 Hongwei Zhang
Hongwei Zhang Yasuo Izumi
Yasuo Izumi- Department of Chemistry, Graduate School of Science, Chiba University, Chiba, Japan
Photocatalytic conversion of CO2 into mainly methane using Pd/TiO2 photocatalyst proceeded faster at 0.80 MPa using water rather than hydrogen as a reductant. The former reaction (CO2 + water) consists of two steps: first, water photosplitting and second, the latter reaction (CO2 + hydrogen). It was paradoxical that total steps proceeded faster than each step based on simple kinetics. To elucidate the reason, Pd and Ti K-edge X-ray absorption fine structure (XAFS) was monitored during CO2 photoconversion using H2 or moisture and the exchange reaction of 13CO2 at Pd/TiO2 surface was also monitored. As a result, the coordination number, N(Ti–O) and N[Ti(–O–)Ti] values, decreased from original values for TiO2 crystalline (6 and 12) to 4.9–5.7 and 9.7–10.6 under CO2 and moisture, respectively, in contrast to significantly smaller decreases under CO2 and H2 and under Ar. The exchange of gas-phase 13CO2 with preadsorbed 12CO2 reached the equilibrium in ~20 h with a rate constant of 0.20 h−1. The reason of the higher activity using water rather than H2 could be explained owing to the oxygen vacancy (Ov) sites as confirmed by XAFS. The reaction of TiO2 surface with water formed Ov sites responsible for water oxidation, specially separated from Pd nanoparticle sites for CO2 reduction. In contrast, Pd nanoparticle sites were competed by CO2 and H species, and the photoconversion of CO2 was suppressed at the elevated pressure of CO2 + H2.
Introduction
Photocatalytic conversion of CO2 into fuels is one of the routes to producing C neutral fuels without a net increase in atmospheric CO2 concentrations associated with fossil-derived alternatives (Izumi, 2013). The reaction includes two steps: water oxidation to O2 followed by CO2 photoconversion to form methane, etc.
We systematically tested the photocatalytic reaction under CO2 + H2 (Table 1A), an essentially identical reaction to Equation (2), for photocatalytic conversion of CO2 into fuels, if it is combined with the water photo-oxidation step (Equation 1) (Kawamura et al., 2017). Based on the formation rates of C-containing products (CH4, CO, and methanol) using Pd/TiO2, water was evidently more reactive (37 μmol-C h−1 g) than H2 (8.6 μmol-C h−1 g) for CO2 photoconversion at the higher reaction pressures, e.g., 0.80 MPa (Table 1C, G). It was paradoxical that a part of the reaction steps, i.e., Equation (2), was slower than total reaction steps, i.e., Equations (1, 2) based on simple kinetics. In this study, the reason was investigated by X-ray absorption fine structure (XAFS) and gas chromatography–mass spectrometry (GCMS).
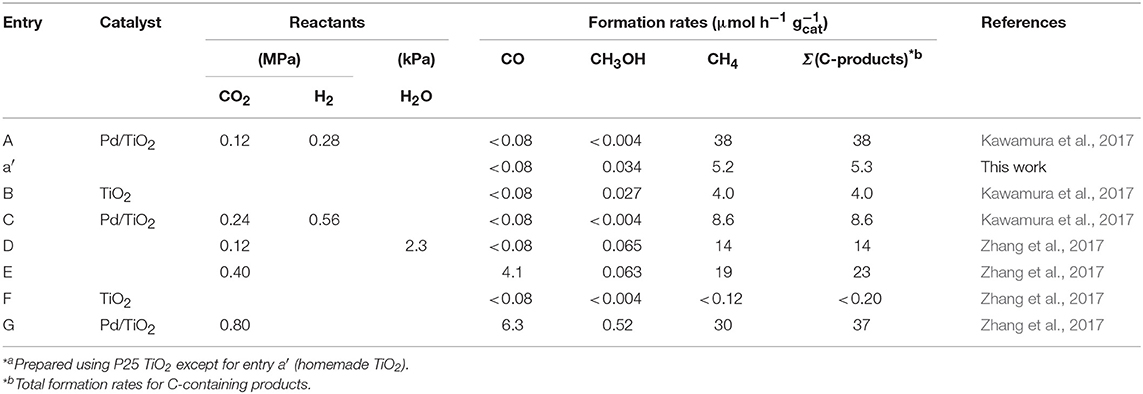
Table 1. Results of photocatalytic tests of CO2 photoconversion for 5 h Using 10 mg of Pd (0.5 weight-%)/TiO2 Photocatalysts*a and TiO2 (P25) at 0.12–0.80 MPa.
