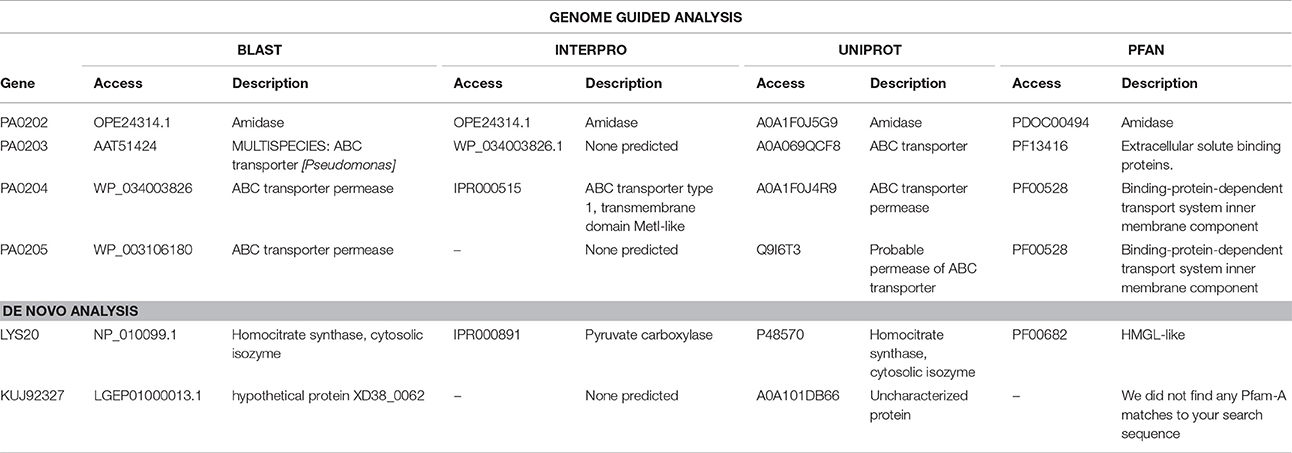
Table 4. Functional categories of down-expressed genes, with statistical significance for the four analyzed databases and their access, in both Analysis: Genome Guided and De novo.
Discussion
Reference-Guided Genome Analysis
Three upregulated genes (PA2691, PA4141, and PA4782) were not functionally characterized in all data banks used and were classified as hypothetical proteins. Other three upregulated genes were related to the transport of metallic ions (PA3521, PA3523, and PA3920) and one gene (PA3574a) was associated with chaperones group linked with copper. The gene (PA4872) was classified as a transcript regulator and gene (phzA2) was related to the phenazine biosynthesis.
Three hyperexpressed genes were related to the efflux system (PA3521, PA3523, and PA3920) and one gene (PA3574a) was related to a copper-chaperone, the CopZ. The PA3920 gene is related to the transport of heavy metals in Pseudomonas sp. and belongs to the P1B-1 subgroup of the carrying family P-type ATPases. In this system, the copper efflux occurs in the presence of ATP, when copper chaperone translocate the cation and link in a carrying protein, moving the complex through the membrane, to outside the bacterial cell (González-Guerrero et al., 2010). The P. aeruginosa LV strain efflux system was triggered by the presence of copper chloride, with hyperexpression of genes related to this mechanism.
The PA4878 gene belongs to a MerR family, which includes regulator proteins involved in the activation of the efflux system, in response to an inductor (Liao et al., 2013). The high expression values of this gene observed in copper-stimulated P. aeruginosa LV strain may be related to a set of mechanisms working to eliminate the metal from within the cell.
The downregulated genes were related to the ABC transporter (PA203), ABC transporter permease (PA204 and PA205) and an amidase (PA202). The ABC transporter system is composed of an outer membrane carrier, a membrane-fusion protein and an outer-membrane porin. These three components assemble into a complex spanning both membranes and providing the conduit for the translocation of unfolded polypeptides, as well as ions and various other substances into bacteria (Morgan et al., 2017). The downregulation of the ABC transporter and permease in the P. aeruginosa LV strain reveals a strategy for blocking the copper influx. Similar data were found by Li et al. (2009) in copper-resistant Pseudomonas spp. The reduction of the amidase gene is related to the decrease of the expression of porins, since they are involved in the entrance of polysaccharides into the bacterial cell.
