 Giulio Rastelli
Giulio Rastelli Luca Pinzi
Luca Pinzi- Department of Life Sciences, University of Modena and Reggio Emilia, Modena, Italy
High-throughput docking is an established computational screening approach in drug design. This methodology enables a rapid identification of biologically active hit compounds, providing an efficient and cost-effective complement or alternative to experimental high-throughput screenings. However, limitations inherent to the methodology make docking results inevitably approximate. Two major Achille's heels include the use of approximated scoring functions and the limited sampling of the ligand-target complexes. Therefore, docking results require careful evaluation and further post-docking analyses. In this article, we will overview our approach to post-docking analysis in virtual screenings. BEAR (Binding Estimation After Refinement) was developed as a post-docking processing tool that refines docking poses by means of molecular dynamics (MD) and then rescores the ligands based on more accurate scoring functions (MM-PB(GB)SA). The tool has been validated and used prospectively in drug discovery applications. Future directions regarding refinement and rescoring in virtual screening are discussed.
Introduction
High-throughput screening (HTS) is a widely used method for the discovery of biologically active hits. However, the high costs and the low hit rates characterizing such experiments often make HTS not affordable for academic labs or small companies (Sliwoski et al., 2014). As a consequence, high-throughput docking screenings represent an attractive alternative (Irwin and Shoichet, 2016). Structure-based virtual screenings (SBVSs) require the knowledge of the three-dimensional structure of the target of interest, as well as the access to large libraries of small molecules available in public databases (Kar and Roy, 2013; Rastelli, 2013). Docking programs generate binding poses of compounds in the active site of a target and evaluate the ligand binding strength by means of scoring functions (Lengauer and Rarey, 1996; Kitchen et al., 2004). Several docking software relying on different algorithms have been developed for virtual screening so far (Rarey et al., 1996; Morris et al., 1998; Friesner et al., 2004; Sánchez-Linares et al., 2012). However, although remarkable improvements have been obtained along the years, several drawbacks and limitations still exist (Huang and Zou, 2010; Rastelli, 2013). First of all, sampling the conformational space accessible to ligand-target complexes in an induced-fit context is a difficult and target-dependent task. To help overcoming such limitations, several in silico strategies including molecular dynamics or induced fit strategies have been introduced (Sherman et al., 2006; Nabuurs et al., 2007; Caporuscio and Rastelli, 2016). Secondly, docking scores and experimental binding affinities usually do not correlate, because screening large numbers of compounds in a reasonable time requires the use of approximate scoring functions. Together, the two effects imply that a variable number of false-positive and false-negative hits populate the ranked lists, which then require careful evaluation and further post-docking analyses. Hence, it has become general opinion that docking results should be improved by means of more rigorous post-docking processing strategies. Several post-processing strategies have been developed to overcome docking limitations over the past decades. In particular, methods based on binding free energy estimations have demonstrated to provide higher hit rates and to be more suitable for ranking cognate ligands in virtual screening (Hou et al., 2011; Genheden and Ryde, 2015; Pu et al., 2017), the predicted binding free energy usually correlating better with experimental data (Brandsdal et al., 2003). One of the first reported energy-based methods is MM-PB(GB)SA, which was developed to more accurately assess the relative free energy of binding for a given macromolecular system from molecular dynamics simulations (Kollman et al., 2000). This method represented a remarkable step forward to the obtainment of in silico predicted binding affinities that are in good agreement with experiments. In fact, it was extensively used to evaluate the free energy of binding for a number of complexes in the last years (Gohlke et al., 2003; Hou and Yu, 2007; Ferri et al., 2009; Yang et al., 