 Ol'ha O. Brovarets'
Ol'ha O. Brovarets' Timothy A. Oliynyk1†
Timothy A. Oliynyk1† Dmytro M. Hovorun
Dmytro M. Hovorun- 1Department of Molecular and Quantum Biophysics, Institute of Molecular Biology and Genetics, National Academy of Sciences of Ukraine, Kyiv, Ukraine
- 2Department of Pharmacology, Bohomolets National Medical University, Kyiv, Ukraine
- 3Department of Molecular Biotechnology and Bioinformatics, Institute of High Technologies, Taras Shevchenko National University of Kyiv, Kyiv, Ukraine
- 4Department of Pathophysiology, Bohomolets National Medical University, Kyiv, Ukraine
For the first time, in this study with the use of QM/QTAIM methods we have exhaustively investigated the tautomerization of the biologically-important conformers of the G*·C* DNA base pair—reverse Löwdin G*·C*(rWC), Hoogsteen G*′·C*(H), and reverse Hoogsteen G*′·C*(rH) DNA base pairs—via the single (SPT) or double (DPT) proton transfer along the neighboring intermolecular H-bonds. These tautomeric reactions finally lead to the formation of the novel G·(rWC), C(rWC), G*′N2·C(rWC), C(H), and G*′N7·C(rH) DNA base mispairs. Gibbs free energies of activation for these reactions are within the range 3.64–31.65 kcal·mol−1 in vacuum under normal conditions. All TSs are planar structures (Cs symmetry) with a single exception—the essentially non-planar transition state TSG*·C*(rWC)↔G+·C−(rWC) (C1 symmetry). Analysis of the kinetic parameters of the considered tautomerization reactions indicates that in reality only the reverse Hoogsteen G*′·C*(rH) base pair undergoes tautomerization. However, the population of its tautomerised state G*′N7·C(rH) amounts to an insignificant value−2.3·10−17. So, the G*·C*(rWC), G*′·C*(H), and G*′·C*(rH) base pairs possess a permanent tautomeric status, which does not depend on proton mobility along the neighboring H-bonds. The investigated tautomerization processes were analyzed in details by applying the author's unique methodology—sweeps of the main physical and chemical parameters along the intrinsic reaction coordinate (IRC). In general, the obtained data demonstrate the tautomeric mobility and diversity of the G*·C* DNA base pair.
Introduction
The study of the tautomerization mechanisms of the hydrogen (H) bonded nucleotide base pair is an important topic of modern quantum biophysics, biochemistry, molecular, and structural biology (Sinden, 1994; Sponer and Lankas, 2006; Alkorta et al., 2018). For over 65 years, this area of research has been under the intense scrutiny of both theoretics and experimentators, since the establishment of the spatial organization of DNA and formulation of the so-called “tautomeric hypothesis of the origin of spontaneous point mutations (transitions and transversions)” (Watson and Crick, 1953a,b; Erdmann et al., 2014) for this biologically important macromolecule—carrier of the genetic information, which is transmitted from generation to generation.
Lately, this tautomeric hypothesis has been experiencing an era of renaissance (Brovarets' and Hovorun, 2018). Thus, for the first time, within the framework of this hypothesis the new structural mechanisms of the tautomerization of pairs of nucleotide bases have been discovered, in which the transition of bases within the base pair into the mutagenic tautomeric form is accompanied by a significant change in the geometry of the base pair itself (Brovarets' and Hovorun, 2009, 2015a,b,c,d,e, 2016, 2018).
However, at the studying of the nature of the mutagenic tautomerization of DNA bases, the researchers limited themselves to the A·T and G·C Watson-Crick base pairs (Löwdin, 1963, 1966; Florian et al., 1994; Gorb et al., 2004; Bertran et al., 2006; Brovarets' and Hovorun, 2014a,b). Now this problem is considered more complex with the involvement of several biologically important conformers of these pairs (Hoogsteen, 1963; Pous et al., 2008; Alvey et al., 2014; Brovarets' and Hovorun, 2014a,b; Nikolova et al., 2014; Acosta-Reyes et al., 2015; Poltev et al., 2016; Zhou, 2016; Szabat and Kierzek, 2017; Ye et al., 2017).
These observations do not only allow to penetrate deeper into the essence of the phenomenon being studied, but also to answer, in particular, on a biologically important question—“Why Nature has exactly chosen the Watson-Crick DNA base pairs as elementary structural units for the construction of genetic material?”
Nowadays, there is an explicit bias to the A·T DNA base pair at the investigations of this type. This is due to a large number of circumstances, which will be outlined and discussed below.
Thus, it is widely known that the classical A·T(WC) Watson-Crick DNA base pair (Brovarets' and Hovorun, 2014b) may acquire different biologically significant conformations with various organization of the three intermolecular H-bonds—reverse Watson-Crick A·T(rWC), Hoogsteen A·T(H), and reverse Hoogsteen A·T(rH), which have been comprehensively studied in the literature (Hoogsteen, 1963; Pous et al., 2008; Alvey et al., 2014; Brovarets' and Hovorun, 2014b; Nikolova et al., 2014; Acosta-Reyes et al., 2015; Poltev et al., 2016; Zhou, 2016; Szabat and Kierzek, 2017; Ye et al., 2017).
Thus, in particular, in previous works (Hoogsteen, 1963; Pous et al., 2008; Brovarets' and Hovorun, 2010, 2014b, 2015f; Brovarets', 2013a,b; Alvey et al., 2014; Nikolova et al., 2014; Acosta-Reyes et al., 2015; Poltev et al., 2016; Zhou, 2016; Szabat and Kierzek, 2017; Ye et al., 2017; Brovarets' et al., 2018a,b,c,d,e,f; Brovarets' and Tsiupa, 2019) by the methods of quantum chemistry we have investigated in details the potential (electronic) energy surface of each of the four biologically important A·T DNA base pairs—Watson-Crick A·T(WC), reverse Watson-Crick A·T(rWC), Hoogsteen A·T(H) and reverse Hoogsteen A·T(rH), leading to the novel conformational or tautomeric states of these base pairs. It was theoretically demonstrated that these A·T(WC/rWC/H/rH) base pairs possess unique ability to perform conformationally-tautomeric transition into the planar wobble A*·T(w), A·(w), and A·T*(w) base mispairs, non-planar wobble A·T(wWC), A·T(wrWC), A·T(wH), and A·T(wrH) base mispairs and incorrect A·T*(), A·(), A·T*(), and A·() base mispairs containing mutagenic tautomers of the DNA bases, and also their interconversions between each other through the non-planar transition states via the structural or conformational rearrangements and intramolecular proton transfer along the intermolecular H-bonds.
In contrast to this, the classical G·C Watson-Crick DNA base pair (Brovarets', 2013b; Brovarets' and Hovorun, 2014a, 2015f) could not acquire different conformations in the main tautomeric state due to the obstacles presented by its geometrical structure. This, however, can be overcome through the G·C(WC) → G*·C*(WC) tautomerisation via the double proton transfer (DPT), according to Löwdin's mechanism (Löwdin, 1963, 1966; Brovarets' and Hovorun, 2014a, 2018). This Löwdin's reaction can proceed over the barrier of tautomerization or under the barrier via the tunneling (Parker and Van Everv, 1971; Boutis, 1992; Al-Khalili and McFadden, 2014; Brovarets' and Hovorun, 2015g; Godbeer et al., 2015; Turaeva and Brown-Kennerly, 2015; Meisner and Kastner, 2016; Roßbach and Ochsenfeld, 2017; McFadden and Al-Khalili, 2018; Pusuluk et al., 2018; Smedarchina et al., 2018; Shekaari and Jafari, 2019; Srivastava, 2019).
At this, the so-called Löwdin G*·C*(WC) base pair with geometry close to Watson-Crick, which is created in such a way, involving mutagenic tautomers of the DNA bases, can acquire similarly to the A·T(WC) base pair different conformations (Brovarets' and Hovorun, 2010, 2015f; Brovarets', 2013a,b; Brovarets' et al., 2018a,b,c,d,e,f; Brovarets' and Tsiupa, 2019)—reverse Löwdin G*·C*(rWC), Hoogsteen G*′·C*(H) and reverse Hoogsteen G*′·C*(rH) (Brovarets', 2013b) (here and below the superscript “*” denotes the rare tautomeric form of the DNA base (Glushenkov and Hovorun, 2016) and “′”—trans-orientation of the OH group). This demonstrates quite unexpected role of the Löwdin's tautomerisation for the conformational variety.
Currently, there is no mention of reverse Löwdin G*·C*(rWC), Hoogsteen G*′·C*(H) and reverse Hoogsteen G*′·C*(rH) conformers or their tautomerisation via the DPT along the intermolecular H-bonds, despite a great number of investigations devoted to this important phenomenon.
In our previous studies we have comperehensively investigated the tautomerisation via the DPT of the canonical A·T(WC) (Brovarets' and Hovorun, 2014b, 2015g) and G·C(WC) (Brovarets' and Hovorun, 2014a) Watson-Crick DNA base pairs, and also of the incorrect DNA base pairs—wobble G·T base pair (Brovarets' et al., 2015), short C·T (Brovarets' and Hovorun, 2013a), C*·C (Brovarets' and Hovorun, 2013b), T*·T (Brovarets' et al., 2014a), H·C (Brovarets' and Hovorun, 2013c; Brovarets' et al., 2013a) and H*·T (Brovarets' and Hovorun, 2013c; Brovarets' et al., 2013a); long A·A* (Brovarets' and Hovorun, 2013d), A·G (Brovarets' et al., 2014b), G·G* (Brovarets' and Hovorun, 2014c), H·H (Brovarets' and Hovorun, 2013c; Brovarets' et al., 2013a), H*·H (Brovarets' and Hovorun, 2013c; Brovarets' et al., 2013b) and H·A (Brovarets' and Hovorun, 2013c; Brovarets' et al., 2014c); Watson-Crick-like A·C* (Brovarets' and Hovorun, 2015h), G*·T (Brovarets' and Hovorun, 2015i), G·Asyn (Brovarets' and Hovorun, 2014d), A*· (Brovarets' and Hovorun, 2014d), A*·Asyn (Brovarets' et al., 2014d), G· (Brovarets' and Hovorun, 2014e), T·2AP*(w) (Brovarets' et al., 2017; Brovarets' and Hovorun, 2019a), and G·2AP*(w) (Brovarets' et al., 2017; Brovarets' and Hovorun, 2019a) base mispairs and protein-DNA complexes (Brovarets' et al., 2012), which we have summarized in our review (Brovarets' and Hovorun, 2019b), devoted to the microstructural mechanisms of the tautomerization by the proton transfer along the neighboring intermolecular H-bonds in 22 biologically important pairs of nucleotide bases in the framework of the author's method, which enable to trace the evolution of the physico-chemical parameters along the intrinsic reaction coordinate (IRC).
In this study, we aim to reapply the approach, which we launched in our previous works (Brovarets' et al., 2018a,b,c,d,e,f; Brovarets' and Tsiupa, 2019) in order to investigate in details the tautomerisation of the reverse Löwdin G*·C*(rWC), Hoogsteen G*′·C*(H), and reverse Hoogsteen G*′·C*(rH) base pairs via PT along the neighboring intermolecular H-bonds.
As a result of the previous investigations, it was established that proton mobility along the intermolecular H-bonds does not change the tautomeric status of the investigated base pairs. Along with this biologically important conclusion, for the first time we have obtained a number of important physical and chemical characteristics. As such, it was documented that tautomerisation of the reverse Löwdin G*·C*(rWC) DNA base pair along the middle H-bond induces analogous SPT along its upper and lower H-bonds. Moreover, for the first time we have described the formation of a dynamically stable base pair with participation of the yilidic form of the purine base, formed through asynchronous DPT and participation of the CH group as proton donor.
Digging deeper into the mechanisms of tautomerisation of the reverse Löwdin G*·C*(rWC), Hoogsteen G*′·C*(H), and reverse Hoogsteen G*′·C*(rH) base pairs, we have carefully obtained sweeps of the physical and chemical parameters that characterize proton mobility along the IRC. Firstly, we have established a monotonic dependence of the base pair's dipole moment along the IRC. Second, it was shown that the SPT processes are characterized by the presence of 6 key points.
Computational Methods
Density Functional Theory Geometry and Vibrational Frequencies Calculations
All calculations of the geometries and harmonic vibrational frequencies of the considered base mispairs and transition states of their conversion have been conducted using Gaussian'09 package (Frisch et al., 2010) at the DFT B3LYP/6-311++G(d,p) level of theory (Lee et al., 1988; Parr and Yang, 1989; Tirado-Rives and Jorgensen, 2008), that has been already applied for analogous systems and approved to give accurate geometrical structures, normal mode frequencies, barrier heights, and characteristics of intermolecular H-bonds (Matta, 2010; Arabi and Matta, 2018; Gatti et al., 2018). We have used a scaling factor equal to 0.9668 in order to correct harmonic frequencies for the investigated base pairs (Brovarets' and Hovorun, 2010, 2015f; Brovarets', 2013a,b; Palafox, 2014; El-Sayed et al., 2015; Brovarets' et al., 2018a,b,c,d,e,f; Brovarets' and Tsiupa, 2019). We have associated structures, which were localized on the potential energy landscape by means of Synchronous Transit-guided Quasi-Newton method (Peng et al., 1996), to the minima or transition state (TS) by the absence or presence of the imaginary frequency in the vibrational spectra of the complexes, respectively. We used standard TS theory (Atkins, 1998) in order to estimate the forward and reverse barriers of the investigated tautomerisation reaction.
IRC Calculations
Reaction pathways have been monitored by following IRC in the forward and reverse directions from each TS using Hessian-based predictor-corrector algorithm for integration (Hratchian and Schlegel, 2005). In such a way we ensure that it was received proper reaction pathway from reactants to products. Further, we have obtained the sweeps of the energetic, polar, and geometric characteristics of the H-bonds and base pairs along the IRC by calculating them at each point of the IRC (Brovarets' et al., 2017, 2018g,h).
Single Point Energy Calculations
In order to take into account electronic correlation effects, we followed geometry optimizations with single point energy calculations using MP2 level of theory (Frisch et al., 1990) and 6-311++G(2df,pd) Pople's basis set of valence triple-ζ quality (Hariharan and Pople, 1973; Krishnan et al., 1980) and aug-cc-pVDZ Dunning's cc-type basis set (Kendall et al., 1992), augmented with polarization and/or diffuse function.
The Gibbs free energy G for all structures was calculated by the formula:
where Eel–electronic energy, while Ecorr–thermal correction.
Interaction Energies Calculations
We have obtained electronic interaction energies Eint at the MP2/6-311++G(2df,pd) level of theory as the difference between the total electronic energy of the base mispair and the electronic energies of the separate monomers. Gibbs free energy of interaction has been obtained using similar approach. At this, we also corrected the interaction energy for the basis set superposition error (BSSE) (Boys and Bernardi, 1970; Gutowski et al., 1986) according to the counterpoise procedure (Sordo et al., 1988; Sordo, 2001).
Estimation of Kinetic Parameters
The time τ99.9% spent for reaching the 99.9% of the equilibrium concentration between the initial and terminal base pairs in the system of reversible first-order forward (kf) and reverse (kr) reactions was estimated by formula (Atkins, 1998):
The lifetime τ of the base pairs was calculated using the formula 1/kr, where the values of the reverse kr and forward kf rate constants for the tautomerisation reactions were calculated as (Atkins, 1998):
where quantum tunneling effects are accounted by Wigner's tunneling correction (Wigner, 1932; Brovarets' and Hovorun, 2014d), that has been successfully used for the DPT reactions (Brovarets' and Hovorun, 2013a,b,c,d, 2014c,d,e, 2015h,i; Brovarets' et al., 2013a,b, 2014a,b,c,d, 2015, 2017):
where kB—Boltzmann's constant, h—Planck's constant, ΔΔGf,r—Gibbs free energy for the forward (f) and reverse (r) tautomerisation reactions, νi—value of the imaginary frequency at the TS of the tautomerisation reaction.