Material and Methods
A sample was prepared via liquid phase reduction (Kawamura et al., 2017) from 1.0 mM aqueous solution of Na2PdCl4 (>98%, Sigma Aldrich) and TiO2 (P25, Degussa; anatase: rutile phase = 4:1, specific surface area 60 m2 g−1) stirred at 290 K for 24 h. Then, 40 mM of NaBH4 aqueous solution was added in a molar ratio of Pd: NaBH4 = 1:8, and the suspension was washed with deionized water (<0.055 μS cm−1) supplied by an RFU424TA system (Advantec) before drying under vacuum at 290 K for 1 h and then filtered. The obtained precipitate was washed with deionized water before drying under vacuum at 290 K for 24 h. The obtained sample was denoted as Pd/TiO2 and the Pd content was 0.5 weight-% as metal.
For comparison, TiO2 was also synthesized in the laboratory. Acid solution (1.2 mL of 5 weight-% H2SO4 aqueous solution) and 1.37 mL of TiCl4 were added into 96 mL of deionized water at 273 K under argon atmosphere and stirred at 900 rotations per minute for 30 min. Then, 28 weight-% ammonia aqueous solution was dropped into the solution at 368 K to adjust the pH 7.0. The solution was stirred at the temperature for 1 h and cooled to 290 K. The obtained suspension was filtered and washed by deionized water (total 500 mL) until no chlorine ions were detected using silver nitrate. The thus-obtained powder was dried at 373 K overnight, calcined at 573 K for 2 h, and white TiO2 powder was obtained. The Pd was loaded on the homemade TiO2 in a similar manner to the liquid phase reduction for P25 as described earlier.
The high-pressure photoconversion tests were performed in homemade stainless steel reactor equipped with quartz windows (Kawamura et al., 2017; Zhang et al., 2017). A mixed CO2/H2 gas or CO2 gas/moisture was introduced into the reactor. A film (10 mg of photocatalyst) in the reactor was irradiated by a 500-W xenon arc lamp (Model OPM2-502, Ushio) for 5 h. The reaction gas was analyzed using packed columns of 13X-S molecular sieves and polyethylene glycol (PEG-6000) supported on Flusin P (GL Sciences, Inc.) set in a gas chromatograph equipped with a thermal conductivity detector (GC-TCD; Model GC-8A, Shimadzu).
The Pd K-edge XAFS spectra were measured at 290 K in the transmission mode in the Photon Factory Advanced Ring at the High Energy Accelerator Research Organization (KEK) on beamline NW10A. An Si (3 1 1) double-crystal monochromator and a Pt-coated focusing cylindrical mirror were inserted into the X-ray beam path. A Piezo transducer was used to detune the X-ray intensity to two-thirds of the maximum to suppress higher harmonics (Izumi et al., 2001; Fujishima et al., 2015). The Pd K-edge absorption energy was calibrated to 24348 eV using the spectrum of a Pd metal foil (Bearden, 1967).
The Ti K-edge XAFS spectra were measured in the transmission mode in the Photon Factory at KEK on beamline 9A, 9C, and 12C. An Si(1 1 1) double-crystal monochromator and a pair of bent conical/cylinder mirrors and/or double flat mirrors were inserted into the X-ray beam path. A Piezo transducer was used to detune to two-thirds of the maximum X-ray intensity to suppress higher harmonics (Izumi et al., 2001; Fujishima et al., 2015). Spectra for the Pd/TiO2 sample (10 mg) diluted by boron nitride were measured under CO2 (100 kPa) and moisture (2.2 kPa), under CO2 (70 kPa) and H2 (30 kPa), or Ar (100 kPa) in the presence/absence of UV–visible light irradiation provided by a 500-W Xe arc lamp (the flux intensity 81.6 mW cm−2) at the beamline (Izumi et al., 2007; Morikawa et al., 2014). The Ti K-edge absorption energy was calibrated to 4964.5 eV using the spectrum of a Ti metal foil (Bearden, 1967). The obtained Pd and Ti K-edge XAFS data were analyzed using XDAP software package using modified Victoreen function, spline function, Fourier transform, and multiple-shell curve fitting (Vaarkamp et al., 2006; Izumi et al., 2009).
Isotope-labeled exchange reaction tests were performed and monitored with GCMS using a JMS-Q1050GC (JEOL) apparatus equipped with a quadruple mass spectrometer. Helium (purity > 99.9999%) was used as the carrier gas. The sampling loop comprised a Pyrex glass system vacuumed using rotary and diffusion pumps (10−6 Pa) and connected to the JMS-Q1050GC using 1.5 m of deactivated fused silica tube (No. 160-2845-10, Agilent; internal diameter 250 μm) maintained at 393 K during the analysis to avoid gas adsorption (Wein et al., 2018).