2012). For example, it was successfully used for identifying residue hot-spots outside the binding interface of the Ras–Raf and Ras–RalGDS protein-protein complexes, discussing also their implications for an allosteric activation of the proteins (Gohlke et al., 2003). More recently, this method was also employed for predicting binding affinities of few inhibitors of HIV-1 protease and to help rationalize drug resistance caused by the mutations on the enzyme binding site (Hou and Yu, 2007). However, it should also be noted that MM-PB(GB)SA results are dependent on the employed parameters and receptor structures used in the calculations (Xu et al., 2013; Sun et al., 2014, 2018; Genheden and Ryde, 2015). More accurate free energy-based methods have also been reported (Brandsdal et al., 2003; Jorgensen and Thomas, 2008; Parenti and Rastelli, 2012; Limongelli et al., 2013; De Vivo et al., 2016). Among them, it is worth mentioning the Free Energy Perturbation (FEP) method, which allows to estimate the free energy of binding of a ligand to a protein by decomposing the system through a series of “alchemical transformations” (Jorgensen and Thomas, 2008; De Vivo et al., 2016). More recently, funnel-metadynamics (FM) methods that use a funnel-shaped potential limiting the sampling space available for a ligand to bind/unbind to a protein have been proposed (Limongelli et al., 2013; De Vivo et al., 2016). However, although these methods demonstrated to accurately estimate ligand binding, they are time-consuming and therefore not suitable for virtual screening rescoring of large databases. With the aim of improving ligand-binding estimations of docking complexes at reasonable computational costs, we developed Binding Estimation After Refinement (BEAR) (Rastelli et al., 2009). BEAR is an automated post-docking tool based on conformational refinement of docking poses with molecular dynamics followed by a more accurate prediction of binding free energies performed with MM-PBSA and MM-GBSA, which take into account desolvation energies (Kuhn et al., 2005; Lyne et al., 2006; Rastelli et al., 2010; Genheden and Ryde, 2015). As it allows accurately rescoring docking poses in reasonable times, BEAR can be considered an efficient tool that could be routinely used for virtual screening. In this article, we will briefly describe the BEAR tool, providing an overview of the validation studies performed so far. Finally, we will describe its prospective applications in drug discovery campaigns and comment on future directions of refinement and rescoring methods.
The BEAR Tool
The BEAR workflow (Figure 1; Rastelli et al., 2009) consists of an initial pre-processing step in which hydrogen atoms are added to the protein, atomic charges (AM1-BCC) are calculated for the docked molecules, and missing force-field parameters are assigned. Then, topologies for the ligand, the protein, and the ligand-protein complex are built. In particular, ligand atom types are assigned according to the Generalized Amber Force Field (GAFF) (Wang et al., 2004), while, the atom types and charges of amino acids are assigned according to the Amber ff03 force field (Duan et al., 2003). The following iterative three steps procedure is based on molecular mechanics (MM) and molecular dynamics (MD) cycles. In particular, an initial MM energy minimization of the whole protein–ligand complex is performed, followed by a short MD simulation where the ligand is allowed to move, and a final re-minimization of the entire complex. All the minimization tasks are performed through 2000 steps without restraints, and with a distance-dependent dielectric constant ε = 4r and a cutoff of 12 Å. The MD simulation is performed at 300 K for 100 ps, with the SHAKE parameter turned on and a time-step of 2.0 fs. This protocol allows evaluating the reliability of the predicted docking complex and to establish potential additional ligand-protein interactions resulting from the structural refinement of the complex, thus obtaining more accurate binding energy predictions. After refinement of the complex, the free energy of binding of the ligand is calculated with the MM-PBSA and MM-GBSA methods. These operations are implemented with the use of AMBER modules (Case et al., 2018). Further details about the BEAR tool are described in Rastelli et al. (2009).
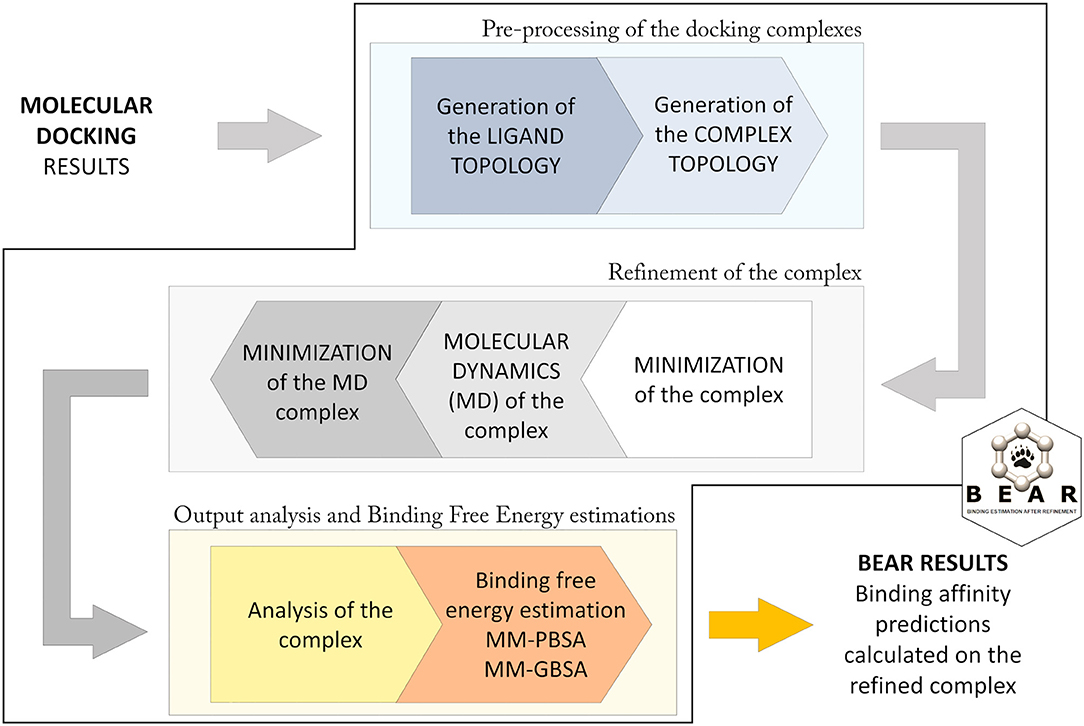
Figure 1. Computational workflow applied in BEAR.
