 Qiaoyu Hu
Qiaoyu Hu Rajeev Prabhakar
Rajeev Prabhakar- Department of Chemistry, University of Miami, Coral Gables, FL, United States
In this study, mechanisms of phosphodiester hydrolysis catalyzed by six di- and tetravalent metal-cyclen (M-C) complexes (Zn-C, Cu-C, Co-C, Ce-C, Zr-C and Ti-C) have been investigated using DFT calculations. The activities of these complexes were studied using three distinct mechanisms: (1) direct attack (DA), (2) catalyst-assisted (CA), and (3) water-assisted (WA). All divalent metal complexes (Zn-C, Cu-C and Co-C) coordinated to the BNPP substrate in a monodentate fashion and activated its scissile phosphoester bond. However, all tetravalent metal complexes (Ce-C, Zr-C, and Ti-C) interacted with BNPP in a bidentate manner and strengthened this bond. The DA mechanism was energetically the most feasible for all divalent M-C complexes, while the WA mechanism was favored by the tetravalent complexes, except Ce-C. The divalent complexes were found to be more reactive than their tetravalent counterparts. Zn-C catalyzed the hydrolysis with the lowest barrier among all M-C complexes, while Ti-C was the most reactive tetravalent complex. The activities of Ce-C and Zr-C, except Ti-C, were improved with an increase in the coordination number of the metal ion. The structural and mechanistic information provided in this study will be very helpful in the development of more efficient metal complexes for this critical reaction.
Introduction
The phosphoester bond [(O=)(RO)(RO)(P-O-R)] is ubiquitous in a wide range of biomolecules such as proteins, nucleic acids, and lipids (Oivanen et al., 1998; Cleland and Hengge, 2006; Neidle, 2008; Kamerlin and Warshel, 2009). For instance, this bond constitutes the backbones of DNA and RNA by connecting the adjacent nucleotides (Sharp, 1985; Robinson et al., 1995; Mikkola et al., 2001; Chandra et al., 2009). It is also present in organophosphorus compounds (OPs) that have been utilized as pesticides and chemical nerve agents (Dubois, 1971; Jeyaratnam, 1990; The, 1998). Thus, the selective hydrolysis of this bond is required in numerous biological and biotechnological applications. In biology, this process has been implicated in DNA repair, post-translational modification of proteins and energy metabolism (Eichler and Lehman, 1977; Sancar and Sancar, 1988; Kia-Ki and Martinage, 1992; Mol et al., 2000). In biotechnology, it is involved in gene sequencing, therapeutics, and bioremediation of pesticides and nerve agents (Gewirtz et al., 1998; Eid et al., 2009; Corda et al., 2014). In nature, three types of phosphoester bonds exist: mono-, di-, and triester (Hadler et al., 2008; Kirby and Nome, 2015). Among them, the phosphodiester bond [(O=)(O−)(RO)(P-O-R)] is exceptionally stable with a half-life of approximately 3 × 107 years at room temperature and a neutral pH (Williams and Wyman, 2001). To hydrolyze this bond at biologically relevant rates, ~1016 times rate-acceleration, nature has devised highly specialized metalloenzymes known as nucleases and phosphoesterases (Wilcox, 1996; Cowan, 1998; Weston, 2005; Fang et al., 2007). Although these enzymes exhibit remarkable activities, they suffer from several limitations such as undesirable selectivity, difficulties in extraction or synthesis, high cost and narrow functional temperature, and pH range (Kövári and Krämer, 1996; Cowan, 2001; Mancin et al., 2012). Therefore, in the last couple of decades, intensive efforts have been made to design small metal complexes as synthetic analogs of natural enzymes for phosphoester hydrolysis (Burstyn and Deal, 1993; Hegg and Burstyn, 1998; Komiyama and Sumaoka, 1998; Blaskó and Bruice, 1999; Williams et al., 1999; Sreedhara and Cowan, 2001; Deck et al., 2002; Mitić et al., 2006; Niittymaki and Lonnberg, 2006; Bonomi et al., 2008; Krauser et al., 2010; Mancin et al., 2012; Daver et al., 2016; Sullivan et al., 2018). These analogs can offer multiple advantages over natural enzymes in terms of cost, size, and functionality (Weston, 2005; Yoji et al., 2006). To advance this goal, among others, several chemically distinct polyazamacrocyclic ligands were synthesized through the Stetter-Richman-Atkins method (Richman and Atkins, 1974; Weisman and Reed, 1996). In particular, 1,4,7,10-tetraazacyclododecane (cyclen, C) and its derivatives containing mononuclear metal complexes have been utilized for phosphodiester and peptide hydrolysis (Figure 1A) (Koike et al., 1994; Shionoya et al., 1994; Hettich and Schneider, 1997; Chae et al., 2005; Fang et al., 2007; Junghun et al., 2007; Subat et al., 2008; Zhang et al., 2014, 2016b). In most studies of phosphodiester hydrolysis, the bis(4-nitrophenyl) phosphate (BNPP) molecule has been used as a model of DNA (Figure 1A). Koike and Kimura (1991) investigated BNPP hydrolysis by the Zn(II)-cyclen (Zn-C) complex and reported the pseudo-first order rate constant of 2.8 × 10−9 s−1 at 35°C and pH 7. This complex provided a 46-fold rate acceleration compared to the background reaction. The Co(III)-cyclen based complexes (cyclen attached to polystyrene or methyl benzoate) also hydrolyzed the phosphodiester bond of DNA and RNA efficiently (Jeung et al., 2001; Delehanty et al., 2005). The polystyrene complex decreased the half-life of supercoiled DNA to 40 min at 4°C, while the one with methyl benzoate promoted the hydrolysis of ~96% of mRNA population within 24 h at 25°C. Furthermore, a Cu(II)-cyclen analog with two pyridine subunits was shown to degrade supercoiled DNA with kcat = 2.31 × 10−3 min−1 under physiological conditions (Li et al., 2007).
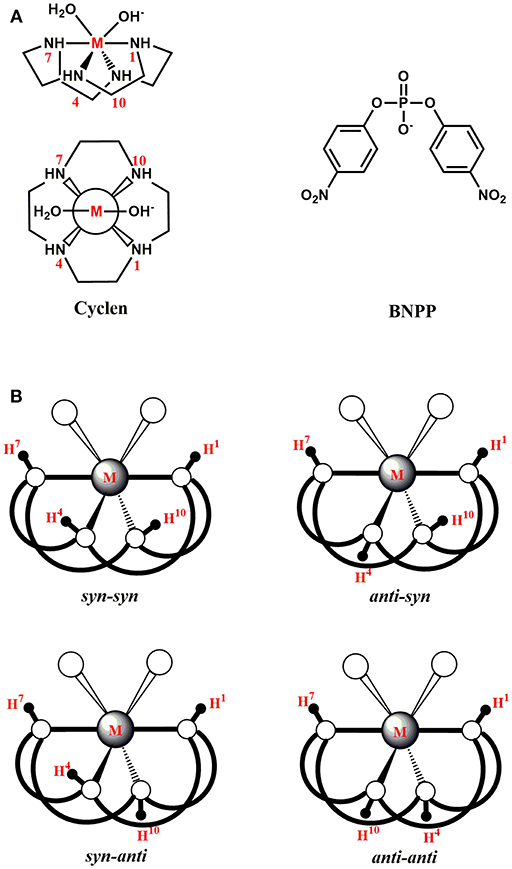
Figure 1. (A) Structures of a metal-cyclen (M-C) complex (side-view and top-view) and the BNPP substrate. (B) Different conformations of a M-C complex.
Additionally, several tri- and tetravalent lanthanides (Eu, La, Zr, and Ce) containing complexes have been reported to hydrolyze the phosphoester bond (Rammo et al., 1996; Baykal et al., 1999; Franklin, 2001; Gómez-Tagle and Yatsimirsky, 2001; Luedtke and Schepartz, 2005). Fanning et al. (2006) synthesized several cyclen based trivalent Eu(III) and La(III) complexes that hydrolyzed 2-hydroxypropyl 4-nitrophenyl phosphate (HPNP, an RNA model compound) within the physiological pH range. Furthermore, tetravalent Ce(IV) in an aqueous solution provided 1011 times rate-acceleration over the background reaction and 20–1000 times over the trivalent lanthanides for DNA hydrolysis (Komiyama et al., 1999). However, the exact nature of the active complexes in this reaction was not known. Nonetheless, based on the X-ray absorption fine structure data, remarkable activity of the Ce complex was proposed to be promoted by a weak covalent interaction between Ce(IV) and the phosphate group of the substrate (Hidemi et al., 1999).
In the proposed mechanism of phosphoester hydrolysis, the metal ion has been suggested to play the following key roles: (1) Lewis acid activation of the substrate, (2) creation of a nucleophile, and (3) generation of a good leaving group of the substrate (Chin, 1991; Bashkin and Jenkins, 1994; Fothergill et al., 1995; Williams et al., 1999; Das et al., 2013). Additionally, it stabilizes the transition states and intermediates by neutralizing their negative charges. To accomplish these functions, a metal ion should possess high Lewis acidity, strong nucleophilicity, redox stability, borderline hardness and low ligand field stabilization energy (Wilkinson et al., 1987; Hegg and Burstyn, 1998). However, an increase in its Lewis acidity causes a decrease in the nucleophilicity and these two effects require the right balance for the optimum reactivity (Koike and Kimura, 1991; Kimura et al., 1995; Bonfá et al., 2003; Coleman et al., 2010).
The metal-cyclen (M-C) complex can exist in equilibrium between several diastereoisomers (sys-syn, anti-syn, syn-anti, and anti-anti) associated with the orientation of protons (H4 or H10) on the nitrogen atoms of the cyclen macrocycle (Figure 1B; Hay and Norman, 1997). The H4 or H10 atoms face the substrate in the syn conformation, while they are located on the opposite side of the substrate in the anti conformation. The NMR data showed that syn-anti conformation of [Co(cyclen)Cl2]+ was more stable than other conformations (Sosa and Tobe, 1985). The X-ray structure of [Co(cyclen)(NO2)2]+ exhibited that this complex also existed in the syn-anti conformation (Iitaka et al., 1974). Additionally, the X-ray structures of both [Co(cyclen)(NH3)2]3+ and [Co(cyclen)(diamine)]3+ (diamine = H2N(CH2)2NH2, H2N(CH2)3NH2) complexes were crystallized in the syn-anti conformation (Clarkson et al., 2000). However, the exact conformation of a M-C complex has been proposed to depend on the nature of the metal ion (Zhang et al., 2014).