A photocatalyst sample (0.100 g) was placed in a quartz photoreactor and evacuated at 295 K for 2 h while being connected to a Pyrex glass circulating system (circulating section 206.1 mL) and rotary and diffusion pumps (10−6 Pa). Then, 13CO2 (0.68 kPa; 13C 99%, 17O 0.1%, 18O 0.7%, chemical purity > 99.9%, Cambridge Isotope Laboratories, Inc.) photoexchange with the photocatalyst surface was monitored at 295 K under irradiation by UV–visible light from a 500 W xenon arc lamp (Model SX-UID502XAM, Ushio). The distance between the exit of the lamp window and the photocatalyst was 18 mm. The exchange with adsorbed 12CO2 at the surface was monitored using a packed column of PEG-6000 supported on a Flusin P (GL Sciences Inc.) set in the JSM-Q1050GC.
The exchange reaction rates were assumed to follow first-order equilibrium kinetics and the rate constants kr and kr′ are for the exchange of gas-phase 13CO2 with adsorbed 12CO2 and gas-phase 12CO2 with adsorbed 13CO2, respectively.
Photocatalytic tests using 13CO2 (2.3 kPa) and H2 (21.7 kPa) were also performed in a similar apparatus and conditions to that of 13CO2 exchange tests.
Results and Discussion
Photoconversion Tests of CO2
First, reported kinetic results are summarized. Both the reactions under CO2 + H2 and under CO2 + moisture critically depended on the pressure of reactants using Pd/TiO2 (Kawamura et al., 2017; Zhang et al., 2017). The formation rates of C-containing products under CO2 + H2 reached a local maximum at 0.40 MPa (0.12 MPa of CO2, 0.28 MPa of H2) and drew a volcanolike dependence (Table 1A, C). In Kawamura et al. (2017), the volcanolike local maximum seems distributed relatively steep based on 13 test data points. Methane was a dominant product under the conditions. In contrast, the formation rates followed a typical Langmuir-type dependence, i.e., they gradually increased as a function of reactants pressure under CO2 + moisture at 0.12–0.80 MPa (Table 1D, E, G) (Zhang et al., 2017). Minor CO and methanol were also produced under the conditions.
Due to the balance of different pressure dependencies between the two types of reactions, H2 was more reactive at lower than 0.40 MPa, whereas water was more reactive at higher than 0.40 MPa as a reductant for CO2. In this context, the major question of this study is the reason for the different pressure dependencies of CO2 photoconversion rates using H2 vs. water.
Photoconversion test was performed at 0.40 MPa of CO2 and H2 using Pd/TiO2 with the help of homemade TiO2 (Table 1a′). Methane was the major product similar to the result using Pd/TiO2 prepared from commercial P25 (Table 1A) (Kawamura et al., 2017). The reason for the difference of the methane formation rates (5.2 μmol h−1 g vs. 38 μmol h−1 g) is not clear, but the crystalline phase (anatase vs. the mixture of anatase and rutile phases) and specific surface area were different. Although the impurity effects for photocatalysts using P25 were suggested (Izumi, 2013; Cybula et al., 2015; Li et al., 2016; Dilla et al., 2017; Grigioni et al., 2017) photoconversion of CO2 into methane was confirmed using Pd/TiO2 prepared by not making use of any organic reagents throughout the synthesis procedure of the photocatalyst (Table 1a′). For this problem, several black tests were reported using Pd/TiO2 (P25), especially the one under UV–visible light and H2 excluding CO2 gas (Zhang et al., 2017). No C-containing products were detected above the detection limit of GC-TCD. Thus, a direct impurity effect is denied as the reason for the rate difference (Table 1A, a′). The indirect effect of gas-phase CO2 on impurity on/in catalyst, leading to the formation of C-containing products under light cannot be denied as the reason for formation rate difference.
The other possibility of formation rate difference is critical pressure dependence of TiO2-based photocatalysts under CO2 + H2 (Kawamura et al., 2017). Due to the critical difference of reactant pressure for the conversion rate maximum (volcano top) for each catalyst, formation rate difference of by a factor of 7.2 times between Pd/TiO2 (P25) and Pd/TiO2 (homemade) may result at a fixed reactant pressure (0.40 MPa).
Active Site Monitoring by XAFS
For the morphological characterization of the photocatalysts, transmission electron microscope images for the Pd/TiO2 catalyst, fresh and used one, after photocatalytic test in CO2 + H2 and cross-sectional scanning electron microscope (SEM) images for fresh Pd/TiO2 were reported in Kawamura et al. (2017). The morphology of the catalyst negligibly changed after the photocatalytic test, but the mean Pd particle size slightly increased starting from 3.1 to 3.9 nm after the photocatalytic test (Kawamura et al., 2017). Cross-sectional SEM images for fresh Pd/TiO2 and used one after photocatalytic test in CO2 + moisture were also reported in Zhang et al. (2017). The color of the top few micrometers of Pd/TiO2 catalyst film (total 11–16 μm) changed after the photocatalytic test (Zhang et al., 2017). In this study, monitoring of local active sites was performed in addition to these morphological studies.
