 Saburo Hosokawa1,2*
Saburo Hosokawa1,2*- 1Elements Strategy Initiative for Catalysts and Batteries, Kyoto University, Kyoto, Japan
- 2Department of Molecular Engineering, Graduate School of Engineering, Kyoto University, Kyoto, Japan
The rare earth-iron mixed oxide (REFeO3) is an attractive material in fields such as electronic, magnetic, and catalytic research. Generally, orthorhombic REFeO3 (o-REFeO3) with a perovskite structure is better known than hexagonal REFeO3 (h-REFeO3), because o-REFeO3 is thermodynamically stable for all RE elements. However, h-REFeO3 has a very interesting crystal structure in which a RE and Fe layer are alternately stacked along the c-axis in the unit cell; nevertheless, synthesis of the h-REFeO3 belonging to metastable phase can be problematic. Fortunately, solution-based synthetic methods like solvothermal or coprecipitation synthesis have recently enabled the selective synthesis of h-REFeO3 and o-REFeO3 with comparative ease. Although the electronic and magnetic properties of h-REFeO3 have typically been evaluated, recent research has also revealed excellent catalytic properties that enable environmental cleanup reactions such as hydrocarbon or CO oxidation. This mini-review introduces a synthetic method for controlling the crystal structure between orthorhombic and hexagonal REFeO3 and the catalytic performance of h-REFeO3-based materials.
Introduction
REFeO3 (RE: rare earth) with a RE/Fe ratio of 1 are largely relevant to the perovskite structure, a structure in which RE ions are basically replaced with 1/4 of cubic close-packed oxygen ions, and Fe ions occupy all octahedral gaps formed from the remaining oxygen ions. REFeO3 having a perovskite structure have been applied in various fields from electronic devices to catalysts. On the other hand, hexagonal REFeO3 is known to exist as a metastable phase, but the synthetic method is very complex. Interestingly, the crystal structure has a unique coordination state for Fe3+ ion and is different from that of the perovskite-type REFeO3; that is, Fe3+ ions in hexagonal structure have trigonal bipyramidal coordination, although Fe3+ ions in α-Fe2O3, Fe3O4, or perovskite-type REFeO3 generally prefer octahedral or tetrahedral coordination. Therefore, hexagonal structure has a potential which provokes an interesting property for the above applied fields. In particular, novel catalysts composed on abundant base metal as Fe ion have been recently desired to replace the use of precious metals, such as Pt or Pd; the catalyst design based on a unique crystal structure is considered to be necessary to dramatically improve the catalytic performance of Fe-based material.
First, the relationship between RE elements and hexagonal structure is introduced, based on a general perovskite structure. Considering that each ion is in contact within the perovskite structure, the following equation is true: (rA + rO) = √2(rB + rO), where rA, rB, and rO represent the ionic radii of the A cation as RE ion, B cation as Fe ion, and oxygen ion, respectively. Therefore, the perovskite structure is generally considered to be a stable phase, when the tolerance factor, t = [(rA + rO)]/[√2(rB + rO)] is close to 1 (Schneider et al., 1961; Woodward, 1997; Li et al., 2004). Regarding the relationship between RE elements and tradition metal ions, the tolerance factor decreases with decreasing ionic radius of the RE element (Kumar et al., 2011); that is, the perovskite structure becomes less stable. However, for REFeO3, the orthorhombic phase is thermodynamically stable for all RE elements, and the trend is different from a general trend observed in REMnO3 (Figure 1A); orthorhombic REMnO3 (o-REMnO3) with a perovskite structure and the space group of Pbnm is preferentially formed for RE elements with large ionic sizes (La–Dy), while hexagonal REMnO3 (h-REMnO3) with P63cm is obtained for RE elements with small ionic sizes (Ho–Lu and Y) (Figures 1B,C). As the ionic size of an Fe3+ ion is almost the same as that of an Mn3+ ion, the difference in the electronic configuration of d orbitals between Fe3+ and Mn3+ ions may contribute to the stability of the hexagonal phase. In other words, because h-REFeO3 is a thermodynamically unstable phase, it is relatively poorly studied compared with o-REFeO3.