Benchmarking Studies
The post-docking tool BEAR has been extensively validated in various test cases. First of all, the MM/MD protocol described above was investigated on a series of aldose reductase inhibitors with notable chemical diversity. Remarkably, the calculated free energies of binding after refinement of ligand-protein complexes resulted to be highly correlated with experimental affinities. This study demonstrated that different classes of aldose reductase inhibitors could be accurately rescored with our procedure (Ferrari et al., 2007). Extensive validations were also made on Plasmodium falciparium dihydrofolate reductase (PfDHFR). These simulations aimed at evaluating the performance of BEAR in virtual screening settings of different size and complexity. Firstly, BEAR performed well in discriminating 14 known inhibitors of PfDHFR from the 1,720 compounds included in the National Cancer Institute diversity database (Rastelli et al., 2009). The achieved performances were clearly superior to those of AutoDock (Morris et al., 1998), demonstrating that rescoring of the predicted docking poses with BEAR heavily improved SBVS results. In a second experiment, enrichment factors (EFs) obtained with BEAR were evaluated by seeding 201 known inhibitors with 7,150 decoys as contained in the DHFR data set of the Directory of Useful Decoys database (Mysinger et al., 2012). Moreover, the same set of ligands was also seeded into the 1.5 million compounds belonging to the lead-like subset of the ZINC database (Irwin et al., 2012), this latter benchmark reflecting a typical virtual screening setting. In both cases, BEAR refinement and rescoring yielded significantly higher EFs compared to docking (Degliesposti et al., 2011). This was also an opportunity for fine-tuning the BEAR parameters, and thus achieving good performances at reasonable computational costs.
The BEAR performance was also assessed on biological targets characterized by flexible binding sites and/or containing water molecules in the binding pocket. Such targets are particularly challenging for SBVS (Elokely and Doerksen, 2013). In fact, certain ligand chemotypes could fit with favorable scores into certain protein conformations but not in others, thus hampering their identification in a virtual screening. To evaluate whether docking into multiple protein conformations (ensemble docking) instead of using a single representative structure would improve BEAR predictions for “difficult” targets (Sgobba et al., 2012), we investigated targets of different families (adenosine deaminase, factor Xa, estrogen receptor, thymidine kinase, aldose reductase, and enoyl ACP reductase). Interestingly, a comparative analysis of the EFs obtained for different proteins and multiple protein conformations revealed that the application of BEAR was able in several cases to yield higher EFs compared to docking. However, in challenging targets such as adenosine deaminase and enoyl ACP reductase, all scoring functions failed in yielding high EFs. This effect was attributed to difficulties in predicting correct ligand binding modes in these two targets. In particular, when the docked pose was completely wrong, for example head-to-tail with respect to the correct binding mode, the MM/MD refinement stage was not enough to turn the binding mode into the correct one. Therefore, the advantage of using MM-PBSA and MM-GBSA in prioritizing active compounds is dependent on the obtainment of correct binding modes, which makes the refinement and rescoring procedures intimately connected.