QTAIM Analysis
Bader's quantum theory of Atoms in Molecules (QTAIM) was used to analyse the electron density distribution (Bader, 1990). The topology of the electron density was analyzed using program package AIMAll (Keith, 2010) with all default options and wave functions obtained at the level of theory used for geometry optimisation. The presence of the (3,−1) bond critical point (BCP), bond path between hydrogen donor and acceptor and positive value of the Laplacian at this BCP (Δρ >0) were considered altogether as criteria for the H-bond formation (Matta and Hernández-Trujillo, 2003; Matta et al., 2006a,b; Matta, 2014; Lecomte et al., 2015; Brovarets' and Pérez-Sánchez, 2016, 2017; Brovarets' et al., 2016).
Energies of the Intermolecular H-Bonds
We calculated the energies of the intermolecular AH···B H-bonds in the base mispairs and TSs and of the sweeps of the H-bond energies by the empirical Espinosa-Molins-Lecomte (EML) formula (Espinosa et al., 1998; Matta et al., 2006b; Mata et al., 2011; Lecomte et al., 2015; Alkorta et al., 2016, 2017) based on the electron density distribution at the (3,−1) BCPs of the H-bonds:
where V(r)—value of a local potential energy at the (3,−1) BCP.
EML fomula has been also used for the estimation of the energy of the non-standard H-bonds CH···O in the stationary points of the base pairs on the hypersurface of their electronic energy.
We evaluated the energies of the classical NH···N/O and OH···O/N intermolecular AH···B H-bonds by the empirical Iogansen's formula (Iogansen, 1999):
where Δν–magnitude of the frequency shift of the stretching mode of the AH H-bonded group involved in the AH···B H-bond relatively the unbound group. We applied partial deuteration in order to minimize the effect of vibrational resonances (Brovarets' and Hovorun, 2014d,e, 2015h,i; Brovarets' et al., 2014d).
The energies of the NH···N and OH···O H-bonds in the TSs containing loosened covalent bridges were calculated by the Nikolaienko-Bulavin-Hovorun formula (Nikolaienko et al., 2012):
where ρ–the electron density at the (3,−1) BCP of the H-bond.
The atom numbering scheme for the DNA bases is conventional (Saenger, 1984).
Results and Their Discussion
In this paper we have investigated in details the tautomerisation processes via the single (SPT) or double (DPT) proton transfer of the G*·C*(rWC), G*′·C*(H), and G*′·C*(rH) base pairs along the neighboring intermolecular H-bonds as their intrinsically inherent property (Figure 1).
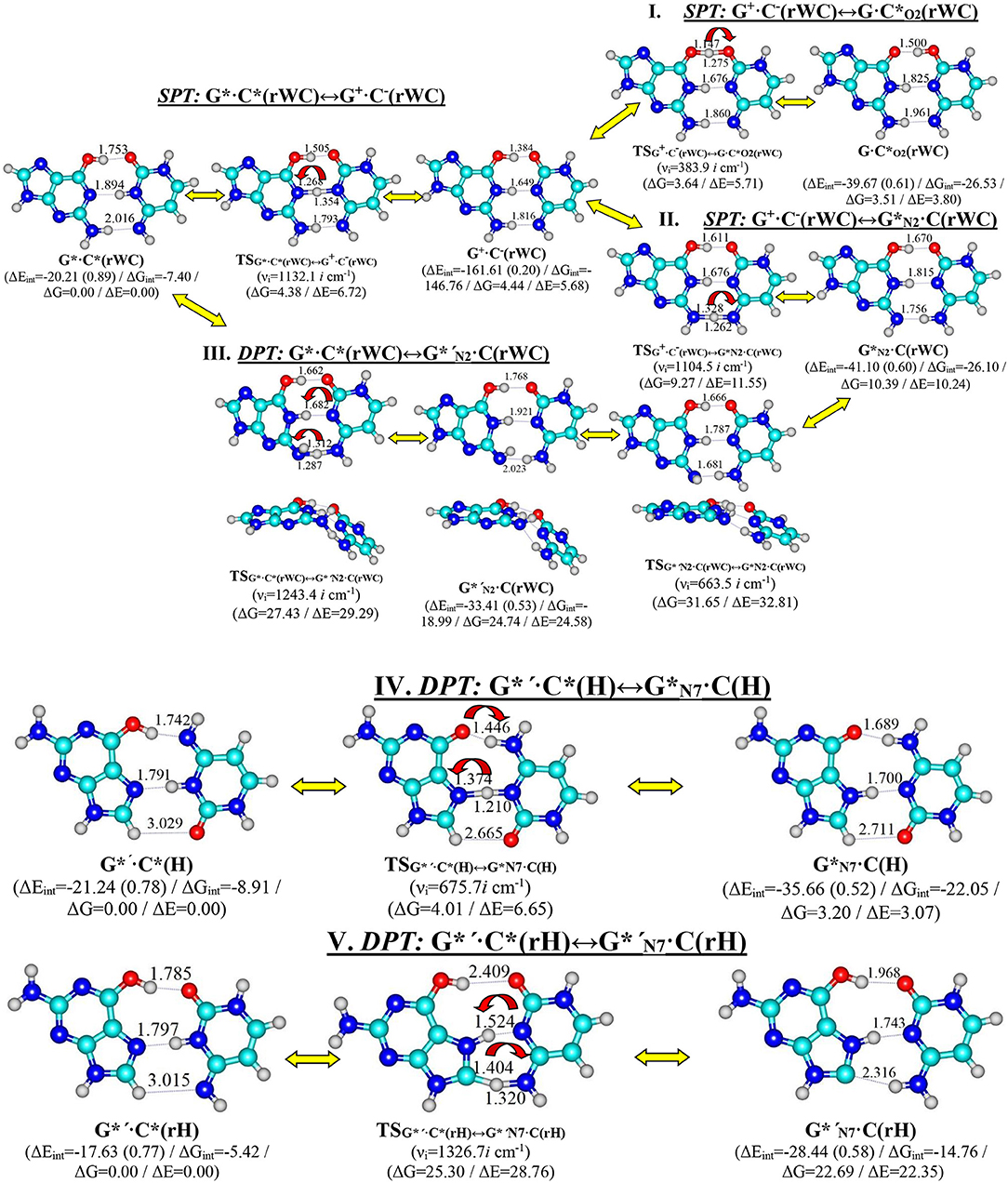
Figure 1. Discovered new reaction pathways of the tautomerization in the reverse Watson-Crick G*·C*(rWC) – I. G*·C*(rWC)↔G+·C−(rWC)↔G·C*O2(rWC), II. G*·C*(rWC)↔G+·C−(rWC)↔G*N2·C(rWC), III. G*·C*(rWC)↔G*′N2·C(rWC), Hoogsteen G*′·C*(H) – IV. G*′·C*(H)↔G*N7·C(H)—and reverse Hoogsteen G*′·C*(rH) – V. G*′·C*(rH)↔G*′N7·C(rH) – DNA base pairs through the single or double proton transfer. Electronic ΔEint and Gibbs free ΔGint energies of the interaction [MP2/6-311++G(2df,pd)//B3LYP/6-311++G(d,p) level of theory, in kcal·mol−1], relative Gibbs free energies ΔG, and electronic energies ΔE [MP2/aug-cc-pVDZ//B3LYP/6-311++G(d,p) level of theory in the continuum with ε = 1 at T = 298.15 in kcal·mol−1] are presented below complexes in brackets. Dotted lines indicate AH···B H-bonds – their lengths H···B are presented in angstroms; carbon atoms are in light-blue, nitrogen—in dark-blue, hydrogen—in gray and oxygen – in red. νi—imaginary frequencies at the TSs of the tautomeric/conformational transitions.
This paper is organized in the following way—firstly, we would discuss the tautomerisation process separately for each base pair and then we would present in details sweeps of the most important physico-chemical parameters along the IRC altogether for all investigated base pairs (Figures 1–12, Tables 1–8).
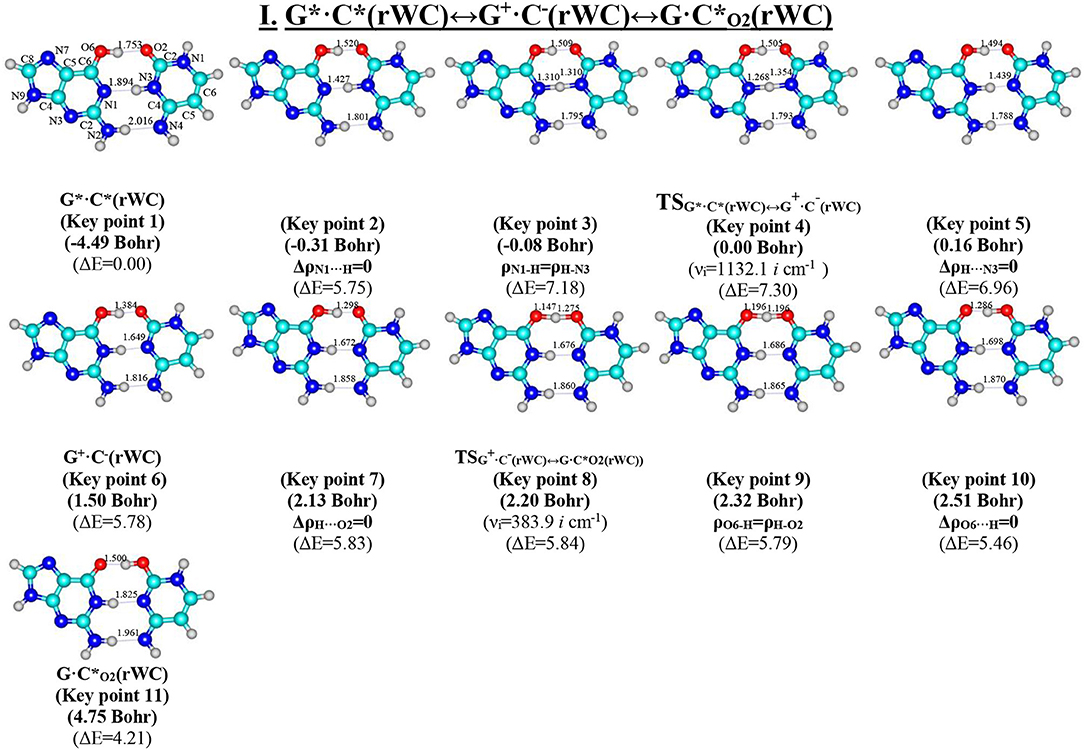
Figure 2. Geometric structures of the 11 key points describing the evolution of the I. G*·C*(rWC)↔G+·C−(rWC)↔G·C*O2(rWC) tautomerisation via the sequential SPT along the IRC obtained at the B3LYP/6-311++G(d,p) level of theory in vacuo. Coordinates of the 11 key points, their relative electronic energies ΔE (in kcal·mol−1 obtained at the B3LYP/6-311++G(d,p) level of theory in vacuum at T = 298.15) and imaginary frequencies νi (cm−1) at the TSs of their interconversions are presented below them in brackets (see Table 3). For more detailed designations see Figure 1.
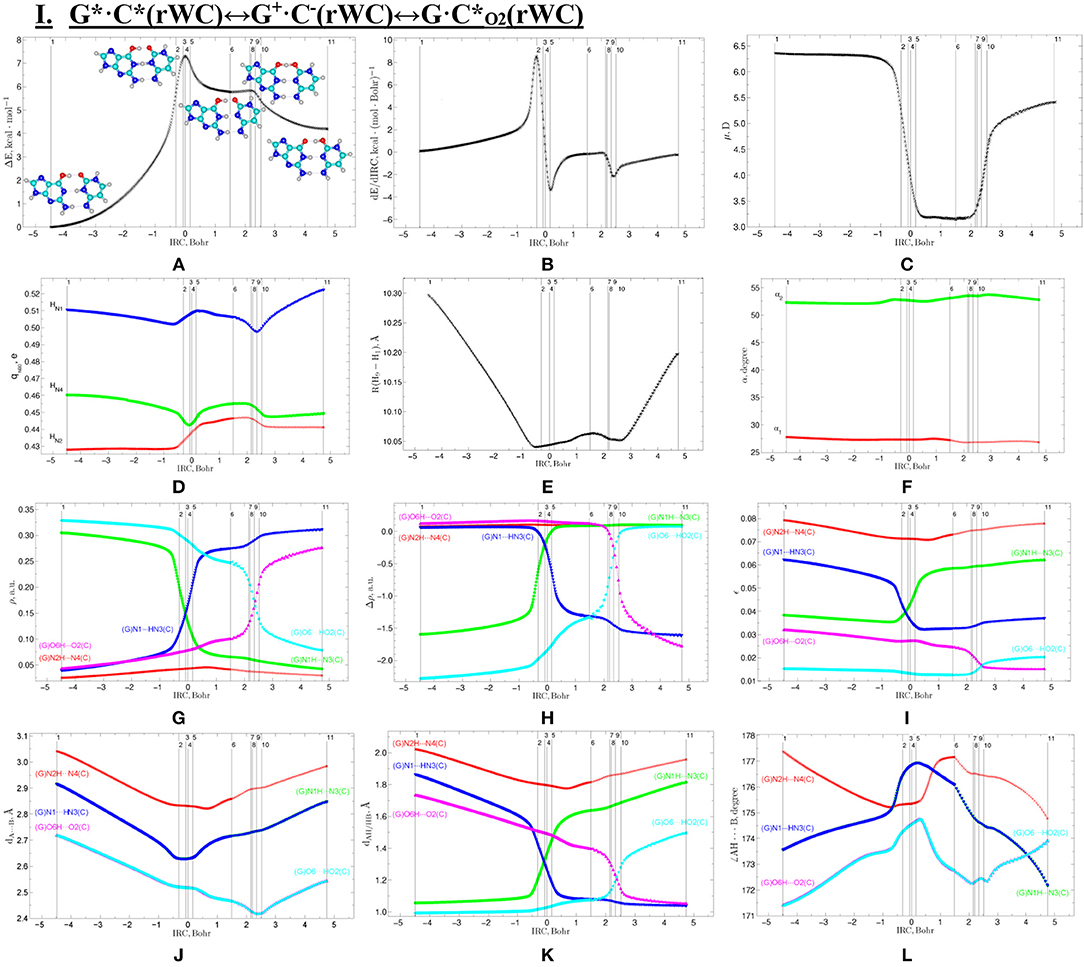
Figure 3. Profiles of: (A) the relative electronic energy ΔE, (B) the first derivative of the electronic energy with respect to the IRC (dE/dIRC), (C) the dipole moment μ, (D) the NBO charges qNBO, (E) the distance R(H1-H9) between the H1 and H9 glycosidic hydrogens, (F) the α1 (∠N1H1(C)H9(G)) and α2 (∠N9H9(G)H1(C)) glycosidic angles, (G) the electron density ρ; (H) the Laplacian of the electron density Δρ, (I) the ellipticity ε at the (3,−1) BCPs, (J) the distance dA···B between the electronegative A and B atoms; (K) the distance dAH/HB between the hydrogen and electronegative A or B atoms and (L) the angle ∠AH···B of the covalent and hydrogen bonds along the IRC of the investigated I. G*·C*(rWC)↔G+·C−(rWC)↔G·C*O2(rWC) tautomerisation via the sequential SPT obtained at the B3LYP/6-311++G(d,p) level of theory in vacuum.