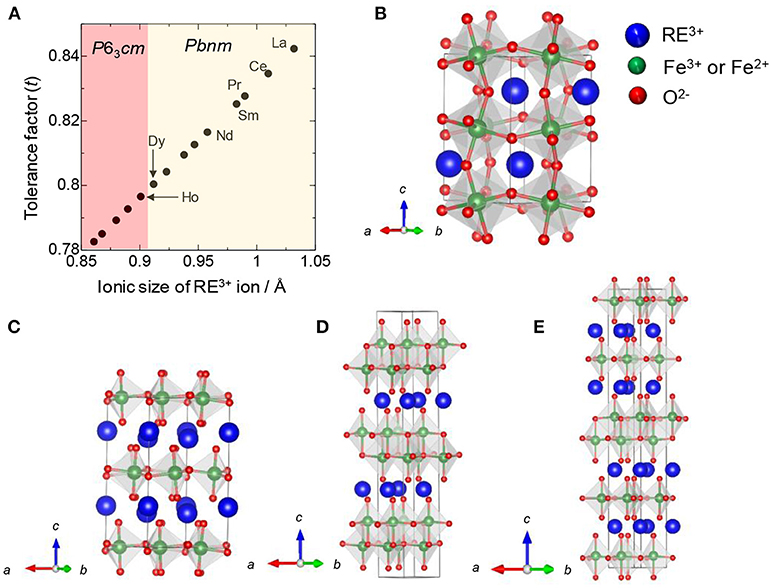
Figure 1. (A) Tolerance factor and stable crystal structure against ionic size of RE in REMnO3, (B) Orthorhombic REFeO3 (o-REFeO3) or o-REMnO3 with the space group of Pbnm, (C) Hexagonal REFeO3 (h-REFeO3) or h-REMnO3 with the space group of P63cm, (D) REFe2O4, and (E) RE2Fe3O7.
By controlling the synthetic route, h-REFeO3 composed from RE ions with a smaller ionic radius than Er or Y have been obtained. The space group of h-REFeO3 has been reported as P63cm with a YMnO3 structure type or P63/mmc with a YAlO3 structure type (Li et al., 2008; Kumar et al., 2009; Magome et al., 2010). The crystal structure belonging to P63cm is very similar to that to P63/mmc, except for a slightly more distorted crystal structure in the former than in the latter. Actually, the oxygen layer has a zigzag structure in the P63cm structure. In either space group, h-REFeO3 has a unique crystal structure other than the coordination state of Fe3+ ion; RE and Fe layers are alternately stacked along the c-axis. REFe2O4 or RE2Fe3O7, represented by the chemical formula REnFen+1O(3n+1), are known as hexagonal phase-related materials (Malaman et al., 1976; Qin et al., 2009; Bourgeois et al., 2012; Lafuerza et al., 2014), and the coordination environment of Fe ions is a trigonal bipyramidal structure similar to the structure of h-REFeO3. REFe2O4 has a hexagonal layer structure with the space group of Rm, which is formed by the alternate stacking of two layers with compositions of REO3/2 and Fe2+Fe3+O5/2 (Figure 1D). The Fe layer consists of two triangular sheets of corner-sharing FeO5 trigonal bipyramids. RE2Fe3O7 can be transcribed to REFe2O4•(REFeO3), and the structures of the REFe2O4•(REFeO3)n phases are described by alternately stacking REFe2O4 and hexagonal REFeO3 blocks along the c-axis direction (Figure 1E).
When h-REFeO3 is viewed as an applied material, its layered structure composed of RE and Fe layers along the c-axis is very attractive for multiple purposes, from electric to catalytic applications. In fact, LuFe2O4+x has shown excellent oxygen storage properties because its hexagonal-related structure has unique structural flexibility accompanied by “topotactic transition” during the oxygen intercalation/de-intercalation process (Hervieu et al., 2014). In this transformation, the original cation ordering in the hexagonal structure is maintained without any intermixing between RE and Fe ions. This mini-review therefore presents the method for controlling the crystal structure of h-REFeO3 and o-REFeO3 and discusses the catalytic properties of h-REFeO3.