The experimentally proposed mechanism, termed direct attack (DA), utilized by metal complexes for the phosphodiester hydrolysis is shown in Figure 2A (Hendry and Sargeson, 1989; Komiyama et al., 1999; Mancin and Tecilla, 2007). In the initial form of the M-C complex (Ri), the metal ion is coordinated to the cyclen macrocycle, a hydroxyl ion and a water molecule (Kim et al., 2009). The protonation states of the hydroxyl ion and water molecule were based on the measured pKa values of the Co-C complex (pKa1 = 5.66 and pKa2 = 8.14) (Kim et al., 2009). According to this mechanism, from Ri, substitution of the metal-bound water molecule by the substrate creates an active complex (R). In the next step, a nucleophilic attack by the metal-bound hydroxyl group on the phosphorus center generates a five-membered phosphorane intermediate (ID). In the final step, the P-OR bond trans to the nucleophile is cleaved to form the final product (P).
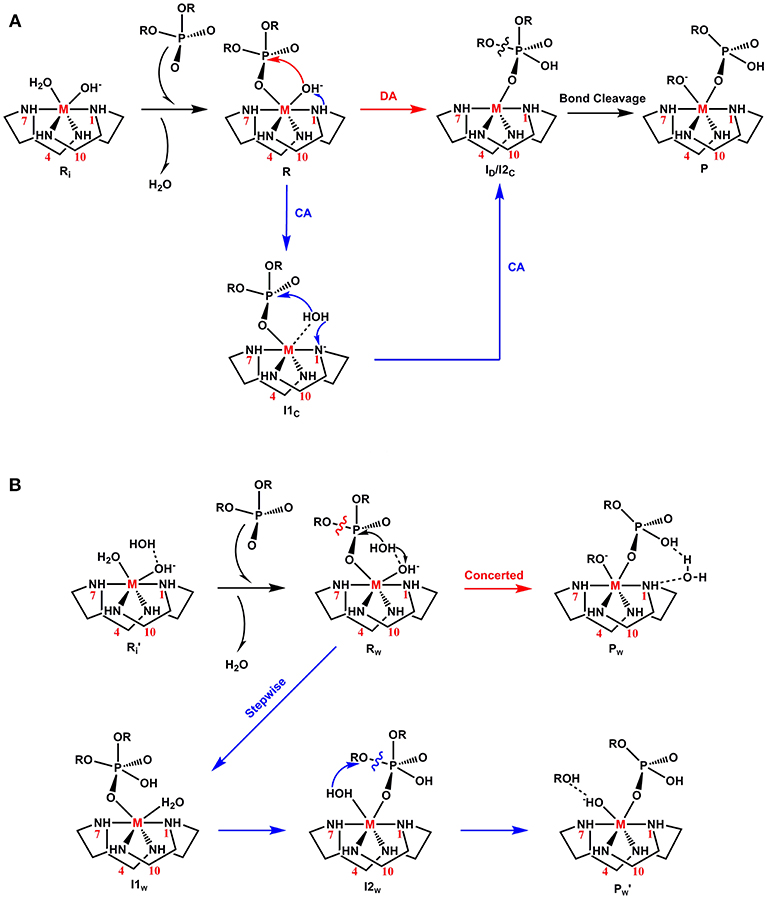
Figure 2. Proposed mechanisms for phosphoester hydrolysis: (A) Direct-attack (DA) and Catalyst-assisted (CA) and (B) Water-assisted (WA).
Recently, based on DFT calculations, another mechanism called catalyst-assisted (CA) was proposed for the Cu-C complex (Figure 2A; Zhang et al., 2016b). According to this mechanism, the metal-bound hydroxide functions as a base and abstracts a proton from the nitrogen atom (N1) of the cyclen to generate a water molecule (I1C). In the next step, the N1 atom acts as a base and accepts the previously donated proton. The hydroxide nucleophile created in this process attacks the electrophilic phosphorus atom of BNPP to form an intermediate (I2C). From I2C, the cleavage of the P-O bond can occur spontaneously and the charged leaving group (RO−) coordinates to the metal ion in the product (P).
Additionally, BNPP hydrolysis could occur through a third mechanism, termed a water assisted (WA) mechanism (Figure 2B; Dal Peraro et al., 2004; Jayasinghe-Arachchige et al., 2019). According to this mechanism, an external water molecule is employed for the nucleophilic attack and/or leaving group departure. After the formation of the reactant (Rw), the metal-bound hydroxide functions as a base and abstracts a proton from a solvent water molecule to generate a free nucleophile (-OH). This hydroxyl nucleophile concomitantly attacks the BNPP substrate. Depending on the nature of the metal ion (di- or tetravalent), this mechanism could also occur in a stepwise manner after this step. In this pathway, the metal-bound water molecule assists the cleavage of the phosphoester bond and creates a neutral leaving group (ROH).
Quite clearly, the metal-bound hydroxyl group play different roles in these mechanisms: (1) nucleophile only (DA mechanism), (2) both base and nucleophile (CA mechanism), and (3) base only (WA mechanism). The rate of this reaction is likely to depend on the stability of the rate-limiting transition state, which is connected with the Lewis acidity of the metal ion and the geometry of the metal-BNPP complex.
Despite the availability of a wealth of experimental and theoretical information, several unresolved issues concerning the exact mechanism, structures and roles of the metal ion still remain. For example, the conformation of the substrate bound M-C complexes (sys-syn, anti-syn, syn-anti, and anti-anti) for different metals (di- and tetravalent) is not known experimentally. The structures (transition states and short-lived intermediates) and energetics of the reaction mechanism for a specific metal ion are also not available. We have addressed all these important issues for a variety of M-C complexes using two sets of metal ions, divalent [Zn(II), Cu(II), and Co(II)] and tetravalent [Ce(IV), Zr(IV), and Ti(IV)], for BNPP hydrolysis through all three (DA, CA, and WA) mechanisms. The available experimental and theoretical information has been fully integrated in these calculations. These results will provide intricate details of the metal assisted phosphodiester hydrolysis and pave the way for the design of the next generation of synthetic metallohydrolases to catalyze this critical reaction.
Computational Details
Method
All Density Functional Theory (DFT) calculations were performed using the Gaussian 09 program package (Frisch et al., 2009). The geometry optimizations of reactants, transition states, intermediates and products were conducted using the B3LYP functional (Becke, 1988, 1993) without any constraints. Mixed basis sets were utilized for the structure optimization and frequency analysis. In particular, the Stuttgart relativistic effective core potential (ECP) basis set (RSC97) (Lee et al., 1988; Dolg et al., 1989) was applied for the metal ions. This is a double zeta basis set that uses 28 core electrons ([Ar]+3d) for the second-row transition metals and the lanthanides and 10 core electrons ([Ne]) for the first row transition metals. The 6-311G(d,p) basis set was used for the O, N and P atoms, while 6-31G was used for C and H atoms (Ditchfield et al., 1971). The final energies of the optimized structures were further improved by performing single point calculations using a bigger triple zeta quality 6-311+G(d,p) basis set for P, O, N, C and H atoms and RSC97 for metal ions. Hessians were calculated at the same level of theory as the optimizations to confirm the nature of the stationary points along the reaction coordinates. The transition states were confirmed to have only one negative eigenvalue corresponding to the reaction coordinates. The intrinsic reaction coordinate (IRC) approach (Ischtwan and Collins, 1988) that connects a transition state to the corresponding minima was utilized. The natural atomic charge for each atom was calculated by natural bond orbital (NBO) analysis using the NBO version 3 (Foster and Weinhold, 1980; Reed and Weinhold, 1983). Solvent effects for water (dielectric constant = 78.39) were calculated utilizing the polarizable continuum model (PCM) using the integral equation formalism variant (IEFPCM), which is a default self-consistent reaction field (SCRF) method (Cancès et al., 1997). The B3LYP energies were compared with the energies calculated using the M06L (Zhao and Truhlar, 2006), MPW1PW91 (Adamo and Barone, 1998) and PBE1PBE (Perdew et al., 1996) functionals. All energy barriers using these functionals were within 2.7–3.6 kcal/mol and provided similar potential energy surfaces (PES). The final energies computed at the B3LYP/6-311+G(d,p) level including zero-point vibrational (unscaled), thermal (298.15 K, 1 atm), entropy corrections (298.15 K) and solvent effects were used to discuss the activities of all M-C complexes. The measured kcat values were converted into activation energy using the Arrhenius equation (k = Ae−Ea/RT, where A is the pre-exponential factor, Ea is the activation energy, R is the gas constant and T is the temperature). It is noteworthy that calculations were performed at room temperature (25°C), while some kcat values were measured at a higher temperature. Due to the temperature dependence of the pre-exponential constant in the Arrhenius equation, it was not possible to accurately estimate the measured barrier at 25°C.
Models
In the calculations, the metal ion was coordinated to the 1,4,7,10-tetraazacyclododecane (cyclen) ligand, a hydroxyl ion and water molecules (Chin et al., 1989; Kim et al., 2009). The number of water molecules was determined by the chemical nature of the metal ion and the underlying mechanism. The overall charge for the Zn-C, Cu-C, and Co-C complexes was 0, while for Ce-C, Zr-C, and Ti-C the charge was +2. Cu-C and Co-C existed in the doublet spin state, while all the other M-C complexes existed in the singlet spin state. BNPP was used as the model of DNA because it contains two nitrophenyl groups which are similar to the deoxyribose rings of DNA.
Results and Discussion
The activities of all six M-C complexes (Zn-C, Cu-C, Co-C, Ce-C, Zr-C, and Ti-C) were investigated using three different mechanisms: (1) direct attack (DA), (2) catalyst-assisted (CA), and (3) water-assisted (WA), Figure 2. Their energetics were compared using the metal-ligand, metal-nucleophile and P-O bond lengths, strain of the cyclen ring, atomic charges and coordination number of metal ions as parameters. The Lewis acidity and nucleophilicity of the metal ions can be qualitatively characterized by the metal-substrate and metal-nucleophile bond lengths (Bertini et al., 1990; Coleman et al., 2010). In this section, the DA, CA and WA mechanisms were first discussed for the divalent metal complexes [Zn-C, Cu-C, and Co-C] followed by for the tetravalent complexes [Ce-C, Zr-C and Ti-C].