The X-ray absorption near-edge structure (XANES) at the Pd K-edge suggested metallic Pd nanoparticles dispersed on TiO2. The extended X-ray absorption fine structure (EXAFS) also did not show significant change during photoreactions under CO2 + H2 or CO2 + moisture based on the Pd–Pd shell, but the coordination number N(Pd–O) decreased from 1.9 for fresh sample to 0.6–0.9 after the photocatalytic tests under CO2 + H2 at 0.024–0.40 MPa (Kawamura et al., 2017) and to 0.8–1.0 under CO2 + moisture at 4.6 kPa−0.40 MPa (Zhang et al., 2017). The decrease of N(Pd–O) values suggested the formation of oxygen vacancy (Ov) sites on/in TiO2 near Pd nanoparticles under the reaction conditions of CO2 photoconversion.
The Ti K-edge XAFS was also investigated under the reaction conditions of CO2 photoconversion with moisture (Figure 1). Under CO2 (100 kPa), moisture (2.2 kPa), and UV–visible light, the change in N(Ti–O) values was monitored (Figure 1A). The changes under light and then under dark were depicted as the Fourier transform in Figure 1C. The intensity of peak for Ti–O interatomic pair at 0.15 nm (phase shift uncorrected) decreased when the light was on (Figure 1C, 5 min), remained almost constant during the light irradiation, and increased to the original when the light was off (Figure 1C, Light off, 0 min). In accordance with the peak intensity change, in comparison with the initial N(Ti–O) value (6.0) under CO2, moisture, and dark, the value significantly decreased to 4.9–5.7 during light irradiation for 3.5 h. At 0.5 h after the light was off, the N(Ti–O) value increased to the original value (6.0).
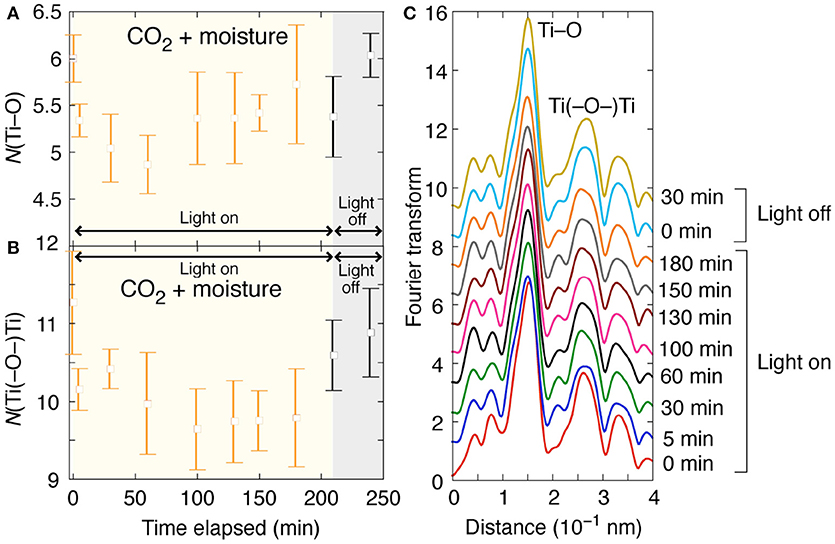
Figure 1. Time course of Ti K-edge EXAFS for Pd/TiO2 photocatalyst (10 mg) under CO2 (100 kPa) and moisture (2.2 kPa) for 3.5 h irradiated by UV–visible light and subsequently for 0.5 h under dark. The changes of (A) N(Ti–O) values, (B) N[Ti(–O–)Ti] values, and (C) Fourier transform of angular wavenumber k3-weighted EXAFS χ function (Zhang et al., 2017).
The peak intensity of Ti(–O–)Ti interatomic pair at 0.26 nm (phase shift uncorrected; Figure 1C) and the N[Ti(–O–)Ti] values followed a similar trend: decrease from the initial 11.3 to 9.7–10.6 during the irradiation of UV–visible light (Figure 1B). Taking the experimental and fit errors into account (Figures 1A,B), the N(Ti–O) and N[Ti(–O–)Ti] values would evaluate the concentration of Ov sites in/on TiO2 (Zhang et al., 2017).