Solvothermal Method
Because h-REFeO3 is a metastable phase, a unique synthesis method is typically required. Among existing methods, a solvothermal method is effective for controlling the hexagonal and orthorhombic phases. In this method, synthesis of inorganic material is achieved by reaction in a liquid medium at high temperatures in a closed vessel (autoclave) (Demazeau, 1999; Cushing et al., 2004). Therefore, the traditional solvothermal method is a hydrothermal method in which water is used as the solvent. Since the last three decades, alcohol or glycol has been used as a solvent in the solvothermal method (Fanelli and Burlew, 1986; Das et al., 2008), with which various metal oxides or mixed oxides with unique morphology or ultrafine nanoparticles have been reported.
Inoue et al. found that h-REFeO3 nanoparticles composed of RE with a smaller ionic radius than Er can be obtained through a non-aqueous solvothermal reaction of RE acetate with Fe acetylacetonate in 1,4-butanediol at 300°C (Figure 2; Inoue et al., 1997). It should be noted that crystal growth in the c-axis direction in h-REFeO3 is drastically suppressed during solvothermal synthesis, resulting in the formation of nanoparticles with a particle size of approximately 20 nm and a well-formed hexagonal plate-like morphology. It was found that h-YbFeO3 and o-YbFeO3 can be controlled by the choice of reaction conditions and the starting material for the solvothermal reaction (Hosokawa et al., 2009). Interestingly, when RE chloride (RE = Sm-Yb) and Fe acetylacetonate are used for the solvothermal reaction in 1,4-butanediol in the presence of hexamethylenediamine, monodisperse particles of o-REFeO3 (crystallite size: about 80 nm) are formed. It is suggested that o-YbFeO3 has a larger crystallite size than solvothermally-synthesized h-YbFeO3, which indicates that crystal nucleation is more difficult for o-YbFeO3 than h-YbFeO3.
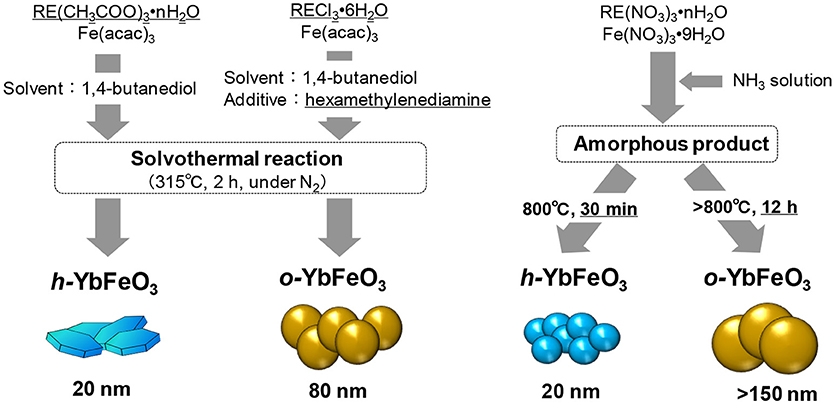
Figure 2. Flow chart showing crystal structure control utilizing the solvothermal method (Left) and coprecipitation method (Right).
High-resolution transmission electron microscopy (TEM) of Yb3Fe4O10 revealed that three or more YbFeO3 layers are inserted between the YbFe2O4 layers (Matsui et al., 1979), suggesting that the REFe2O4 phase with Fe3+ and Fe2+ species may act as a nucleus for h-REFeO3. The result indicates that the existence of Fe2+ ions plays an important role in the formation of h-REFeO3. The solvothermal reaction in 1,4-butanediol causes reductive conditions because of the reducing ability of glycol; that is, Fe2+ species formed in the reaction system seem to contribute to the formation of h-REFeO3. On the other hand, because the incorporation of amine into the solvothermal reaction suppresses the reduction from Fe3+ to Fe2+ ions owing to the chelating ability of hexamethylenediamine, o-YbFeO3 must be obtained. In addition, the reducibility of Fe3+ species is unsurprisingly also considered to depend on whether the ligand of the starting material is acetate or chloride.