The starting point of all these mechanisms was the BNPP substrate bound structure of the M-C complexes. The syn-syn conformation was found to be the energetically most stable for all six metals (Figure 1B). The other conformations were 1.8–27.9 kcal/mol higher in energy. The relative stability of the syn-syn structure could be due to its lower strain computed as the sum of the N1-M-N7 and N4-M-N10 angles (Figure 1). This conformation possessed the largest angle (204.22–255.99°), and least strain, in comparison to the other three conformers. Additionally, the H4 and H10 atoms of the cyclen formed hydrogen bonds with the phosphate group of BNPP to provide extra stabilization to this conformation.
Phosphodiester Hydrolysis by Divalent Metal-Cyclen (M-C) Complexes
The divalent Zn, Cu, and Co ions are known to form stable complexes with a common coordination number of six for the phosphodiester and peptide hydrolysis (Holm et al., 1996; Berreau, 2006; Jang and Suh, 2008; Chei et al., 2011). Therefore, the BNPP substrate could only be singly coordinated to these metal ions (P-O-M mode), and the remaining coordination sites were occupied by four nitrogen atoms (N1, N4, N7, and N10) of the cyclen ligand and one hydroxyl group. In this section, for the sake of clarity, all three mechanisms for Zn-C were discussed in detail followed by comparisons with the Cu-C and Co-C complexes.
Direct Attack (DA) Mechanism
In the reactant (RZn in Figure 3) of Zn-C, one phosphoryl oxygen (O1) atom of BNPP was bound to the Zn(II) ion, while the other one (O2) interacted with the -N10H group of the cyclen through a hydrogen bond. This metal-substrate coordination elongated the scissile P-O4 bond of BNPP by 0.04 Å in comparison to this bond in its free form (P-O4 = 1.64 Å in Figure 3). In RZn, the Lewis acidity of the Zn ion played a key role in the activation of the P-O4 bond. The interaction between the p orbital of the oxygen atom with the d orbital of the Zn atom promoted this activation (Figure S1). In the first step, the Zn-bound -OHHH nucleophile directly attacked the electrophilic P atom of the substrate to generate a five-membered trigonal bipyramidal phosphorane intermediate (). This process occurred with a barrier of 20.7 kcal/mol and in the optimized transition state () the Zn-OHHH and P-O4 bonds, trans to the nucleophile, became substantially longer by 0.55 and 0.09 Å, respectively (Figure 3 and Table S1). was endergonic by 15.5 kcal/mol from RZn and the P-O4 bond was significantly activated but not completely broken in this intermediate (P-O4 = 1.82 Å). However, this bond was cleaved in the next step and the nitrophenolate (-OC6H4NO2) group was released. In the transition state () for this process, the P-O4 bond was significantly elongated to 1.97 Å (Figure 3). The negatively charged nitrophenolate group generated in this process was coordinated to the Zn ion in the product (PZn). PZn was computed to be exergonic by 19.2 kcal/mol from RZn. The overall barrier (20.7 kcal/mol) for this mechanism was somewhat underestimated in comparison to the measured barrier of 29.3 kcal/mol (computed from the kcat value using the Arrhenius equation) for BNPP hydrolysis by Zn-C (Koike and Kimura, 1991).
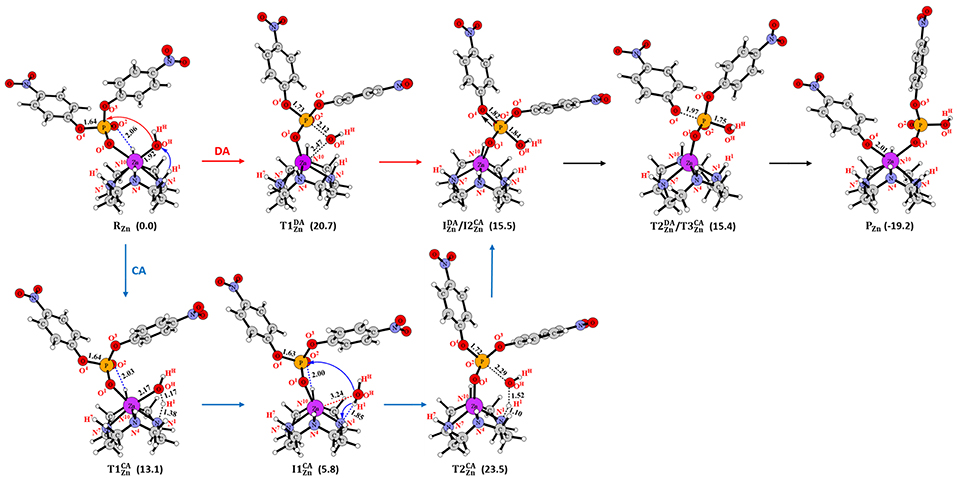
Figure 3. Structures (in Å) and energies (in kcal/mol) in the DA and CA mechanisms for Zn-C.
The overall energetics of this mechanism for Cu-C and Co-C were quite comparable to that of Zn-C (Figures S2, S3). However, the barrier (22.0 and 23.4 kcal/mol) and endergonicity (17.7 and 17.2 kcal/mol) of the rate-limiting first step were slightly higher for Cu-C and Co-C, respectively, in comparison to Zn-C. This difference could be due to the greater strength of the Zn-bound nucleophile in RZn i.e., longer Zn-OH distance and higher charge on the OH atom (Tables S1, S2).
These results suggested that both Lewis acidity and nucleophilicity of the metal center controlled the energetics of this mechanism. Zn-C was found to be slightly more reactive than the Cu-C and Co-C complexes.
Catalyst-Assisted (CA) Mechanism
The catalyst-assisted (CA) mechanism (Figure 3; Zhang et al., 2016b) started with the same reactant (RZn) as the DA mechanism. In the first step, the Zn-bound hydroxyl group of RZn functioned as a base and abstracted the H1 proton from the cyclen ring to form a water molecule (OHHHH1). This process occurred with a barrier of 13.1 kcal/mol and the Zn-OH bond was extended by 0.25 Å in the optimized transition state () in comparison to the corresponding distance in RZn. The intermediate () formed in this process was 5.8 kcal/mol endergonic from RZn (Figure 3). In , the water molecule was not coordinated to the Zn ion and associated with the N1 atom of the cyclen ligand through a hydrogen bond. The strain of the cyclen ring and the acidity of the -N1H1 group controlled the energetics of this step. The sum of the N1-M-N7 and N4-M-N10 angles (238.6° in RZn) was increased by 15.3° in (253.9°) i.e., less strain in . However, a high charge (−0.77e) on the N1 atom of the cyclen ligand also lowered the acidity of the H1 atom. From , the cyclen ligand directly participated in the mechanism by creating a nucleophile and regenerating the ligand through activation of the previously generated water molecule (OHHHH1 in Figure 3). In this step, the N1 atom of the ligand abstracted the H1 proton concomitantly with the attack of the -OHHH nucleophile to the electrophilic P atom of BNPP. This concerted process, through transition state (), led to the creation of the phosphorane intermediate () that was endergonic by 15.5 kcal/mol (Figure 3). From RZn, this step occurred with a barrier of 23.5 kcal/mol and found to be the rate-limiting step of the CA mechanism. At , both DA and CA mechanisms merged and led to the generation of the common product (PZn in Figure 3).
The structures of the reactants for Cu-C and Co-C (RCu and RCo, respectively) were quite similar to RZn (Figures S2, S3). However, the barrier of the first step for Cu-C and Co-C was lowered by 1.4 and 4.4 kcal/mol, respectively, in comparison to the barrier for Zn-C. Additionally, the phosphorane intermediate for these systems was found to be more stable by 4.8 and 4.3 kcal/mol, respectively (Figures S2, S3). These energy differences were likely to be caused by lower strain of the cyclen ring i.e., 238.6°, 245.0°, and 256.0° For RZn, RCu, and RCo, respectively. Additionally, a lower charge on the N1 atom increased the acidity of the -N1H1 group of Cu-C and Co-C and made this process energetically more favorable (Table S2). The computed barrier of the next rate-determining step for Cu-C and Co-C (24.3 and 24.6 kcal/mol, respectively) was slightly higher than the barrier for Zn-C (23.5 kcal/mol). Here, due to the higher basicity of the N1 atom in Zn-C, the proton transfer occurred with a lower barrier.
These results suggested that the CA mechanism was energetically less favorable than the DA mechanism for all divalent metals. Due to the direct involvement of the cyclen ligand, the strain of the cyclen ring and acidity of the N1H1 group determined the energetics of the CA mechanism. Similar to the DA mechanism, Zn-C was more reactive than Cu-C and Co-C.
Water-Assisted (WA) Mechanism
The major difference between the DA and WA mechanisms is that in the latter, the metal-bound hydroxide played the role of a base and created a nucleophile through activation of an external water molecule (Figure 2B). In the reactant () of Zn-C, the Zn-bound hydroxyl group (-OHHH) interacted with an external water molecule (HOwHw) through a hydrogen bond (Figure 4A). This interaction elongated the Zn-OHHH bond by 0.05 Å in comparison to RZn. From , the Zn-bound hydroxyl (-OHHH) abstracted the Hw proton of the external water molecule and generated the free -OwH nucleophile that concomitantly attacked the BNPP substrate and cleaved the P-O4 bond. This concerted process occurred through transition state () with a barrier of 20.9 kcal/mol (Figure 4A and Table 1). In the product (), the negatively charged -OC6H4NO2 group created by the nucleophilic attack coordinated to the Zn ion with the release of the water molecule (HwOHHH). was 13.9 kcal/mol exergonic from . The strength of the hydroxyl nucleophile generated from an external water was weaker than that of a metal-bound nucleophile. However, quite surprisingly, the barrier for the WA mechanism (20.9 kcal/mol) was quite comparable to the one computed for the DA mechanism (20.7 kcal/mol). That could be due to the extra stability of the five-membered transition state () formed in the former, in comparison to the four-membered transition state () created in the latter. For Cu-C and Co-C the barrier for the WA mechanism was also slightly higher by 2.2 and 1.0 kcal/mol, respectively, than for the DA mechanism (Figures 4B,C). However, the barrier for the WA mechanism for Cu-C and Co-C was 3.3 and 3.5 kcal/mol, respectively higher than the barrier for Zn-C i.e., 24.2 and 24.4 kcal/mol, respectively (Figure 4). This difference was likely to be due to the stronger basicity of the Zn-OHHH group among all three complexes. It was caused by the longer M-OH distance and higher charge on the OH atom in Zn-C (Tables S1, S2). Additionally, in contrast to the singlet spin state of Zn in Zn-C, both Cu and Co existed in the doublet spin state in Cu-C and Co-C. That might also be the reason for the similar energetics of Cu-C and Co-C.