The phenazine family are nitrogenous heterocycle compounds with redox activity, produced by fluorescent pseudomonads. The phenazines are small molecules that can penetrate different types of cells, including prokaryotes. Many phenazines present antimicrobial activity, suppressing pathogenic microorganisms of humans, animals and plants, also playing an important role in the survival strategies of bacteria in the environment (Chen et al., 2014; Briard et al., 2015; Dasgupta et al., 2015; Garrison et al., 2016; Morrison et al., 2016). The P. aeruginosa LV strain is a great producer of phenazine carboxylic acid (PCA).
The biosynthesis of phenazine in P. aeruginosa is realized by two seven-gene operons, phz1 (phzA1B1C2D1E1F1G1) and phz2 (phzA2B2C2D2E2F2G2). According to Li et al. (2011), there is a combined regulation of these two operons in the biosynthesis of PCA. The first one (phzA2-G2 gene) produces small amount of PCA and the second one (the phzA1-G1 gene) produces a large amount of PCA. The expression of two operons working together optimizes the PCA biosynthesis, through a positive feedback mechanism. The operon phz2 produces a basal amount of PCA, self-inducing the same operon to keep the biosynthesis process and triggering the transcription of operon phz1, amplifying PCA production.
In the present study, the hyperexpression of phzA2 gene belonging of phz2 operon was observed. Among the nine-upregulated genes identified, the phzA2 is the only one related to a biosynthesis pathway. The proposition is that the augmented expression of phzA2 gene stimulates the production of PCA and, once there is PCA in the cytoplasm, it activates its own biosynthesis process.
Many studies carried out in the Microbial Ecology Laboratory demonstrated that, in absence of copper, the P. aeruginosa LV strain produces pyoverdine (PYO) from PCA, suggesting that the PCA is a precursor for PYO biosynthesis. However, when copper is present, the LV strain produces PCA and the PYO formed helps the bacteria to bioremediate the copper, forming the organometallic compound (Figures S1–S6) with C, Cu, S and O (Figure S9) and N (Figure S10). Possibly, the three hypothetical proteins detected are involved in the biosynthesis of PYO, using PCA as precursor (Figure 3). Further studies will be carried out to better understand such process and to confirm this hypothesis.
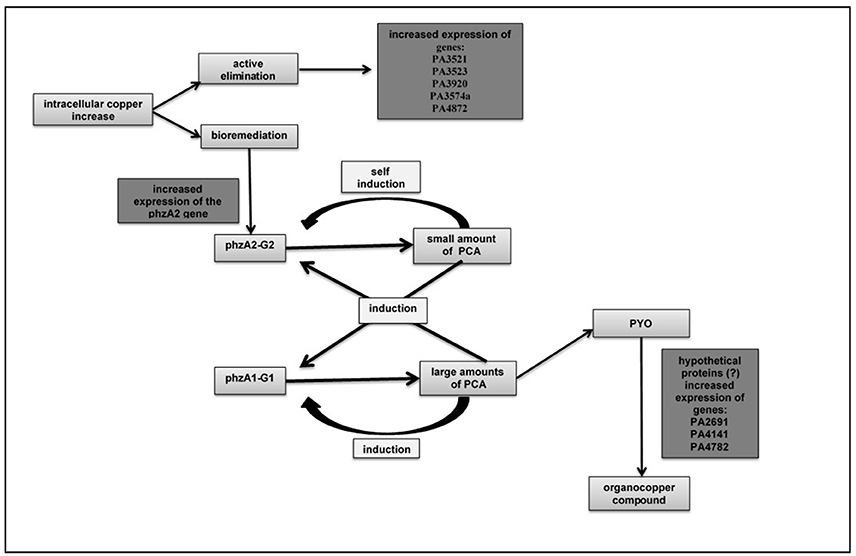
Figure 3. An integrated relationship of the hyperexpressed genes when there is an increase of copper in the culture medium and the possible hypotheses of its expulsion: active elimination and bioremediation.
