 Prasanta K. Dash
Prasanta K. Dash Rhitu Rai
Rhitu Rai Ajay K. Mahato
Ajay K. Mahato Kishor Gaikwad
Kishor Gaikwad Nagendra K. Singh
Nagendra K. SinghIntroduction
Drought is a global phenomenon that affects productivity of all field crops. Comparatively, flax is prone to drought stress and concomitant yield penalty. Owing to vagaries of climate change, erratic monsoon and global warming; drought research received significant attention in model as well as field crops. While, priority crops like rice, wheat, corn, and canola witnessed significant advances in drought research (Aprile et al., 2009; Hayano-Kanashiro et al., 2009; Lenka et al., 2011; Zhang et al., 2014), limited impetus has been accomplished in an industrially important crop flax/linseed (Linum usitatissimum; Gupta and Dash, 2015). Recently, transcriptome data for legumes such as pigeonpea (Cajanus cajan) and its wild relative C scaraboides have been available (Nigam et al., 2017) for translational research. Flax, a dual purpose crop grown for fiber and seed oil, entered genomics research with decoding of its genome in 2012 (Wang et al., 2012). Since then genomic resources in flax are accumulating (Dash et al., 2014, 2015; Gupta et al., 2017; Shivaraj et al., 2017) to accelerate its varietal improvement program. Of late, development of high-throughput RNA sequencing revolutionized analysis of eukaryotic transcriptomes (Wang et al., 2009) and facilitated elucidating pathways and mapping of novel genes. Globally, meager genomic information are available in flax for translational research with information only on related Linum species like L. bienne, L. grandiflorum, and L. leonii (Johnson et al., 2012), In a recent study, fiber development in flax was elucidated using the RNA-Seq information generated in L. usitatissumum (Zhang and Deyholos, 2016). Thus, there is an urgent need among Linum research community to saturate genomic information on drought, salt, cold, and heat-stress mechanisms operative in flax genotypes grown across the globe. In this endeavor, transcriptome analysis of a moderately drought tolerant flax cultivar T-397 was accomplished to delineate biochemical pathway and genes operative in imparting tolerance to drought in this cultivar of Indian flax.
To accomplish translational research for drought tolerance in flax, no genomic data are available from any drought tolerant cultivar. Also, information related to gene expression profiling in flax is limited. This is the first report of high resolution transcriptome data from a moderately drought tolerant flax cultivar of an Indian origin. This dataset will be a cardinal genomic resource in annotating and understanding the genes and the intrinsic pathways involved in drought tolerance in flax. The data will further help to identify transcripts that are detectable under normal growing conditions encompassing different stages of flax growth as well as identifying genes involved in warding off drought at vegetative as well as reproductive stages in flax. It can also be used for gene discovery and/or comparative transcriptome analysis with the other related species. Besides being a useful resource for delineating the molecular basis of drought tolerance in flax, the inherent information can be used by plant breeders in flax breeding strategies.
Experimental Design, Materials, and Methods
Plant Materials
Seeds of linseed variety T-397 were obtained from project coordinating unit (Linseed), Kanpur, India. Seeds were sown in 25 cm diameter plastic pots filled with mixture of peat soil, vermiculite, and river sand in 1:1:1 ratio. The seed-laden pots were kept in dark for 3 days. After germination, the pots were shifted to tissue culture room having 12 h photoperiod and 24°C/18°C (day/night) temperature. Shoot tissues were collected from 20 d old five independent plants with three biological replications while bud and inflorescence tissues were collected in a similar manner after onset of flowering (60 d). Same tissue types from 15 independent plants were pooled to represent a homogenous sample. All the tissues were frozen in liquid nitrogen before storing at −80°C.
Total RNA Extraction, Quality Check, and Sequencing
Total RNA was isolated using 500 mg of shoot, flower, and bud separately using commercially available RNA isolation kit (Qiagen). Extracted RNA was treated with TURBO DNA-free™ kit to get rid of chromosomal DNA contamination. Quality of the isolated RNA was checked by 1% denaturing agarose gel electrophoresis and quantified by using a Nanodrop. Equal amount of total RNA having A260/280 ratio >2.0 were supplied to service provider for library preparation. Illumina TruSeq RNA sample preparation kit was used to prepare the cDNA librarry. Four micro-gram of total RNA was used to isolate polyA mRNA using oligodT coupled magnetic beads. Subsequently, the mRNA from three different samples were pooled, fragmented and cDNA was synthesized using random primers and reverse transcriptase (Super-Script II). The double stranded cDNA after an end repair process (Klenow fragment, T4 polynucleotide kinase and T4 polymerase), was ligated to Illumina paired end (PE) adaptors. The Library was enriched using 15 cycles of PCR, purified and diluted to a final concentration of 4 nM and run at a concentration of 9 pM on MiSeq Instrument (Illumina, USA) using MiSeq Reagent Kit v2 (300 Cycles) with 2 × 150 PE sequencing.