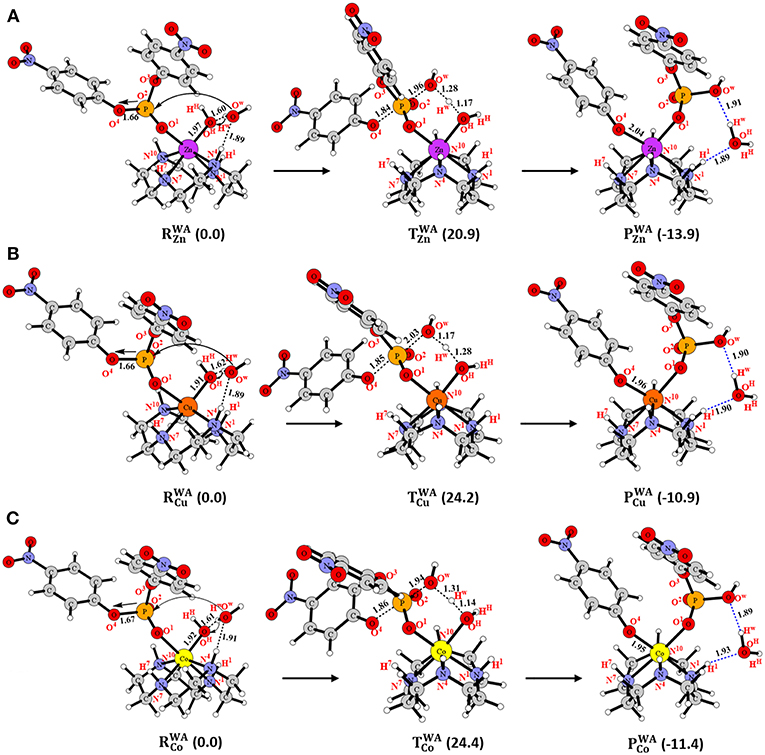
Figure 4. Structures (in Å) and energies (in kcal/mol) in the WA mechanism for (A) Zn-C, (B) Cu-C and (C) Co-C.
According to these results, the basicity of the M-OH group influenced the energetics of the WA mechanism. Additionally, energetics of all three mechanisms (DA, CA, and WA) were quite comparable for all divalent metal complexes. Furthermore, Zn-C was found to be more reactive than Cu-C and Co-C for all three mechanisms.
Phosphodiester Hydrolysis by Tetravalent Metal-Cyclen (M-C) Complexes
The tetravalent metals (Ce, Zr, and Ti) form complexes with higher coordination numbers 6–12, than the divalent metals (Zn, Cu, and Co) with coordination numbers 5–6. Among tetravalent metals, Ce can form complexes with coordination numbers 7–12, while Zr and Ti with 6–8 (Komiyama et al., 1999; Bonomi et al., 2010). Here, due to the difference in their coordination number, all three DA, CA, and WA mechanisms are first discussed for Ce-C, followed by for Zr-C and Ti-C.
Direct Attack (DA) Mechanism for the Ce-C Complex
The activity of Ce-C was studied using three different coordination numbers (7–9). Due to the steric hindrance, complexes with higher coordination numbers (10–12) could not be optimized. In the reactant (RCe) with coordination number 7, BNPP was coordinated to the Ce ion through the O1 and O2 atoms in the bidentate form. In contrast, BNPP binding to the divalent metals occurred in the monodentate fashion. As a result, the scissile P-O4 bond became stronger by 0.03 Å in Ce-C (Figure S5). In RCe, all metal-ligand distances (Table S3) were significantly longer than those in the reactant of Zn-C (Table S1). The excellent hydrolytic activity of the Ce ion in aqueous solution was reported to be due to the hybridization of the 4f orbitals of Ce with the 2p orbitals of the coordinated oxygen atoms of the substrate (Komiyama, 2016). However, Ce can form complexes with different coordination numbers in the solution, and the actual active complex in the previous study was not known. Here, substrate-bound mononuclear Ce-C complex was not found to activate the P-O4 bond (Figure S4). The direct nucleophilic attack of the metal-bound hydroxide (-OHHH) to BNPP occurred with a barrier of 39.1 kcal/mol (Figure S5), which was almost twice the barrier computed for Zn-C. The reason for this significantly higher barrier was the change of Ce-C from a hepta-coordinated (coordination number 7) RCe to an unfavorable hexa-coordinated (coordination number 6) . From , the P-O4 bond was completely broken with a small barrier of 2.1 kcal/mol and the separated nitrophenolate (-OC6H4NO2) and phosphate [-(O)2P(OC6H4NO2)OH] groups were coordinated to the Ce(IV) ion in the product (PCe in Figure S5).
This mechanism was further studied by including extra water as a ligand (coordination number of Ce = 8). It was also suggested in our previous study (Zhang et al., 2017) that an increased coordination number of the metal ion enhanced the peptidase activity of the Zr(IV) azacrown ether complex [Zr-(NO2)(OHH)(H2O)n]. In the reactant (R'Ce) with coordination number 8 (Figure S6), the additional Ce-bound water formed a hydrogen bond with BNPP. From R'Ce, the barrier for the nucleophilic attack in the rate-determining first step was lowered slightly by 1.4 kcal/mol i.e., 37.7 kcal/mol from R'Ce. The inclusion of the second water molecule (coordination number of Ce = 9) further lowered the barrier for this step by 1.5 kcal/mol i.e., 36.2 kcal/mol from the reactant (R”Ce in Figure S7). This slight reduction of barrier upon increasing the coordination number of Ce (7–9) could be caused by a slight increase in the metal-nucleophile (Ce-OH) distance and a decrease in the charge on the Ce atom (Tables S3, S4). This indicates the provision of a stronger nucleophile in the complexes with a higher coordination number.
These results suggested that, in comparison to the divalent metals, the lower activity of all Ce-C complexes with different coordination numbers (7–9) was caused by the strengthening of the scissile phosphoester bond and weaker nucleophilicity of the hydroxyl nucleophile. However, the reactivity of the Ce-C complex was slightly enhanced with an increase in the coordination number (7–9) of the Ce ion.
Catalyst-Assisted (CA) Mechanism for the Ce-C Complex
In this mechanism, from RCe, an abstraction of the H1 proton of the cyclen ring by the Ce-bound hydroxyl (-OHHH) took place with a barrier of 26.9 kcal/mol (Figure S5). Similar to the DA mechanism, the barrier for this process was 13.8 kcal/mol higher than the barrier for Zn-C (Figure 3). The metal-nucleophile (Ce-OH) distance was significantly elongated from 2.04 Å to 2.33 and 2.50 Å in the transition state () and the intermediate (), respectively, in comparison to RCe. In the next step, the reverse transfer of the H1 proton from the Ce-bound water molecule (HHOHH1) to the N1 atom of the cyclen and simultaneous nucleophilic attack of the hydroxide (-OHHH) to BNPP generated a phosphorane intermediate (). This synchronous process occurred in the rate-limiting step with a barrier of 46.8 kcal/mol and was endergonic by 35.5 kcal/mol from RCe. After its formation, the product (PCe) was generated through the cleavage of the P-O4 bond (Figure S5).
The inclusion of an additional water molecule to this complex increased the coordination number of Ce to 8 and lowered the barrier for the rate-limiting step by 2.3 kcal/mol i.e., 44.5 kcal/mol from the corresponding reactant (R'Ce in Figure S6). The inclusion of the second water molecule (coordination number of Ce = 9) further lowered the barrier only by 0.5 kcal/mol i.e., 44.0 kcal/mol from the reactant (R”Ce in Figure S7). This lowering of the barrier could be attributed to an increase in the Ce-OHHH (nucleophile) distance (Table S3) and a reduction in charge of the Ce atom (Table S4).
Similar to divalent metal-complexes, the CA mechanism was found to be energetically less favorable than the DA mechanism and the energetics of this mechanism improved slightly with an increase in the coordination number of Ce.
Water-Assisted (WA) Mechanism for the Ce-C Complex
The reactant () of the WA mechanism was similar to RCe, except for an external water molecule that was hydrogen bonded to the Ce-bound hydroxyl (-OHHH) and BNPP (Figure S8). In the first step the nucleophile (-O1wH), generated through the abstraction of a proton (H1w) by the Ce-OHHH group simultaneously attacked BNPP. This process took place with a barrier of 43.5 kcal/mol and led to the creation of the phosphorane intermediate (). In , the scissile P-O4 bond was substantially activated to 1.81 Å but not completely broken. In the next step, this bond was cleaved with a small barrier of 2.2 kcal/mol i.e., 44.3 kcal/mol from . The nitrophenolate group bound product () was computed to be exergonic by 4.7 kcal/mol from .
As observed previously, the inclusion of an additional water molecule (coordination number of Ce = 8) lowered the barrier for the rate-limiting second step by 2.8 kcal/mol i.e., 41.5 kcal/mol from the corresponding reactant (, Figure S9). The addition of the second water molecule (coordination number of Ce = 9) further lowered this barrier by 4.5 kcal/mol i.e., 37.0 kcal/mol from the reactant ( in Figure S10). This lowering in the barrier (44.3 > 41.5 > 37.0 kcal/mol) with an increase in the coordination number of Ce was due to the following factors. possessed the longest Ce-OH distance (2.05 Å) followed by (1.99 Å) and (1.98 Å), Table S3. Additionally, the charge on the Ce atom followed the order > > (Table S4). These differences indicated that the basicity of the metal-bound hydroxide in was greater than its basicity in and .
The reactivity of Ce-C was enhanced with an increase in the coordination number (7-9) of the Ce atom. The CA mechanism was substantially less favorable, and the WA and DA mechanisms were quiet comparable for Ce-C. Based on these results, the CA mechanism was not explored for Zr-C and Ti-C in the next section.
