 Haneen Mansoor
Haneen Mansoor William L. Harrigan
William L. Harrigan Keith A. Lehuta
Keith A. Lehuta Kevin R. Kittilstved
Kevin R. Kittilstved- Department of Chemistry, University of Massachusetts Amherst, Amherst, MA, United States
We demonstrate a low-temperature reduction method for exhibiting fine control over the oxidation state of substitutional Mn ions in strontium titanate (SrTiO3) bulk powder. We employ NaBH4 as the chemical reductant that causes significant changes in the oxidation state and oxygen vacancy complexation with Mn2+ dopants at temperatures <350°C where lattice reduction is negligible. At higher reduction temperatures, we also observe the formation of Ti3+ in the lattice by diffuse-reflectance and low-temperature electron paramagnetic resonance (EPR) spectroscopy. In addition to Mn2+, Mn4+, and the Mn2+ complex with an oxygen vacancy, we also observe a sharp resonance in the EPR spectrum of heavily reduced Mn-doped SrTiO3. This sharp signal is tentatively assigned to surface superoxide ion that is formed by the surface electron transfer reaction between Ti3+ and O2. The ability to control the relative amounts of various paramagnetic defects in SrTiO3 provides many possibilities to study in a model system the impact of tunable dopant-defect interactions for spin-based electronic applications or visible-light photocatalysis.
Introduction
The oxide SrTiO3 is a classic perovskite-type member of the valuable ABO3 semiconductor family. The promising properties such as a large tunable dielectric constant, structural phase transitions, superior charge storage capacity and tunable electronic structure have made SrTiO3 an exciting candidate for a wide range of multifunctional applications (Weaver, 1959; Faughnan, 1971; Mattheiss, 1972; Müller and Burkard, 1979; Kamalasanan et al., 1993). Although in ambient conditions it exhibits a wide band gap and low electron mobility, introducing impurity dopants and intrinsic defects radically influence the conductivity and optical properties of the host material (Wild et al., 1973; Kozuka et al., 2010). The function of a semiconductor is intimately related to the chemistry and physics of native and targeted defects. The rich defect chemistry enabled by native oxygen vacancies (VO) in semiconductors such as SrTiO3, PbTiO3, and BaTiO3 has been correlated with numerous functions including ferroelectricity, visible-light photocatalysis, and multiferroics. These VO defects can donate up to two electrons to the host lattice. Transition metal dopants may also impart additional functionality that result from partially-filled d-orbitals. For example, Cr3+ dopants, and Rh3+ dopants in SrTiO3 can reduce protons to generate H2 gas using visible light that creates an oxidized dopant ion and a conduction band electron, ecb (Ishii et al., 2004; Sasaki et al., 2009; Kato et al., 2013). However, undesirable defects such as Cr6+ can form to maintain charge neutrality, but limit the photochemical efficiency by serving as a trap for the ecb. These types of high-valent defects can be removed by either post-synthetic annealing under reducing atmospheres (Zuo et al., 2010; Tan et al., 2014; Lehuta and Kittilstved, 2016), co-doping the host lattice with additional n-type dopants (Chan et al., 1981; Kato and Kudo, 2002; Wang et al., 2014), irradiating with UV light (Wang et al., 2006), or applying a large electrical bias (La Mattina et al., 2008). Of these, the only potentially “green” reduction source could be UV irradiation from the sun. However, we note that the reported photoreduction step using a 400 W Hg-lamp in Cr:SrTiO3 powders was of the order of tens of hours. The realization of a fast, low-energy method to modulate the oxidation state of transition-metal dopants in SrTiO3 and related metal oxide semiconductors could impact various fields such as visible-light photocatalysis, sensing, and spin-based electronics. To this end, recent studies on the photodoping of colloidal Cr:SrTiO3 nanocrystals show promise (Harrigan and Kittilstved, 2018).
We previously studied the effect of a relatively low-temperature NaBH4 reduction reaction on the oxidation state of Cr dopants in SrTiO3 and related Sr2TiO4 bulk powders (Lehuta and Kittilstved, 2016; Lehuta et al., 2017). In those studies, we observed an order of magnitude increase in the Cr3+ concentration by EPR spectroscopy that we attributed to the reduction of EPR-silent high-valent Cr4+ and Cr6+ ions. The increase in the Cr3+ concentration in n-type SrTiO3 presents an interesting scenario where the Cr3+ ion has a dual role of being an electron donor and a paramagnetic ion (S = 3/2). In addition, these observed changes are quantitatively reversible upon annealing the powders in air.
An isoelectronic analog of Cr3+ is Mn4+, which is known to also occupy the Ti4+-site in SrTiO3. Although additional defects are required to maintain charge neutrality in Cr3+:SrTiO3, Mn4+ in the B-site of SrTiO3 is an isovalent dopant. Amongst the transition-metal doped oxides, Mn:SrTiO3 has recently received extensive attention due to its complex and unique behavior than intrinsic SrTiO3. The concurrent doping of Mn and oxygen vacancies in SrTi1−xMnxO3−δ is reported to promote ferromagnetic ordering, dielectric permittivity and possible metallic behavior (Savinov et al., 2008; Choudhury et al., 2011, 2013; Middey et al., 2012; Thanh et al., 2014). These observations make nonstoichiometric Mn:SrTiO3 a highly attractive candidate for spin-based electronics applications. Although not completely understood, the results are attributed to the interplay of redox-active Mn ions and the intrinsic charge compensating defects. In this regard, quantitative research is challenging due to a lack of experimental control over the interactions, and the complexity of Mn ions present in multiple oxidation states. Herein we report on the nature of the oxidation state of Mn ions and associated defect centers in bulk Mn:SrTiO3 powders. We utilized various dopant-specific spectroscopic probes to elucidate the Mn oxidation state including EPR and diffuse-reflectance spectroscopies. We extend the use of NaBH4 as a solid-state reductant to monitor changes in the three, unique Mn-related species as well as oxygen-related defects and “self-doped” Ti3+ ions. Comparison to other studies of reduced Mn:SrTiO3 and noticeable absences of certain EPR-active Mn-centers is also discussed. We also observe a new signal in reduced samples that we attribute to superoxide anions, .