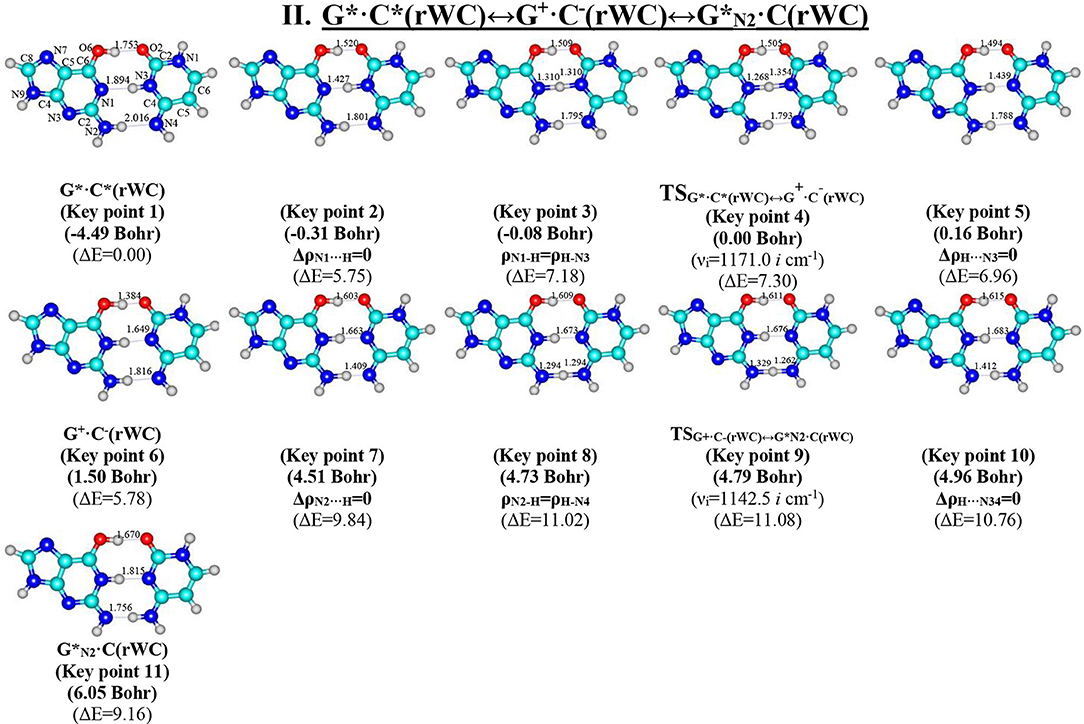
Figure 4. Geometric structures of the 11 key points describing the evolution of the II. G*·C*(rWC)↔G+·C−(rWC)↔G*N2·C(rWC) tautomerisation via the sequential SPT along the IRC obtained at the B3LYP/6-311++G(d,p) level of theory in vacuo (see Table 4). For more detailed designations see Figure 2.
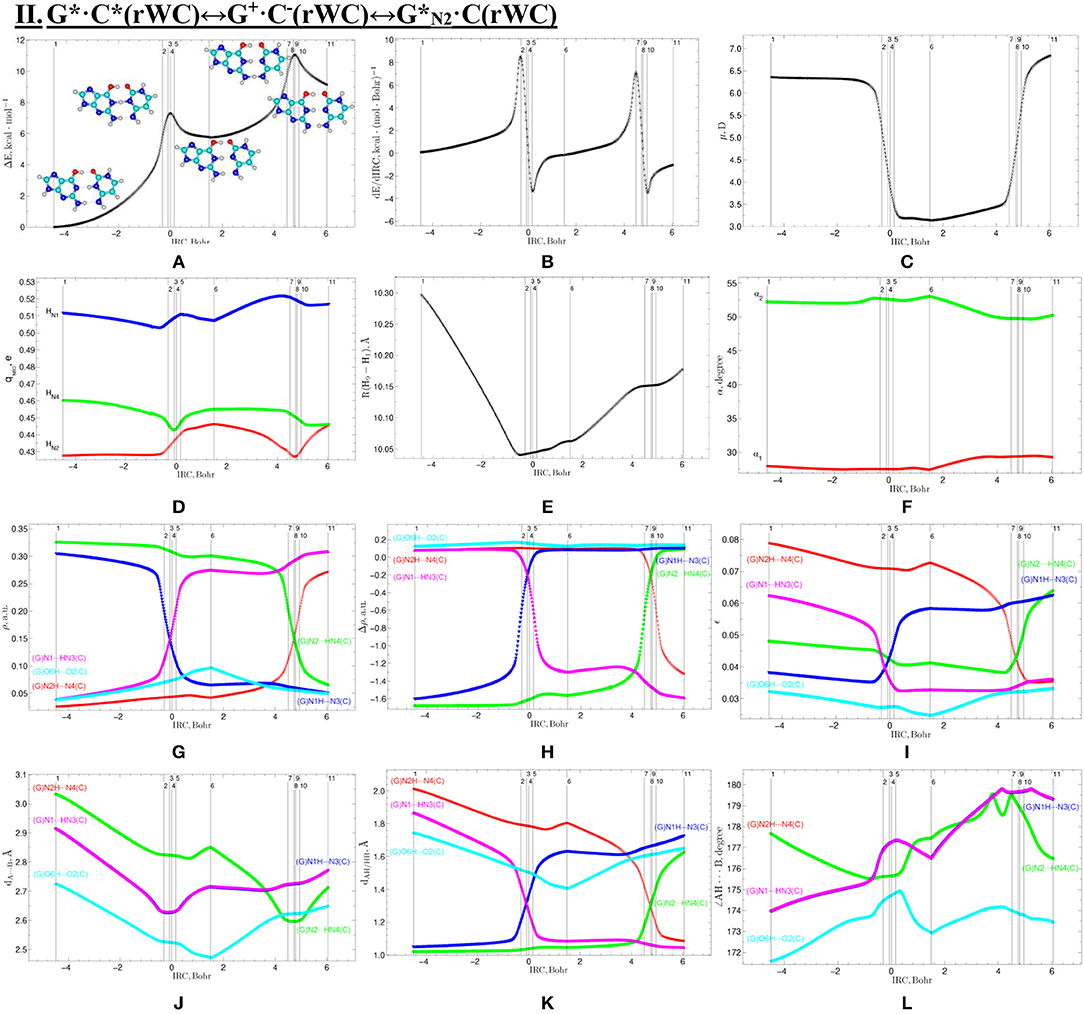
Figure 5. Profiles of physico-chemical parameters of the investigated II. G*·C*(rWC)↔G+·C−(rWC)↔G*N2·C(rWC) tautomerisation via the sequential SPT obtained at the B3LYP/6-311++G(d,p) level of theory in vacuum. For more detailed designations see Figure 3.
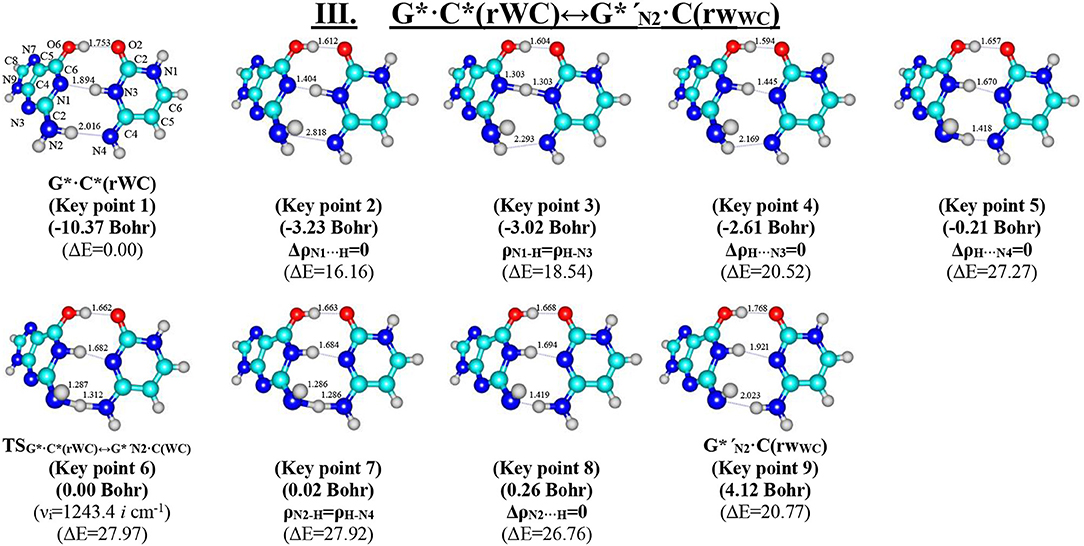
Figure 6. Geometric structures of the 9 key points describing the evolution of the III. G*·C*(rWC)↔G*′N2·C(rWC) tautomerisation via the DPT along the IRC obtained at the B3LYP/6-311++G(d,p) level of theory in vacuo (see Table 5). For more detailed designations see Figure 2.
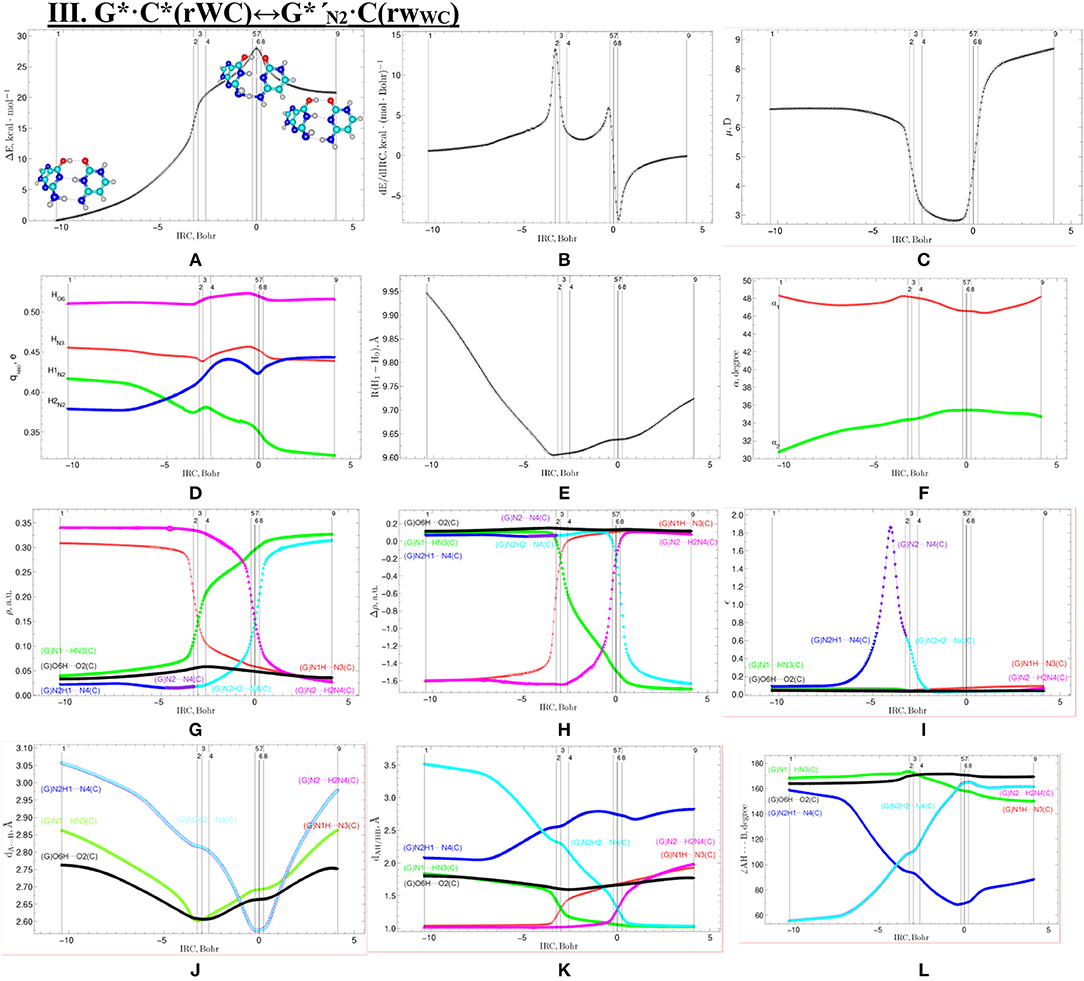
Figure 7. Profiles of the physico-chemical parameters of the investigated III. G*·C*(rWC)↔G*′N2·C(rWC) tautomerisation via the DPT obtained at the B3LYP/6-311++G(d,p) level of theory in vacuum. For more detailed designations see Figure 3.
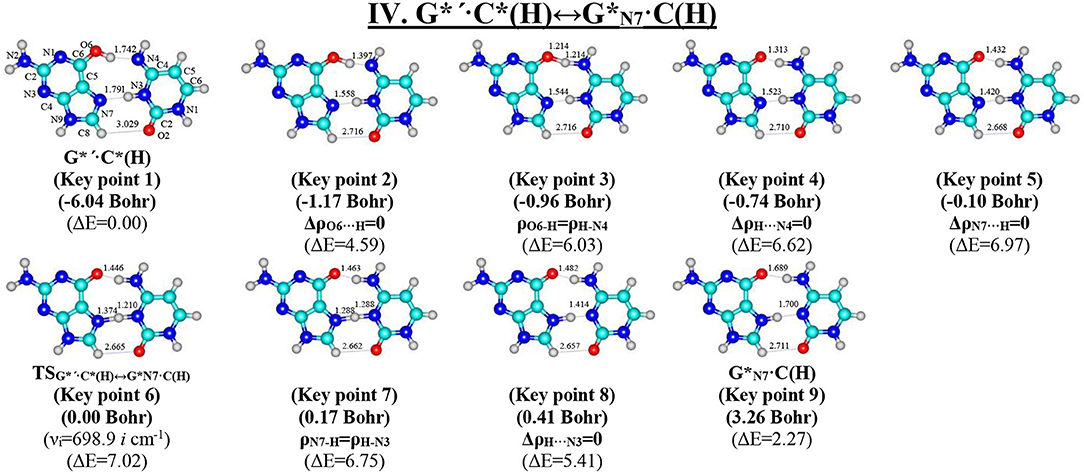
Figure 8. Geometric structures of the 9 key points describing the evolution of the IV. G*′·C*(H)↔G*N7·C(H) tautomerisation via the DPT along the IRC obtained at the B3LYP/6-311++G(d,p) level of theory in vacuo (see Table 6). For more detailed designations see Figure 2.
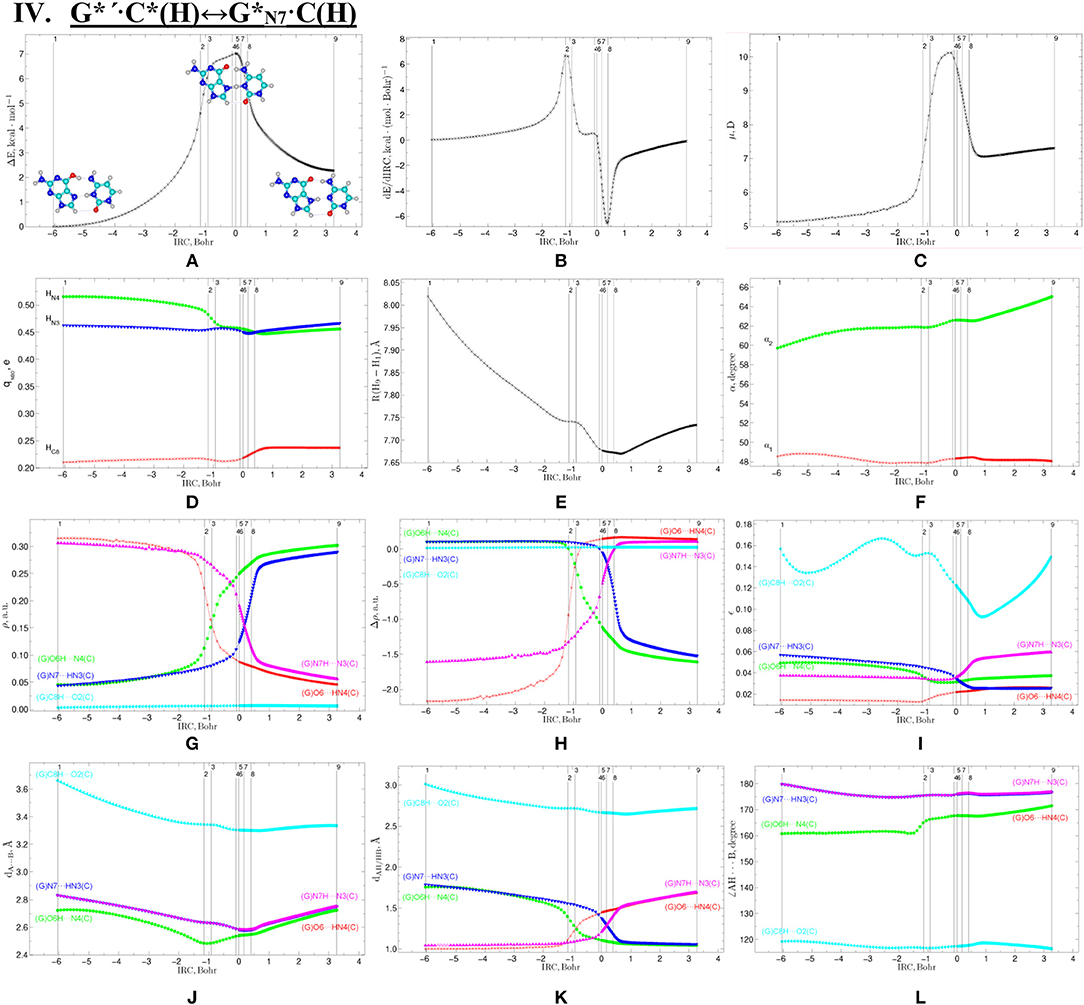
Figure 9. Profiles of the physico-chemical parameters of the investigated IV. G*′·C*(H)↔G*N7·C(H) tautomerisation via the DPT obtained at the B3LYP/6-311++G(d,p) level of theory in vacuum. For more detailed designations see Figure 3.