De novo Assembly
In this analysis, twelve upregulated genes and two downregulated genes were identified. In relation to upregulated genes, one gene was not functionally characterized and was classified as a hypothetical protein. Seven upregulated genes were related to increased protein synthesis (RPII, MaK, agaA, PA474, atg4, TIM21, mug8) and two genes related to oxidative stress (FAM120A and AFUA_3G00590). The PA2853 gene was related to an outer membrane lipoprotein (OprI) and the BAV0296 gene (glutamyl-tRNA reductase) was related to the synthesis of tetrapyrroles compounds. From the downregulated genes, one was related to homocitrate synthase, LYS20, and one was an uncharacterized protein, KUJ92327.
Copper is a cofactor for many important enzymes, in eukaryotes and prokaryotes and it is essential for almost all organisms. However, the excess copper can also be harmful due to its redox activity and the ability of binding to sites of other metals, particularly iron-sulfur clusters, leads to an increased microbial metabolism, related to the stress generated by the ion inside the organism (Vita et al., 2016). Seven genes related to increased protein synthesis were detected in this study. From those, five can be detected in prokaryotes: the RPII gene (a RNA-polymerase); MaK (protein kinase) related to many cellular processes, including metabolism, transcription, cell cycle progression, cytoskeletal rearrangement and cell movement, apoptosis, and differentiation; agaA (carbohydrate binding module family 6), which binds to specific ligands, such as cell-surface-attached carbohydrate substrates for galactose oxidase and sialidase; PA474 (preprotein translocase subunit SecG), involved in protein export and translocation and atg4 (peptidase family C54), a proteolytic enzyme that hydrolyses peptide bonds using the thiol group of the cysteine residue as a nucleophile.
Another two genes may be related to increased protein synthesis and cell division, TIM21 and mug8. These genes have been described in eukaryotes and are responsible for the translocation of transit peptide-containing proteins across the mitochondrial membrane and role in bud formation, respectively. This may help the bacteria to maintain the cellular division in the presence of cooper.
The gene FAM120A (oxidative stress-associated Src activator), a critical component of the oxidative stress-induced survival signaling in human, and AFUA_3G00590 (asp-hemolysin), expressed during bacterial sporulation, were also detected. Furthermore, the OprI, the major outer membrane lipoprotein of P. aeruginosa, presented increased expression (Basto et al., 2014). The upregulation of these genes can be explained. High levels of copper are toxic to bacteria and lead to cell damage, by generating reactive oxygen species, affecting the function of proteins or inactivating the enzymes (Li et al., 2009) and hence those overexpressed proteins act on protecting the cell against such damage.
The tetrapyrrole compounds are molecules that have four rings of the pyrrole type, generally linked together by single-atom bridges, between the alpha positions of the five-membered pyrrole rings. They function as a metal-binding cofactor in many important enzymes, proteins and pigments, such as heme, chlorophyll, cobalamine (vitamin B12), siroheme and cofator F430 (Schulze et al., 2006; Schalk and Cunrath, 2016) may suggest its participation in cooper insertion in the bioactive compound produced by LV strain. The Glutamyl-tRNA reductase is an important enzyme involved in this process. The super-expression of this enzyme detected in our study reaffirms our hypothesis that the compound with antimicrobial activity comes from the biosynthesis of pigments produced by P. aeruginosa strain, since the increase of tetrapyrrole can be related to the binding of copper on these pigments.
The overexpressed genes detected were different in the two methods applied in this study. Still, most of them are related to the stress in the cell caused by copper and its elimination. Furthermore, in the reference-guided genome analysis, an increase in phzA2 gene expression was found (related to phenazine biosynthesis) whilst an increased expression of the BAV0296 gene was detected in the de novo analysis (related to the production of tetrapyrrole compounds). Although the results are different, there is a complementarity between them, since the increase in expression of these genes, related to the pigments produced by the P. aeruginosa strain, can also be related to the production of the organometallic compound with antimicrobial activity, as a mechanism of bioremediation of intracellular copper. Additional studies are being carried out with these genes to confirm such hypothesis.