Materials and Methods
Chemicals
TiO2 (>99.5%, Aeroxide P25 powder, Acros Organics), Sr(NO3)2 (>99%, Acros Organics), Mn(NO3)2·4H2O (analytical grade, Acros Organics), NaBH4 (≥98%, white powder, MP Biomedical), MgO (Fisher Science Education), and ethanol (200 proof, ACS/USP grade, Pharmco-Aaper) were used as received.
Synthesis of Bulk Mn-Doped SrTiO3
Bulk powders of SrTi1−xMnxO3−δ (abbreviated Mn:SrTiO3) were synthesized by a conventional solid-state reaction method, where x is the nominal concentration of Mn (x = 0.001) and δ is the concentration of oxygen vacancies. Briefly, Sr(NO3)2, Mn(NO3)2·4H2O, and TiO2 were mixed in the desired stoichiometry and ground with a mortar and pestle for about 10 min. The mixture was then transferred to a porcelain combustion boat and placed in the center of a tube furnace inside a quartz insert. The reaction mixture was heated in air for 6 h at 1,000°C, reground for 10 min, then heated again for an additional 16 h at 1,000°C.
NaBH4 Reductions and Reoxidation
Chemical reductions of the bulk powders were carried out using a modified version of reduction previously described by our group for Cr:SrTiO3 (Lehuta and Kittilstved, 2016). For each reduction, an amount of powder was mixed in a 1:1 mole ratio with NaBH4 using a mortar and pestle for 5 min and then placed in a porcelain combustion boat in the middle of a quartz insert in a tube furnace. The atmosphere in the quartz insert was continuously purged by a controlled flow of Ar gas monitored by a rotameter (Matheson 7300). The samples were heated at temperatures ranging from 300 to 425°C in 25°C increments under Ar flow for 30 min. After reducing, samples were cooled under Ar to room temperature, washed and centrifuged alternately with deionized water and ethanol to ensure complete removal of NaBH4. After washing, samples were dried in an oven at 100°C for 2 h.
Reoxidation was performed by aerobically annealing the reduced samples until the physical color of the sample reversed to Mn:SrTiO3 as-prepared sample.
Characterization
Powder X-ray diffraction (XRD) patterns were collected at room temperature using a Bragg-Brentano configuration with Cu K-α source (Rigaku SmartLab SE Diffraction System with cross-beam optics and D/Tex 250 Ultra 1D Si strip detector). X-band quantitative EPR spectra were collected at room temperature in 4 mm quartz EPR tubes (Wilmad-Glass) in a double rectangular resonator cavity (Bruker Elexsys E-500 with ER 4105DR cavity). Room temperature quantitative EPR spectra were collected consecutively on chemically perturbed samples (either oxidized or reduced) and an as-prepared sample using identical sample placement and instrument settings (Eaton et al., 2010). The resonance field positions in the EPR spectra for each paramagnetic Mn center were simulated using the “resfields” function in EasySpin using the reported EPR parameters from literature and referenced below (Stoll and Schweiger, 2006). Low-temperature X-band EPR spectra were measured at 77 K on powders using the perpendicular mode of a dual-mode resonator cavity with a quartz finger dewar insert (Bruker Elexsys E-500 with ER-4116 cavity) ensuring the sample height exceeded the cavity height for quantitative analysis. Diffuse-reflectance spectra were collected with an integrating sphere (Ocean Optics ISP-REF) coupled by fiber optics to a CCD-based spectrophotometer (Ocean Optics USB2000+ VIS-NIR). The optical density between the absorption minimum and the absorption at 320 nm was adjusted by diluting the powders with MgO.
Results and Discussion
The room temperature powder XRD patterns of as-prepared and reduced (Tred = 425°C) Mn:SrTiO3 are shown in Figure 1. All samples designated Mn:SrTiO3 contain nominally 0.1% Mn content. Both the as-prepared and reduced samples indicate the presence of the cubic phase of SrTiO3 (Mitchell et al., 2000). However, a clear increase in the lattice parameter is observed after reduction. This result is consistent with other observations and has been attributed to both changes in ionic size and electronic effects after reduction of the lattice (Janotti et al., 2012). For example, the reduction of both Mn ions (Mn4+ → Mn2+) and lattice cations (Ti4+ → Ti3+) would result in larger ions leading to lattice expansion (Shannon, 1976). No appreciable secondary phases were observed after low-temperature chemical reduction despite clear spectroscopic changes in the samples (vide infra).
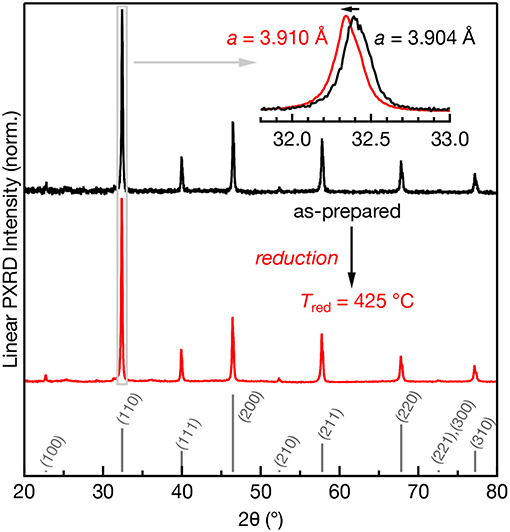
Figure 1. Powder XRD patterns of 0.1% Mn:SrTiO3 before (black) and after Tred = 425°C (red). The calculated pattern for SrTiO3 (cubic phase) is shown in gray as sticks (Yamanaka et al., 2002). Inset: expanded region of the (110) diffraction peak clearly showing a shift to lower 2θ after Tred = 425°C.