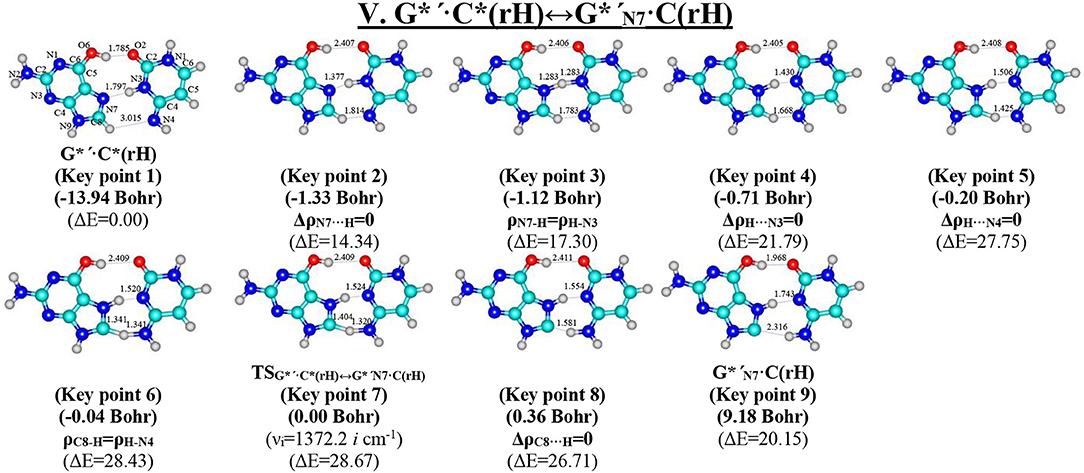
Figure 10. Geometric structures of the 9 key points describing the evolution of the V. G*′·C*(rH)↔G*′N7·C(rH) tautomerisation via the DPT along the IRC obtained at the B3LYP/6-311++G(d,p) level of theory in vacuo (see Table 7). For more detailed designations see Figure 2.
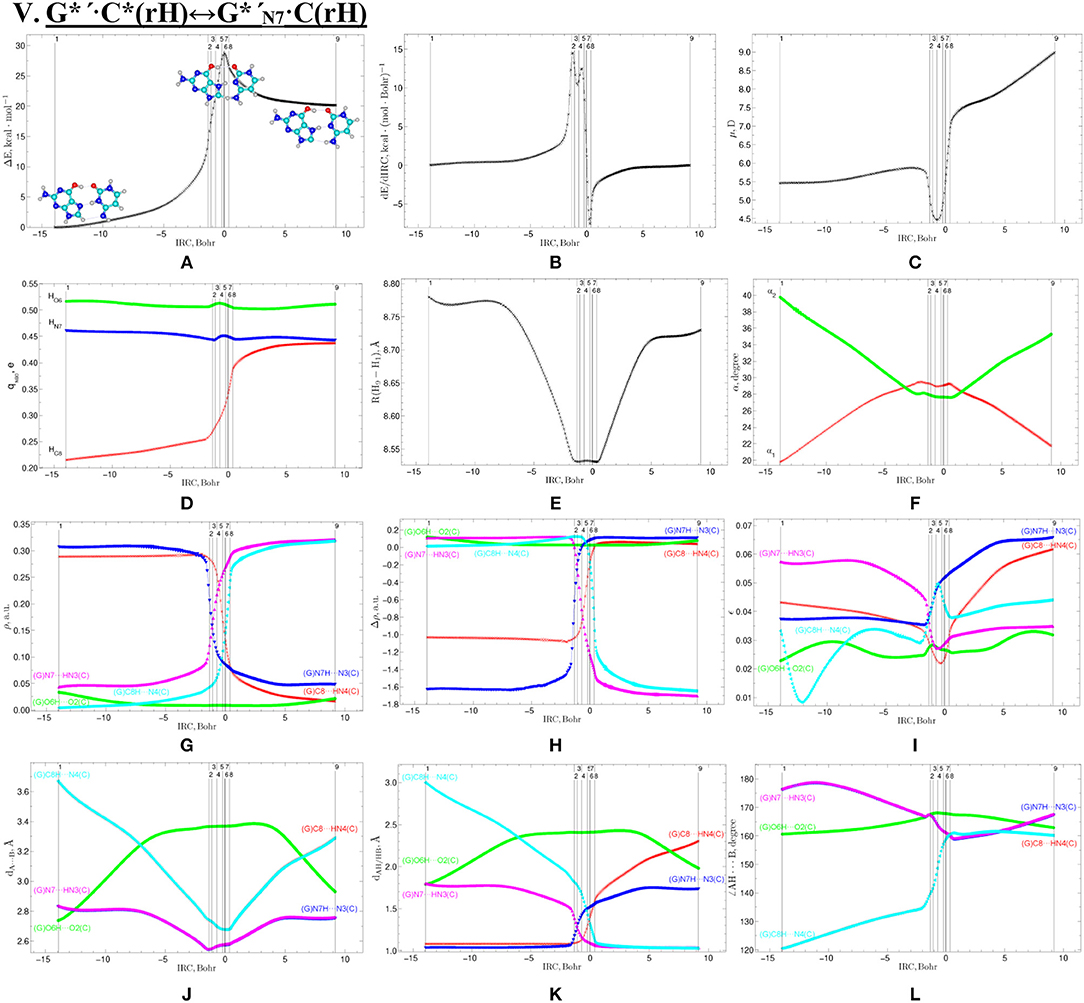
Figure 11. Profiles of the physico-chemical parameters of the investigated V. G*′·C*(rH)↔G*′N7·C(rH) tautomerisation via the DPT obtained at the B3LYP/6-311++G(d,p) level of theory in vacuum. For more detailed designations see Figure 3.
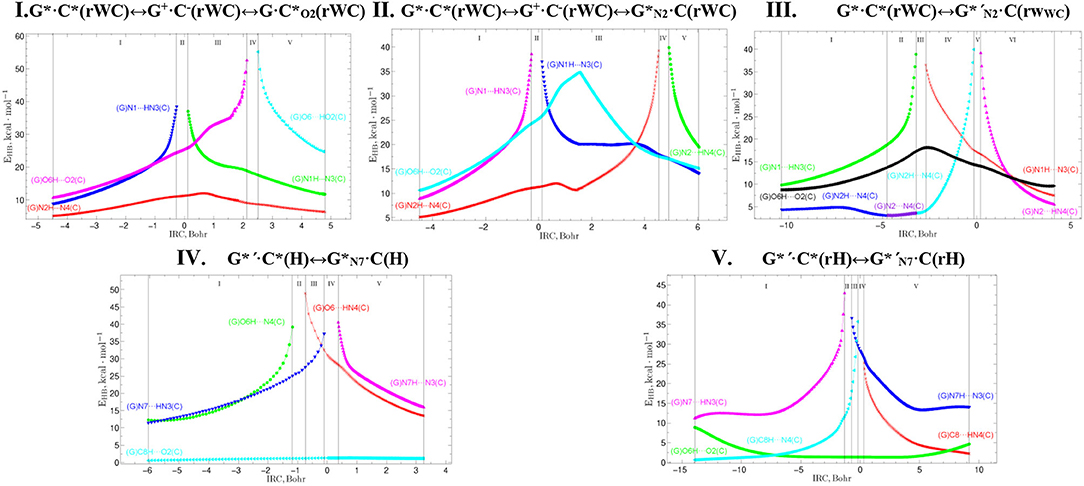
Figure 12. Profiles of the energy of the intermolecular H-bonds EHB estimated by the EML formula at the (3,−1) BCPs along the IRC of the investigated tautomerisations via the SPT or DPT obtained at the B3LYP/6-311++G(d,p) level of theory (see Table 3).
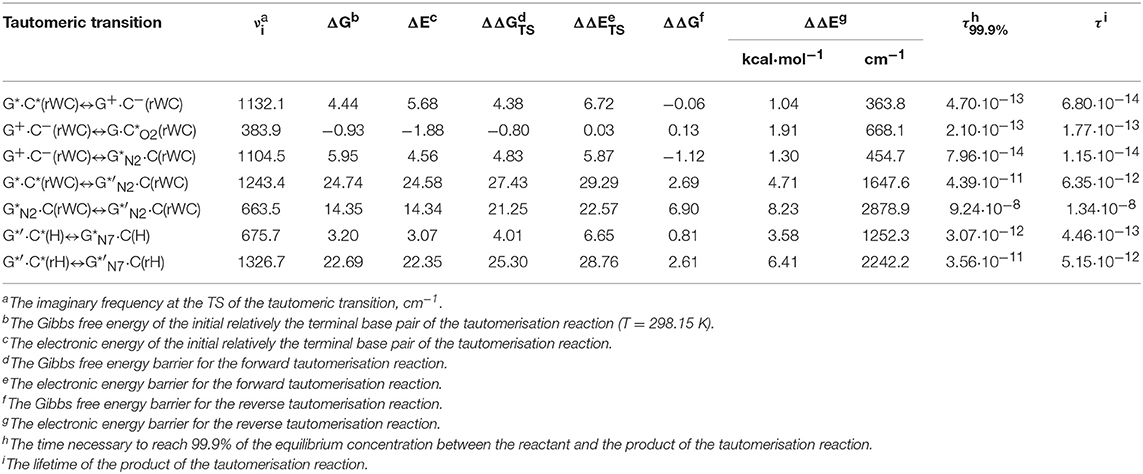
Table 1. Energetic (in kcal·mol−1) and kinetic (in s) characteristics of the tautomerisation of the G*·C*(rWC), G*′·C*(H), and G*′·C*(rH) DNA base pairs via the SPT or DPT obtained at the MP2/aug-cc-pVDZ//B3LYP/6-311++G(d,p) level of QM theory in the continuum with ε = 1 under normal conditions (see Figure 1).
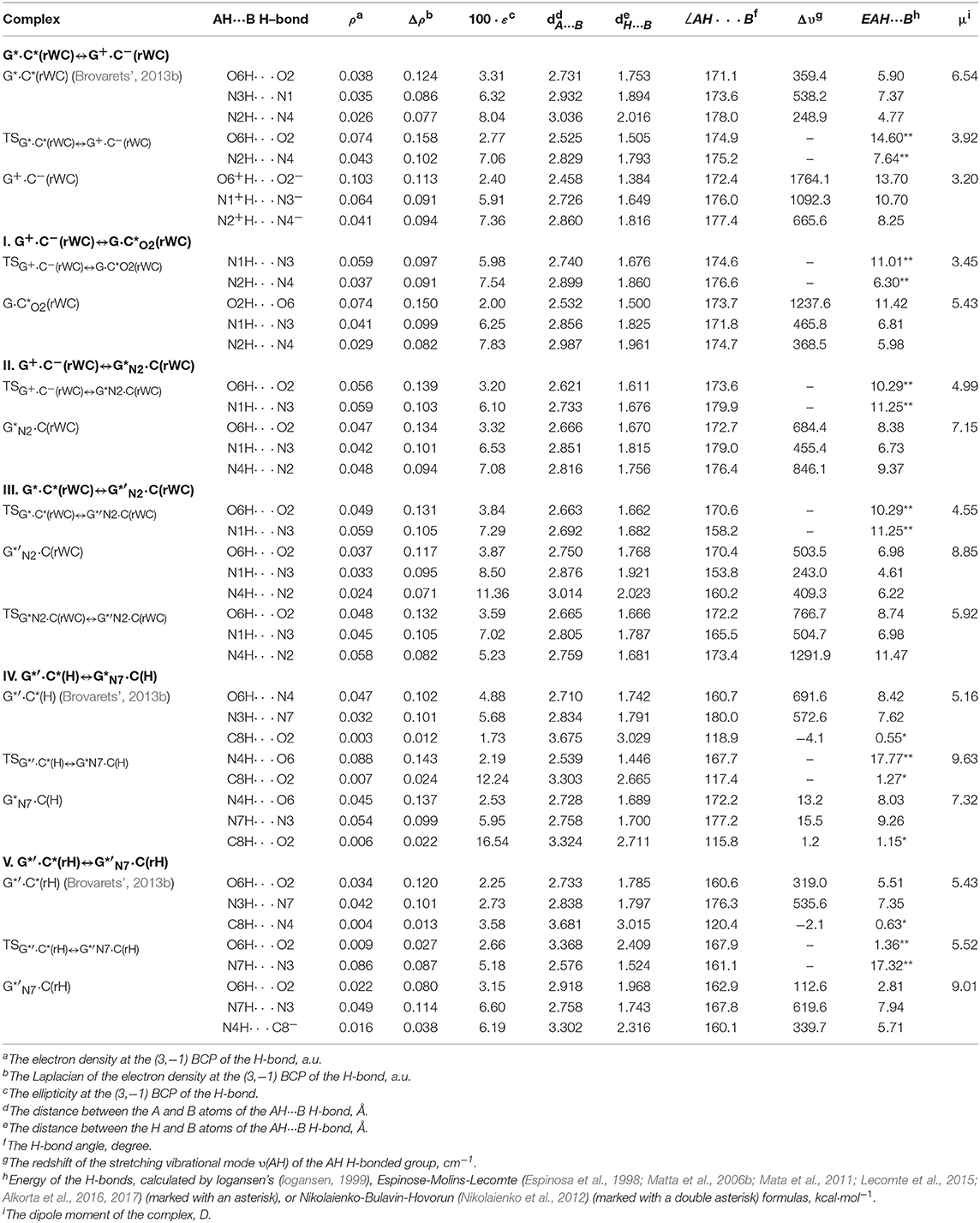
Table 2. Electron-topological, geometrical, and energetic characteristics of the intermolecular H-bonds in the investigated DNA base pairs in rare tautomeric forms and TSs of their tautomerization via the SPT or DPT obtained at the B3LYP/6-311++G(d,p) level of QM theory (ε = 1) (see Figure 1).