This non-aqueous solvothermal method enables the crystallization of REFeO3 nanoparticles below 100 nm without calcination. Therefore, it is promising to control the crystal structure, shape, and particle size by adjusting the additives and solvent in the solvothermal system.
Coprecipitation and Sol-gel Method
General synthetic routes such as the coprecipitation method inevitably involve a calcination process for the synthesis of metal oxide materials, and it is difficult to synthesize the metastable phase. Therefore, h-REFeO3 is often synthesized by optimizing the crystallization process. For example, h-REFeO3 is synthesized by introducing solutions containing metal salts into a high-temperature inductively coupled plasma (ICP) (Mizoguchi et al., 1996). An instant crystallization under the ICP atmosphere yields h-REFeO3 nanoparticles sized 20–50 nm. Kumar et al. reported that the hexagonal phase is synthesized by rapidly cooling a melt of the composition REFeO3 under controlled oxygen pressure (Vijaya Kumar et al., 2008). This method requires special equipment in which the precursor is suspended on a nozzle fitted to an oxygen gas jet, thereby preventing heterogeneous nucleation that occurs on the walls of the container. As a result, h-LuFeO3 with high crystallinity is obtained under an oxygen partial pressure of 1 × 105 Pa.
On the other hand, in the case of h-REFeO3 synthesis from an amorphous precursor obtained by coprecipitation method in which ammonia water is added immediately to an aqueous solution containing RE and Fe ions (Nishimura et al., 2013), h-REFeO3 for RE ions with a smaller ionic radius than Er can be easily obtained under precisely controlled calcination conditions. For an amorphous precursor with the composition of YbFeO3, an exothermic peak without weight change is observed in the temperature range slightly below 800°C, by thermal gravimetric-differential thermal analysis. Therefore, the sample calcined in air at 700°C for 30 min is amorphous, and the formation of pure-phase h-YbFeO3 is confirmed by calcining the amorphous product at 800°C for 30 min (Figure 2). Note that, when the retention time at 800°C is extended to 12 h, o-YbFeO3 is obtained as the main product. These results strongly suggest that the exothermic peak is attributed not only to crystallization from an amorphous to hexagonal structure but also a phase transition from a hexagonal structure to orthorhombic structure. In other words, the hexagonal and orthorhombic phase can be kinetically classified by adjusting the heat retention time in the calcination process. In this manner, because a subtle change in calcination conditions leads to the formation of h-REFeO3 from an amorphous product, the synthesis of h-REFeO3 is much more difficult than that of o-REFeO3.
Similar crystallization behavior is also observed for a precursor containing organic species synthesized by a citric acid-assisted sol-gel process (Zhang et al., 2012). For an amorphous precursor with the composition of YFeO3, the crystallization temperature of h-YFeO3 is 700°C. This is lower than that of h-YbFeO3 (800°C); therefore, the crystallization temperature depends on the ionic radius of RE ions. Interestingly, doping Pd on h-YFeO3 via the sol-gel method stabilizes the hexagonal phase, and the phase transformation to the orthorhombic phase hardly proceeds even by calcination at 1,000°C (Li et al., 2008).