The electronic structure of Mn:SrTiO3 is dependent on the nature of the Mn-ion speciation (i.e., oxidation state(s) and first-coordination sphere). Mn4+ has a d3 electronic configuration yielding a 4A2g ground state when substituted at the Ti4+-site in SrTiO3. The physical appearance of the Mn:SrTiO3 as-prepared (oxidized) powders is off-white and gradually turns to black with increased reduction temperature as shown in Figure 2A. The black appearance of SrTiO3 has been previously observed and indicates reduction in the SrTiO3 lattice resulting in self-trapped electrons localized at Ti3+ sites (Tan et al., 2014; Lehuta and Kittilstved, 2016). The diffuse-reflectance spectra corroborates the assignment of the black color to excitations from Ti3+ to conduction band states also referred to as a metal-to-metal charge transfer (MMCT) transition in the near-IR region (Khomenko et al., 1998). In the Mn:SrTiO3 powder this MMCT appears at Tred between 350 and 375°C. The sub-bandgap tailing absorption at ~2.9 eV has been assigned to excitations from the valence band to VO's with different charge states (Mitra et al., 2012). With increasing Tred, the relative intensity of the VO-related transitions decreases and disappears at Tred = 375°C and is consistent with electron accumulation in the VO states. Both spectral changes observed here for Mn:SrTiO3 with increasing Tred are similar to our recent study on chemically-reduced Cr:SrTiO3 (Lehuta and Kittilstved, 2016).
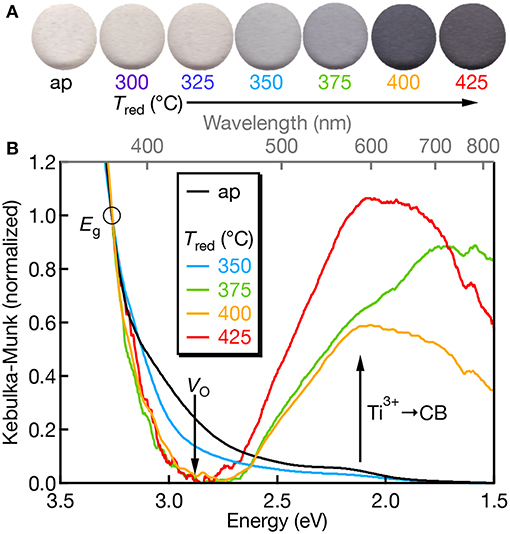
Figure 2. (A) Scanned color images of Mn:SrTiO3 powders as a function of Tred. (B) Diffuse-reflectance spectra of Mn:SrTiO3 as a function of reduction temperature (as-prepared = black; Tred at 350°C (blue), 375°C (green), 400°C (orange), and 425°C (red), respectively). Data were normalized at 380 nm (3.26 eV; denoted by the circle) after setting the lowest y-value in the spectrum as the “zero.” The Tred = 400°C spectrum was smoothed for presentation.
We also do not observe Mn-centered transitions from the diffuse-reflectance spectra which we attribute to either (1) low concentrations of Mn3+ or Mn4+, which have spin-allowed transitions in the visible, or (2) the Mn ions are primarily in their +2 oxidation state, which only has spin-forbidden transitions when the d-electrons order in the high-spin configuration (S = 5/2, 6A1g ground state).
EPR-active species involving Mn ions in the +2, +3, and +4 oxidation states in SrTiO3 have previously been reported (Müller, 1959; Serway et al., 1977; Azamat et al., 2012). However, Mn3+ exhibits large zero-field splitting due to the S = 2 electronic spin state and thus, it is EPR-silent at conventional X-band frequencies (Azamat et al., 2012). The room temperature quantitative X-band EPR spectra of the Mn-doped SrTiO3 samples are shown in Figure 3 as a function of reduction temperatures ranging from Tred = 300–425°C. The as-prepared sample consists of two sets of sextet peaks. In accordance with the reported g-value and hyperfine splitting constant (A) of Mn4+ in SrTiO3, the main sextet in the as-prepared sample is assigned to Mn4+ substituting for Ti4+ with an isotropic g = 1.996 and |A| = 69.4 × 10−4 cm−1 (Müller, 1959). The second and much weaker set of sextets is somewhat occluded by the Mn4+ transitions, but the low-field resonances agree well with substitutional Mn2+ at the Ti4+ site in SrTiO3 with g = 2.004 and |A| = 82.30 × 10−4 cm−1 (Azzoni et al., 2000; Choudhury et al., 2013). Despite reports of both axial Mn2+- and Mn3+- complexes in Mn:SrTiO3, we do not observe these complexes in the as-prepared Mn:SrTiO3 sample. Hence, only the substitutional Mn4+ and Mn2+ species in an octahedral oxide crystal field co-exist in the as-prepared Mn:SrTiO3 powders.