Regarding the down regulated gene LYS20, related to homocitrate synthase, it is known that this enzyme participates in lysine biosynthesis and pyruvate metabolism. The homocitrate synthase catalyzes the first step in lysine biosynthesis in many fungi and certain Archaea (Bulfer et al., 2009). The down expression of this gene can be related to a decreased cell anabolism, caused by cellular stress derived from excess intracellular copper.
Conclusion
The gene expression analysis of the P. aeruginosa LV strain, in presence of copper chloride, suggests that the excess of copper forces the microorganism to eliminate it through different pathways. The first way of elimination is related to the hyperexpression of efflux systems, capable of expelling the metal into the periplasmic space or to the extracellular environment. A second hypothesis of ion elimination is directly related to the synthesis pathway of the bioactive compound, where the microorganism is able to bioremediate the accumulated copper inside the cell. This elimination may be associated with a deviation of the metabolic route of pigments produced by the strain, possibly bioremediating the copper and forming the compound with antimicrobial activity. The downregulation of ABC transporter genes (related to ion transport) can be a mechanism to avoid the excess of copper influx to bacteria, which would guarantee the microorganism survival. Studies are being designed and carried out to confirm the hypotheses suggested in the present study.
Author Contributions
BG, ET, SY, AdO, and GA designed the study protocol, and participated in its design and coordination. BG and ET validated and performed the experiment. AdC performed the Pseudomonas aeruginosa LV strain sequencing. UP performed and interpreted de Novo analysis. AS and MN produced and purified the compounds produced by the LV strain. BG, UP, AS, AC, and GA contributed to drafting the manuscript and/or critically revising the paper and intellectual content.
Funding
The authors are grateful to CNPq and CAPES for the financial support to this study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would also like to thank the Laboratory of Spectroscopy—ESPEC UEL/FINEP for help with the NMR experiments, the Laboratory of Analysis of Materials and Molecules—LAMM UEL/FINEP for help with the Mass experiments, Biotechnology and Molecular Markers Laboratory in the Universidade Federal de Minas Gerais for help with the sequencing analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fchem.2017.00066/full#supplementary-material
References
Bassetti, M., Merelli, M., Temperoni, C., and Astilean, A. (2013). New antibiotics for bad bugs: where are we? Ann. Clin. Microbiol. Antimicrobiol. 28, 12–22. doi: 10.1186/1476-0711-12-22
Basto, A. P., Morais, J., Marcelino, E., Leitão, A., and Santos, D. M. (2014). An eficiente depyrogenation method for recombinant bacterial outer membrane lipoproteins. Protein. Expr. Purif. 98, 10–17. doi: 10.1016/j.pep.2014.02.012
Briard, B., Bomme, P., Lechner, B. E., Mislin, G. L., Lair, V., Prévost, M. C., et al. (2015). Pseudomonas aeruginosa manipulates redox and iron homesostasis of its microbiota partner Aspergillus fumigatus via phenazines. Scie. Rep. 10, 8220–8225. doi: 10.1038/srep08220