Two control monitoring tests were also performed: (i) using H2 instead of moisture as reductant for CO2 and (ii) using only argon. First, under CO2 (70 kPa), H2 (30 kPa), and UV–visible light, the changes in peak intensity of Ti–O and Ti(–O–)Ti interatomic pair at 0.15 nm and 0.26 nm (phase shift uncorrected), respectively (Figure 2E) and the resultant N(Ti–O) and N[Ti(–O–)Ti] values were monitored (Figures 2A,B). The peak intensity of Ti–O interatomic pair decreased at 75 min of light irradiation, but no clear correlation between light irradiation and the Ti–O peak intensity was found. The peak intensity of Ti(–O–)Ti interatomic pair did not change much. In accordance with the peak intensity changes, in comparison with the initial value (5.9) under CO2, H2, and dark, the N(Ti–O) value varied, but within a small range of 5.6–5.8 during light irradiation for 125 min. At 50 min after the light was off, the N(Ti–O) value increased to the original value (6.0). In comparison with the monitoring under CO2 and moisture (starting from 6.0 to 4.9–5.7; Figure 1A), the decrease in N(Ti–O) value was effectively smaller under CO2 and H2 (Δ = 0.1–0.3) during photoirradiation. The N[Ti(–O–)Ti] value remained between 12 and 11.7 under the irradiation of UV–visible light and negligibly changed after the light was off: 12–11.2 (Figure 2B). Under CO2 and moisture, the decrease was greater: from 11.3 to 9.7–10.6 by the effect of light (Figure 1B).
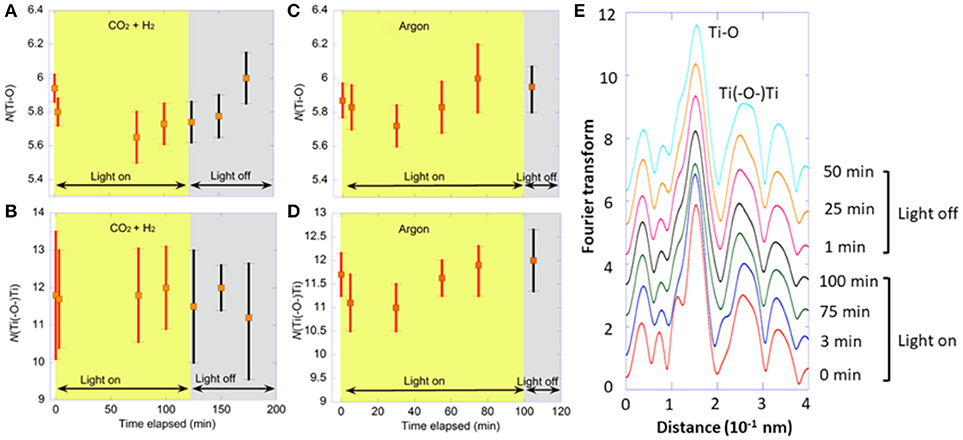
Figure 2. Time course of Ti K-edge (A–D) EXAFS for Pd/TiO2 photocatalyst (10 mg). (A,B) Under CO2 (70 kPa) and H2 (30 kPa) for 125 min irradiated by UV–visible light and subsequently for 75 min under dark. (C,D) Under Ar (100 kPa) for 100 min irradiated by UV–visible light and subsequently for 20 min under dark. The changes of (A,C) N(Ti–O) values and (B,D) N[Ti(–O–)Ti] values. (E) Fourier transform of angular wavenumber k3-weighted EXAFS χ function under CO2 (70 kPa) and H2 (30 kPa).
Under Ar (100 kPa) and UV–visible light, the N(Ti–O) value changed between 5.7 and 6.0 during light irradiation for 100 min. At 5 min after the light was off, the N(Ti–O) value remained at 5.9 (Figure 2C). The changes in N[Ti(–O–)Ti] values were also minimal under Ar: 12–11 throughout the monitoring test under light/dark (Figure 2D). These differences in N(Ti–O) and N[Ti(–O–)Ti] values were because more Ov sites were formed under the photoreduction of CO2 using moisture rather than H2 or Ar.
Monitoring of 13CO2 Exchange by GCMS
The 13CO2 exchange reaction test was performed using Pd–TiO2 (P25) photocatalyst. The amount of 13CO2 gas gradually decreased over 20 h by the exchange with 12CO2 on Pd–TiO2 sample that was preadsorbed from air (Figure 3A). The changes followed equilibrium kinetics as listed in Equations (3–6), and the formation of 12CO2 (n: mol) in gas phase was well fitted by Equation (6′) (Figure 3A).
Initial 0.657 μmol is the impurity (1.18 mol%) included in the introduced 13CO2 gas (0.68 kPa). The value (1.80 μmol) obtained by the fit of Equation (6′) to the data corresponds to 0.18 molecules of CO2 adsorbed per nm2 of Pd–TiO2 that was an acceptable value after the evacuation at 295 K for 2 h as pretreatment. The sum of rate constants kr and kr′ corresponds to total rate constant (Equation 6) to reach the equilibrium 13CO2 and 12CO2 (0.20 h−1).