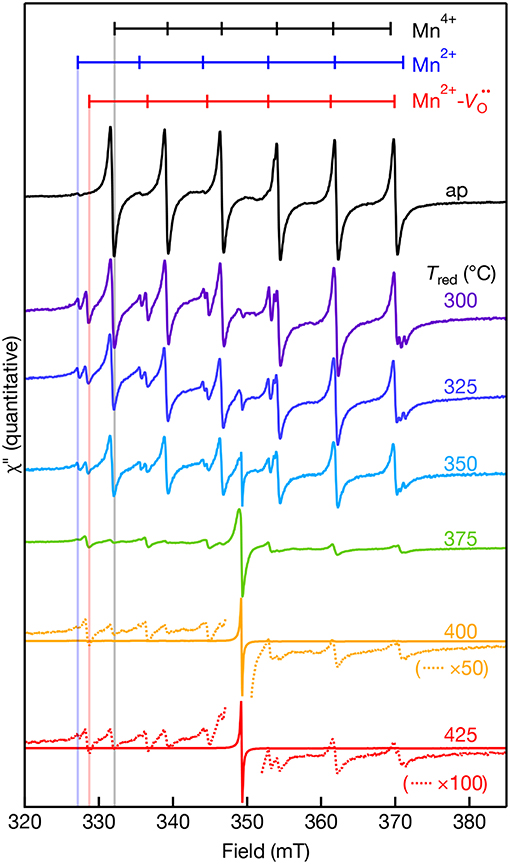
Figure 3. Room temperature X-band EPR spectra of 0.1% Mn-doped SrTiO3 bulk powder before (as-prepared, black) and after NaBH4 reduction for 30 min at various temperatures (300–425°C). The dotted lines in the 400 and 425°C spectra are selected regions of the spectra that are multiplied (×) by 50 and 100, respectively. The calculated resonance fields for the hyperfine-split transitions of Mn4+ (black), Mn2+- (red), and Mn2+ (blue) in SrTiO3 are shown as vertical markers. The calculated resonance fields were simulated for each Mn center observed in the EPR with EasySpin (see characterization). Data were collected on the same day in a double-resonator cavity equipped with an as-prepared sample in the 2nd resonator and used to standardize sample intensities.
After chemical reduction with NaBH4 under Ar(g) at Tred = 300°C, a new, third set of transitions are detected near the Mn2+ lines. Concomitant with the appearance of this new set of peaks is a decrease in the intensity of Mn4+ lines and an increase in the relative intensity of Mn2+. The new set of lines agree well with the report of a substitutional Mn2+ center at the Ti4+-site coupled to a doubly ionized oxygen vacancy (Serway et al., 1977). The reported EPR parameters of this Mn2+- complex includes a large axial component to the zero-field splitting (D = 0.544 cm−1), g|| = 2.003, and |A| = 76 × 10−4 cm−1. We were unable to detect any transitions at lower or higher magnetic fields likely from the low relative concentration and low nominal concentration of Mn in the lattice. The Mn2+- complex forms at low temperatures before reduction of the SrTiO3 lattice at Tred < 375°C (see Figure 2). One possible mechanism to explain the formation of this complex could be that oxygen vacancies may diffuse through the lattice and localize in the vicinity of Mn4+ substitutional sites at low temperatures. This work demonstrates that mild reduction at only Tred = 300–325°C is sufficient to form the Mn2+- complex in bulk powders. This result contrasts with the high temperature reductions above 825°C previously used to create these centers in Mn:SrTiO3 (Blazey et al., 1983; Kutty et al., 1986). In addition, we observe the co-existence of Mn4+ and the Mn2+- complex in the same EPR spectra at every Tred. This does not agree with previous single crystal studies where the Mn2+- complex was only observed when the Mn4+ lines were fully removed upon reduction in 5% hydrogen for 3 h at 1,000°C (Serway et al., 1977).
A new single feature centered at B0 ~ 350 mT (g ~ 2.003) with no associated hyperfine structure was also observed in the EPR spectra after reduction. This feature increases in spectral intensity and also narrows with increasing Tred. This feature is similar to a feature observed in Cr:SrTiO3 at Tred > 375°C (Lehuta and Kittilstved, 2016), but has a significantly larger relative intensity compared to the dopant EPR signal in the Mn:SrTiO3 sample with the same nominal dopant concentration. This feature is tentatively assigned as the EPR-active superoxide anion () adsorbed on the surface of SrTiO3 and could form via a surface reaction between Ti3+ and oxygen (Bykov et al., 2013; Harrigan et al., 2016). Table 1 below summarizes the EPR spectral parameters for previously reported Mn species in SrTiO3 in the as-prepared and reduced Mn:SrTiO3.
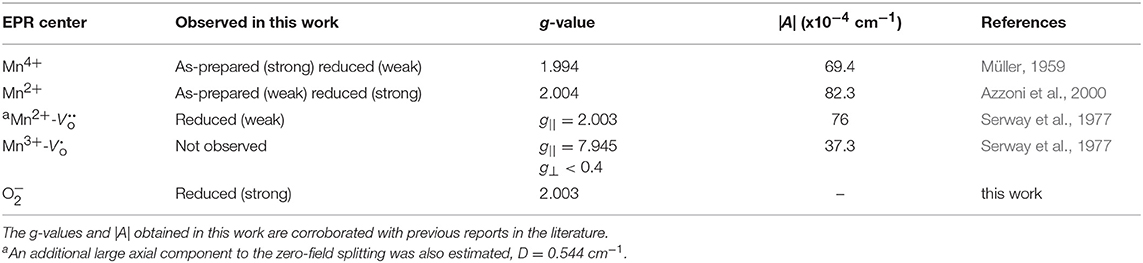
Table 1. Summary of EPR parameters for the Mn centers observed in the as-prepared and chemically treated samples reduced at various low temperatures.