Direct Attack (DA) Mechanism for the Zr-C and Ti-C Complexes
Zr and Ti have been reported to prefer different coordination numbers i.e., 8 and 7, respectively (Luong et al., 2016; Zhang et al., 2016a; Assi et al., 2017). In the reactant (RZr) with coordination number 8, an external water molecule was directly coordinated to the Zr ion (Figure S11). In comparison to R'Ce (the reactant of Ce-C with the same coordination number), all metal-ligand distances, except P-O4, were substantially shorter in RZr (Tables S3, S5). From RZr, the Zr bound -OHHH nucleophile attacked BNPP with a barrier of 40.3 kcal/mol, which was 2.6 kcal/mol higher than the one (37.7 kcal/mol) computed for Ce-C. A weaker nucleophile (shorter Zr-OH distance by 0.05 Å) and increase in the charge of Zr (by 0.15e) raised the barrier for this step. The intermediate () formed in this step was endergonic by 28.3 kcal/mol from RZr. However, the P-O4 bond in was substantially stronger (by 0.12 Å) than in the Ce-C case (Figures S11, S6). Due to the extra stability of this bond, unlike the mechanism for Ce-C, a complete cleavage of this bond required the assistance of metal-bound water in the next step. From , the Zr-bound water donated a proton to the O4 atom and cleaved the P-O4 bond. The splitting of this bond occurred with a barrier of 8.8 kcal/mol from i.e., 37.1 kcal/mol from RZr (Figure S11). In the product (), the neutral nitrophenol group was hydrogen bonded to the Zr-hydroxyl moiety and it was exergonic by 9.9 kcal/mol. The removal of a water ligand from this complex (coordination number of Zr = 7) raised the barrier for the rate-limiting first step by 2.4 kcal/mol (Figure S12).
Since Ti prefers coordination number 7, the geometry of the reactant (RTi) of Ti-C was different from the reactant of Zr-C. In RTi (Figure S13), an external water molecule, instead of directly coordinating to the metal ion, was bridged through hydrogen bonding between the cyclen ring and BNPP. All metal-ligand distances in RTi were substantially shorter than the corresponding distances in RZr (Table S5), while the P-O4 bond distance (P-O4 = 1.57 Å) remained unchanged. From RTi, the nucleophilic attack took place with a barrier of 39.0 kcal/mol. This barrier was slightly (1.3 kcal/mol) lower than the one computed for Zr-C. The intermediate () formed in this step was endergonic by 24.9 kcal/mol from RTi. The cleavage of the P-O4 bond using the Ti-bound water molecule took place with a barrier of 11.5 kcal/mol from i.e., 36.4 kcal/mol from RTi (Figure S13). The product () in which the neutral nitrophenol group was associated with the metal-bound hydroxyl through hydrogen bonding was exergonic by 10.7 kcal/mol. The inclusion of a water ligand to this complex (coordination number of Ti = 8) raised the barrier for the rate-limiting step by 9.0 kcal/mol i.e., 48.0 kcal/mol from the corresponding reactant (Figure S14).
Zr-C and Ti-C showed higher activity with different coordination numbers i.e., 8 and 7 for Zr and Ti, respectively. They also required assistance of an external water, unlike Ce-C, for the complete cleavage of the P-O bond. However, both Zr-C (coordination number = 8) and Ti-C (coordination number = 7) were found to be less active than Ce-C (with coordination number 9) for the DA mechanism (Table 1).
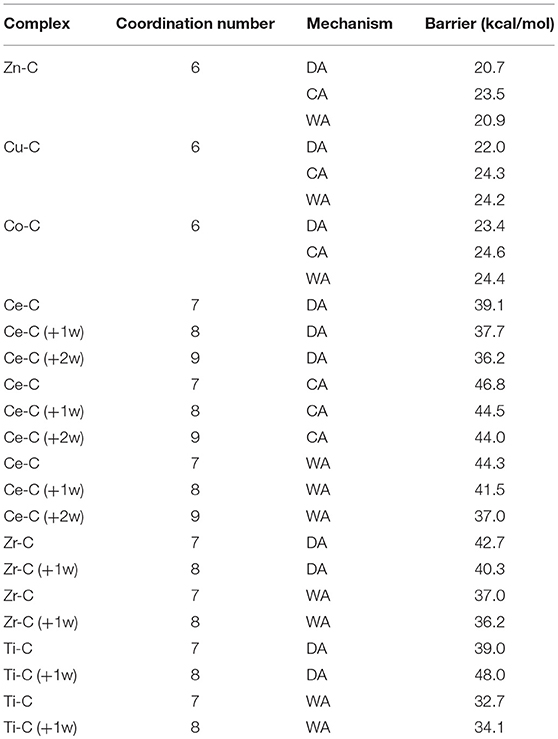
Table 1. Computed energy barrier in the rate-limiting step for all M-C complexes.
Water-Assisted (WA) Mechanism for the Zr-C and Ti-C Complexes
In the reactant () of Zr-C, an external water molecule was hydrogen bonded between the Zr-OHHH and BNPP (Figure S15). In this mechanism, the Zr-bound hydroxyl functioned as a base and created a hydroxyl (-O1wH1w) nucleophile from the external water molecule that concomitantly attacked the electrophilic P atom of BNPP. This synchronous step took place with a barrier of 28.8 kcal/mol (Figure S15). The barrier for this step was significantly (6.0 kcal/mol) lower than the barrier for Ce-C. The intermediate () formed in this step was 22.3 kcal/mol endergonic from . As observed for the previous DA mechanism, the scissile P-O4 bond in was activated but still quite strong (1.62 Å) i.e., 0.13 Å stronger than for Ce-C. The complete cleavage of this bond also needed the assistance of a metal-bound water molecule in the next step. The intermediate reoriented itself and created another 3.0 kcal/mol endergonic intermediate () in which the water molecule was located in a position to protonate the O4 atom of BNPP. From , this water molecule donated its proton and cleaved the P-O4 bond with a barrier of 10.9 kcal/mol. The final product () was 33.6 kcal/mol exergonic from (Figure S15). The removal of a water molecule in this complex (coordination number of Zr = 7) slightly raised the rate-limiting barrier by 0.8 kcal/mol (Figure S16).
The reactant () of Ti-C (for coordination number of Ti = 7) was structurally similar to (Figure S17). However, all metal-ligand distances in the former were shorter than the corresponding distances in the latter (Table S5). From , proton abstraction from the external water molecule occurred with a barrier of 32.7 kcal/mol (Figure S17). The barrier in this nucleophilic attack step was 3.9 kcal/mol higher than the one computed for Zr-C. As discussed previously, this increase was due to a shorter M-OH bond distance (by 0.19 Å) and lower charge on the OH atom (by 0.22e) in (Tables S5, S6). However, the phosphorane intermediate () in this step was 7.2 kcal/mol more favorable than in the Zr-C case. i.e., 15.1 kcal/mol endergonic from . The presence of a stronger hydrogen bond provided extra stability to this complex. Similar to Zr-C, the newly formed Ti-bound water molecule in reoriented itself between the cyclen ring and BNPP to create another intermediate . This intermediate was 5.7 kcal/mol higher in energy than . From , the transfer of the H1w proton to the O4 atom led to the splitting of the P-O4 bond. This process took place with a barrier of 11.5 kcal/mol from . In the product (), the released nitrophenol group was hydrogen bonded to the metal-bound hydroxyl and it was 14.8 kcal/mol exergonic from . The addition of a water molecule in this complex (coordination number of Ti = 8) slightly increased the barrier in the rate-limiting step by 1.4 kcal/mol (Figure S18).
These results suggest that the WA mechanism was energetically more favorable than the DA mechanism for both Zr-C and Ti-C. Among these two complexes, Ti-C was found to be more reactive than Zr-C.
Conclusions
In this DFT study, phosphodiester hydrolysis by metal-cyclen (M-C) complexes using both divalent [Zn(II), Cu(II) and Co(II)] and tetravalent [Ce(IV), Zr(IV), and Ti(IV)] metals were investigated. The reactivities of all six M-C complexes (Zn-C, Cu-C, Co-C, Ce-C, Zr-C and Ti-C) for BNPP hydrolysis were studied using three different mechanisms: (1) direct attack (DA), (2) catalyst-assisted (CA), and (3) water-assisted (WA). Their energetics were compared using the metal-ligand, metal-nucleophile and P-O bond lengths, strain of the cyclen ring, atomic charges and coordination number of metal ions as parameters. The potential energy surface diagrams (PES) of all these mechanisms for the divalent and tetravalent complexes are shown in Figures 5, 6, respectively.
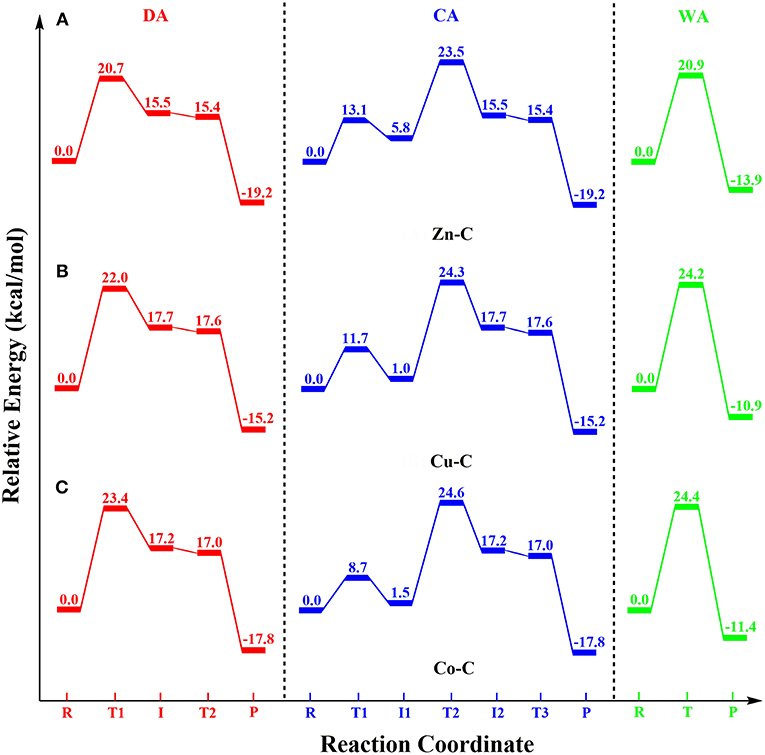
Figure 5. Potential energy surface diagrams for the divalent M-C complexes: (A) Zn-C, (B) Cu-C and (C) Co-C.