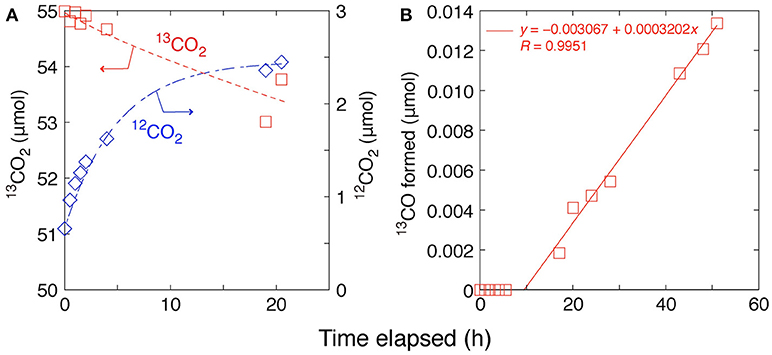
Figure 3. Time courses of (A) 13CO2 (0.68 kPa) photoexchange reaction with preadsorbed 12CO2 on Pd/TiO2 (P25) and (B) photocatalytic reduction test in 13CO2 (2.3 kPa) and H2 (21.7 kPa) on Pd/TiO2 (homemade).
As a comparison, if we assume that the dispersion of mean 3.1 nm Pd nanoparticles (0.50 weight-%) over TiO2 observed by transmission electron microscopy (Kawamura et al., 2017) is ~50% (Kip et al., 1987) and the conversion to C-containing products (methane, CO, and methanol) proceeded on the surface of the Pd atoms starting from CO2, the formation rate of C-containing products (37 μmol h−1 g) under 0.80 MPa of CO2 and moisture (Table 1G) corresponds to the turnover frequency (TOF) = 1.6 h−1. This comparison is contradictory because the early adsorption equilibrium of CO2 (0.20 h−1 based on Equation 6′) is slower than CO2 photoreduction to methane via much more difficult reaction steps (1.6 h−1). However, the exchange rate (0.20 h−1) observed under 13CO2 (initial 0.68 kPa) in this study was by a couple of times higher compared with the formation rate of C-containing products under initial 3.4 kPa of CO2 + 1.2 kPa of moisture (0.9 μmol h−1 g) (Zhang et al., 2017) corresponding to the TOF = 0.04 h−1. The CO2 exchange was reported using isotope labeled Ti18O2 irradiated by XeCl excimer laser (308 nm) to form C18O2 and 16OC18O (Civiš et al., 2011).
Furthermore, stability test of photocatalytic reaction using 13CO2 (2.3 kPa) + H2 (21.7 kPa) and Pd/TiO2 was performed for more than 50 h (Figure 3B). To avoid the possibility of impurity effects, Pd/TiO2 (homemade) was used and a byproduct 13CO was monitored by GCMS rather than methane that may be produced from impurity in TiO2 (Izumi, 2013; Cybula et al., 2015; Li et al., 2016; Dilla et al., 2017; Grigioni et al., 2017). In accordance with the rate of 13CO2 exchange reaction (Figure 3A), 13CO started to be formed at ~10 h of reaction and constantly formed for more than 50 h (Figure 3B). As a byproduct, the formation rate was low 0.0032 μmol h−1 g corresponding to the TOF 0.00014 h−1. Nevertheless, the stability test confirmed the availability of Pd/TiO2 photocatalyst for more than 50 h of photoreaction test.
Thus, we should be careful in the isotope tracing tests as 12CO2 could be included derived from preadsorbed 12CO2 in the reactant 13CO2 and 12C-products could be included by the photocatalytic conversion.
The Pd/TiO2 catalyst used in this study is advantageous for CO2 photoconversion irradiated by visible light (Kawamura et al., 2017; Zhang et al., 2017). The photocatalytic performance was compared with TiO2 (P25) in 0.40 MPa of CO2 + H2 (Table 1A, B) or CO2 + moisture (Table 1E, F). The formation rates of total C-containing products were 11% and <0.9% using TiO2 in comparison with corresponding values using Pd/TiO2, respectively. As TiO2 is mostly activated by UV light based on the band gap value (3.2 and 3.0 eV for anatase and rutile phases, respectively) (Izumi, 2013), the increased photoactivity using Pd/TiO2 could be the effects of localized surface plasmon resonance of Pd nanoparticles owing to visible light irradiation (Kawamura et al., 2017; Zhang et al., 2017).
Thus, the answer to the question as to why water was more reactive than H2 in photocatalytic CO2 conversion at higher pressures is that water forms hydroxy radical under the irradiation of UV–visible light (Equation 7) and then Ov sites are formed (Equation 8), but not by using H2 or Ar. The exchange of CO2 (Figure 3A) and/or the elimination of C–O bond to form CO are also suggested over Ov sites of Au–Cu–TiO2 (Neatu et al., 2014), MgO–Pt–TiO2 (Xie et al., 2014), TiO2 (Liu et al., 2012, 2016; Li et al., 2017), and theoretical models of defective anatase (Ji and Luo, 2016), anatase (1 0 1), and small TiO2 cluster (Lee and Kanai, 2012).