More recently, BEAR was also applied to screen ligands of G-protein coupled receptors (GPCRs) with known crystal structure, namely β2-adrenergic (β2), adenosine A2A (A2A), dopamine D3 (D3), and histamine H1 (H1) receptors (Anighoro and Rastelli, 2013). Results were analyzed in terms of the ability to recognize known antagonists from decoys, as well as to predict correct binding modes. In all cases except for A2A, significant or dramatic improvements of EFs were obtained after the application of BEAR. A2A was challenging because antagonists participated to an extended water-mediated hydrogen bond network. Interestingly, explicit consideration of a suitable number of these structural waters significantly improved the predictions. This finding is in line with the fact that MM-PB(GB)SA calculations do not explicitly consider water molecules mediating ligand-protein interactions, and binding mode predictions heavily depend on the presence of bridging water molecules participating to hydrogen bond networks. For all GPCRs, a more accurate account of desolvation effects, such as the one performed by MM-PBSA, is important to accurately predict the affinity of the protonated biogenic amines. We also found that five known H1 and D3 receptor antagonists were top-scored and ranked well in each of the two target screenings, prospecting for the first time the utility of post-docking tools in multi-target drug design (Anighoro et al., 2014). Indeed, as the free energy of binding calculated by BEAR allows to more accurately predicting the affinity of ligands for their target(s) with a reasonable computer time investment, we envision that in silico strategies embedding this tool can be useful to allow the identification of ligands with the desired multi-target profiles.
Prospective Validations
The BEAR workflow has also been implemented in the computing GRID infrastructure EGEE, as part of the WISDOM (Wide in silico DOcking on Malaria) initiative against malaria (Kasam et al., 2009). Then, it was deployed to perform virtual screenings against antimalarial drug targets. One massive data challenge was performed on Plasmepsin II, an aspartic protease involved in the metabolism of P. falciparum (Degliesposti et al., 2009). In this work, BEAR was used to refine and rescore the 5,000 top-scoring compounds docked with FlexX (Rarey et al., 1996). Then, the final step of candidates' selection was performed on the top 200 compounds resulting from both MM-PBSA and MM-GBSA ranked lists. Interestingly, an analysis of the BEAR ranked lists, together with an inspection of the protein-ligand complexes and a similarity-based clustering of the ligands allowed selecting 30 compounds belonging to 5 different chemotypes as potential Plasmepsin II inhibitors. Remarkably, 26 of them were active, resulting in an impressive hit rate of 87%, and some of the compounds displayed nanomolar inhibitory activity.
More recently, BEAR was successfully applied in a virtual screening campaign that allowed the identification of the first-in-class allosteric inhibitors of CDK2 (Rastelli et al., 2014). In this work, around 600.000 commercially available compounds were screened against a crystal structure of CDK2 with an open type III allosteric pocket, by using AutoDock for docking and BEAR for post-docking analyses. The adopted virtual screening protocol led to the identification of 7 allosteric ligands of CDK2, providing a hit rate of 20%. Interestingly, the most potent compound was able to selectively inhibit CDK2-mediated Retinoblastoma phosphorylation, confirming that its mechanism of action is fully compatible with a selective inhibition of CDK2 phosphorylation in cells. Moreover, some of these ligands inhibited the proliferation of MDA-MB231 and ZR-75-1 breast cancer cells with IC50 values in the low micromolar range (Rastelli et al., 2014).