The quantitative EPR spectra measured using the double resonator cavity shown in Figure 3 was analyzed and the relative intensity of the observed EPR centers is shown in Figure 4 as a function of Tred. Compared to the as-prepared Mn4+ signal intensity (defined as 1), a gradual decrease in the Mn4+ and Mn2+ EPR signals is observed with similar correlations in their temperature dependences. The signal for Mn2+ is not detected for Tred ≥ 375°C. In contrast, the EPR intensity of the Mn2+- complex shows little change and is more intense than the Mn4+ and Mn2+ EPR signals for Tred ≥ 375°C. The intensity of the Mn2+- complex drops by an order of magnitude after increasing Tred from 375 to 400°C. At the highest temperature, Tred = 425°C, the EPR intensity of the ion is nearly 3 orders of magnitude more intense than the Mn2+- complex, indicative of substantial surface defects. Studies to identify the nature of this defect center are currently underway.
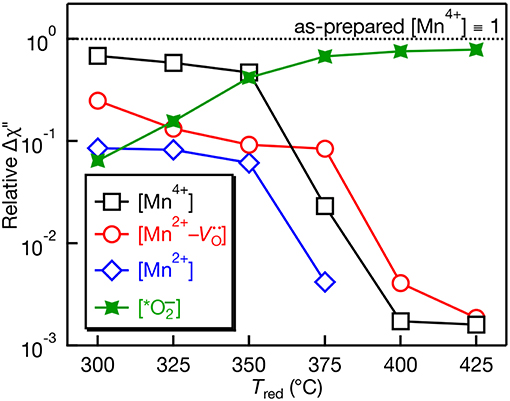
Figure 4. Relative EPR peak intensities plotted on a log scale as a function of NaBH4 reduction temperature (Tred) for Mn:SrTiO3. The peak intensities are normalized vs. the intensity of the Mn4+ EPR signal in the as-prepared sample. Quantitative measurements performed using a dual-cavity EPR resonator (see characterization section).
The EPR spectra of the Mn:SrTiO3 powders after preparation, after Tred = 400°C, and after aerobic reoxidation at Tair = 500°C for ~1 h are shown in Figure 5. The observed changes in the EPR spectra of the reduced samples revert to the as-prepared EPR spectrum by aerobically annealing the sample. The process of forming Mn2+- complex in the reduced samples is thus reversible. However, elevated temperatures and longer reoxidation times were required in contrast with the chemical reductions. Since the Mn2+- complex is a charge-neutral complex in the lattice, it is expected to be at least metastable. The apparently slower reoxidation kinetics compared to reduction kinetics suggest a metastable complex.
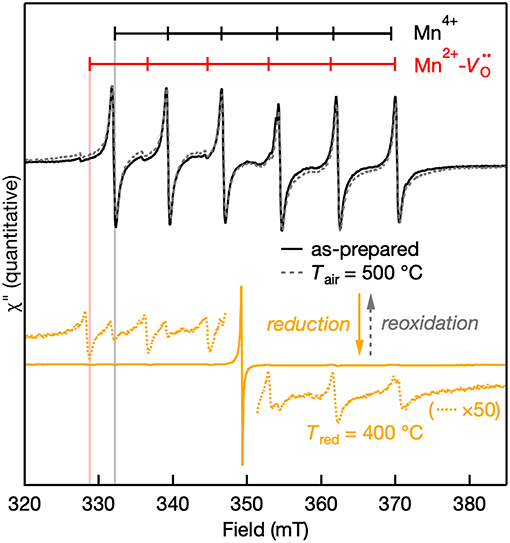
Figure 5. Quantitative reversibility of the EPR signal of Mn:SrTiO3 bulk powder as-prepared (solid black line), after Tred at 400°C (solid orange line), and after Tair at 500°C for ~1 h (gray dotted line). The weak Mn4+ and Mn2+ features after NaBH4 reduction are still observed in the 50× scaled spectra (orange dotted lines).
The 300 K (room temperature) and 77 K (liquid N2) EPR spectra of the Mn centers in the as-prepared and Tred = 300°C powders are shown in Figure 6. Two things are revealed from the EPR spectra of both as-prepared and lightly-reduced Mn:SrTiO3 samples: (1) there is no evidence of self-trapped electrons at Ti3+ sites in the lattice based on the 77 K spectra, and (2) the EPR intensity of Mn2+ completely disappears at 77 K. At low temperature, the intensity of Mn4+ is pronounced following the typical Boltzmann statistics. In contrast, the Mn2+ EPR signal completely disappears at 77 K in these two samples. These results agree with a previous magnetic susceptibility and EPR study of the Mn2+ signal vanishing, where the behavior was attributed to increased antiferromagnetic interactions between adjacent Mn2+ ions with decreasing temperature (Azzoni et al., 2000). This explanation cannot be extended to describe the EPR signal of the Mn2+- complex, which does not disappear at 77 K in the Tred = 300°C sample. To confirm the behavior of EPR signals as a function of temperature, the EPR spectrum of the reduced sample at 300 K was repeated after cooling it to 77 K and the entire spectrum is nearly identical.