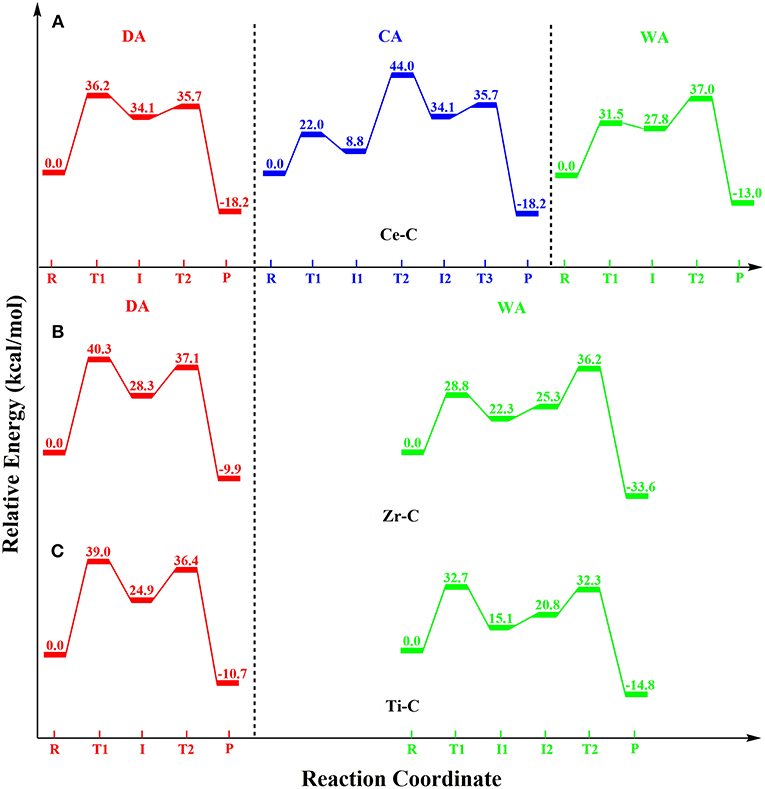
Figure 6. Potential energy surface diagrams for the tetravalent M-C complexes: (A) Ce-C (coordination number = 9), (B) Zr-C (coordination number = 8), and (C) Ti-C (coordination number = 7).
For all divalent metal complexes (Zn-C, Cu-C, and Co-C), the binding of the BNPP substrate in the monodentate fashion activated its scissile phosphoester bond (P-O4) by ~0.04 Å. Their energetics were controlled by distinct chemical factors: nucleophilicity of the metal center in the DA mechanism; basicity of the N1 atom of the cyclen ring in the CA mechanism; and basicity of the metal bound hydroxyl group in the WA mechanism. The DA mechanism was found to be energetically most favorable for all these complexes. Among the divalent complexes, Zn-C was more reactive than Cu-C and Co-C for all three mechanisms (Figure 5, Table 1).
On the other hand, the binding of BNPP to the tetravalent metal complexes (Ce-C, Zr-C, and Ti-C) in the bidentate manner strengthened its P-O4 bond by ~0.03 Å. The computed barriers for these complexes were substantially higher than the barriers for their divalent counterparts for all three mechanisms (Table 1). Unlike the DA mechanism for divalent M-C complexes, the WA mechanism was energetically most favorable for Zr-C and Ti-C. On the other hand, energetics of both DA and WA mechanisms were comparable for Ce-C. The activities of Ce-C and Zr-C improved with an increase in the coordination number (7-9) of the metal ion for all three mechanisms, while Ti-C exhibited the opposite trend (Table 1). In comparison to Ce-C, both Zr-C and Ti-C required additional assistance for the complete cleavage of the P-O4 bond. Ce-C exhibited the highest activity with a coordination number of Ce = 9, Zr-C with a coordination number of Zr = 8 and Ti-C with a coordination number of Ti = 7. However, among all tetravalent complexes, Ti-C was found to be the most reactive (barrier = 32.7 kcal/mol using the WA mechanism) followed by Ce-C and Zr-C (Figure 6, Table 1).
These results have provided detailed structural, mechanistic and kinetic information regarding the activities of a wide range of M-C complexes. They will pave the way for the design of efficient synthetic metallohydrolases for applications in biology, biotechnology and medicine.
Author Contributions
QH performed most of the DFT calculations and analyzed them. He also wrote the first draft of the manuscript and made figures and tables. VJ-A performed some DFT calculations and analyzed them. She also helped with the writing of the draft. JZ started the project and performed initial DFT calculations. RP designed and supervised the project. He also analyzed the results and edited the manuscript.
Funding
This material is based upon work supported by the grant from the National Science Foundation (Grant Number CHE-1664926) to RP.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Computational resources from the Center for Computational Science (CCS) at the University of Miami are greatly acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00195/full#supplementary-material
References
Adamo, C., and Barone, V. (1998). Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: the mPW and mPW1PW models. J. Chem. Phys. 108, 664–675. doi: 10.1063/1.475428
Assi, H., Mouchaham, G., Steunou, N., Devic, T., and Serre, C. (2017). Titanium coordination compounds: from discrete metal complexes to metal–organic frameworks. Chem. Soc. Rev. 46, 3431–3452. doi: 10.1039/C7CS00001D
Bashkin, J. K., and Jenkins, L. A. (1994). The role of metals in the hydrolytic cleavage of DNA and RNA. Comments Inorg. Chem. 16, 77–93. doi: 10.1080/02603599408035852
Baykal, U., Akkaya, M. S., and Akkaya, E. U. (1999). A novel lanthanide complex with remarkable phosphodiester transesterification activity and DNA-conjugatable functionality. J. Incl. Phenom. Macrocycl. Chem. 35, 311–315. doi: 10.1023/A:1008111418731
Becke, A. D. (1988). Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 38, 3098–3100. doi: 10.1103/PhysRevA.38.3098
Becke, A. D. (1993). Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. doi: 10.1063/1.464913
Berreau, L. M. (2006). “Kinetic and mechanistic studies of the reactivity of Zn–OHn (n = 1 or 2) species in small molecule analogs of zinc-containing metalloenzymes,” in Advances in Physical Organic Chemistry, ed J. P. Richard (Cambridge, MA: Academic Press), 79–181.
Bertini, I., Luchinat, C., Rosi, M., Sgamellotti, A., and Tarantelli, F. (1990). pKa of zinc-bound water and nucleophilicity of hydroxo-containing species. Ab initio calculations on models for zinc enzymes. Inorg. Chem. 29, 1460–1463. doi: 10.1021/ic00333a004
Blaskó, A., and Bruice, T. C. (1999). Recent studies of nucleophilic, general-acid, and metal ion catalysis of phosphate diester hydrolysis. Acc. Chem. Res. 32, 475–484. doi: 10.1021/ar980060y
Bonfá, L., Gatos, M., Mancin, F., Tecilla, P., and Tonellato, U. (2003). The ligand effect on the hydrolytic reactivity of Zn(II) complexes toward phosphate diesters. Inorg. Chem. 42, 3943–3949. doi: 10.1021/ic034139x
Bonomi, R., Scrimin, P., and Mancin, F. (2010). Phosphate diesters cleavage mediated by Ce(IV) complexes self-assembled on gold nanoparticles. Org. Biomol. Chem. 8, 2622–2626. doi: 10.1039/b926916a
Bonomi, R., Selvestrel, F., Lombardo, V., Sissi, C., Polizzi, S., Mancin, F., et al. (2008). Phosphate Diester and DNA hydrolysis by a multivalent, nanoparticle-based catalyst. J. Am. Chem. Soc. 130, 15744–15745. doi: 10.1021/ja801794t
Burstyn, J. N., and Deal, K. A. (1993). Selective catalytic hydrolysis of a simple phosphodiester by a macrocyclic copper(II) complex. Inorg. Chem. 32, 3585–3586. doi: 10.1021/ic00069a005
Cancès, E., Mennucci, B., and Tomasi, J. (1997). A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 107, 3032–3041. doi: 10.1063/1.474659
Chae, P. S., Kim, M.-S., Jeung, C.-S., Lee, S. D., Park, H., Lee, S., et al. (2005). Peptide-cleaving catalyst selective for peptide deformylase. J. Am. Chem. Soc. 127, 2396–2397. doi: 10.1021/ja044043h
Chandra, M., Sachdeva, A., and Silverman, S. K. (2009). DNA-catalyzed sequence-specific hydrolysis of DNA. Nat. Chem. Biol. 5, 718–720. doi: 10.1038/nchembio.201
Chei, W. S., Ju, H., and Suh, J. (2011). New chelating ligands for Co(III)-based peptide-cleaving catalysts selective for pathogenic proteins of amyloidoses. J. Biol. Inorg. Chem. 16, 511–519. doi: 10.1007/s00775-010-0750-y
Chin, J. (1991). Developing artificial hydrolytic metalloenzymes by a unified mechanistic approach. Acc. Chem. Res. 24, 145–152. doi: 10.1021/ar00005a004
Chin, J., Banaszczyk, M., Jubian, V., and Zou, X. (1989). Cobalt(III) complex-promoted hydrolysis of phosphate diesters: comparison in reactivity of rigid cis-diaquo(tetraaza)cobalt(III) complexes. J. Am. Chem. Soc. 111, 186–190. doi: 10.1021/ja00183a029
Clarkson, A. J., Buckingham, D. A., Rogers, A. J., Blackman, A. G., and Clark, C. R. (2000). Kinetic origin of the chelate effect. Base hydrolysis, H-exchange reactivity, and structures of syn,anti-[Co(cyclen)(NH3)2]3+ and syn,anti-[Co(cyclen)(diamine)]3+ Ions (diamine = H2N(CH2)2NH2, H2N(CH2)3NH2). Inorg. Chem. 39, 4769–4775. doi: 10.1021/ic000325g
Cleland, W. W., and Hengge, A. C. (2006). Enzymatic mechanisms of phosphate and sulfate transfer. Chem. Rev. 106, 3252–3278. doi: 10.1021/cr050287o
Coleman, F., Hynes, M. J., and Erxleben, A. (2010). Ga(III) complexes as models for the M(III) site of purple acid phosphatase: ligand effects on the hydrolytic reactivity toward bis(2,4-dinitrophenyl) phosphate. Inorg. Chem. 49, 6725–6733. doi: 10.1021/ic100722w
Corda, D., Mosca, M. G., Ohshima, N., Grauso, L., Yanaka, N., and Mariggi,ò, S. (2014). The emerging physiological roles of the glycerophosphodiesterase family. FEBS J. 281, 998–1016. doi: 10.1111/febs.12699
Cowan, J. A. (1998). Metal activation of enzymes in nucleic acid biochemistry. Chem. Rev. 98, 1067–1088. doi: 10.1021/cr960436q
Cowan, J. A. (2001). Chemical nucleases. Curr. Opin. Chem. Biol. 5, 634–642. doi: 10.1016/S1367-5931(01)00259-9
Dal Peraro, M., Llarrull, L. I., Rothlisberger, U., Vila, A. J., and Carloni, P. (2004). Water-assisted reaction mechanism of monozinc β-lactamases. J. Am. Chem. Soc. 126, 12661–12668. doi: 10.1021/ja048071b
Das, P., Chandar, N. B., Chourey, S., Agarwalla, H., Ganguly, B., and Das, A. (2013). Role of metal ion in specific recognition of pyrophosphate ion under physiological conditions and hydrolysis of the phosphoester linkage by alkaline phosphatase. Inorg. Chem. 52, 11034–11041. doi: 10.1021/ic401243h
Daver, H., Das, B., Nordlander, E., and Himo, F. (2016). Theoretical study of phosphodiester hydrolysis and transesterification catalyzed by an unsymmetric biomimetic dizinc complex. Inorg. Chem. 55, 1872–1882. doi: 10.1021/acs.inorgchem.5b02733
Deck, K. M., Tseng, T. A., and Burstyn, J. N. (2002). Triisopropyltriazacyclononane Copper(II): an efficient phosphodiester hydrolysis catalyst and DNA cleavage agent. Inorg. Chem. 41, 669–677. doi: 10.1021/ic0107025
Delehanty, J. B., Stuart, T. C., Knight, D. A., Goldman, E. R., Thach, D. C., Bongard, J. E., et al. (2005). RNA hydrolysis and inhibition of translation by a Co(III)–cyclen complex. RNA 11, 831–836. doi: 10.1261/rna.7156805
Ditchfield, R., Hehre, W. J., and Pople, J. A. (1971). Self-consistent molecular-orbital methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 54, 724–728. doi: 10.1063/1.1674902
Dolg, M., Stoll, H., and Preuss, H. (1989). Energy-adjusted ab initio pseudopotentials for the rare earth elements. J. Chem. Phys. 90, 1730–1734. doi: 10.1063/1.456066
Dubois, K. P. (1971). The toxicity of organophosphorus compounds to mammals. Bull. World Health Organ. 44, 231–240.