h-REFeO3, which is obtained by the calcination of a precursor synthesized by the solution method, often comprises fine particles with a crystallite diameter below 50 nm. On the other hand, o-REFeO3, which is obtained by phase transition from h-REFeO3, has a much larger particle size than h-REFeO3. For example, the particle size of h-YbFeO3 synthesized by the coprecipitation method is approximately 20 nm, whereas that of o-YbFeO3 is above 150 nm. A nucleation and growth mechanisms has been reported to proceed in a phase transition from γ-Al2O3 or θ-Al2O3 to α-Al2O3 (Dynys and Halloran, 1982; Bagwell et al., 2001): that is, rapid crystal growth of α-Al2O3 simultaneously progresses with the formation of crystal nuclei of α-Al2O3, resulting in the formation of α-Al2O3 with a very large particle size. Similar to the phenomena for α-Al2O3, crystal nucleation and rapid crystal growth of o-REFeO3 must proceed during the phase transition from h-REFeO3 to o-REFeO3. These results imply that h-REFeO3 nanoparticles can be synthesized by the coprecipitation or sol-gel method, but o-REFeO3 with a particle size below 100 nm is difficult to synthesize for RE ions with a smaller ionic radius than Er.
Catalytic Performance of h-REFeO3
o-REFeO3 has been applied as a catalyst material in various fields including the purification of volatile organic compound etc. (Ciambelli et al., 2001; Barbero et al., 2006). Applications of h-REFeO3 have been rare, except for in magnetic or electronic fields, until about 15 years ago. However, recent applications have emerged in the field of catalysts (Kurzman et al., 2011; Ismael et al., 2017). For example, h-YFeO3 has been reported to show a higher catalytic activity than o-YFeO3 for the photodecomposition of methyl orange under visible light irradiation (> 420 nm) (Zhang et al., 2012). As h-YFeO3 (1.94 eV) has a narrower band gap than o-YFeO3 (2.43 eV), the band structure of h-YFeO3 may be contributed to the high catalytic activity. h-YFeO3 with the unique band structure is also demonstrated to be suitable as a photoanode for solar water splitting (Guo et al., 2017).
The catalytic performance of h-REFeO3 as an oxidation catalyst has actively been investigated. When CH4 oxidations over h-YbFeO3 and o-YbFeO3 obtained by the solvothermal reaction are evaluated (Hosokawa et al., 2011), the h-YbFeO3 catalysts shows extremely high combustion activity compared with o-YbFeO3. The T50 value, at which the catalyst exhibits 50% CH4 conversion, is 475°C for h-YbFeO3, while that for o-YbFeO3 550°C. The surface area of the o-YbFeO3 catalyst significantly decreases during calcination at 800°C in catalyst preparation; that is, the surface area of o-YbFeO3 synthesized by the solvothermal method is 26 m2g−1 and that of o-YbFeO3 after calcination is 6 m2g−1. Interestingly, the surface area of the h-YbFeO3 catalyst (approximately 30 m2g−1) does not change significantly during the calcination, and the well-formed hexagonal plate shape is maintained. This result suggests that the surface energy of o-YbFeO3 is higher than that of h-YbFeO3. In other words, because h-YbFeO3 synthesized by the solvothermal method has high thermal stability, high catalytic activity must be achieved.
The h-YFe1−xPdxO3−δ catalyst synthesized by the sol-gel method has been reported to show high catalytic activity (T50 = 100°C) for CO oxidation under excess O2 concentration (1,000 ppm CO and 10% O2), comparable to that of a Pd/Al2O3 catalyst with high surface area (Li et al., 2008). The ionic Pd species in the hexagonal lattice contributes to the catalytic activity. Lu et al. also reported that a Pd catalyst supported on h-YFeO3 is more effective for CH4 oxidation than Pd/h-YMnO3 or Pd/o-LaFeO3, and that the Pd/YFeO3 catalyst maintains a high catalytic activity for CH4 oxidation even after aging treatment at 900°C despite the phase transformation to o-YFeO3 (Lu et al., 2014a,b, 2015). In these cases, as the Pd species is adequately anchored with the h-YFeO3 support, the catalyst seems to have high catalytic activity.