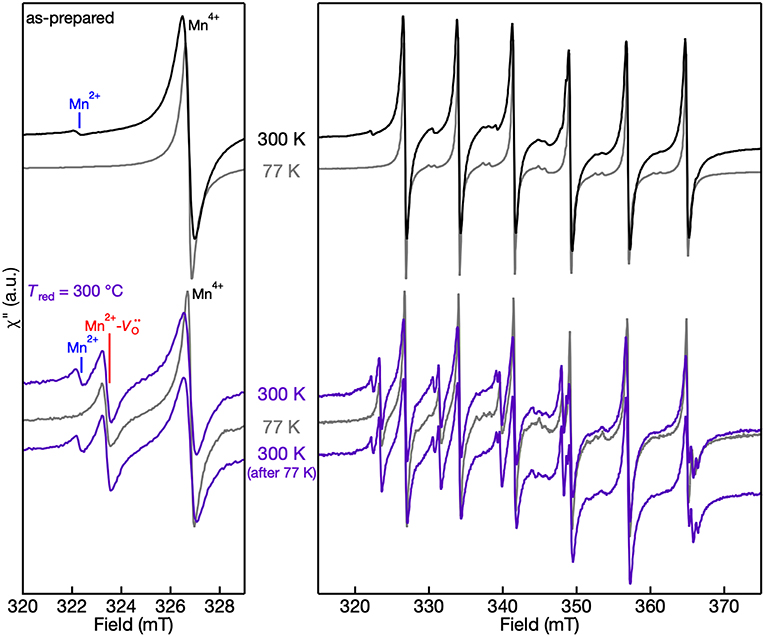
Figure 6. 300 K and 77 K EPR spectra of Mn:SrTiO3 powders as-prepared (top) and after Tred = 300°C (bottom). (left) Spectra in the region of the lowest field transitions within the hyperfine structure and (right) full spectrum. The temperatures at which the spectra were collected are labeled in the figure. Spectra are normalized to account for the increased EPR intensity with decreasing temperature.
The cryogenic EPR measurements were also collected for samples reduced above 350°C to reveal the effect of Ti3+ defects on the EPR spectra that are observed in the diffuse-reflectance spectra shown in Figure 2. Figure 7 shows the 300 K and 77 K EPR spectra of Tred = 375°C. The paramagnetic Ti3+ defects are not observed in the EPR performed at 300 K due to fast spin-lattice relaxation but are promptly observed at 77 K (Lehuta and Kittilstved, 2016; Harrigan and Kittilstved, 2018). At 77 K, the Tred = 375°C sample is dominated by a broad and intense asymmetric Ti3+ lattice defect centered at g = 1.94. The appearance of this fast-relaxing defect, however, has no apparent effect on the linewidth of the Mn-centers nor the single line that we tentatively assign to surface-adsorbed ions. We recently showed that linewidth and relaxation-dynamics of substitutional Cr3+ ions in SrTiO3 powders and colloidal nanocrystals can be significantly altered when Ti3+ defects are present in the lattice through a near-resonant cross-relaxation process (Lehuta and Kittilstved, 2016; Harrigan and Kittilstved, 2018). This same behavior is not observed for any of the Mn-centers in the reduced SrTiO3 powder.
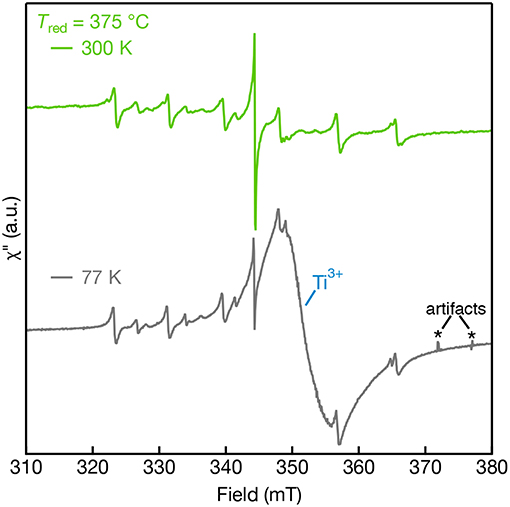
Figure 7. EPR spectra of Mn:SrTiO3 bulk powders reduced at Tred = 375°C measured at 300 K (top) and 77 K (bottom). Sharp features in the 77 K spectra are attributed to artifacts resulting from bubbles that arise in the liquid nitrogen finger dewar.
Conclusions
A low-temperature chemical reduction technique has been implemented for tunability of the Mn dopant oxidation states and the related intrinsic defects in bulk Mn:SrTiO3. We employed a myriad of structural and spectroscopic techniques on samples subjected to a systematic chemical reduction. Both isotropic Mn4+ and Mn2+ species were identified in the as-prepared powders. Following the thermal reduction, the samples exhibited a continuous decrease in Mn4+ EPR signal and an increase in the Mn2+ intensity, accompanied by the introduction of a Mn2+- complex. We demonstrate that our chemical treatment at merely Tred = 300–325°C generates sufficient driving force to significantly reduce the intensity of the octahedral Mn4+ and Mn2+ dopants and form the Mn2+- complex. All the Mn peaks showed distinctive changes at low-temperature in the EPR that are readily reversible upon warming back the samples. Reductions at 375°C and above generated significant concentrations of Ti3+ defects that were confirmed by diffuse-reflectance and low-temperature EPR spectroscopy. All the observed perturbations in the reduced samples are entirely reversible by aerobic annealing at elevated temperatures. We also observe an intense spectral feature in the EPR spectrum in heavily-reduced Mn:SrTiO3 powders that we attribute to ions at the surface. This fast and effective strategy offers a general low-temperature reduction process that allows tunability and control over the rich dopant-defect chemistry in transition-metal doped SrTiO3 materials.
Data Availability
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
HM and KL carried out the experiments, data analysis and interpretation, and edited the manuscript. WH contributed to the interpretation of the results and edited the manuscript. HM and KK contributed to the data analysis and interpretation and wrote the manuscript.