Eichler, D. C., and Lehman, I. R. (1977). On the role of ATP in phosphodiester bond hydrolysis catalyzed by the recBC deoxyribonuclease of Escherichia coli. J. Biol. Chem. 252, 499–503.
Eid, J., Fehr, A., Gray, J., Luong, K., Lyle, J., Otto, G., et al. (2009). Real-time DNA sequencing from single polymerase molecules. Science 323, 133–138. doi: 10.1126/science.1162986
Fang, Y.-G., Zhang, J., Chen, S.-Y., Jiang, N., Lin, H.-H., Zhang, Y., et al. (2007). Chiral multinuclear macrocyclic polyamine complexes: synjournal, characterization and their interaction with plasmid DNA. Biorg. Med. Chem. 15, 696–701. doi: 10.1016/j.bmc.2006.10.057
Fanning, A.-M., Plush, S. E., and Gunnlaugsson, T. (2006). Tuning the properties of cyclen based lanthanide complexes for phosphodiester hydrolysis; the role of basic cofactors. Chem. Commun. 36, 3791–3793. doi: 10.1039/B609923H
Foster, J. P., and Weinhold, F. (1980). Natural hybrid orbitals. J. Am. Chem. Soc. 102, 7211–7218. doi: 10.1021/ja00544a007
Fothergill, M., Goodman, M. F., Petruska, J., and Warshel, A. (1995). Structure-energy analysis of the role of metal ions in phosphodiester bond hydrolysis by DNA polymerase I. J. Am. Chem. Soc. 117, 11619–11627. doi: 10.1021/ja00152a001
Franklin, S. J. (2001). Lanthanide-mediated DNA hydrolysis. Curr. Opin. Chem. Biol. 5, 201–208. doi: 10.1016/S1367-5931(00)00191-5
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09. Wallingford CT: Gaussian, Inc.
Gewirtz, A. M., Sokol, D. L., and Ratajczak, M. Z. (1998). Nucleic acid therapeutics: state of the art and future prospects. Blood 92, 712–736.
Gómez-Tagle, P., and Yatsimirsky, A. K. (2001). Phosphodiester hydrolysis by lanthanide complexes of bis-tris propane. Inorg. Chem. 40, 3786–3796. doi: 10.1021/ic0010205
Hadler, K. S., Tanifum, E. A., Yip, S. H.-C., Mitić, N., Guddat, L. W., Jackson, C. J., et al. (2008). Substrate-promoted formation of a catalytically competent binuclear center and regulation of reactivity in a glycerophosphodiesterase from enterobacter aerogenes. J. Am. Chem. Soc. 130, 14129–14138. doi: 10.1021/ja803346w
Hay, R. W., and Norman, P. R. (1997). The kinetics and mechanism of aquation and base hydrolysis of the cis-[Co(cyclen)Cl2]+ cation (cyclen = 1,4,7,10-tetraazacyclododecane). Transition Met. Chem. 22, 248–253. doi: 10.1023/A:1018460307204
Hegg, E. L., and Burstyn, J. N. (1998). Toward the development of metal-based synthetic nucleases and peptidases: a rationale and progress report in applying the principles of coordination chemistry. Coord. Chem. Rev. 173, 133–165. doi: 10.1016/S0010-8545(98)00157-X
Hendry, P., and Sargeson, A. M. (1989). Metal ion promoted phosphate ester hydrolysis. Intramolecular attack of coordinated hydroxide ion. J. Am. Chem. Soc. 111, 2521–2527. doi: 10.1021/ja00189a025
Hettich, R., and Schneider, H.-J. (1997). Cobalt(III) polyamine complexes as catalysts for the hydrolysis of phosphate esters and of DNA. A measurable 10 million-fold rate increase1. J. Am. Chem. Soc. 119, 5638–5647. doi: 10.1021/ja964319o
Hidemi, S., Masahiko, I., and Koji, M. (1999). Extended x-ray absorption fine structure study on the cerium(IV)-induced DNA hydrolysis: implication to the roles of 4f orbitals in the catalysis. Appl. Phys. Lett. 74, 460–462. doi: 10.1063/1.123036
Holm, R. H., Kennepohl, P., and Solomon, E. I. (1996). Structural and functional aspects of metal sites in biology. Chem. Rev. 96, 2239–2314. doi: 10.1021/cr9500390
Iitaka, Y., Shina, M., and Kimura, E. (1974). Crystal structure of dinitro(1,4,7,10-tetraazacyclododecane)cobalt(III) chloride. Inorg. Chem. 13, 2886–2891. doi: 10.1021/ic50142a023
Ischtwan, J., and Collins, M. A. (1988). Determination of the intrinsic reaction coordinate: comparison of gradient and local quadratic approximation methods. J. Chem. Phys. 89, 2881–2885. doi: 10.1063/1.454992
Jang, S. W., and Suh, J. (2008). Proteolytic activity of Cu(II) complex of 1-Oxa-4,7,10-triazacyclododecane. Org. Lett. 10, 481–484. doi: 10.1021/ol702860h
Jayasinghe-Arachchige, V. M., Hu, Q., Sharma, G., Paul, T. J., Lundberg, M., Quinonero, D., et al. (2019). Hydrolysis of chemically distinct sites of human serum albumin by polyoxometalate: a hybrid QM/MM (ONIOM) study. J. Comput. Chem. 40, 51–61. doi: 10.1002/jcc.25528
Jeung, C. S., Kim, C. H., Min, K., Suh, S. W., and Suh, J. (2001). Hydrolysis of plasmid DNA catalyzed by Co(III) complex of cyclen attached to polystyrene. Bioorg. Med. Chem. Lett. 11, 2401–2404. doi: 10.1016/S0960-894X(01)00439-5
Jeyaratnam, J. (1990). Acute pesticide poisoning: a major global health problem. World. Health. Stat. Q. 43, 139–144.