Preprocessing of Raw Reads
Raw reads were filtered with Q20 quality trimming (removal of low quality reads with average quality score <20 and trimming of low quality bases from the end of reads). The quality filtering was performed to remove adapter sequences with sequence pre-processing Trimmomatic v0.30 (Bolger et al., 2014) software with following parameters: sliding window length of 20, leading and trailing threshold quality value of 20. After trimming, reads with read length less than 40 bp were discarded.
De novo Transcriptome Assembly
High quality Illumina raw reads with phred score ≥25 were used for assembly. The de novo assembly of these processed reads was accomplished using Trinity assembler (version r2014-07-17) (Grabherr et al., 2011) with the following parameters: Jellyfish Memory of 300G, minimum contig length of 1,000 base pair and heap-space of 7 (Marçais and Kingsford, 2011). Subsequently, the assembly statistics were obtained using custom perl script (Bradnam et al., 2013). The assembled transcripts were further clustered into non-redundant unigene set using CD-HIT-EST software with default parameters (% similarity >95%). The quality of assembled unigenes transcripts was evaluated by mapping the total high quality reads to final unigene set number of reads to assembled transcripts using Bowtie2 (Li and Durbin, 2009).
Results
The transcriptome assembly has turned out to be a method of choice for the discovery and characterization of novel transcripts involved in diverse pathways in eukaryotes (Wang et al., 2009; Surget-Groba and Montoya-Burgos, 2010). Availability of next generation sequencing technology has expedited identification and characterization of genes based on transcriptome approach in several crops. As a first step toward identifying drought specific genes, RNA-seq library in flax was prepared from the pooled samples of shoot, bud, and inflorescence. A total of 47,004,561 paired end reads were obtained after quality filtering. The cleand reads were obtained by filtering raw reads by removing adapters, low-quality, and ambiguous reads. Pre-assembly statistics of raw reads along with distribution of contigs and contigs length is presented in Table 1. The mapping statistics of high quality filtered data obtained are presented in Table 2. The GC content of the assembled data was 44.94% while 55.06% was AT content.
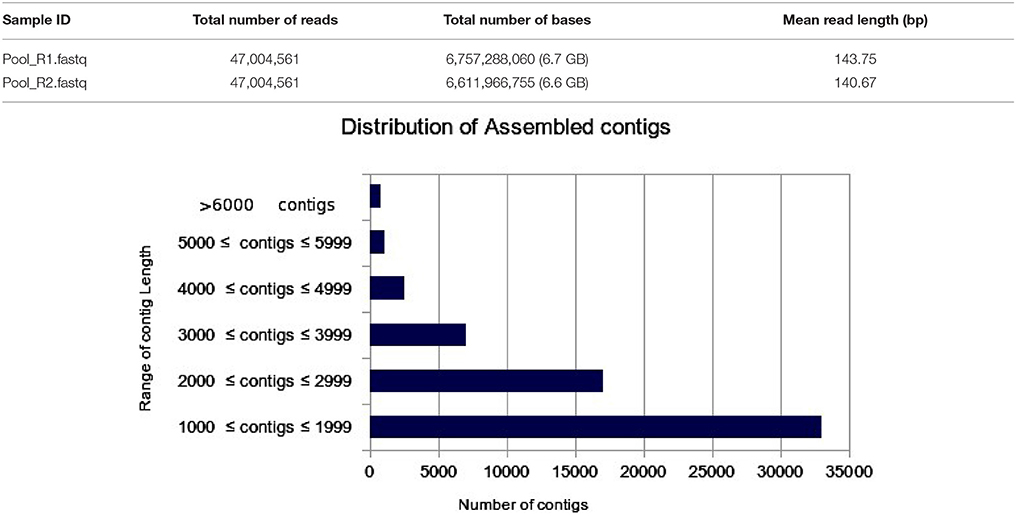
Table 1. Pre-assembly statistics of raw reads along with bar chart representing the distribution of assembled contigs with number of contigs in the X-axis and Range of contig length in the Y-axis.