Funding
This work was supported by the National Science Foundation (NSF:DMR-1747593). The acquisition of the X-ray powder diffractometer was made possible through the National Science Foundation Major Research Instrumentation Program (NSF:CHE-1726578).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Azamat, D. V., Badalyan, A. G., Dejneka, A., Trepakov, V. A., Jastrabik, L., and Frait, Z. (2012). High-frequency electron paramagnetic resonance investigation of Mn3+ centers in SrTiO3. J. Phys. Chem. Solids 73, 822–826. doi: 10.1016/j.jpcs.2012.02.009
Azzoni, C. B., Mozzati, M. C., Paleari, A., Massarotti, V., Bini, M., and Capsoni, D. (2000). Magnetic evidence of different environments of manganese ions in Mn-substituted strontium titanate. Solid State Commun. 114, 617–622. doi: 10.1016/s0038-1098(00)00121-6
Blazey, K. W., Cabrera, J. M., and Müller, K. A. (1983). Oxygen vacancy-transition metal-ion impurity association in SrTiO3. Solid State Commun. 45, 903–906. doi: 10.1016/0038-1098(83)90332-0
Bykov, I., Makarova, M., Trepakov, V., Dejneka, A., Yurchenko, L., Yurchenko, L., et al. (2013). Intrinsic and impurity defects in chromium-doped SrTiO3 nanopowders: EPR and NMR study. Phys. Status Solidi B 250, 821–824. doi: 10.1002/pssb.201200871
Chan, N.-H., Sharma, R. K., and Smyth, D. M. (1981). Nonstoichiometry in SrTiO3. J. Electrochem. Soc. 128, 1762–1769. doi: 10.1149/1.2127727
Choudhury, D., Mukherjee, S., Mandal, P., Sundaresan, A., Waghmare, U. V., Bhattacharjee, S., et al. (2011). Tuning of dielectric properties and magnetism of SrTiO3 by site-specific doping of Mn. Phys. Rev. B 84:125124. doi: 10.1103/PhysRevB.84.125124
Choudhury, D., Pal, B., Sharma, A., Bhat, S. V., and Sarma, D. D. (2013). Magnetization in electron- and Mn-doped SrTiO3. Sci. Rep. 3:1433. doi: 10.1038/srep01433
Eaton, G. R., Eaton, S. S., Barr, D. P., and Weber, R. T. (2010). Quantitative EPR. Vienna: Spring-Verlag/Wien. doi: 10.1007/978-3-211-92948-3
Faughnan, B. W. (1971). Photochromism in transition-metal-doped SrTiO3. Phys. Rev. B 4, 3623–3636. doi: 10.1103/PhysRevB.4.3623
Harrigan, W. L., and Kittilstved, K. R. (2018). Reversible modulation of the Cr3+ spin dynamics in colloidal SrTiO3 nanocrystals. J. Phys. Chem. C 122, 26652–26657. doi: 10.1021/acs.jpcc.8b08680
Harrigan, W. L., Michaud, S. E., Lehuta, K. A., and Kittilstved, K. R. (2016). Tunable electronic structure and surface defects in chromium-doped colloidal SrTiO3−δ nanocrystals. Chem. Mater. 28, 430–433. doi: 10.1021/acs.chemmater.6b00049
Ishii, T., Kato, H., and Kudo, A. (2004). H2 evolution from an aqueous methanol solution on SrTiO3 photocatalysts codoped with chromium and tantalum ions under visible light irradiation. J. Photochem. Photobiol. A Chem. 163, 181–186. doi: 10.1016/S1010-6030(03)00442-8
Janotti, A., Jalan, B., Stemmer, S., and Van de Walle, C. G. (2012). Effects of doping on the lattice parameter of SrTiO3. Appl. Phys. Lett. 100:262104. doi: 10.1063/1.4730998
Kamalasanan, M. N., Deepak Kumar, N., and Chandra, S. (1993). Structural, optical, and dielectric properties of sol-gel derived SrTiO3 thin films. J. Appl. Phys. 74, 679–686. doi: 10.1063/1.355230
Kato, H., and Kudo, A. (2002). Visible-light-response and photocatalytic activities of TiO2 and SrTiO3 photocatalysts codoped with antimony and chromium. J. Phys. Chem. B 106, 5029–5034. doi: 10.1021/jp0255482
Kato, H., Sasaki, Y., Shirakura, N., and Kudo, A. (2013). Synthesis of highly active rhodium-doped SrTiO3 powders in Z-scheme systems for visible-light-driven photocatalytic overall water splitting. J. Mater. Chem. A 1, 12327–12333. doi: 10.1039/c3ta12803b
Khomenko, V. M., Langer, K., Rager, H., and Fett, A. (1998). Electronic absorption by Ti3+ ions and electron delocalization in synthetic blue rutile. Phys. Chem. Miner. 25, 338–346. doi: 10.1007/s002690050124
Kozuka, Y., Hikita, Y., Bell, C., and Hwang, H. Y. (2010). Dramatic mobility enhancements in doped SrTiO3 thin films by defect management. Appl. Phys. Lett. 97:012107. doi: 10.1063/1.3457994
Kutty, T. R. N., Gomathi Devi, L., and Murugaraj, P. (1986). The change in oxidation state of Mn ions in semiconducting BaTiO3 and SrTiO3 around the phase transition temperatures. Mater. Res. Bull. 21, 1093–1102. doi: 10.1016/0025-5408(86)90225-4
La Mattina, F., Bednorz, J. G., Alvarado, S. F., Shengelaya, A., and Keller, H. (2008). Detection of charge transfer processes in Cr-doped SrTiO3 single crystals. Appl. Phys. Lett. 93:022102. doi: 10.1063/1.2959059