The reactions 7 and 8 proceeded as water oxidation over TiO2 spatially separated from CO2 reduction sites on Pd for Equation (2). Thus, the effective redox reaction of Equations (1, 2) proceeded using water. Both the reactions 1 and 2 would proceed competitively on Pd under CO2 + H2, and the efficiency followed a volcano-like dependence as a function of partial pressures of reactants (Table 1A, B) (Kawamura et al., 2017).
Conclusions
The formation of Ov sites was monitored by means of Ti K-edge EXAFS using Pd/TiO2. The coordination number around Ti atoms clearly demonstrated the effective formation of Ov sites under CO2, moisture, and UV–visible light in contrast to the much smaller population of Ov sites under CO2, H2, and UV–visible light. This difference explained the effective red-ox site separation for water oxidation via ·OH radicals and Ov sites and CO2 reduction over Pd nanoparticle sites under CO2, moisture, and UV–visible light. The exchange of 13CO2 with preadsorbed 12CO2 over Pd/TiO2 reached the equilibrium in ~20 h. This result suggested the earlier step of CO2 photoconversion, but we need to be aware that 12C-products are formed via photocatalytic reaction tests under 13CO2.
Author Contributions
This study was planned by YI, most of the experiments and analyses were performed by HZ, and this manuscript was written together.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful for the financial supports from the Grant-in-Aid for Scientific Research C (17K05961, 26410204) from the Japan Society for the Promotion of Science and Leading Research Promotion Program (2015–) from the Institute for Global Prominent Research, Chiba University. X-ray absorption experiments were conducted under the approval of the Photon Factory Proposal Review Committee (2016G577, 2015G586, 2014G631).
References
Civiš, S., Ferus, M., Kubát, P., Zukalová, M., and Kavan, L. (2011). Oxygen-isotope exchange between CO2 and solid Ti18O2. J. Phys. Chem. C 115, 11156–11162. doi: 10.1021/jp201935e
Cybula, A., Klein, M., and Zaleska, A. (2015). Methane formation over TiO2-based photocatalysts: reaction pathways. Appl. Catal. B 164, 433–442. doi: 10.1016/j.apcatb.2014.09.038
Dilla, M., Schlögl, R., and Strunk, J. (2017). Photocatalytic CO2 reduction under continuous flow high-purity conditions: quantitative evaluation of CH4 formation in the steady state. Chem. Cat. Chem. 9, 696–704. doi: 10.1002/cctc.201601218
Fujishima, Y., Okamoto, S., Yoshiba, M., Itoi, T., Kawamura, S., Yoshida, Y., et al. (2015). Photofuel cell comprising titanium oxide and bismuth oxychloride (BiO1−xCl1−y) photocatalysts that uses acidic water as a fuel. J. Mater. Chem. A 3, 8389–8404. doi: 10.1039/C4TA06824F
Grigioni, I., Dozzi, M. V., Bernareggi, M., Chiarello, G. L., and Selli, E. (2017). Photocatalytic CO2 reduction vs. H2 production: the effects of surface carbon-containing impurities on the performance of TiO2-based photocatalysts. Catal. Today 281, 214–220. doi: 10.1016/j.cattod.2016.05.040
Izumi, Y. (2013). Recent advances in the photocatalytic conversion of carbon dioxide to fuels with water and/or hydrogen using solar energy and beyond. Coord. Chem. Rev. 257, 171–186. doi: 10.1016/j.ccr.2012.04.018
Izumi, Y., Itoi, T., Peng, S., Oka, K., and Shibata, Y. (2009). Site structure and photocatalytic role of sulfur or nitrogen-doped titanium oxide with uniform mesopores under visible light. J. Phys. Chem. C 113, 6706–6718. doi: 10.1021/jp810817y
Izumi, Y., Kiyotaki, F., Nagamori, H., and Minato, T. (2001). Site-selective XAFS spectroscopy tuned to surface active sites of copper catalysts. J. Electro. Spectrosc. Relat. Phenomen. 119, 193–199. doi: 10.1016/S0368-2048(01)00292-4
Izumi, Y., Konishi, K., Obaid, D. M., Miyajima, T., and Yoshitake, H. (2007). X-Ray absorption fine structure combined with X-ray fluorescence spectroscopy. monitoring of vanadium sites in mesoporous titania, excited under visible light by selective detection of vanadium Kβ5, 2 fluorescence. Anal. Chem. 79, 6933–6940. doi: 10.1021/ac070427p