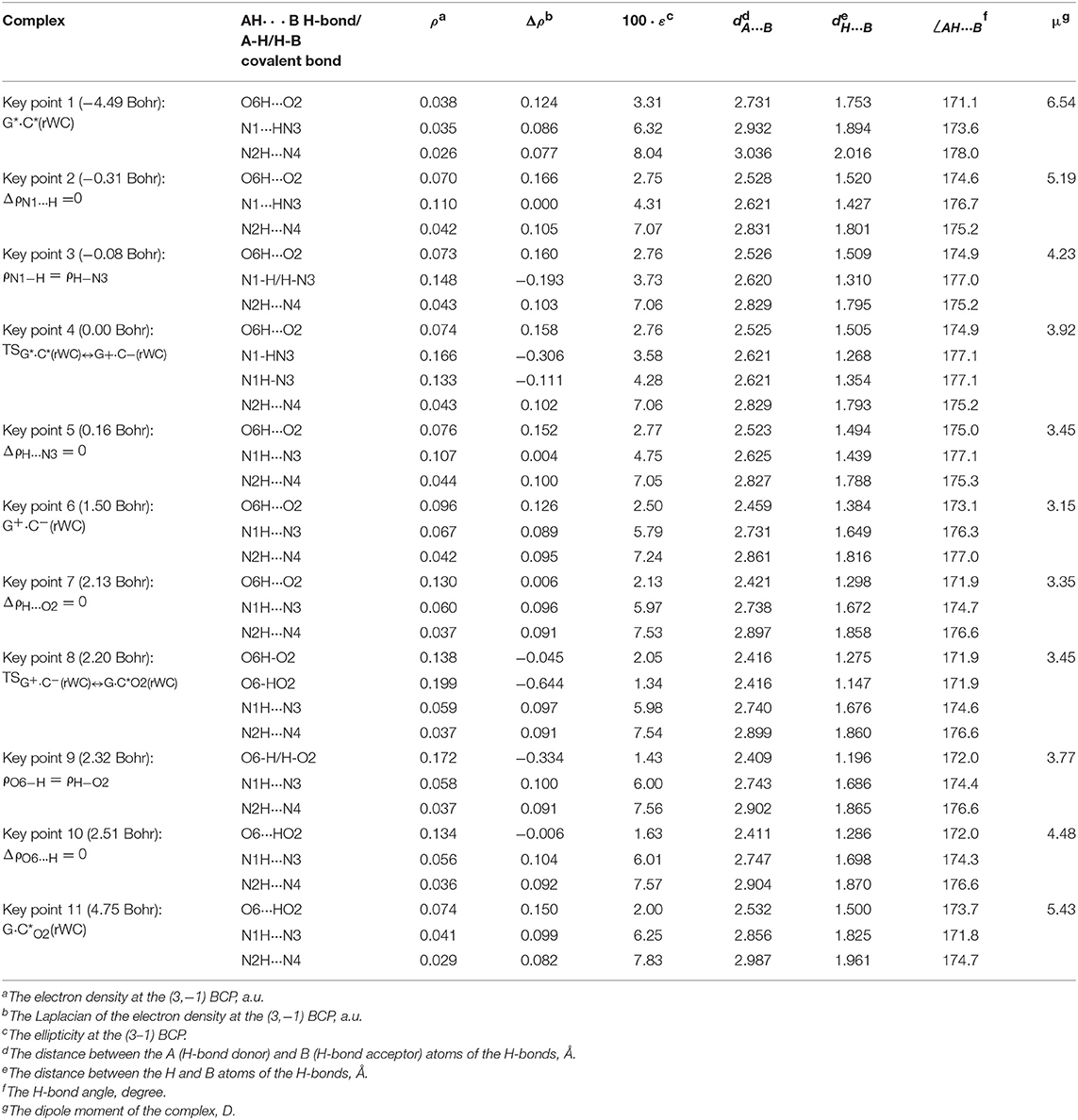
Table 3. Electron-topological and structural characteristics of the specific intermolecular bonds revealed in the 11 key points and the polarity of the latters along the IRC of the I. G*·C*(rWC)↔G+·C−(rWC)↔G·C*O2(rWC) tautomerisation obtained at the B3LYP/6-311++G(d,p) level of theory in vacuum (see Figure 2).
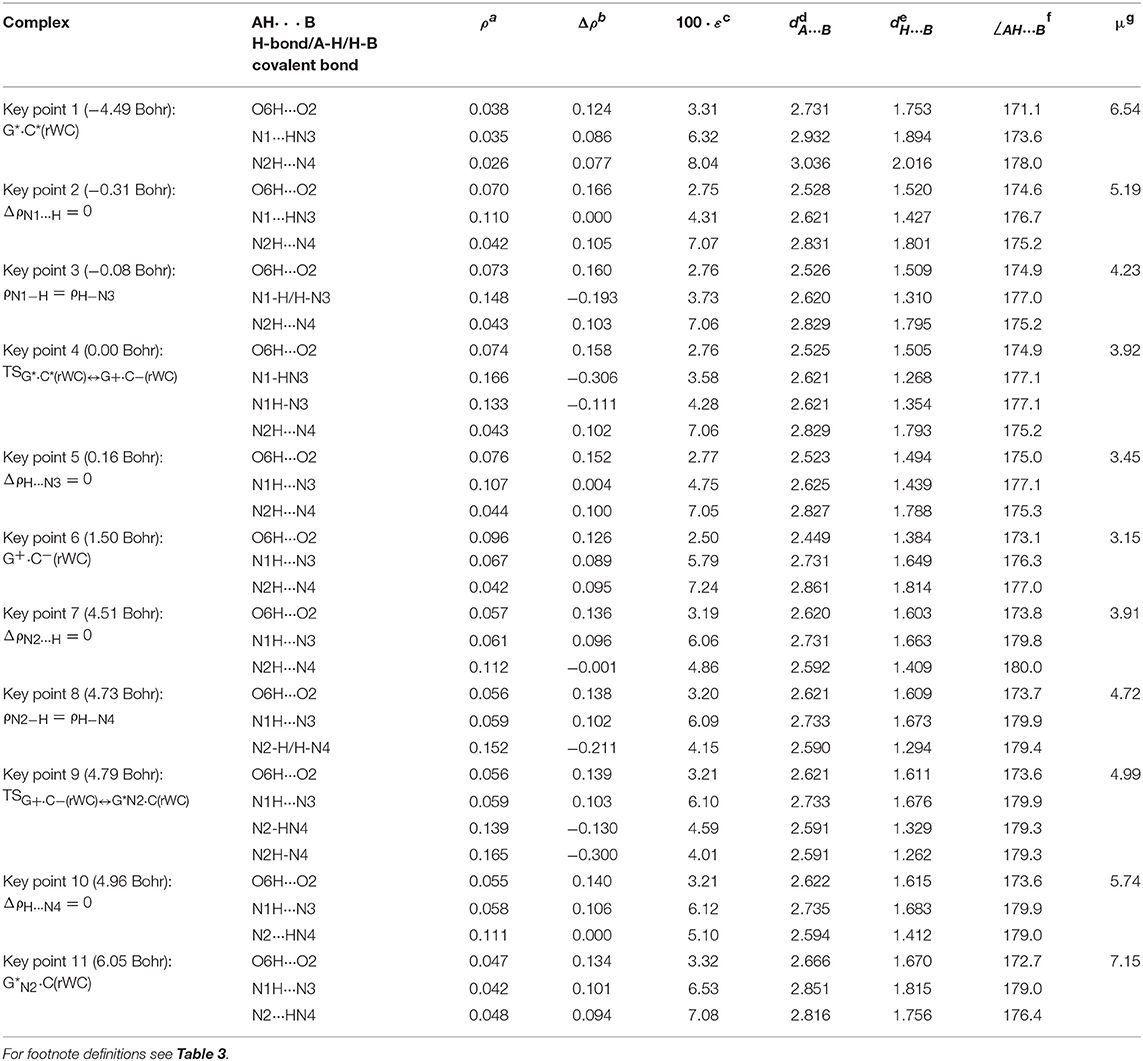
Table 4. Electron-topological and structural characteristics of the specific intermolecular bonds revealed in the 11 key points and the polarity of the latters along the IRC of the II. G*·C*(rWC)↔G+·C−(rWC)↔G*N2·C(rWC) tautomerisation obtained at the B3LYP/6-311++G(d,p) level of theory in vacuum (see Figure 4).
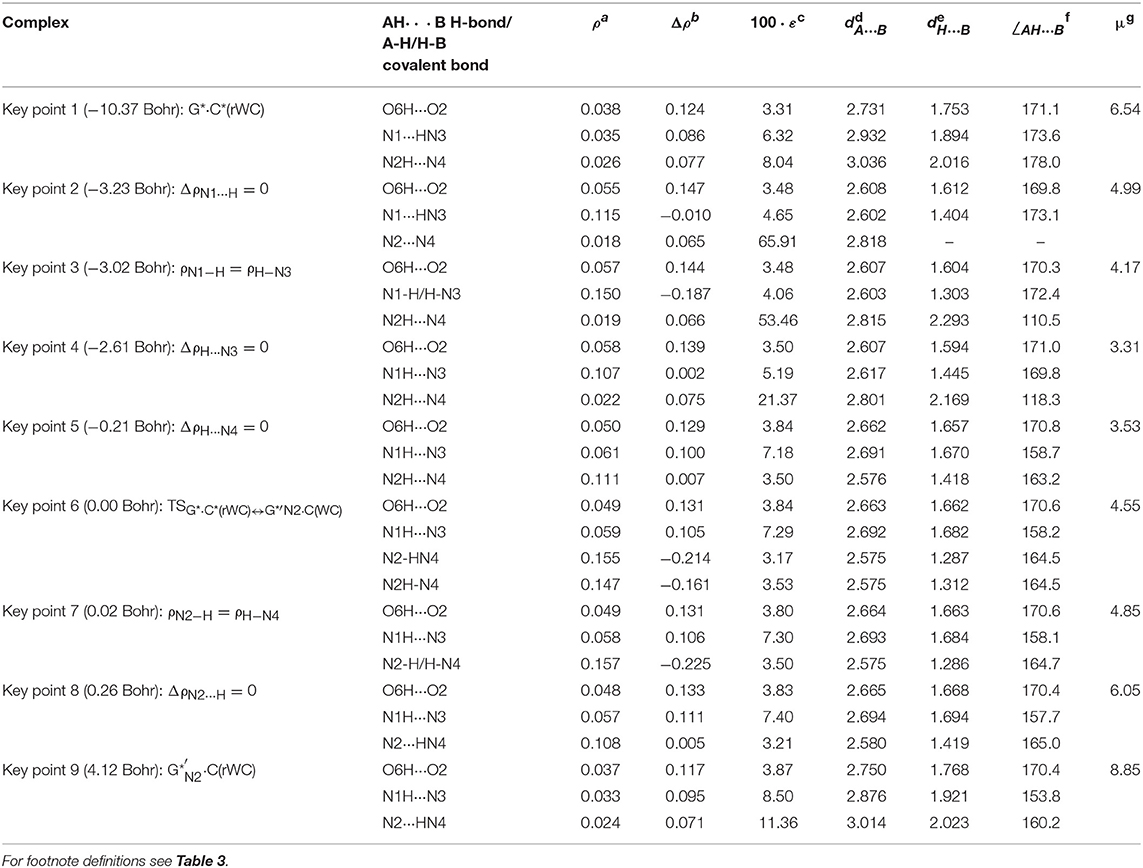
Table 5. Electron-topological and structural characteristics of the specific intermolecular bonds revealed in the 9 key points and the polarity of the latters along the IRC of the III. G*·C*(rWC)↔G*′N2·C(rWC) tautomerisation obtained at the B3LYP/6-311++G(d,p) level of theory in vacuum (see Figure 6).
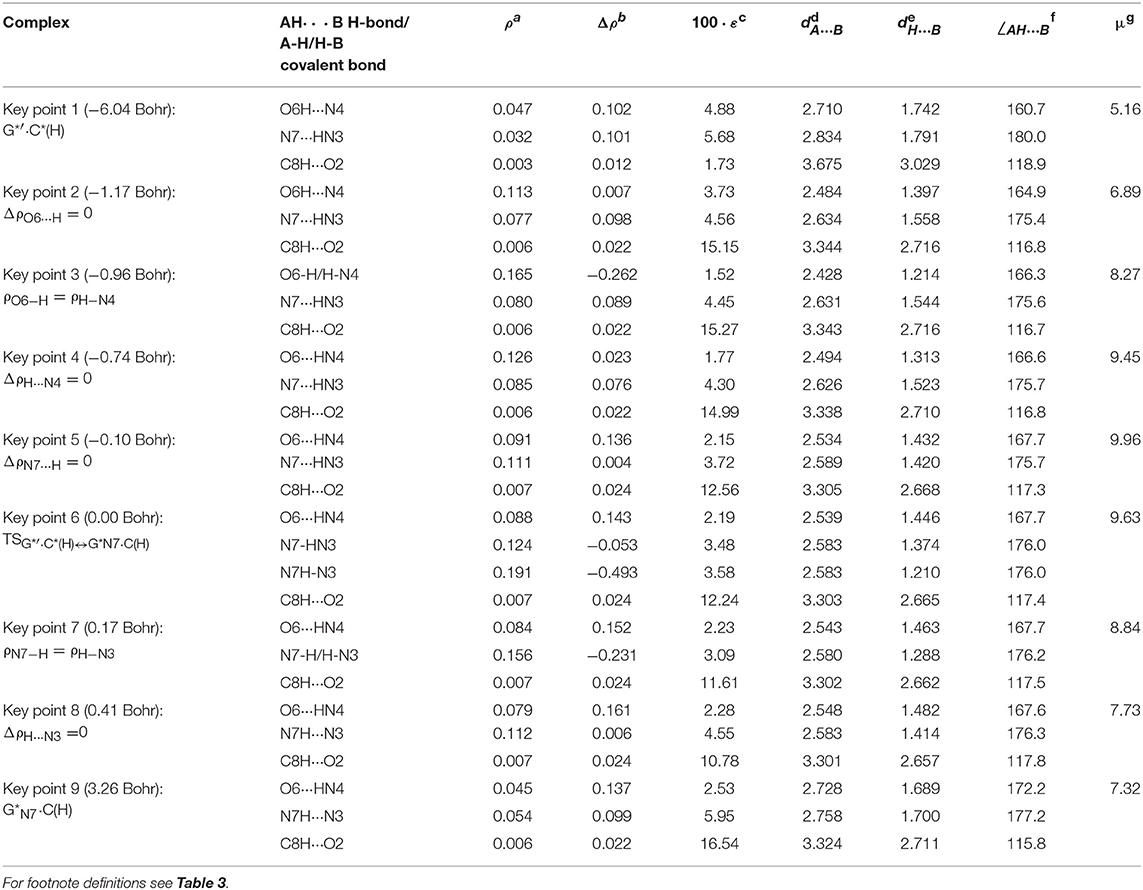
Table 6. Electron-topological and structural characteristics of the specific intermolecular bonds revealed in the 9 key points and the polarity of the latters along the IRC of the IV. G*′·C*(H)↔G*N7·C(H) tautomerisation obtained at the B3LYP/6-311++G(d,p) level of theory in vacuum (see Figure 8).
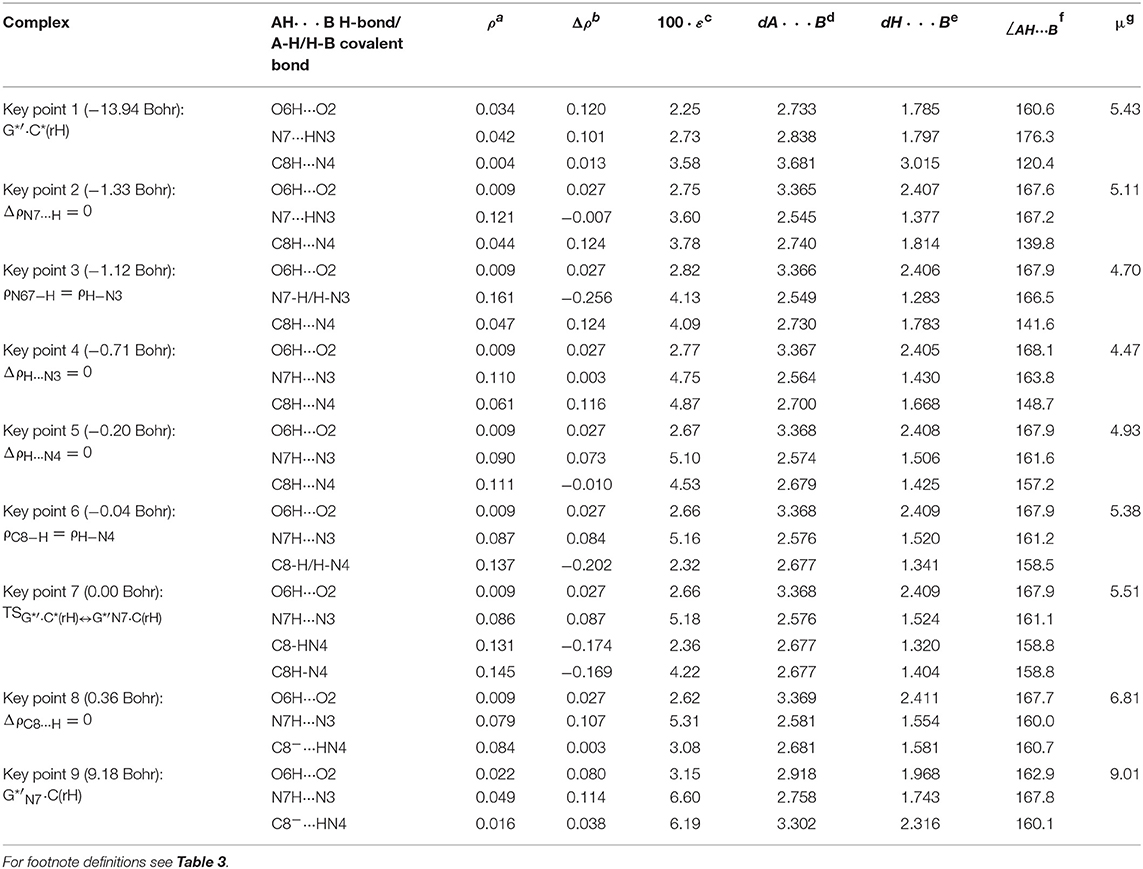
Table 7. Electron-topological and structural characteristics of the specific intermolecular bonds revealed in the 9 key points and the polarity of the latters along the IRC of the V. G*′·C*(rH)↔G*′N7·C(rH) tautomerisation obtained at the B3LYP/6-311++G(d,p) level of theory in vacuum (see Figure 10).