Furthermore, to develop a noble-metal free h-YbFeO3-based catalyst with high catalytic activity, transition metal-modified h-YbFeO3 catalysts are synthesized by solvothermal methods (Hosokawa et al., 2016). Mn modification by the solvothermal method dramatically improves the catalytic activity of h-YbFeO3 itself for CO oxidation in which a reaction gas composed of 5,000 ppm CO and 5,000 ppm O2 is introduced on catalyst bed. The catalytic activity (T50 = 120°C) of Mn-modified h-YbFeO3 (Mn-h-YbFeO3) with the composition of YbFe0.6Mn0.4O3 exceeds that (T50 = 134°C) of the noble metal Pd/Al2O3 catalyst.
Conclusion
Conventionally, a special method has been required for the synthesis of h-REFeO3 due to its metastable nature; however, h-REFeO3 can now be easily synthesized by precisely controlling the calcination process even when employing the coprecipitation or sol-gel methods. h-REFeO3-based materials have been demonstrated to be applicable for catalyst materials, as well as magnetic or electronic materials. I believe that excellent catalyst materials will evolve from catalyst designs focusing on the unique crystal structure of h-REFeO3 or hexagonal-related materials (REnFen+1O(3n+1)).
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author sincerely thanks Masashi Inoue, Professor Emeritus at Kyoto University, for his invaluable discussion on hexagonal REFeO3 materials.
References
Bagwell, R. B., Messing, G. L., and Howell, P. R. (2001). The formation of α-Al2O3 from θ-Al2O3: the relevance of a “critical size” and: diffusional nucleation or “synchro-shear”? J. Mater. Sci. 36, 1833–1841. doi: 10.1023/A:1017545213590
Barbero, B. P., Gamboa, J. A., and Cadus, L. E. (2006). Synthesis and characterization of La1−xCaxFeO3 perovskite-type oxide for total oxidaiton of volatile organic compound. Appl. Catal. B Environ. 65, 21–30. doi: 10.1016/j.apcatb.2005.11.018
Bourgeois, J., Hevieu, M., Poienar, M., Abakumov, A. M., Elkaim, E., Sougrati, M. T., et al. (2012). Evidence of oxygen-dependent modulation in LuFe2O4. Phys. Rev. B 85:064102. doi: 10.1103/PhysRevB.85.064102
Ciambelli, P., Cimino, S., Rossi, S., Lisi, L., Minelli, G., Porta, P., et al. (2001). AFeO3 (A = La, Nd, Sm) and LaFe1−xMgxO3 perovskites as methane combustion and CO oxidation catalysts: structual, redox and catalytic properties. Appl. Catal. B Environ. 29, 239–250. doi: 10.1016/S0926-3373(00)00215-0
Cushing, B. L., Kolesnichenko, V. L., and O'Connor, C. J. (2004). Recent advances in liquid-phase syntheses of inorganic nanoparticles. Chem. Rev. 104, 3893–3946. doi: 10.1021/cr030027b
Das, S., Chauduri, S., and Maji, S. (2008). Ethanol-water mediated solvothermal synthesis of cube and pyramid shaped nanostructured tin oxide. J. Phys. Chem. C 112, 6213–6219. doi: 10.1021/jp800612v
Demazeau, G. (1999). Solvothermal processes: a route to the stabilization of new materials. J. Mater. Chem. 9, 15–18. doi: 10.1039/a805536j
Dynys, F. W., and Halloran, J. W. (1982). Alpha-alumina formation in alum-derived gamma-alumina. J. Am. Ceram. Soc. 65, 442–448. doi: 10.1111/j.1151-2916.1982.tb10511.x
Fanelli, A. J., and Burlew, J. (1986). Preparation of fine alumina powder in alcohol. J. Am. Ceram. Soc. 69, C174–C175. doi: 10.1111/j.1151-2916.1986.tb04828.x