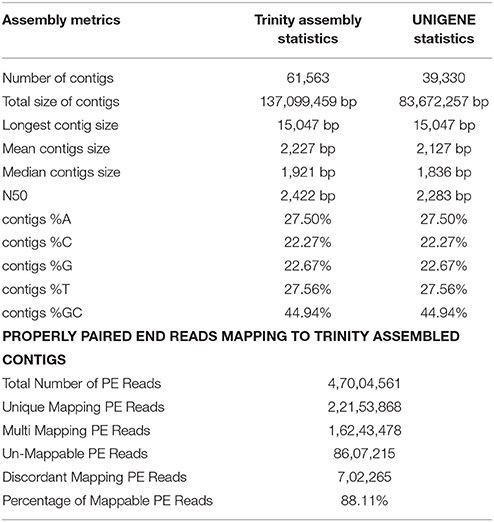
Table 2. Statistics of de novo assembly and UniGenes along with mapping statistics of L. usitatissimum transcriptome.
The filtered data assembled using Trinity software (Grabherr et al., 2011) resulted in 61,563 transcript contigs with N50 value of 2.4 kb, with average length of 1.9 kb. The transcripts were further analyzed using Cluster Database at High Identity with -EST i.e. CD-HIT_EST software (Nakasugi et al., 2013). A total of 61,563 transcripts were clustered into 39,330 UniGene's using CD-HIT-EST (Li and Godzik, 2006; Nakasugi et al., 2013). The clustered UniGene's has average length of 1.8 kb with N50 value of 2.2 kb and. Subsequently, 52,678 CDS were identified using transdecoder from 39,330 UniGene's. The statistics of transcriptome assembly and UniGene prediction are represented in Table 2. Assessment of transcriptome assembly was accomplished using unigenes formed after clustering. Subsequently, Bowtie2 was used to align the HQ reads on unigenes and the mapping statistics are represented in Table 2. Our result revealed that out of 47,004,561 PE reads 88.11% are mapable pairs reflecting the high quality of the de novo generated assembly. While, 22,153,568 are unique mapping reads; 16,243,478 reads mapped to multiple sites and 8,607,215 reads were un-mapable. The discordant/unmapable reads most likely correspond to low expressed transcripts/unsatisfactory coverage or are of aberrant nature.
Link to Deposited Data and Information to the User
Transcriptome profile of L. usitatissimum was generated from the polyA-enriched cDNA library prepared by pooling equal amount of total RNA extracted from shoot, flower and buds separately. The short reads were filtered, processed, assembled, and analyzed as described in the previous section. The complete data from the current study was submitted at NCBI under the BioProject ID PRJNA338739. The raw data for this project were deposited at SRA database at NCBI with the accession number SRR4034646 (http://www.ncbi.nlm.nih.gov/sra/SRR4034646). Users can download and reuse the data for research purpose with an acknowledgment and by quoting this paper as reference to the data.
Conclusion
This is the first report of a transcriptome dataset of an Indian flax cultivar T-397. We used next-generation RNA sequencing of leaf, shoot, bud, and flowering inflorescence to identify the genes involved in intrinsic drought tolerance in this cultivar. The data can be further used for identifying SSR loci and markers to be used in flax improvement program specific to drought tolerance.
Author Contributions
PD and RR conceived the study, performed the experiments, analyzed the data with help from AM, KG, and NS. PD wrote the manuscript with input from all authors. All authors read and approved the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Rohini Sreevathsa for critical reading of the manuscript. Work was carried out under ICAR-NPTC and in-house RPP theme.
References
Aprile, A., Mastrangelo, A. M., De Leonardis, A. M., Galiba, G., Roncaglia, E., Ferrari, F., et al. (2009). Transcriptional profiling in response to terminal drought stress reveals differential responses along the wheat genome. BMC Genomics 10:279. doi: 10.1186/1471-2164-10-279
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bradnam, K. R., Fass, J. N., Alexandrov, A., Baranay, P., Bechner, M., Birol, I., et al. (2013). Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. Gigascience 2:10. doi: 10.1186/2047-217X-2-10
Dash, P. K., Cao, Y., Jailani, A. K., Gupta, P., Venglat, P., Xiang, D., et al. (2014). Genome-wide analysis of drought induced gene expression changes in flax (Linum usitatissimum). GM Crops & Food 5, 106–119. doi: 10.4161/gmcr.29742
Dash, P., Gupta, P., and Rai, R. (2015). Hydroponic method of halophobic response elicitation in flax (Linum usitatissimum) for precise down-stream gene expression studies. Int. J. Trop. Agric. 33, 1079–1085.