Final Remarks
Although many progresses have been made in molecular docking, limitations deriving from the use of rigid protein conformations and of approximate scoring functions often impair virtual screening results. Therefore, docking results require careful evaluation and further post-docking analyses. BEAR is a post-processing tool that performs binding free energy estimations after MM and MD refinement of docking complexes. Our previous studies demonstrated that BEAR performed well in a number of benchmarking investigations, as well as in discovering biologically active hits in different prospective virtual screening campaigns. Moreover, as it not computationally demanding as other free energy-based methods, it constitutes a reasonable compromise to obtain accurate rescoring of ligands at reasonable computational costs. One might argue that the application of more accurate workflows would require longer computing times with respect to docking. This is especially true considering the recent contributions provided by high performance computing systems to molecular docking, which enable the screening of millions of compounds in a reasonable time (Perez-Sanchez and Wenzel, 2011; Guerrero et al., 2012; Dong et al., 2018). However, future advances in hardware and software will help circumventing such limitation (De Vivo et al., 2016; Wang et al., 2018). Moreover, further advances in our ability to correctly estimate entropies of binding, which are usually not considered in the calculations, will certainly improve post-docking tools, and binding free energy predictions in general. The implementation of enhanced sampling MD protocols in post-docking protocols is another possibility that may enable a more efficient sampling of ligand-protein complexes. Because free energy predictions are heavily dependent on correct binding modes, this may have dramatic consequences on our ability to predict active ligands in virtual screenings. Another interesting question is how to further increase hit rates while enabling post-docking tools to identify significantly vs. moderately active hits. This is an important aspect that would make the subsequent hit-to-lead optimization much easier. Exponential consensus ranking approaches such as the one developed by Palacio-Rodríguez et al. (2019) could be of help, for example to favorably exploit both MM-PBSA and MM-GBSA ranked lists, which generally differ.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
GR conceived and wrote the study. LP contributed in writing and editing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Anighoro, A., Bajorath, J. J., and Rastelli, G. (2014). Polypharmacology: challenges and opportunities in drug discovery. J. Med. Chem. 57, 7874–7887. doi: 10.1021/jm5006463
Anighoro, A., and Rastelli, G. (2013). Enrichment factor analyses on G-protein coupled receptors with known crystal structure. J. Chem. Inf. Model. 53, 739–743. doi: 10.1021/ci4000745
Brandsdal, B. O., Österberg, F., Almlöf, M., Feierberg, I., Luzhkov, V. B., and Åqvist, J. B. (2003). Free energy calculations and ligand binding in protein simulations. Adv. Protein Chem. 66, 123–158. doi: 10.1016/S0065-3233(03)66004-3
Caporuscio, F., and Rastelli, G. (2016). Exploiting computationally derived out-of-the-box protein conformations for drug design. Future Med. Chem. 8, 1887–1897. doi: 10.4155/fmc-2016-0098
Case, D. A., Ben-Shalom, I. Y., Brozell, S. R., Cerutti, D. S., Cheatham, T. E. III., Cruzeiro, V. W. D., et al. (2018). AMBER 2018. University of California, San Francisco, CA. Available online at: http://www.ambermd.org/ (accessed April 15, 2019).
De Vivo, M., Masetti, M., Bottegoni, G., and Cavalli, A. (2016). Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 59, 4035–4061. doi: 10.1021/acs.jmedchem.5b01684
Degliesposti, G., Kasam, V., Da Costa, A., Kang, H. K., Kim, N., Kim, D. W., et al. (2009). Design and discovery of plasmepsin II inhibitors using an automated workflow on large-scale grids. ChemMedChem 4, 1164–1173. doi: 10.1002/cmdc.200900111
Degliesposti, G., Portioli, C., Parenti, M. D., and Rastelli, G. (2011). BEAR, a novel virtual screening methodology for drug discovery. J. Biomol. Screen. 16, 129–133. doi: 10.1177/1087057110388276
Dong, D., Xu, Z., Zhong, W., and Peng, S. (2018). Parallelization of molecular docking: a review. Curr. Top. Med. Chem. 18, 1015–1028. doi: 10.2174/1568026618666180821145215