Guo, Y., Zhang, N., Huang, H., Li, Z., and Zou, Z. (2017). A novel wide-spectrum response hexagonal YFeO3 photoanode for solar water splitting. RSC Adv. 7, 18418–18420. doi: 10.1039/C6RA28390J
Hervieu, M., Guesdon, A., Bourgeois, J., Elkaim, E., Poienar, M., Damay, F., et al. (2014). Oxygen storage capacity and structural flexibility of LuFe2O4+x (0 ≤ x ≤ 0.5). Nat. Mater. 13, 74–80. doi: 10.1038/nmat3809
Hosokawa, S., Jeon, H.-J., and Inoue, M. (2011). Thermal stabilities of hexagonal and orthorhombic YbFeO3 synthesized by solvothermal method and their catalytic activities for methane combustion. Res. Chem. Intermed. 37, 291–296. doi: 10.1007/s11164-011-0251-9
Hosokawa, S., Jeon, H.-J., Iwamoto, S., and Inoue, M. (2009). Synthesis of rare earth iron-mixed oxide nanoparticles by solvothermal methods. J. Am. Ceram. Soc. 92, 2847–2853. doi: 10.1111/j.1551-2916.2009.03295.x
Hosokawa, S., Tada, R., Shibano, T., Matsumoto, S., Teramura, K., and Tanaka, T. (2016). Promoter effect of Pd species on Mn oxide catalysts supported on rare-earth-iron mixed oxide. Catal. Sci. Technol. 6, 7868–7874. doi: 10.1039/C6CY01462C
Inoue, M., Nishikawa, T., Nakamura, T., and Inui, T. (1997). Glycothermal reaction of rare-earth acetate and iron acetylacetonate: formation of hexagonal REFeO3. J. Am. Ceram. Soc. 80, 2157–2160. doi: 10.1111/j.1151-2916.1997.tb03103.x
Ismael, M., Elhaddad, E., Taffa, D., and Wark, M. (2017). Synthesis of phase pure hexagonal YFeO3 perovskite as efficient visible light active photocatalyst. Catalysts 7:326. doi: 10.3390/catal7110326
Kumar, M. S. V., Higaki, N., Kuribayashi, K., Hibiya, T., and Yoda, S. (2011). Formation of orthorhombic and multiferroic hexagonal phases from an undercooled RMnO3 (R = Rare-Earth Element) melt using a containerless technique. J. Am. Ceram. Soc. 94, 281–288. doi: 10.1111/j.1551-2916.2010.04042.x
Kumar, M. S. V., Kuribayashi, K., and Kitazono, K. (2009). Effect of oxygen partial pressure on the formation of metastable phases from an undercooled YbFeO3 melt using an aerodynamic levitator. J. Am. Ceram. Soc. 92, 903–910. doi: 10.1111/j.1551-2916.2009.02974.x
Kurzman, J. A., Li, J., Schladt, T. D., Parra, C. R., Ouyang, X., Davis, R., et al. (2011). Pd2+/Pd0 redox cycling in hexagonal YMn0.5Fe0.5O3: implications for catalysis by PGM-substituted complex oxides. Inorg. Chem. 50, 8073–8084. doi: 10.1021/ic200455a
Lafuerza, S., García, J., Subías, G., Blasco, J., and Cuartero, V. (2014). Strong local lattice instability in hexagonal ferrites RFe2O4 (R = Lu,Y,Yb) revealed by x-ray absorption spectroscopy. Phys. Rev. B 89:045129. doi: 10.1103/PhysRevB.89.045129
Li, C., Soh, K. C. K., and Wu, P. (2004). Formability of ABO3 perovskites. J. Alloys Compd. 372, 40–48. doi: 10.1016/j.jallcom.2003.10.017
Li, J., Singh, U. G., Schladt, T. D., Stalick, J. K., Scott, S. L., and Seshadri, R. (2008). Hexagonal YFe1−xPdxO3−δ: nonperovskite host compounds for Pd2+ and their catalytic activity for CO oxidation. Chem. Mater. 20, 6567–6576. doi: 10.1021/cm801534a
Lu, Y., Keav, S., Maegli, A. E., Weidenkaff, A., and Ferri, D. (2015). Pd loading and structure of flame-made Pd/YFeO3±δ. Top. Catal. 58, 910–918. doi: 10.1007/s11244-015-0459-9