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gupta, P., and Dash, P. K. (2015). Precise method of in situ drought stress induction in flax (Linum usitatissimum) for RNA isolation towards down-stream analysis. Ann. Agric. Res. 36, 1–8.
Gupta, P., Saini, R., and Dash, P. K. (2017). Origin and evolution of group XI secretory phospholipase A2 from flax (Linum usitatissimum) based on phylogenetic analysis of conserved domains. 3Biotech 7, 216–225. doi: 10.1007/s13205-017-0790-x
Hayano-Kanashiro, C., Calderón-Vázquez, C., Ibarra-Laclette, E., Herrera-Estrella, L., and Simpson, J. (2009). Analysis of gene expression and physiological responses in three Mexican maize landraces under drought stress and recovery irrigation. PLoS ONE 4:e7531. doi: 10.1371/journal.pone.0007531
Johnson, M. T. J., Carpenter, E. J., Tian, Z., Bruskiewich, R., Burris, J. N., Carrigan, C. T., et al. (2012). Evaluating methods for isolating total RNA and predicting the success of sequencing phylogenetically diverse plant transcriptomes. PLoS ONE 7:e50226. doi: 10.1371/journal.pone.0050226
Lenka, S. K., Katiyar, A., Chinnusamy, V., and Bansal, K. C. (2011). Comparative analysis of drought-responsive transcriptome in Indica rice genotypes with contrasting drought tolerance. Plant Biotechnol. J. 9, 315–327. doi: 10.1111/j.1467-7652.2010.00560.x
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Nakasugi, K., Crowhurst, R. N., Bally, J., Wood, C. C., Hellens, R. P., and Waterhouse, P. M. (2013). De novo transcriptome sequence assembly and analysis of RNA silencing genes of Nicotiana benthamiana. PLoS ONE 8:e59534. doi: 10.1371/journal.pone.0059534
Nigam, D., Saxena, S., Ramakrishna, G., Singh, A., Singh, N. K., and Gaikwad, K. (2017). De novo assembly and Characterization of Cajanus scarabaeoides (L.) thouars transcriptome by paired-end sequencing. Front. Mol. Biosci. 4:48. doi: 10.3389/fmolb.2017.00048
Shivaraj, S. M., Deshmukh, R. K., Rai, R., Bélanger, R., Agrawal, P. K., and Dash, P. K. (2017). Genome-wide identification, characterization, and expression profile of aquaporin gene family in flax (Linum usitatissimum). Sci. Rep. 7:46137. doi: 10.1038/srep46137
Surget-Groba, Y., and Montoya-Burgos, J. I. (2010). Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 20, 1432–1440. doi: 10.1101/gr.103846.109
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Wang, Z., Hobson, N., Galindo, L., Zhu, S., Shi, D., McDill, J., et al. (2012). The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 72, 461–473. doi: 10.1111/j.1365-313X.2012.05093.x
Zhang, H., Yang, B., Liu, W.-Z., Li, H., Wang, L., Wang, B., et al. (2014). Identification and characterization of CBL and CIPK gene families in canola (Brassica napus L.). BMC Plant Biol. 14:8. doi: 10.1186/1471-2229-14-8
Keywords: flax/linseed, RNAseq/transcriptome, drought stress, de novo assembly, Illumina MiSeq
Citation: Dash PK, Rai R, Mahato AK, Gaikwad K and Singh NK (2017) Transcriptome Landscape at Different Developmental Stages of a Drought Tolerant Cultivar of Flax (Linum usitatissimum). Front. Chem. 5:82. doi: 10.3389/fchem.2017.00082
Received: 31 July 2017; Accepted: 29 September 2017;
Published: 09 November 2017.
Edited by:
Raju Datla, National Research Council Canada, CanadaReviewed by:
Panagiotis Kalaitzis, Mediterranean Agronomic Institute of Chania, GreeceTahira Fatima, Purdue University, United States
Copyright © 2017 Dash, Rai, Mahato, Gaikwad and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Prasanta K. Dash, cGRhc0BucmNwYi5vcmc=