Lehuta, K. A., Haldar, A., Zhou, D., and Kittilstved, K. R. (2017). Spectroscopic study of the reversible chemical reduction and reoxidation of substitutional Cr ions in Sr2TiO4. Inorg. Chem. 56, 9177–9184. doi: 10.1021/acs.inorgchem.7b01210
Lehuta, K. A., and Kittilstved, K. R. (2016). Reversible control of the chromium valence in chemically reduced Cr-doped SrTiO3 bulk powders. Dalton. Trans. 45, 10034–10041. doi: 10.1039/c6dt00706f
Mattheiss, L. F. (1972). Effect of the 110°K Phase transition on the SrTiO3 conduction bands. Phys. Rev. B 6, 4740–4753. doi: 10.1103/PhysRevB.6.4740
Middey, S., Meneghini, C., and Ray, S. (2012). Evidence of oxygen-vacancy-induced ferromagnetic order in single crystal Mn-doped SrTiO3. Appl. Phys. Lett. 101:042406. doi: 10.1063/1.4738785
Mitchell, R. H., Chakhmouradian, A. R., and Woodward, P. M. (2000). Crystal chemistry of perovskite-type compounds in the tausonite-loparite series, (Sr1–2xNaxLax)TiO3. Phys. Chem. Miner. 27, 583–589. doi: 10.1007/s002690000103
Mitra, C., Lin, C., Robertson, J., and Demkov, A. A. (2012). Electronic structure of oxygen vacancies in SrTiO3 and LaAlO3. Phys. Rev. B 86:155105. doi: 10.1103/PhysRevB.86.155105
Müller, K., and Burkard, H. (1979). SrTiO3: an intrinsic quantum paraelectric below 4 K. Phys. Rev. B 19, 3593–3602. doi: 10.1103/PhysRevB.19.3593
Müller, K. A. (1959). Electron paramagnetic resonance of manganese IV in SrTiO3. Phys. Rev. Lett. 2, 341–343. doi: 10.1103/PhysRevLett.2.341
Sasaki, Y., Nemoto, H., Saito, K., and Kudo, A. (2009). Solar water splitting using powdered photocatalysts driven by Z-schematic interparticle electron transfer without an electron mediator. J. Phys. Chem. C 113, 17536–17542. doi: 10.1021/jp907128k
Savinov, M., Trepakov, V. A., Syrnikov, P. P., Železný, V., Pokorný, J., Dejneka, A., et al. (2008). Dielectric properties of Mn doped SrTiO3. J. Phys. Cond. Matter 20:095221. doi: 10.1088/0953-8984/20/9/095221
Serway, R. A., Berlinger, W., Müller, K. A., and Collins, R. W. (1977). Electron paramagnetic resonance of three manganese centers in reduced SrTiO3. Phys. Rev. B 16, 4761–4768. doi: 10.1103/PhysRevB.16.4761
Shannon, R. D. (1976). Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A32, 751–767. doi: 10.1107/S0567739476001551
Stoll, S., and Schweiger, A. (2006). EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55. doi: 10.1016/j.jmr.2005.08.013
Tan, H., Zhao, Z., Zhu, W. B., Coker, E. N., Li, B., Zheng, M., et al. (2014). Oxygen vacancy enhanced photocatalytic activity of pervoskite SrTiO3. ACS Appl. Mater. Interfaces 6, 19184–19190. doi: 10.1021/am5051907
Thanh, T. D., Phan, T. L., Oanh, L. M., Minh, N. V., Lee, J. S., and Yu, S. C. (2014). Influence of Mn doping on the crystal structure, and optical and magnetic properties of SrTiO3 compounds. IEEE Trans. Magn. 50, 1–4. doi: 10.1109/TMAG.2014.2304562
Wang, D., Ye, J., Kako, T., and Kimura, T. (2006). Photophysical and photocatalytic properties of SrTiO3 doped with Cr cations on different sites. J. Phys. Chem. B 110, 15824–15830. doi: 10.1021/jp062487p
Wang, Q., Hisatomi, T., Ma, S. S. K., Li, Y., and Domen, K. (2014). Core/shell structured La- and Rh-codoped SrTiO3 as a hydrogen evolution photocatalyst in Z-scheme overall water splitting under visible light irradiation. Chem. Mater. 26, 4144–4150. doi: 10.1021/cm5011983
Weaver, H. E. (1959). Dielectric properties of single crystals of SrTiO3 at low temperatures. J. Phys. Chem. Solids 11, 274–277. doi: 10.1016/0022-3697(59)90226-4
Wild, R. L., Rockar, E. M., and Smith, J. C. (1973). Thermochromism and electrical conductivity in doped SrTiO3. Phys. Rev. B 8, 3828–3835. doi: 10.1103/PhysRevB.8.3828
Yamanaka, T., Hirai, N., and Komatsu, Y. (2002). Structure change of Ca1−xSrxTiO3 perovskite with composition and pressure. Am. Miner. 87, 1183–1189. doi: 10.2138/am-2002-8-917
Keywords: electron paramagnetic resonance, oxidation state, manganese, strontium titanate, inorganic materials
Citation: Mansoor H, Harrigan WL, Lehuta KA and Kittilstved KR (2019) Reversible Control of the Mn Oxidation State in SrTiO3 Bulk Powders. Front. Chem. 7:353. doi: 10.3389/fchem.2019.00353
Received: 19 February 2019; Accepted: 29 April 2019;
Published: 22 May 2019.
Edited by:
Luís D. Carlos, University of Aveiro, PortugalReviewed by:
Emre Erdem, Sabanci University, TurkeyHelene Serier-Brault, UMR6502 Institut des Matériaux Jean Rouxel (IMN), France
Copyright © 2019 Mansoor, Harrigan, Lehuta and Kittilstved. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin R. Kittilstved, a2l0dGlsc3R2ZWRAY2hlbS51bWFzcy5lZHU=