Duan, Y., Wu, C., Chowdhury, S., Lee, M. C., Xiong, G., Zhang, W., et al. (2003). A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012. doi: 10.1002/jcc.10349
Elokely, K. M., and Doerksen, R. J. (2013). Docking challenge: protein sampling and molecular docking performance. J. Chem. Inf. Model. 53, 1934–1945. doi: 10.1021/ci400040d
Ferrari, A. M., Degliesposti, G., Sgobba, M., and Rastelli, G. (2007). Validation of an automated procedure for the prediction of relative free energies of binding on a set of aldose reductase inhibitors. Bioorg. Med. Chem. 15, 7865–7877. doi: 10.1016/j.bmc.2007.08.019
Ferri, N., Corsini, A., Bottino, P., Clerici, F., and Contini, A. (2009). Virtual screening approach for the identification of New Rac1 inhibitors. J. Med. Chem. 52, 4087–4090. doi: 10.1021/jm8015987
Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., et al. (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749. doi: 10.1021/jm0306430
Genheden, S., and Ryde, U. (2015). The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10, 449–461. doi: 10.1517/17460441.2015.1032936
Gohlke, H., Kiel, C., and Case, D. A. (2003). Insights into protein–protein binding by binding free energy calculation and free energy decomposition for the Ras–Raf and Ras–RalGDS complexes. J. Mol. Biol. 330, 891–913. doi: 10.1016/S0022-2836(03)00610-7
Guerrero, G. D., Perez-S'nchez, H. E., Cecilia, J. M., and Garcia, J. M. (2012). “Parallelization of virtual screening in drug discovery on massively parallel architectures,” in 2012 20th Euromicro International Conference on Parallel, Distributed and Network-based Processing (Garching), 588–595. doi: 10.1109/PDP.2012.26
Hou, T., Wang, J., Li, Y., and Wang, W. (2011). Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 32, 866–877. doi: 10.1002/jcc.21666
Hou, T., and Yu, R. (2007). Molecular dynamics and free energy studies on the wild-type and double mutant HIV-1 protease complexed with amprenavir and two amprenavir-related inhibitors: mechanism for binding and drug resistance. J. Med. Chem. 50, 1177–1188. doi: 10.1021/jm0609162
Huang, S.-Y., and Zou, X. (2010). Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 11, 3016–3034. doi: 10.3390/ijms11083016
Irwin, J. J., and Shoichet, B. K. (2016). Docking screens for novel ligands conferring new biology. J. Med. Chem. 59, 4103–4120. doi: 10.1021/acs.jmedchem.5b02008
Irwin, J. J., Sterling, T., Mysinger, M. M., Bolstad, E. S., and Coleman, R. G. (2012). ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 52, 1757–1768. doi: 10.1021/ci3001277
Jorgensen, W. L., and Thomas, L. L. (2008). Perspective on free-energy perturbation calculations for chemical equilibria. J. Chem. Theory Comput. 4, 869–876. doi: 10.1021/ct800011m
Kar, S., and Roy, K. (2013). How far can virtual screening take us in drug discovery? Expert Opin. Drug Discov. 8, 245–261. doi: 10.1517/17460441.2013.761204
Kasam, V., Salzemann, J., Botha, M., Dacosta, A., Degliesposti, G., Isea, R., et al. (2009). WISDOM-II: screening against multiple targets implicated in malaria using computational grid infrastructures. Malar. J. 8:88. doi: 10.1186/1475-2875-8-88
Kitchen, D. B., Decornez, H., Furr, J. R., and Bajorath, J. (2004). Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discov. 3, 935–949. doi: 10.1038/nrd1549
Kollman, P. A., Massova, I., Reyes, C., Kuhn, B., Huo, S., Chong, L., et al. (2000). Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897. doi: 10.1021/ar000033j
Kuhn, B., Gerber, P., Schulz-Gasch, T., and Stahl, M. (2005). Validation and use of the MM-PBSA approach for drug discovery. J. Med. Chem. 48, 4040–4048. doi: 10.1021/jm049081q
Lengauer, T., and Rarey, M. (1996). Computational methods for biomolecular docking. Curr. Opin. Struct. Biol. 6, 402–406. doi: 10.1016/S0959-440X(96)80061-3
Limongelli, V., Bonomi, M., and Parrinello, M. (2013). Funnel metadynamics as accurate binding free-energy method. Proc. Natl. Acad. Sci. U.S.A. 110, 6358–6363. doi: 10.1073/pnas.1303186110
Lyne, P. D., Lamb, M. L., and Saeh, J. C. (2006). Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J. Med. Chem. 49, 4805–4808. doi: 10.1021/jm060522a
Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K., et al. (1998). Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19, 1639–1662.