Junghun, S., Ho, Y. S., Gyum, K. M., Keunhong, J., Young, A. J., Myoung-Soon, K., et al. (2007). Cleavage agents for soluble oligomers of amyloid β peptides. Angew. Chem. Int. Ed. 46, 7064–7067. doi: 10.1002/anie.200702399
Kamerlin, S. C. L., and Warshel, A. (2009). On the energetics of ATP hydrolysis in solution. J. Phys. Chem. B 113, 15692–15698. doi: 10.1021/jp907223t
Kia-Ki, H., and Martinage, A. (1992). Post-translational chemical modification(S) of proteins. Int. J. Biochem. 24, 19–28. doi: 10.1016/0020-711X(92)90225-P
Kim, H. M., Jang, B., Cheon, Y. E., Suh, M. P., and Suh, J. (2009). Proteolytic activity of Co(III) complex of 1-oxa-4,7,10-triazacyclododecane: a new catalytic center for peptide-cleavage agents. J. Biol. Inorg. Chem. 14, 151–157. doi: 10.1007/s00775-008-0434-z
Kimura, E., Kodama, Y., Koike, T., and Shiro, M. (1995). Phosphodiester hydrolysis by a New Zinc(II) macrocyclic tetraamine complex with an alcohol pendant: elucidation of the roles of Ser-102 and Zinc(II) in Alkaline Phosphatase. J. Am. Chem. Soc. 117, 8304–8311. doi: 10.1021/ja00137a002
Kirby, A. J., and Nome, F. (2015). Fundamentals of phosphate transfer. Acc. Chem. Res. 48, 1806–1814. doi: 10.1021/acs.accounts.5b00072
Koike, T., and Kimura, E. (1991). Roles of zinc(II) ion in phosphatases. A model study with zinc(II)-macrocyclic polyamine complexes. J. Am. Chem. Soc. 113, 8935–8941. doi: 10.1021/ja00023a048
Koike, T., Takamura, M., and Kimura, E. (1994). Role of Zinc(II) in .beta.-Lactamase II: a model study with a Zinc(II)-macrocyclic tetraamine (1,4,7,10-Tetraazacyclododecane, Cyclen) Complex. J. Am. Chem. Soc. 116, 8443–8449. doi: 10.1021/ja00098a002
Komiyama, M. (2016). Design of highly active Ce(IV) catalysts for DNA hydrolysis and their applications. Chem. Lett. 45, 1347–1355. doi: 10.1246/cl.160786
Komiyama, M., and Sumaoka, J. (1998). Progress towards synthetic enzymes for phosphoester hydrolysis. Curr. Opin. Chem. Biol. 2, 751–757. doi: 10.1016/S1367-5931(98)80113-0
Komiyama, M., Takeda, N., and Shigekawa, H. (1999). Hydrolysis of DNA and RNA by lanthanide ions: mechanistic studies leading to new applications. Chem. Commun. 1443–1451. doi: 10.1039/A901621J
Kövári, E., and Krämer, R. (1996). Rapid phosphodiester hydrolysis by an ammonium-functionalized Copper(II) complex. A model for the cooperativity of metal ions and NH-acidic groups in phosphoryl transfer enzymes. J. Am. Chem. Soc. 118, 12704–12709. doi: 10.1021/ja962806y
Krauser, J. A., Joshi, A. L., and Kady, I. O. (2010). Metal complexes of modified cyclen as catalysts for hydrolytic restriction of plasmid DNA. J. Inorg. Biochem. 104, 877–884. doi: 10.1016/j.jinorgbio.2010.04.007
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789. doi: 10.1103/PhysRevB.37.785
Li, Y., Lu, X.-M., Sheng, X., Lu, G.-Y., Shao, Y., and Xu, Q. (2007). DNA cleavage promoted by Cu2+ complex of cyclen containing pyridine subunit. J. Incl. Phenom. Macrocycl. Chem. 59, 91–98. doi: 10.1007/s10847-007-9299-y
Luedtke, N. W., and Schepartz, A. (2005). Lanthanide-mediated phosphoester hydrolysis and phosphate elimination from phosphopeptides. Chem. Commun. 43, 5426–5428. doi: 10.1039/B510123A
Luong, T. K. N., Mihaylov, T. T., Absillis, G., Shestakova, P., Pierloot, K., and Parac-Vogt, T. N. (2016). Phosphate ester bond hydrolysis promoted by lanthanide-substituted keggin-type polyoxometalates studied by a combined experimental and density functional theory approach. Inorg. Chem. 55, 9898–9911. doi: 10.1021/acs.inorgchem.6b01802
Mancin, F., Scrimin, P., and Tecilla, P. (2012). Progress in artificial metallonucleases. Chem. Commun. 48, 5545–5559. doi: 10.1039/C2CC30952A
Mancin, F., and Tecilla, P. (2007). Zinc(ii) complexes as hydrolytic catalysts of phosphate diester cleavage: from model substrates to nucleic acids. New J. Chem. 31, 800–817. doi: 10.1039/B703556J
Mikkola, S., Kaukinen, U., and Lönnberg, H. (2001). The effect of secondary structure on cleavage of the phosphodiester bonds of RNA. Cell Biochem. Biophys. 34, 95–119. doi: 10.1385/cbb:34:1:95
Mitić, N., Smith, S. J., Neves, A., Guddat, L. W., Gahan, L. R., and Schenk, G. (2006). The catalytic mechanisms of binuclear metallohydrolases. Chem. Rev. 106, 3338–3363. doi: 10.1021/cr050318f
Mol, C. D., Izumi, T., Mitra, S., and Tainer, J. A. (2000). DNA-bound structures and mutants reveal abasic DNA binding by APE1 DNA repair and coordination. Nature 403, 451–456. doi: 10.1038/35000249
Neidle, S. (2008). “The building-blocks of DNA and RNA,” in Principles of Nucleic Acid Structure, ed S. Neidle. (New York, NY: Academic Press), 20–37.
Niittymaki, T., and Lonnberg, H. (2006). Artificial ribonucleases. Org. Biomol. Chem. 4, 15–25. doi: 10.1039/b509022a
Oivanen, M., Kuusela, S., and Lönnberg, H. (1998). Kinetics and mechanisms for the cleavage and isomerization of the phosphodiester bonds of RNA by brønsted acids and bases. Chem. Rev. 98, 961–990. doi: 10.1021/cr960425x
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi: 10.1103/PhysRevLett.77.3865
Rammo, J., Hettich, R., Roigk, A., and Schneider, H.-J. (1996). Catalysis of DNA cleavage by lanthanide complexes with nucleophilic or intercalating ligands and their kinetic characterization. Chem. Commun. 105–107. doi: 10.1039/CC9960000105
Reed, A. E., and Weinhold, F. (1983). Natural bond orbital analysis of near-Hartree–Fock water dimer. J. Chem. Phys. 78, 4066–4073. doi: 10.1063/1.445134
Richman, J. E., and Atkins, T. J. (1974). Nitrogen analogs of crown ethers. J. Am. Chem. Soc. 96, 2268–2270. doi: 10.1021/ja00814a056
Robinson, H., Jung, K.-E., Switzer, C., and Wang, A. H. J. (1995). DNA with 2'-5' phosphodiester bonds forms a duplex structure in the A-type conformation. J. Am. Chem. Soc. 117, 837–838. doi: 10.1021/ja00107a036
Sancar, A., and Sancar, G. B. (1988). DNA repair enzymes. Annu. Rev. Biochem 57, 29–67. doi: 10.1146/annurev.bi.57.070188.000333
Sharp, P. A. (1985). On the origin of RNA splicing and introns. Cell 42, 397–400. doi: 10.1016/0092-8674(85)90092-3
Shionoya, M., Ikeda, T., Kimura, E., and Shiro, M. (1994). Novel “Multipoint” molecular recognition of nucleobases by a New Zinc(II) complex of acridine-pendant cyclen (Cyclen = 1,4,7,10-Tetraazacyclododecane). J. Am. Chem. Soc. 116, 3848–3859. doi: 10.1021/ja00088a021
Sosa, M. E., and Tobe, M. L. (1985). Proton exchange and base hydrolysis of syn,anti-cis-dichloro(1,4,7,10-tetra-azacyclododecane)cobalt(III) cations. J. Chem. Soc. Dalton Trans. 475–477. doi: 10.1039/DT9850000475
Sreedhara, A., and Cowan, J. A. (2001). Catalytic hydrolysis of DNA by metal ions and complexes. J. Biol. Inorg. Chem. 6, 337–347. doi: 10.1007/s007750100209
Subat, M., Woinaroschy, K., Gerstl, C., Sarkar, B., Kaim, W., and Konig, B. (2008). 1,4,7,10-tetraazacyclododecane metal complexes as potent promoters of phosphodiester hydrolysis under physiological conditions. Inorg. Chem. 47, 4661–4668. doi: 10.1021/ic702413q
Sullivan, K. P., Yin, Q., Collins-Wildman, D. L., Tao, M., Geletii, Y. V., Musaev, D. G., et al. (2018). Multi-tasking POM Systems. Front Chem. 6:365. doi: 10.3389/fchem.2018.00365
The, L. (1998). Organophosphorus compounds: good, bad, and difficult. Lancet 352:499. doi: 10.1016/S0140-6736(98)21033-6
Weisman, G. R., and Reed, D. P. (1996). A new synjournal of cyclen (1,4,7,10-Tetraazacyclododecane). J. Org. Chem. 61, 5186–5187. doi: 10.1021/jo9606665
Weston, J. (2005). Mode of Action of Bi- and trinuclear zinc hydrolases and their synthetic analogues. Chem. Rev. 105, 2151–2174. doi: 10.1021/cr020057z
Wilkinson, G., Gillard, R. D., and Mccleverty, J. A. (1987). Comprehensive Coordination Chemistry: The Synjournal, Reactions, Properties & Applications of Coordination Compounds. Oxford: Pergamon Press.
Williams, N. H., Takasaki, B., Wall, M., and Chin, J. (1999). Structure and nuclease activity of simple dinuclear metal complexes: quantitative dissection of the role of metal ions. Acc. Chem. Res. 32, 485–493. doi: 10.1021/ar9500877
Williams, N. H., and Wyman, P. (2001). Base catalysed phosphate diester hydrolysis. Chem. Commun. 1268–1269. doi: 10.1039/B103317B
Yoji, Y., Akihiko, U., Akira, W., Hiroyuki, A., and Makoto, K. (2006). Chemical-reaction-based site-selective DNA cutter for PCR-free gene manipulation. Chem. Bio. Chem. 7, 673–677. doi: 10.1002/cbic.200500402
Zhang, G., Liu, C., Long, D.-L., Cronin, L., Tung, C.-H., and Wang, Y. (2016a). Water-soluble pentagonal-prismatic titanium-oxo clusters. J. Am. Chem. Soc. 138, 11097–11100. doi: 10.1021/jacs.6b06290
Zhang, T., Sharma, G., Paul, T. J., Hoffmann, Z., and Prabhakar, R. (2017). Effects of ligand environment in Zr(IV) assisted peptide hydrolysis. J. Chem. Inf. Model. 57, 1079–1088. doi: 10.1021/acs.jcim.6b00781
Zhang, T., Zhu, X., and Prabhakar, R. (2014). Peptide hydrolysis by metal-cyclen complexes and their analogues: insights from theoretical studies. Organometallics 33, 1925–1935. doi: 10.1021/om400903r
Zhang, X., Liu, X., Phillips, D. L., and Zhao, C. (2016b). Mechanistic insights into the factors that influence the DNA nuclease activity of mononuclear facial copper complexes containing hetero-substituted cyclens. ACS Catal. 6, 248–257. doi: 10.1021/acscatal.5b01735
Keywords: phosphodiester hydrolysis, metal-cyclen complexes, di- and tetravalent metal ions, reaction mechanisms, density functional theory (DFT)
Citation: Hu Q, Jayasinghe-Arachchige VM, Zuchniarz J and Prabhakar R (2019) Effects of the Metal Ion on the Mechanism of Phosphodiester Hydrolysis Catalyzed by Metal-Cyclen Complexes. Front. Chem. 7:195. doi: 10.3389/fchem.2019.00195
Received: 19 December 2018; Accepted: 14 March 2019;
Published: 05 April 2019.
Edited by:
Soumyajit Roy, Indian Institute of Science Education and Research Kolkata, IndiaReviewed by:
Jean-Claude Georges Bunzli, École Polytechnique Fédérale de Lausanne, SwitzerlandMaria Letizia Di Pietro, University of Messina, Italy
Copyright © 2019 Hu, Jayasinghe-Arachchige, Zuchniarz and Prabhakar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajeev Prabhakar, cnByQG1pYW1pLmVkdQ==