Table 8. Patterns of the specific intermolecular interactions including AH···B H-bonds and loosened A-H-B covalent bridges that sequentially replace each other along the IRC of the investigated tautomerisations via the SPT or DPT obtained at the B3LYP/6-311++G(d,p) level of theory (see Figure 12).
Tautomerisation of the Reverse Löwdin G*·C*(rWC) Base Pair via the SPT: I. G*·C*(rWC)↔G+·C−(rWC)↔G·(rWC) and II. G*·C*(rWC)↔G+·C−(rWC)↔C(rWC)
For the first time we have discovered three local minima on the hypersurface of the electronic energy of the G*·C*(rWC) base pair corresponding to the high-energy tautomerised G·(rWC), G+·C−(rWC), and C(rWC) base pairs (Figure 1, Table 1). All of them are stabilized by the participation of three intermolecular H-bonds, among which the upper O6H…O2/O2H…O6 H-bonds are the strongest (Table 2).
In fact, the tautomerisation of the G*·C*(rWC) base pair with relative Gibbs free energy ΔG = 0.00 kcal·mol−1 starts from the single transfer of the proton localized at the N3 nitrogen atom of the C base to the N1 nitrogen atom of the G base along the intermolecular H-bond (C)N3H…N1(G). This G*·C*(rWC)↔G+·C−(rWC) tautomerization process occurs via the TSG*·C*(rWC)↔G+·C−(rWC) (Cs symmetry) (ΔG = 4.38 kcal·mol−1) containing N1-H-N3 covalent bridge and further proceeds through the intermediate—tight ion pair G+·C−(rWC) (ΔG = 4.44 kcal·mol−1) (Cs symmetry), which is the point of bifurcation. By the way, it should be noted that this is the first case of the reliable fixation of the ionic pair of bases, formed as a result of the SPT along the intermediate molecular H-bond, which is involved in its stabilization. Similar attempts to localize such structures for the A·T(WC) and G·C(WC) DNA base pairs didn't lead to result.
Further the G*·C*(rWC)↔G+·C−(rWC) tautomerisation reaction proceeds according two scenarios via the proton transfer along:
- the (G)O6H….O2(C) H-bond through the TSG+·C−(rWC)↔G·C*O2(rWC) (ΔG = 3.64 kcal·mol−1; Cs symmetry) with O6-H-O2 covalent bridge leading to the G·(rWC) product (Cs symmetry);
- the (G)N2H….N4(C) H-bond through the TSG+·C−(rWC)↔G*N2·C(rWC) (ΔG = 9.27 kcal·mol−1; Cs symmetry) N2-H-N4 covalent bridge leading to the ·C(rWC) product (Cs symmetry).
It attracts attention that electronic ΔEint and Gibbs free ΔGint energies of the interaction for the tautomerised G·(rWC) (ΔEint = −39.67/ΔGint = −26.53) and ·C(rWC) (ΔEint = −41.10/ΔGint = −26.10) base mispairs exceed the values for the initial G*·C*(rWC) base mispair (ΔEint = −20.21/ΔGint = −7.40) and also canonical G·C(WC) base pair (ΔEint = −29.28/ΔGint = −15.97 kcal·mol−1) (Brovarets' and Hovorun, 2014a).
Moreover, it was revealed that all three formed high-energy complexes—the G+·C−(rWC) ion pair and G·(rWC), ·C(rWC) tautomers of the Löwdin G*·C*(rWC) base pair are dynamically unstable structures, since for them the zero energy of the vibrations, which frequency become imaginary at the TS of tautomerisation, significantly exceeds the reverse electronic barrier (363.8, 668.1, and 454.7 cm−1) (Table 1). Moreover, Gibbs free energies of the reverse barrier for the G*·C*(rWC)↔G+·C−(rWC) and G+·C−(rWC)↔·C(rWC) tautomeric transformations are negative (−0.06 and −1.12 kcal·mol−1, accordingly) and for the G+·C−(rWC)↔G·(rWC)—it is less (0.13 kcal·mol−1) than kT (0.62 kcal·mol−1 under normal conditions) (Table 1).
So, in fact the Löwdin G*·C*(rWC) base pair does not tautomerise to the novel G·(rWC) and ·C(rWC) base mispairs via the SPT along the intermolecular H-bonds. However, despite this verdict, obtained data can be useful as an analogy or even as a heuristic push at the investigation of the tautomerisation mechanisms of the H-bonded complexes of any nature.
Tautomerisation of the Reverse Löwdin G*·C*(rWC) Base Pair via the DPT: III. G*·C*(rWC)↔G*′N2·C(rWC)
We have also detected the unusual tautomerisation of the reverse Lowdin's G*·C*(rWC) DNA base mispair via the asynchronous [with a level of asynchrony 3.49 Bohr (Brovarets' and Hovorun, 2019b)] concerted DPT to the G*′N2·C(rWC) DNA base mispair with trans-oriented N2H imino group of the G DNA base, in which participates the protons at the N3(C) and N2(G) nitrogen atoms moving in opposite directions. Unusual nature of this process consists in the fact that the transitions of the protons from N3(C) to N1(G) and from N2(G) to N4(C) along the intermolecular H-bonds provokes the rotation of the NH2 amino group of the G base into the trans-position relatively the neighboring double C2N3(G) bond. As a results, this G*·C*(rWC)↔G*′N2·C(rWC) tautomerisation reaction proceeds through the substantially non-planar intermediate – G+′N2·C−(rWC) ion pair with non-planar NH2 amino group (C1 symmetry), the substantially non-planar TSG*·C*(rWC)↔G*′N2·C(rWC) (C1 symmetry), and substantially non-planar product of this reaction—the G*′N2·C(rWC) DNA base mispair (C1 symmetry). This DNA base mispair is stabilized by the participation of the three intermolecular H-bonds—(G)O6H…O2(C), (G)N1H…N3(C), and (C)N4H…N2(G) and represents itself quite stable structure (ΔEint = −33.41/ΔGint = −18.99 kcal·mol−1). Its characteristic structural feature is the substantial non-flatness (∠N1C6C2N3 = 23.3°; ∠C2N2N4C4 = 53.8°) and the exit from the plane of the purine ring of the O6H (∠HO6C6N1 = 10.8°), N1H (∠HN1C2N3 = -157.4°), and N2H (∠HN2C2N3 = -167.6°) external groups with the trans-orientation relative to the neighboring bond C2N3.
Moreover, we have revealed that the C(rWC) and G*′N2·C(rWC) DNA base pairs interconvert via the conformational rotation of the N2H imino group around the C2N2 bond through the Gibbs freee energy barrier 31.65 kcal·mol−1 (Figure 1, Tables 1–2).
Tautomerisation of the Hoogsteen G*′·C*(H) Base Pair via the Classical DPT: IV. G*′·C*(H)↔C(H)
The tautomerisation of the Hoogsteen G*′·C*(H) base pair (ΔG = 0.00 kcal·mol−1; Cs symmetry) is possible via one-single way—through the asynchronous DPT (the values of the asynchronity consists 1.58 Bohr) (for more details see further discussion and Figure 8) along two intermolecular antiparallel lower (G)O6H…N4(C) and (C)N3H…N7(G) H-bonds through the TSG*′·C*(H)↔G*N7·C(H) (ΔG = 4.01 kcal·mol−1; Cs symmetry) connected by the N7-H-N3 covalent bridge with further formation of the dynamically-unstable C(H) base mispair with small lifetime τ = 4.46·10−13 s (ΔG = 3.20 kcal·mol−1; Cs symmetry). The G*′·C*(H)↔C(H) tautomerisation starts from the initial transfer of the hydrogen atom localized at the O6 oxygen atom of the G*′ DNA base to the N4 nitrogen atom of the C* DNA base within the G*′·C*(H) DNA base pair and then through the G+·C−(H) Hoogsteen base pair via the proton transfer from the N3 nitrogen atom to the N7 nitrogen atom leading to the formation of the C(H) DNA base mispair by the participation of the rare tautomer and canonical C DNA base.
Notably, that initial and final structures involved in this tautomerisation process are stabilized by the participation of three intermolecular H-bonds (Table 2), one of which (G)C8H…O2(C) is non-standard with energy 1.15 kcal·mol−1.
Notably, electronic (ΔEint = −35.66) and Gibbs free (ΔGint = −22.05 kcal·mol−1) energies of the interaction for the terminal C(H) base mispair exceed the values for the initial base mispair (ΔEint = −21.24/ΔGint = −8.91 kcal·mol−1). At this, total energies of the H-bonds make a great contribution to the electronic interaction energy−78.1% for the G*′·C*(H) DNA base mispair and 51.7% for the C(H) DNA base mispair.
All low-frequency intermolecular vibrations of the C(H) base mispair, which periods are in the range 8.06·10−13-1.16·10−12 s, can't develop during its lifetime. This situation is typical for the structures, which are deprived of dynamic stability (Brovarets' and Hovorun, 2019b).
So, in this case in fact the G*′·C*(H) base pair does not tautomerise via the DPT similarly to the previous G*·C*(rWC) base pair.
Tautomerisation of the Reverse Hoogsteen G*′·C*(rH) Base Pair via the DPT by the Participation of the C8H(G*′) Group: V. G*′·C*(rH)↔G*′N7·C(rH)
The G*′·C*(rH) base pair differs from two previous ones, since it tautomerises via the asynchronous DPT (with the value of asynchronity 1.69 Bohr) along the intermolecular antiparallel (C)N3H…N7(G) and (G)C8H…N4(C) H-bonds with further formation of the yilidic form G*′N7 of the G DNA base (Govorun et al., 1995a,b; Kondratyuk et al., 2000), which is characterized by the transferred proton of the C8H group to the neighboring N7 nitrogen atom. The G*′·C*(rH)↔G*′N7·C(rH) tautomerisation proceeds via the initial transfer of the proton localized at the N3 nitrogen atom of C base to the N7 nitrogen atom of G base through the formation of the G+′N7·C− ion pair followed by further proton transfer localized at the C8 carbon atom of G*′ base to the N4 nitrogen atom of C* base. Notably, that TS of this process—TSG*′·C*(rH)↔G*′N7·C(rH)–has planar structure (Cs symmetry) and contains C8-H-N4 covalent bridge, which angle is 158.8°.
This process is become possible due to the fact that G base, from one side, is CH-acid (Kondratyuk et al., 2000) and from the other—it is able to transfer into the zwitterionic tautomer—so-called yilidic form (Govorun et al., 1995a,b; Kondratyuk et al., 2000). Analysis of the obtained data (Table 1) evidences that G*′N7·C(rH) tautomer is dynamically stable structure with quite long lifetime (τ = 5.15·10−12 s). Characteristically, that all 6 low-frequency intermolecular vibrations of the G*′N7·C(rH) base mispair, which period are in the range 6.83·10−13-1.51·10−12 s, can develop during this lifetime.
This is the first case (Brovarets' et al., 2018f), when the product of the tautomerization of the H-bonded base pair by the participation of the yilidic purine base is dynamically stable structure. However, formed G*′N7·C(rH) base pair has low population−2.3·10−17 under normal conditions, which complicates the understanding of its biological role.
At the same time, such structures represent a considerable interest from a theoretical point of view, in particular, they contain unusual (C)N4H…C8−(G) H-bond (Table 2), which supplements existing data about the nature of the H-bonding in the pairs of DNA/RNA bases (Brovarets' et al., 2014e).
Profiles of the Physico-Chemical Parameters of the Investigated SPT and DPT Tautomerisations
We have investigated in details the mechanisms of the abovementioned processes of the tautomerisation of the reverse Lowdin's G*·C*(rWC), Hoogsteen G*′·C*(H), and reverse Hoogsteen G*′·C*(rH) base pairs via the PT along the intermolecular H-bonds. Tautomerisations proceed in a synchronous concerted manner via the stepwise SPT in the case of the I. G*·C*(rWC)↔G+·C−(rWC)↔G·(rWC) and II. G*·C*(rWC)↔G+·C−(rWC)↔C(rWC) reactions, while in an asynchronous concerted manner via the DPT in the case of the III. G*·C*(rWC)↔G*′N2·C(rWC), IV. G*′·C*(H)↔·C(H) and V. G*′·C*(rH)↔G*′N7·C(rH) reactions (Figures 2, 4, 6, 8, 10, Tables 3–7).
We have established following regularities of the general character for the obtained sweeps of the most important physico-chemical parameters along the IRC.
The widths of the reaction zone of the investigated reactions starting from the reagent and till the product are almost the same for the I. G*·C*(rWC)↔G·(rWC) (9.24); II. G*·C*(rWC)↔·C(rWC) (10.54) and IV. G*′·C*(H)↔·C(H) (9.30 Bohr), while these widths are significantly larger for the III. G*·C*(rWC)↔G*′N2·C(rWC) (14.49) and V. G*′·C*(rH)↔G*′N7·C(rH) (23.12 Bohr) reactions (Figures 2, 4, 6, 8, 10).
All tautomerisation processes are dipole-active, since they are followed by the significant change of the dipole moment μ of the tautomerising base pair (Figures 3C, 5C, 7C, 9C, 11C). These dependencies are U-like with maximal and minimal values located in the transition state zone for the I. G*·C*(rWC)↔G·(rWC) SPT (values of μ change in the range: 3.15–6.54); II. G*·C*(rWC)↔·C(rWC) SPT (3.15–7.15); III. G*·C*(rWC)↔G*′N2·C(rWC) DPT (2.81–8.84) and V. G*′·C*(rH)↔G*′N7·C(rH) DPT (4.47–9.01 D) reactions, while it is reverse with maximal values at the transition state zone for the IV. G*′·C*(H)↔·C(H) reaction (5.16–10.12 D) DPT (Figures 3C, 5C, 7C, 9C, 11C).
Tautomerisation process does not change the configuration of the base pair—complexes slightly compress on several dozens of Angstrom at the zone of the TSs of tautomerisation. TSs demonstrate significant deviations from a plane in the case, when steric conflicts arise between the interacting exocyclic groups (Figures 2, 4, 6, 8, 10).
According to the authors' conception (Brovarets' and Hovorun, 2013a,b,c,d, 2014c,d, 2015h,i; Brovarets' et al., 2013a,b, 2014a,b,c, 2015), it was introduced key points, namely 11 key points were obtained in the cases of the I. G*·C*(rWC)↔G·(rWC) and II. G*·C*(rWC)↔·C(rWC) SPT along the IRC in contrast to the processes of the III. G*·C*(rWC)↔G*′N2·C(rWC), IV. G*′·C*(H)↔·C(H) and V. G*′·C*(rH)↔G*′N7·C(rH) DPT, for which 9 key points have been localized.
At this, it was obtained typical crossings of the profiles for the electron density ρ, the Laplacian of the electron density Δρ and the distance dAH/HB between the hydrogen and electronegative A or B atoms for the H-bonds involved in the tautomerisation, notifying the equalization of these parameters. They occur at the 3rd and 9th key points in the case of the reaction I, 3rd and 8th—for the reaction II, 3rd and 7th—for the reactions III and IV and 3rd and 6th—for the reaction V (Figures 3G,H,K, 5G,H,K, 7G,H,K, 9G,H,K, 11G,H,K).