Mysinger, M. M., Carchia, M., Irwin, J. J., and Shoichet, B. K. (2012). Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 55, 6582–6594. doi: 10.1021/jm300687e
Nabuurs, S. B., Wagener, M., and de Vlieg, J. (2007). A flexible approach to induced fit docking. J. Med. Chem. 50, 6507–6518. doi: 10.1021/jm070593p
Palacio-Rodríguez, K., Lans, I., Cavasotto, C. N., and Cossio, P. (2019). Exponential consensus ranking improves the outcome in docking and receptor ensemble docking. Sci. Rep. 9:5142. doi: 10.1038/s41598-019-41594-3
Parenti, M.D., and Rastelli, G. (2012). Advances and applications of binding affinity prediction methods in drug discovery. Biotechnol. Adv. 30, 244–250. doi: 10.1016/j.biotechadv.2011.08.003
Perez-Sanchez, H., and Wenzel, W. (2011). Optimization methods for virtual screening on novel computational architectures. Curr. Comput. Aided. Drug Des. 7, 44–52. doi: 10.2174/157340911793743565
Pu, C., Yan, G., Shi, J., and Li, R. (2017). Assessing the performance of docking scoring function, FEP, MM-GBSA, and QM/MM-GBSA approaches on a series of PLK1 inhibitors. Medchemcomm 8, 1452–1458. doi: 10.1039/c7md00184c
Rarey, M., Kramer, B., Lengauer, T., and Klebe, G. (1996). A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 261, 470–489. doi: 10.1006/jmbi.1996.0477
Rastelli, G. (2013). Emerging topics in structure-based virtual screening. Pharm. Res. 30, 1458–1463. doi: 10.1007/s11095-013-1012-9
Rastelli, G., Anighoro, A., Chripkova, M., Carrassa, L., and Broggini, M. (2014). Structure-based discovery of the first allosteric inhibitors of cyclin-dependent kinase 2. Cell Cycle 13, 2296–2305. doi: 10.4161/cc.29295
Rastelli, G., Degliesposti, G., Del Rio, A., and Sgobba, M. (2009). Binding estimation after refinement, a new automated procedure for the refinement and rescoring of docked ligands in virtual screening. Chem. Biol. Drug Des. 73, 283–286. doi: 10.1111/j.1747-0285.2009.00780.x
Rastelli, G., Del Rio, A., Degliesposti, G., Sgobba, M., Rio, A., Del Degliesposti, G., et al. (2010). Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 31, 797–810. doi: 10.1002/jcc.21372
Sánchez-Linares, I., Pérez-Sánchez, H., Cecilia, J. M., and García, J. M. (2012). High-throughput parallel blind virtual screening using BINDSURF. BMC Bioinformatics 13:S13. doi: 10.1186/1471-2105-13-S14-S13
Sgobba, M., Caporuscio, F., Anighoro, A., Portioli, C., and Rastelli, G. (2012). Application of a post-docking procedure based on MM-PBSA and MM-GBSA on single and multiple protein conformations. Eur. J. Med. Chem. 58, 431–440. doi: 10.1016/j.ejmech.2012.10.024
Sherman, W., Beard, H. S., and Farid, R. (2006). Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 67, 83–84. doi: 10.1111/j.1747-0285.2005.00327.x
Sliwoski, G., Kothiwale, S., Meiler, J., and Lowe, E. W. (2014). Computational methods in drug discovery. Pharmacol. Rev. 66, 334–395. doi: 10.1124/pr.112.007336
Sun, H., Duan, L., Chen, F., Liu, H., Wang, Z., Pan, P., et al. (2018). Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 20, 14450–14460. doi: 10.1039/C7CP07623A
Sun, H., Li, Y., Shen, M., Tian, S., Xu, L., Pan, P., et al. (2014). Assessing the performance of MM/PBSA and MM/GBSA methods. 5. Improved docking performance using high solute dielectric constant MM/GBSA and MM/PBSA rescoring. Phys. Chem. Chem. Phys. 16, 22035–22045. doi: 10.1039/C4CP03179B
Wang, C., Greene, D., Xiao, L., Qi, R., and Luo, R. (2018). Recent developments and applications of the MMPBSA method. Front. Mol. Biosci. 4:87. doi: 10.3389/fmolb.2017.00087
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., Case, D. A., Wang, J., et al. (2004). Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174. doi: 10.1002/jcc.20035
Xu, L., Sun, H., Li, Y., Wang, J., and Hou, T. (2013). Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J. Phys. Chem. B 117, 8408–8421. doi: 10.1021/jp404160y
Keywords: docking, post-docking, virtual screening, molecular dynamics, BEAR, binding free energy
Citation: Rastelli G and Pinzi L (2019) Refinement and Rescoring of Virtual Screening Results. Front. Chem. 7:498. doi: 10.3389/fchem.2019.00498
Received: 23 April 2019; Accepted: 28 June 2019;
Published: 11 July 2019.
Edited by:
Simone Brogi, University of Pisa, ItalyReviewed by:
Demet Akten, Kadir Has University, TurkeyHoracio Pérez-Sánchez, Catholic University San Antonio of Murcia, Spain
Tingjun Hou, Zhejiang University, China
Copyright © 2019 Rastelli and Pinzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giulio Rastelli, Z2l1bGlvLnJhc3RlbGxpQHVuaW1vcmUuaXQ=