Bulfer, S. L., Scott, E. M., Couture, J. F., Pillus, L., and Trievel, R. C. (2009). Crystal structure and functional analysis of Homocitrate Synthase, an essential enzyme in Lysine biosynthesis. J. Biol. Chem. 51, 35769–35780. doi: 10.1074/jbc.M109.046821
Butler, M. S., Blaskovich, M. A., and Cooper, M. A. (2013). Antibiotics in the clinical pipeline in 2013. J. Antibiot. 66, 571–591. doi: 10.1038/ja.2013.86
Cardozo, V. F., Oliveira, A. G., Nishio, E. K., Perugini, M. R., Andrade, C. G., Silveira, W. D., et al. (2013). Antibacterial activity of extracellular compounds produced by a Pseudomonas strain against methicillin-resistant Staphylococcus aureus (MRSA) strain. Ann. Clin. Microbiol. Antimicrob. 17, 12–24. doi: 10.1186/1476-0711-12-12
Carlet, J., Jarlier, V., Harbarth, S., Voss, A., Goossens, H., Pittet, D., et al. (2012). Ready for a world without antibiotics? The pensières antibiotic resistance call to action. Antimicrob. Resist. Infect. Control. 1, 11–24. doi: 10.1186/2047-2994-1-11
Chen, A. L., Dolben, D. E. F., Okegbe, C., Harty, C. E., Golub, Y., Thao, S., et al. (2014). Candida albicans ethanol stimulates Pseudomonas aeruginosa WspR-controlled biofilm formation as part of a cyclic relationship involving phenazines. PLoS Pathog. 10:e1004480. doi: 10.1371/journal.ppat.1004480
Dasgupta, D., Kumar, A., Mukhopadyay, B., and Sengupta, T. K. (2015). Isolation of phenazine 1,6-di-carboxylic acid from Pseudomonas aeruginosa strain HRW.1-S3 and its role in biofilm-mediated crude oil degradation and cytotoxicity against bacterial and cancer cells. Appl. Microbiol. Biotechnol. 99, 8653–8665. doi: 10.1007/s00253-015-6707-x
De Oliveira, A. G., Spago, F. R., Simionato, A. S., Navarro, M. O., Da Silva, C. S., Barazetti, A. R., et al. (2016). Bioactive organocopper compound from Pseudomonas aeruginosa inhibits the grow of Xanthomonas citri subsp. citri. Front. Microbiol. 7:113. doi: 10.3389/fmicb.2016.00113
Depoorter, E., Bull, M. J., Peeters, C., Coenye, T., Vandamme, P., and Mahenthiralingam, E. (2016). Burkholderia: an update on taxonomy and biotechnological potential as antibiotic producers. Appl. Microbiol. Biotechnol. 100, 5215–5229. doi: 10.1007/s00253-016-7520-x
Fernandes, P. (2015). The global challenge of new classes of antibacterial agents: an industry perspective. Curr. Opin. Pharmacol. 24, 7–11. doi: 10.1016/j.coph.2015.06.003
Garrison, A. T., Abouelhassan, Y., Norwood, V. M. IV., Kallifidas, D., Bai, F., Nguyen, M. T., et al. (2016). Structure-Activity relationships of a diverse class of halogenated phenazines that targets persistent, antibiotic-tolerant bacterial biofilms and Mycobacterium tuberculosis. J. Med. Chem. 59, 3808–3825. doi: 10.1021/acs.jmedchem.5b02004
González-Guerrero, M., Raimunda, D., Cheg, X., and Aegüello, J. M. (2010). Distinct functional roles of homologous Cu+ efflux ATPases in Pseudomonas aeruginosa. Mol. Microbiol. 78, 1246–1258. doi: 10.1111/j.1365-2958.2010.07402.x
Kerbauy, G., Vivan, A. C., Simões, G. C., Simionato, A. S., Pelisson, M., Vespero, E. C., et al. (2016). Effect of metalloantibiotic produced by Pseudomonas aeruginosa on Klebsiella pneumoniae Carbapenemase (KPC) – producing K. pneumoniae. Cur. Pharm. Biotec. 17, 389–397. doi: 10.2174/138920101704160215171649
Li, K., Pidatala, R. R., and Ramakrishna, W. (2009). Mutational, proteomic and metabolomic analysis of a plant growth promoting copper-resistant Pseudomonas spp. J. Biol. Chem. 51, 35769–35780.