One and the same regularity is observed for the dE/dIRC function in all cases of tautomerisations—with two local maxima and two local minima achieved at the TS zone (Figures 3B, 5B, 7B, 9B, 11B).
Also it was observed five patterns for the energy EHB of the intermolecular H-bonds, estimated by the EML method (Espinosa et al., 1998; Matta et al., 2006b; Mata et al., 2011; Lecomte et al., 2015; Alkorta et al., 2016, 2017), along the IRC for the I, II, IV and V reactions and four patterns—for the III reaction (Figure 12, Table 8). These sweeps allow to estimate numerically the cooperativity of the neighboring H-bonds according to the methodology, proposed by us earlier (Brovarets' and Hovorun, 2019b). It was established the general pattern—the anti-parallel H-bonds amplify each other and parallel—weaken each other (Turaeva and Brown-Kennerly, 2015). Moreover, some of the dependencies of the energy EHB of the intermolecular H-bonds exist during entire IRC, such as (G)N2H…N4(C) for the I. G*·C*(rWC)↔G·(rWC) reaction, (G)O6H…O2(C) for the II. G*·C*(rWC)↔·C(rWC) reaction, (G)O6H…O2(C) for the III. G*·C*(rWC)↔G*′N2·C(rwWC) reaction, (G)C8H…O2(C) (its energy remains almost unchangeable during the IRC) for the G*′·C*(H)↔·C(H) reaction and (G)O6H…O2(C) for the G*′·C*(rH)↔G*′N7·C(rH) reaction. In the case of the III. G*·C*(rWC)↔G*′N2·C(rwWC) reaction, some of the H-bonds transform into the van der Waals contact and then to another H-bonds during the IRC: (G)N2H…N4(C) H-bond → (G)N2…N4(C) vdW contact → (G)N2H…N4(C) H-bond for the III. G*·C*(rWC)↔G*′N2·C(rwWC) reaction (Figure 12). For more details according the patterns refer to Table 8.
Finally, we would like to note some general regularities, which are characteristic for all without exclusion processes of tautomerisation.
Thus, in the vast majority of cases base pairs are plane symmetric structures during the entire PT and DPT tautomerization processes along the IRC, despite the ability of the DNA bases for the out-of-plane bending (Govorun et al., 1992; Hovorun et al., 1999; Nikolaienko et al., 2011), excluding two mentioned above cases, when there are deviation from the planarity—III. G*·C*(rWC)↔G*′N2·C(rWC) (Figure 1).
Interestingly, the total energy of the intermolecular H-bonds only partially contributes to the electron energy of the monomers interactions among all without any exceptions H-bonded structures investigated in this work (see Figures 1, 12). This result is in a good accordance with generalized literature data (Brovarets' and Hovorun, 2014e, 2019b).
Conclusions
For the first time the tautomerisation of the biologically-important conformers of the G*·C* DNA base pair - reverse Löwdin G*·C*(rWC), Hoogsteen G*′·C*(H) and reverse Hoogsteen G*′·C*(rH) pairs - was investigated and thoroughly analyzed. It was found out that the G*·C*(rWC)↔G+·C−(rWC)↔G·(rWC) and G*·C*(rWC)↔G+·C−(rWC)↔C(rWC) tautomerization processes occur via the two-stage sequential SPT via dynamically-unstable zwitterion-like G+·C−(rWC) intermediate, while the G*·C*(rWC)↔G*′N2·C(rwWC), G*′·C*(H)↔·C(H), and G*′·C*(rH)↔G*′N7·C(rH) tautomerization processes occur through the one-stage DPT. At this, proton transfer along the non-canonical (G*)CH…N(C*) H-bond is accompanied by the significant deviation of the C-H-N bridge at the TS.
Obtained data evidence that among the G*·C*(rWC), G′·C*(H) and G*′·C*(rH) base pairs only the tautomerisation of the latest of them lead to the formation of the dynamically stable G*′N7·C(rH) base pair with lifetime 5.15 ps with a miserable population 2.3·10−17.
Moreover, it was revealed that the I. G*·C*(rWC)↔G+·C−(rWC)↔G·(rWC) and II. G*·C*(rWC)↔G+·C−(rWC)↔C(rWC) tautomerization reactions proceed in a synchronous concerted manner via the stepwise SPT, while the III. G*·C*(rWC)↔G*′N2·C(rWC), IV. G*′·C*(H)↔C(H), and V. G*′·C*(rH)↔G*′N7·C(rH) reactions occur in an asynchronous concerted manner via the DPT.
We have also established dependencies of the most important physico-chemical parameters along the IRC enabling to understand more precisely the inherent nature of the investigated processes.
Data Availability
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
OB analysis and preparation of the current literature survey, discussion of the strategy of the current investigation, study conception and design, acquisition of data, drafting of manuscript analysis and interpretation of data, performance of calculations, discussion of the obtained data, preparation of the numerical data for Tables 1–8, graphical materials for Figures 1–12, and text of the manuscript. TO preparation of the numerical data for Tables 1–8 and graphical materials for Figures 1–12. DH study conception, critical revision of manuscript, proposition of the task of the investigation, discussion of the obtained data, and preparation of the text of the manuscript. All authors were involved in the proofreading of the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Authors sincerely grateful for technical support to Dr. Ivan Voiteshenko (Taras Shevchenko National University of Kyiv) and computational facilities of joint computer cluster of SSI Institute for Single Crystals of the National Academy of Sciences of Ukraine (NASU) and Institute for Scintillation Materials of the NASU incorporated into Ukrainian National Grid. DrSci OB expresses sincere gratitude to the U.S.-Ukraine Foundation (USUF) Biotech Initiative for a travel grant (2018 Emerging Biotech Leader of Ukraine; https://www.usukraine.org/biotechnology-initiative/), enabling to participate in the 63rd Annual Meeting of the Biophysical Society BPS'2019 (Baltimore, Maryland, March 2–6, 2019; https://www.biophysics.org/2019meeting#/; https://bioukraine.org/news/emerging-biotech-leader-olha-brovarets-attends-63rd-biophysical-society-meeting-in-baltimore/; https://bioukraine.org/news/emerging-leader-olha-brovarets-shares-her-us-experience-with-bionity-student-biotech-club/).
References
Acosta-Reyes, F. J., Alechaga, E., Subirana, J. A., and Campos, J. L. (2015). Structure of the DNA duplex d(ATTAAT)2 with Hoogsteen hydrogen bonds. PLoS ONE 10:e0120241. doi: 10.1371/journal.pone.0120241
Al-Khalili, J., and McFadden, J. (2014). Life on the Edge: the Coming of Age of Quantum Biology. London: Bantam Press.
Alkorta, I., Mata, I., Molins, E., and Espinosa, E. (2016). Charged versus neutral hydrogen-bonded complexes: is there a difference in the nature of the hydrogen bonds? Chem. Eur. J. 22, 9226–9234. doi: 10.1002/chem.201600788
Alkorta, I., Mata, I., Molins, E., and Espinose, E. (2018). Energetic, topological and electric field analyses of cation-cation nucleic acid interactions in Watson-Crick disposition. ChemPhysChem 20, 148–158. doi: 10.1002/cphc.201800878
Alkorta, I., Thacker, J. C. R., and Popelier, P. L. A. (2017). An interacting quantum atom study of model SN2 reactions (X–·CH3X, X = F, Cl, Br, and I). J. Comput. Chem. 39, 546–556. doi: 10.1002/jcc.25098
Alvey, H. S., Gottardo, F. L., Nikolova, E. N., and Al-Hashimi, H. M. (2014). Widespread transient Hoogsteen base-pairs in canonical duplex DNA with variable energetics. Nat. Comm. 5, 4786–4794. doi: 10.1038/ncomms5786
Arabi, A., and Matta, C. F. (2018). Adenine–thymine tautomerization under the influence of strong homogeneous electric fields. Phys. Chem. Chem. Phys. 20, 12406–12412. doi: 10.1039/C8CP01122B
Bertran, J., Blancafort, L., Noguera, M., and Sodupe, M. (2006). “Proton transfer in DNA base pairs,” in Computational Studies of RNA and DNA, eds J. Šponer and F. Lankaš (Dordrecht: Springer), 411–432. doi: 10.1007/978-1-4020-4851-3_16
Boutis, T. (1992). Proton Transfer in Hydrogen Bonded Systems. New York, NY: Plenum. doi: 10.1007/978-1-4615-3444-0
Boys, S. F., and Bernardi, F. (1970). The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 19, 553–566. doi: 10.1080/00268977000101561
Brovarets', O. O. (2013a). Structural and energetic properties of the four configurations of the A·T and G·C DNA base pairs. Ukr. Biochem. J. 85, 104–110. doi: 10.15407/ubj85.04.104
Brovarets', O. O. (2013b). Under what conditions does G·C Watson-Crick DNA base pair acquire all four configurations characteristic for A·T Watson-Crick DNA base pair? Ukr. Biochem. J. 85, 98–103. doi: 10.15407/ubj85.04.098
Brovarets', O. O., and Hovorun, D. M. (2009). Physicochemical mechanism of the wobble DNA base pairs Gua·Thy and Ade·Cyt transition into the mismatched base pairs Gua*·Thy and Ade·Cyt* formed by the mutagenic tautomers. Ukr. Bioorg. Acta. 8, 12–18.
Brovarets', O. O., and Hovorun, D. M. (2010). Quantum-chemical investigation of tautomerization ways of Watson-Crick DNA base pair guanine-cytosine. Ukr. Biochem. J. 82, 55–60.
Brovarets', O. O., and Hovorun, D. M. (2013a). Atomistic understanding of the C·T mismatched DNA base pair tautomerization via the DPT: QM and QTAIM computational approaches. J. Comput. Chem. 34, 2577–2590. doi: 10.1002/jcc.23412
Brovarets', O. O., and Hovorun, D. M. (2013b). Atomistic nature of the DPT tautomerisation of the biologically important C·C* DNA base mispair containing amino and imino tautomers of cytosine: a QM and QTAIM approach. Phys. Chem. Chem. Phys. 15, 20091–20104. doi: 10.1039/c3cp52644e
Brovarets', O. O., and Hovorun, D. M. (2013c). Prototropic tautomerism and basic molecular principles of hypoxanthine mutagenicity: an exhaustive quantum-chemical analysis. J. Biomol. Struct. Dynam. 31, 913–936. doi: 10.1080/07391102.2012.715041
Brovarets', O. O., and Hovorun, D. M. (2013d). DPT tautomerization of the long A·A* Watson-Crick base pair formed by the amino and imino tautomers of adenine: combined QM and QTAIM investigation. J. Mol. Model. 19, 4223–4237. doi: 10.1007/s00894-013-1880-2
Brovarets', O. O., and Hovorun, D. M. (2014a). Why the tautomerization of the G·C Watson–Crick base pair via the DPT does not cause point mutations during DNA replication? QM and QTAIM comprehensive analysis. J. Biomol. Struct. Dynam. 32, 1474–1499. doi: 10.1080/07391102.2013.822829
Brovarets', O. O., and Hovorun, D. M. (2014b). Can tautomerisation of the A·T Watson-Crick base pair via double proton transfer provoke point mutations during DNA replication? A comprehensive QM and QTAIM analysis. J. Biomol. Struct. Dynam. 32, 127–154. doi: 10.1080/07391102.2012.755795
Brovarets', O. O., and Hovorun, D. M. (2014c). How the long G·G* Watson-Crick DNA base mispair comprising keto and enol tautomers of the guanine tautomerises? The results of the QM/QTAIM investigation. Phys. Chem. Chem. Phys. 6, 15886–15899. doi: 10.1039/C4CP01241K
Brovarets', O. O., and Hovorun, D. M. (2014d). DPT tautomerisation of the G·Asyn and A*· DNA mismatches: A QM/QTAIM combined atomistic investigation. Phys. Chem. Chem. Phys. 16, 9074–9085. doi: 10.1039/C4CP00488D
Brovarets', O. O., and Hovorun, D. M. (2014e). Does the G· DNA mismatch containing canonical and rare tautomers of the guanine tautomerise through the DPT? A QM/QTAIM microstructural study. Mol. Phys. 112, 3033–3046. doi: 10.1080/00268976.2014.927079
Brovarets', O. O., and Hovorun, D. M. (2015a). Wobble↔Watson-Crick tautomeric transitions in the homo-purine DNA mismatches: a key to the intimate mechanisms of the spontaneous transversions. J. Biomol. Struct. Dynam. 33, 2710–2715. doi: 10.1080/07391102.2015.1077737
Brovarets', O. O., and Hovorun, D. M. (2015b). Novel physico-chemical mechanism of the mutagenic tautomerisation of the Watson–Crick-like A·G and C·T DNA base mispairs: a quantum-chemical picture. RSC Adv. 5, 66318–66333. doi: 10.1039/C5RA11773A
Brovarets', O. O., and Hovorun, D. M. (2015c). A novel conception for spontaneous transversions caused by homo-pyrimidine DNA mismatches: a QM/QTAIM highlight. Phys. Chem. Chem. Phys. 17, 21381–21388. doi: 10.1039/C5CP03211C
Brovarets', O. O., and Hovorun, D. M. (2015d). Tautomeric transition between wobble A·C DNA base mispair and Watson-Crick-like A·C* mismatch: miscrostructural mechanism and biological significance. Phys. Chem. Chem. Phys. 17, 15103–15110. doi: 10.1039/C5CP01568E
Brovarets', O. O., and Hovorun, D. M. (2015e). How many tautomerisation pathways connect Watson-Crick-like G*·T DNA base mispair and wobble mismatches? J. Biomol. Struct. Dynam. 33, 2297–2315. doi: 10.1080/07391102.2015.1046936
Brovarets', O. O., and Hovorun, D. M. (2015f). New structural hypostases of the A·T and G·C Watson-Crick DNA base pairs caused by their mutagenic tautomerisation in a wobble manner: a QM/QTAIM prediction. RSC Adv. 5, 99594–99605. doi: 10.1039/C5RA19971A
Brovarets', O. O., and Hovorun, D. M. (2015g). Proton tunneling in the A·T Watson-Crick DNA base pair: myth or reality? J. Biomol. Struct. Dynam. 12, 2716–2720. doi: 10.1080/07391102.2015.1092886
Brovarets', O. O., and Hovorun, D. M. (2015h). The physicochemical essence of the purine·pyrimidine transition mismatches with Watson-Crick geometry in DNA: A·C* versa A*·C. A QM and QTAIM atomistic understanding. J. Biomol. Struct. Dynam. 33, 28–55. doi: 10.1080/07391102.2013.852133
Brovarets', O. O., and Hovorun, D. M. (2015i). The nature of the transition mismatches with Watson-Crick architecture: the G*·T or G·T* DNA base mispair or both? A QM/QTAIM perspective for the biological problem. J. Biomol. Struct. Dynam. 33, 925–945. doi: 10.1080/07391102.2014.924879
Brovarets', O. O., and Hovorun, D. M. (2016). By how many tautomerisation routes the Watson-Crick-like A·C* DNA base mispair is linked with the wobble mismatches? A QM/QTAIM vision from a biological point of view. Struct. Chem. 27, 119–131. doi: 10.1007/s11224-015-0687-4
Brovarets', O. O., and Hovorun, D. M. (2018). “Renaissance of the tautomeric hypothesis of the spontaneous point mutations in DNA: new ideas and computational approaches,” in Mitochondrial DNA - New Insights, ed H. Seligmann (London: IntechOpen, 31–55.