Ji, Y., and Luo, Y. (2016). New mechanism for photocatalytic reduction of CO2 on the anatase TiO2(1 0 1) surface: the essential role of oxygen vacancy. J. Am. Chem. Soc. 138, 15896–15902. doi: 10.1021/jacs.6b05695
Kawamura, S., Zhang, H., Tamba, M., Kojima, T., Miyano, M., Yoshida, Y., et al. (2017). Efficient volcano-type dependence of photocatalytic CO2 conversion into methane using hydrogen at reaction pressures up to 0.80 MPa. J. Catal. 345, 39–52. doi: 10.1016/j.jcat.2016.10.024
Kip, B. J., Duivenvoorden, F. B. M., Koningsberger, D. C., and Prins, R. (1987). Determination of metal particle size of highly dispersed Rh, Ir, and Pt catalysts by hydrogen chemisorption and EXAFS. J. Catal. 105, 26–38. doi: 10.1016/0021-9517(87)90005-4
Lee, D., and Kanai, Y. (2012). Role of four-fold coordinated titanium and quantum confinement in CO2 reduction at titania surface. J. Am. Chem. Soc. 134, 20266–20269. doi: 10.1021/ja309871m
Li, J., Zhang, M., Guan, Z., Li, Q., He, C., and Yang, J. (2017). Synergistic effect of surface and bulk single-electron-trapped oxygen vacancy of TiO2 in the photocatalytic reduction of CO2. Appl. Catal. B 206, 300–307. doi: 10.1016/j.apcatb.2017.01.025
Li, K., Peng, B., and Peng, T. (2016). Recent advances in heterogeneous photocatalytic CO2 conversion to solar fuels. ACS Catal. 6, 7485–7527. doi: 10.1021/acscatal.6b02089
Liu, L., Jiang, Y., Zhao, H., Chen, J., Cheng, J., Yang, K., et al. (2016). Engineering coexposed {0 0 1} and {1 0 1} facets in oxygen-deficient TiO2 nanocrystals for enhanced CO2 photoreduction under visible light. ACS Catal. 6, 1097–1108. doi: 10.1021/acscatal.5b02098
Liu, L., Zhao, H., Andino, J. M., and Li, Y. (2012). Photoatalytic CO2 reduction with H2O on TiO2 nanocrystals: comparison of anatase, rutile, and brookite polymorphs and exploraiton of surface chemistry. ACS Catal. 2, 1817–1828. doi: 10.1021/cs300273q
Morikawa, M., Ahmed, N., Yoshida, Y., and Izumi, Y. (2014). Photoconversion of carbon dioxide in zinc–copper–gallium layered double hydroxides: the kinetics to hydrogen carbonate and further to CO/methanol. Appl. Catal. B 144, 561–569. doi: 10.1016/j.apcatb.2013.07.065
Neatu, S., Maciá-Agulló, J. A., Concepción, P., and Garcia, H. (2014). Gold–copper nanoalloys supported on TiO2 as photocatalysts for CO2 reduction by water. J. Am. Chem. Soc. 136, 15969–15976. doi: 10.1021/ja506433k
Vaarkamp, M., Linders, H., and Koningsberger, D. (2006). XDAP Version 2.2.7, XAFS Services International. Woudenberg.
Wein, L. A., Zhang, H., Urushidate, K., Miyano, M., and Izumi, Y. (2018). Optimized photoreduction of CO2 exclusively into methanol utilizing liberated reaction space in layered double hydroxides comprising zinc, copper, and gallium. Appl. Surf. Sci. 447, 687–696. doi: 10.1016/j.apsusc.2018.04.046
Xie, S., Wang, Y., Zhang, Q., Deng, W., and Wang, Y. (2014). MgO- and Pt-promoted TiO2 as an efficient photocatalyst for the preferential reduction of carbon dioxide in the presence of water. ACS Catal. 4, 3644–3653. doi: 10.1021/cs500648p
Keywords: CO2, oxygen vacancy, X-ray absorption fine structure, gas chromatography–mass spectrometry, 13CO2
Citation: Zhang H and Izumi Y (2018) Why Is Water More Reactive Than Hydrogen in Photocatalytic CO2 Conversion at Higher Pressures? Elucidation by Means of X-Ray Absorption Fine Structure and Gas Chromatography–Mass Spectrometry. Front. Chem. 6:408. doi: 10.3389/fchem.2018.00408
Received: 03 July 2018; Accepted: 20 August 2018;
Published: 27 September 2018.
Edited by:
Junguang Tao, Hebei University of Technology, ChinaReviewed by:
Ellen B. Stechel, Arizona State University, United StatesFederica Valentini, Università di Roma Tor Vergata, Italy
Copyright © 2018 Zhang and Izumi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yasuo Izumi, eWl6dW1pQGZhY3VsdHkuY2hpYmEtdS5qcA==