Li, Y., Du, X., Lu, Z. J., Wu, D., Zhao, Y., Ren, B., et al. (2011). Regulatory feedback loop of two phz gene clusters through 5'-untranslated regions in Pseudomonas sp. M18. PLoS ONE 6:e19413. doi: 10.1371/journal.pone.0019413
Liao, J., Schurr, M. J., and Sauer, K. (2013). The MerR-like regulator BrlR confers biofilm tolerance by activating multidrug efflux pumps in Pseudomonas aeruginosa biofilm. J. Bacteriol. 195, 3352–3362. doi: 10.1128/JB.00318-13
Morgan, J. L., Acheson, J. F., and Zimmer, J. (2017). Structure of Type-1 secretion system ABC-transporter. Structure 3, 522–529. doi: 10.1016/j.str.2017.01.010
Morrison, C. K., Novinscaka, A., Gadkar, V. J., Joly, D. L., and Filion, M. (2016). Complete genome sequence of Pseudomonas fluorescens LBUM636, a strain with biocontrol capabilities against late blight of potato. Genome Announc. 26, 4–7. doi: 10.1128/genomeA.00446-16
Newman, D. J., and Cragg, G. M. (2007). Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 70, 461–477. doi: 10.1021/np068054v
Robinson, M. D., and Smyth, G. K. (2008). Small-sample estimation of negative binomial dispersion with applications to SAGE data. Bioestatistics 9, 321–332. doi: 10.1093/biostatistics/kxm030
Rundramurthy, G. R., Swamy, M. K., Sinniah, U. R., and Ghasemzadeh, A. (2016). Nanoparticles: alternatives against drug-resistant pathogenic microbes. Molecules 27, 21–28. doi: 10.3390/molecules21070836
Schalk, I. J., and Cunrath, O. (2016). An overview of biological metal uptake pathways in Pseudomonas aeruginosa. Environ. Microbiol. 18, 3227–3246. doi: 10.1111/1462-2920.13525
Schulze, J. O., Schubert, W. D., Moser, J., Jaahn, D., and Heinz, D. W. (2006). Evolutionary relationship between initial enzymes of tetrapyrrole biosynthesis. J. Mol. Biol. 5, 1212–1220. doi: 10.1016/j.jmb.2006.02.064
Simionato, A. S., Navarro, M. O. P., Jesus, M. L. A., Barazetti, A. R., Silva, C. S., Simoes, G. C., et al. (2017). The effect of phenazine-1-carboxylic acid on mycelial growth of botrytis cinerea produced by Pseudomonas aeruginosa LV strain. Front. Microbiol. 8:1102. doi: 10.3389/fmicb.2017.01102
Stierle, A. A., and Stierle, D. B. (2015). Bioactive secondary metabolites produced by the fungal endophytes of conifers. Nat. Prod. Commum. 10, 1671–1682.
Thi, Q. V., Tran, V. H., Mai, H. D., Le, C. V., Hong, M. L. T., Murphy, B. T, et al. (2016). Secondary metabolites from an Actinomycete from Vietnam's East Sea. Nat. Prod. Commum. 11, 401–404.
Tjaden, B. (2015). De Novo assembly of bacterial transcriptomes from RNA-Seq data. Genome Biol. 16:1. doi: 10.1186/s13059-014-0572-2
Keywords: bioactive compounds, RNA-seq, antibiotic, Organometallic Antibiotic
Citation: Gionco B, Tavares ER, de Oliveira AG, Yamada-Ogatta SF, do Carmo AO, Pereira UdP, Chideroli RT, Simionato AS, Navarro MOP, Chryssafidis AL and Andrade G (2017) New Insights about Antibiotic Production by Pseudomonas aeruginosa: A Gene Expression Analysis. Front. Chem. 5:66. doi: 10.3389/fchem.2017.00066
Received: 19 July 2017; Accepted: 31 August 2017;
Published: 15 September 2017.
Edited by:
Maria Luisa Mangoni, Sapienza Università di Roma, ItalyReviewed by:
Ashootosh Tripathi, University of Michigan, United StatesFrancisco Solano, Universidad de Murcia, Spain
Copyright © 2017 Gionco, Tavares, de Oliveira, Yamada-Ogatta, do Carmo, Pereira, Chideroli, Simionato, Navarro, Chryssafidis and Andrade. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Galdino Andrade, YW5kcmFkZWdAdWVsLmJy