Lu, Y., Keav, S., Marchionni, V., Chiarello, G. L., Pappacena, A., Di Michiel, M., et al. (2014a). Ageing induced improvement of methane oxidation activity of Pd/YFeO3. Catal. Sci. Technol. 4, 2919–2931. doi: 10.1039/C4CY00289J
Lu, Y., Michalow, K. A., Matam, S. K., Winkler, A., Maegli, A. E., Yoon, S., et al. (2014b). Methane abatement under stoichiometric conditions on perovskite-supported palladium catalysts prepared by flame spray synthesis. Appl. Catal. B Environ. 144, 631–643. doi: 10.1016/j.apcatb.2013.08.001
Magome, E., Moriyoshi, C., Kuroiwa, Y., Masuno, A., and Inoue, H. (2010). Noncentrosymmetric structure of LuFeO3 in metastable state. Jpn. J. Appl. Phys. 49:09ME06. doi: 10.1143/JJAP.49.09ME06
Malaman, B., Tanniere, N., Courtoas, A., and Protas, J. (1976). Structure cristalline de la phase Yb2Fe3O7. Acta Cryst. B 32, 749–752. doi: 10.1107/S0567740876003919
Matsui, Y., Kato, K., Kimizuka, N., and Horiuchi, S. (1979). Structure image of Yb3Fe4O10 by a 1 MV high-resolution electron microscope. Acta Cryst. B 35, 561–564. doi: 10.1107/S0567740879004155
Mizoguchi, Y., Onodera, H., Yamauchi, H., Kagawa, M., Syono, Y., and Hirai, T. (1996). Mossbauer spectra and magnetic susceptibilities of ultrafine hexagonal RFeO3 (R = Eu, Yb) particles formed by the spray inductively coupled plasma technique. Mater. Sci. Eng. A 217/218, 164–166. doi: 10.1016/S0921-5093(96)10339-7
Nishimura, T., Hosokawa, S., Masuda, Y., Wada, K., and Inoue, M. (2013). Synthesis of metastable rare-earth–iron mixed oxide with the hexagonal crystal structure. J. Solid State Chem. 197, 402–407. doi: 10.1016/j.jssc.2012.08.056
Qin, Y. B., Yang, H. X., Zhang, Y., Tian, H. F., Ma, C., Zhao, Y. G., et al. (2009). The effect of Mg doping on the structural and physical properties of LuFe2O4 and Lu2Fe3O7. J. Phys. Condens. Matter. 21:015401. doi: 10.1088/0953-8984/21/1/015401
Schneider, S. J., Roth, R. S., and Waring, J. L. (1961). Solid state reactions involving oxides of trivalent cations. J. Res. Nat. Bur. Stand. A 65, 345–374. doi: 10.6028/jres.065A.037
Vijaya Kumar, M. S., Nagashio, K., Hibiya, T., and Kuribayashi, K. (2008). Formation of hexagonal metastable phases from an undercooled LuFeO3 melt in an atmosphere with low oxygen partial pressure. J. Am. Ceram. Soc. 91, 806–812. doi: 10.1111/j.1551-2916.2007.02200.x
Woodward, P. M. (1997). Octahedral tilting in perovskites. II. structure stabilizing forces. Acta Cryst. B 53, 44–46.
Keywords: solvothermal method, coprecipitation method, catalyst, perovskite structure, hexagonal structure
Citation: Hosokawa S (2019) Hexagonal Rare Earth-Iron Mixed Oxides (REFeO3): Crystal Structure, Synthesis, and Catalytic Properties. Front. Chem. 7:8. doi: 10.3389/fchem.2019.00008
Received: 19 July 2018; Accepted: 07 January 2019;
Published: 28 January 2019.
Edited by:
Nobuhito Imanaka, Osaka University, JapanReviewed by:
Roberto Nisticò, Politecnico di Torino, ItalyTilo Söhnel, The University of Auckland, New Zealand
Shinji Tamura, Osaka University, Japan
Copyright © 2019 Hosokawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saburo Hosokawa, aG9zb2thd2FAc2NsLmt5b3RvLXUuYWMuanA=