Brovarets', O. O., and Hovorun, D. M. (2019a). Key microstructural mechanisms of the 2-aminopurine mutagenicity: results of extensive quantum-chemical research. J. Biomol. Struct. Dyn. 37, 2716–2732. doi: 10.1080/07391102.2018.1495577
Brovarets', O. O., and Hovorun, D. M. (2019b). Atomistic mechanisms of the double proton transfer in the H-bonded nucleobase pairs: QM/QTAIM computational lessons. J. Biomol. Struct. Dyn. 37, 1880–1907. doi: 10.1080/07391102.2018.1467795
Brovarets', O. O., and Pérez-Sánchez, H. E. (2016). Whether the amino-imino tautomerism of 2-aminopurine is involved into its mutagenicity? Results of a thorough QM investigation. RSC Adv. 110, 108255–108264. doi: 10.1039/C6RA24277D
Brovarets', O. O., and Pérez-Sánchez, H. E. (2017). Whether 2-aminopurine induces incorporation errors at the DNA replication? A quantum-mechanical answer on the actual biological issue. J. Biomol. Struct. Dynam. 35, 3398–3411. doi: 10.1080/07391102.2016.1253504
Brovarets', O. O., Pérez-Sánchez, H. E., and Hovorun, D. M. (2016). Structural grounds for the 2-aminopurine mutagenicity: a novel insight into the old problem of the replication errors. RSC Adv. 6, 99546–99557. doi: 10.1039/C6RA17787E
Brovarets', O. O., and Tsiupa, K. S. (2019). Novel conformationally-tautomeric properties of the biologically important A·T DNA base pairs. Biophys. J. 116:75a. doi: 10.1016/j.bpj.2018.11.446
Brovarets', O. O., Tsiupa, K. S., Dinets, A., and Hovorun, D. M. (2018d). Unexpected routes of the mutagenic tautomerization of the T nucleobase in the classical A·T DNA base pairs: a QM/QTAIM comprehensive view. Front. Chem. 6:532. doi: 10.3389/fchem.2018.00532
Brovarets', O. O., Tsiupa, K. S., and Hovorun, D. M. (2018a). Non-dissociative structural transitions of the Watson-Crick and reverse Watson-Crick A·T DNA base pairs into the Hoogsteen and reverse Hoogsteen forms. Sci. Repts. 8:10371. doi: 10.1038/s41598-018-28636-y
Brovarets', O. O., Tsiupa, K. S., and Hovorun, D. M. (2018b). Surprising conformers of the biologically important A·T DNA base pairs: QM/QTAIM proofs. Front. Chem. 6:8. doi: 10.3389/fchem.2018.00008
Brovarets', O. O., Tsiupa, K. S., and Hovorun, D. M. (2018c). Unexpected A·T(WC)↔A·T(rWC)/A·T(rH) and A·T(H)↔A·T(rH)/A·T(rWC) conformational transitions between the classical A·T DNA base pairs: a QM/QTAIM comprehensive study. Int. J. Quant. Chem. 118:e25674. doi: 10.1002/qua.25692
Brovarets', O. O., Tsiupa, K. S., and Hovorun, D. M. (2018e). Novel pathway for mutagenic tautomerization of classical A·T DNA base pairs via sequential proton transfer through quasi-orthogonal transition states: a QM/QTAIM investigation. PLoS ONE. 13:e0199044. doi: 10.1371/journal.pone.0199044
Brovarets', O. O., Tsiupa, K. S., and Hovorun, D. M. (2018f). The A·T(rWC)/A·T(H)/A·T(rH) ↔ A·T*(rwWC)/A·T*(wH)/A·T*(rwH) mutagenic tautomerization via sequential proton transfer: a QM/QTAIM study. RSC Adv. 8, 13433–13445. doi: 10.1039/C8RA01446A
Brovarets', O. O., Voiteshenko, I., Pérez-Sánchez, H. E., and Hovorun, D. M. (2018h). A QM/QTAIM detailed look at the Watson-Crick↔wobble tautomeric transformations of the 2-aminopurine·pyrimidine mispairs. J. Biomol. Struct. Dynam. 36, 1649–1665. doi: 10.1080/07391102.2017.1331864
Brovarets', O. O., Voiteshenko, I. S., and Hovorun, D. M. (2018g). Physico-chemical profiles of the wobble↔Watson-Crick G*·2AP(w)↔G·2AP(WC) and A·2AP(w)↔A*·2AP(WC) tautomerisations: A QM/QTAIM comprehensive survey. Phys. Chem. Chem. Phys. 20, 623–636. doi: 10.1039/C7CP05139E
Brovarets', O. O., Voiteshenko, I. S., Pérez-Sánchez, H. E., and Hovorun, D. M. (2017). A QM/QTAIM research under the magnifying glass of the DPT tautomerisation of the wobble mispairs involving 2-aminopurine. New J. Chem. 41, 7232–7243. doi: 10.1039/C7NJ00717E
Brovarets', O. O., Yurenko, Ye. P., Dubey, I. Y., and Hovorun, D. M. (2012). Can DNA-binding proteins of replisome tautomerize nucleotide bases? Ab initio model study. J. Biomol. Struct. Dynam. 29, 1101–1109. doi: 10.1080/07391102.2011.672624
Brovarets', O. O., Yurenko, Y. P., and Hovorun, D. M. (2014e). Intermolecular CH···O/N H-bonds in the biologically important pairs of natural nucleobases: a thorough quantum-chemical study. J. Biomol. Struct. Dynam. 32, 993–1022. doi: 10.1080/07391102.2013.799439
Brovarets', O. O., Zhurakivsky, R. O., and Hovorun, D. M. (2013a). The physico-chemical “anatomy” of the tautomerisation through the DPT of the biologically important pairs of hypoxanthine with DNA bases: QM and QTAIM perspectives. J. Mol. Model. 19, 4119–4137. doi: 10.1007/s00894-012-1720-9
Brovarets', O. O., Zhurakivsky, R. O., and Hovorun, D. M. (2013b). The physico-chemical mechanism of the tautomerisation via the DPT of the long Hyp*·Hyp Watson-Crick base pair containing rare tautomer: a QM and QTAIM detailed look. Chem. Phys. Lett. 578, 126–132. doi: 10.1016/j.cplett.2013.05.067
Brovarets', O. O., Zhurakivsky, R. O., and Hovorun, D. M. (2014a). Structural, energetic and tautomeric properties of the T·T*/T*·T DNA mismatch involving mutagenic tautomer of thymine: a QM and QTAIM insight. Chem. Phys. Lett. 592, 247–255. doi: 10.1016/j.cplett.2013.12.034
Brovarets', O. O., Zhurakivsky, R. O., and Hovorun, D. M. (2014b). Is the DPT tautomerisation of the long A·G Watson-Crick DNA base mispair a source of the adenine and guanine mutagenic tautomers? A QM and QTAIM response to the biologically important question. J. Comput. Chem. 35, 451–466. doi: 10.1002/jcc.23515
Brovarets', O. O., Zhurakivsky, R. O., and Hovorun, D. M. (2014c). A QM/QTAIM microstructural analysis of the tautomerisation via the DPT of the hypoxanthine·adenine nucleobase pair. Mol. Phys. 112, 2005–2016. doi: 10.1080/00268976.2013.877170
Brovarets', O. O., Zhurakivsky, R. O., and Hovorun, D. M. (2014d). Does the tautomeric status of the adenine bases change upon the dissociation of the A*·Asyn Topal-Fresco DNA mismatch? A combined QM and QTAIM atomistic insight. Phys. Chem. Chem. Phys. 16, 3715–3725. doi: 10.1039/c3cp54708f
Brovarets', O. O., Zhurakivsky, R. O., and Hovorun, D. M. (2015). DPT tautomerisation of the wobble guanine·thymine DNA base mispair is not mutagenic: QM and QTAIM arguments. J. Biomol. Struct. Dynam. 33, 674–689. doi: 10.1080/07391102.2014.897259
El-Sayed, A. A., Tamara Molina, A., Alvarez-Ros, M. C., and Palafox, M. A. (2015). Conformational analysis of the anti-HIV Nikavir prodrug: comparisons with AZT and thymidine, and establishment of structure-activity relationships/tendencies in other 6′-derivatives. J. Biomol. Struct. Dynam. 33, 723–748. doi: 10.1080/07391102.2014.909743
Erdmann, V. A., Markiewicz, W. T., and Barciszewski, J. (2014). Chemical Biology of Nucleic Acids. Heidelberg: Springer.
Espinosa, E., Molins, E., and Lecomte, C. (1998). Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 285, 170–173. doi: 10.1016/S0009-2614(98)00036-0
Florian, J., Hrouda, V., and Hobza, P. (1994). Proton transfer in the adenine-thymine base pair. J. Am. Chem. Soc. 116, 1457–1460. doi: 10.1021/ja00083a034
Frisch, M. J., Head-Gordon, M., and Pople, J. A. (1990). Semi-direct algorithms for the MP2 energy and gradient. Chem. Phys. Lett. 166, 281–289. doi: 10.1016/0009-2614(90)80030-H
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2010). GAUSSIAN 09 (Revision, B.01). Wallingford, CT: Gaussian Inc.
Gatti, C., Macetti, G., Boyd, R. J., and Matta, C. F. (2018). An electron density source-function study of DNA base pairs in their neutral and ionized ground states. J. Comput. Chem. 39, 1112–1128. doi: 10.1002/jcc.25222
Glushenkov, A. N., and Hovorun, D. M. (2016). Complete set of H-bonded homoassociates of 9-methylguanine by the participation of mutagenic tautomers: quantum-mechanical study. Repts. Natl. Acad. Sci. Ukraine. 3, 98–106. doi: 10.15407/dopovidi2016.03.098
Godbeer, A. D., Al-Khalili, J. S., and Stevenson, P. D. (2015). Modelling proton tunnelling in the adenine-thymine base pair. Phys. Chem. Chem. Phys. 7, 13034–13044. doi: 10.1039/C5CP00472A
Gorb, L., Podolyan, Y., Dziekonski, P., Sokalski, W. A., and Leszczynski, J. (2004). Double-proton transfer in adenine–thymine and guanine–cytosine base pairs. A post Hartree–Fock ab initio study. J. Am. Chem. Soc. 126, 10119–10129. doi: 10.1021/ja049155n
Govorun, D. M., Danchuk, V. D., Mishchuk Ya, R, Kondratyuk, I. V., Radomsky, N. F., and Zheltovsky, N. V. (1992). AM1 calculation of the nucleic acid bases structure and vibrational spectra. J. Mol. Struct. 267, 99–103. doi: 10.1016/0022-2860(92)87016-O
Govorun, D. M., Kondratyuk, I. V., and Zheltovsky, N. V. (1995a). About physico-chemical mechanisms of hydrogen-tritium exchange of purine nucleotide bases C8H8 groups with water. Biopol. Cell. 11, 24–28. doi: 10.7124/bc.0003E9
Govorun, D. M., Kondratyuk, I. V., and Zheltovsky, N. V. (1995b). Prototropic molecular-zwitterion tautomerism of purine. Biopol. Cell. 11, 45–50. doi: 10.7124/bc.000402
Gutowski, M., Van Lenthe, J. H., Verbeek, J., Van Duijneveldt, F. B., and Caahalasinski, G. (1986). The basis set superposition error in correlated electronic structure calculations. Chem. Phys. Lett. 124, 370–375. doi: 10.1016/0009-2614(86)85036-9
Hariharan, P. C., and Pople, J. A. (1973). The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta. 28, 213–222. doi: 10.1007/BF00533485
Hoogsteen, K. (1963). The crystal and molecular structure of a hydrogen-bonded complex between 1-methylthymine and 9-methyladenine. Acta Cryst. 16, 907–916. doi: 10.1107/S0365110X63002437
Hovorun, D. M., Gorb, L., and Leszczynski, J. (1999). From the nonplanarity of the amino group to the structural nonrigidity of the molecule: a post-Hartree-Fock ab initio study of 2-aminoimidazole. Int. J. Quantum. Chem. 75, 245–253. doi: 10.1002/(SICI)1097-461X(1999)75:3<245::AID-QUA14>3.0.CO;2-0
Hratchian, H. P., and Schlegel, H. B. (2005). “Finding minima, transition states, and following reaction pathways on ab initio potential energy surfaces,” in Theory and applications of computational chemistry: The first 40 years, eds C. E. Dykstra, G. Frenking, K. S. Kim, and G. Scuseria (Amsterdam: Elsevier), 195–249. doi: 10.1016/B978-044451719-7/50053-6
Iogansen, A. V. (1999). Direct proportionality of the hydrogen bonding energy and the intensification of the stretching ν(XH) vibration in infrared spectra. Spectrochim. Acta Part A 55, 1585–1612. doi: 10.1016/S1386-1425(98)00348-5
Kendall, R. A., Dunning, T. H. Jr, and Harrison, R. J. (1992). Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 96, 6796–6806. doi: 10.1063/1.462569
Kondratyuk, I. V., Samijlenko, S. P., Kolomiets,', I. M, and Hovorun, D. M. (2000). Prototropic molecular–zwitterionic tautomerism of xanthine and hypoxanthine. J. Mol. Struct. 523, 109–118. doi: 10.1016/S0022-2860(99)00385-3
Krishnan, R., Binkley, J. S., Seeger, R., and Pople, J. A. (1980). Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 72, 650–654. doi: 10.1063/1.438955
Lecomte, C., Espinosa, E., and Matta, C. F. (2015). On atom–atom ‘short contact' bonding interactions in crystals. IUCr J. 2, 161–163. doi: 10.1107/S2052252515002067
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 37, 785–789. doi: 10.1103/PhysRevB.37.785
Löwdin, P-O. (1963). Proton tunneling in DNA and its biological implications. Rev. Mod. Phys. 35, 724–732. doi: 10.1103/RevModPhys.35.724
Löwdin, P-O. (1966). “Quantum genetics and the aperiodic solid: some aspects on the biological problems of heredity, mutations, aging, and tumors in view of the quantum theory of the DNA molecule,” in Advances in Quantum Chemistry, : ed P.-O. Löwdin (New York, NY; London: Academic Press), 213–360. doi: 10.1016/S0065-3276(08)60076-3
Mata, I., Alkorta, I., Espinosa, E., and Molins, E. (2011). Relationships between interaction energy, intermolecular distance and electron density properties in hydrogen bonded complexes under external electric fields. Chem. Phys. Lett. 507, 185–189. doi: 10.1016/j.cplett.2011.03.055
Matta, C. F. (2010). How dependent are molecular and atomic properties on the electronic structure method? Comparison of Hartree-Fock, DFT, and MP2 on a biologically relevant set of molecules. J. Comput. Chem. 31, 1297–1311. doi: 10.1002/jcc.21417
Matta, C. F. (2014). Modeling biophysical and biological properties from the characteristics of the molecular electron density, electron localization and delocalization matrices, and the electrostatic potential. J. Comput. Chem. 35, 1165–1198. doi: 10.1002/jcc.23608
Matta, C. F., Castillo, N., and Boyd, R. J. (2006a). Atomic contributions to bond dissociation energies in aliphatic hydrocarbons. J. Chem. Phys. 125;204103. doi: 10.1063/1.2378720
Matta, C. F., Castillo, N., and Boyd, R. J. (2006b). Extended weak bonding interactions in DNA: π-stacking (base-base), base-backbone, and backbone-backbone interactions. J. Phys. Chem. B 110, 563–578. doi: 10.1021/jp054986g
Matta, C. F., and Hernández-Trujillo, J. (2003). Bonding in polycyclic aromatic hydrocarbons in terms of the electron density and of electron delocalization. J. Phys. Chem. 107, 7496–7504. doi: 10.1021/jp034952d
McFadden, J., and Al-Khalili, J. (2018). The origins of quantum biology. Proc. R Soc. 474:20180674. doi: 10.1098/rspa.2018.0674
Meisner, J., and Kastner, J. (2016). Atom tunnelling in chemistry. Angew. Chem. Int/ Ed. 55, 5400–5413. doi: 10.1002/anie.201511028
Nikolaienko, T. Y., Bulavin, L. A., and Hovorun, D. M. (2011). How flexible are DNA constituents? The quantum-mechanical study. J. Biomol. Struct. Dynam. 29, 563–575. doi: 10.1080/07391102.2011.10507406
Nikolaienko, T. Y., Bulavin, L. A., and Hovorun, D. M. (2012). Bridging QTAIM with vibrational spectroscopy: the energy of intramolecular hydrogen bonds in DNA-related biomolecules. Phys. Chem. Chem. Phys. 14, 7441–7447. doi: 10.1039/c2cp40176b
Nikolova, E. N., Zhou, H., Gottardo, F. L., Alvey, H. S., Kimsey, I. J., and Al-Hashimi, H. M. (2014). A historical account of Hoogsteen base pairs in duplex DNA. Biopolymers 99, 955–968. doi: 10.1002/bip.22334
Palafox, M. A. (2014). Molecular structure differences between the antiviral nucleoside analogue 5-iodo-2‘-deoxyuridine and the natural nucleoside 2'-deoxythymidine using MP2 and DFT methods: conformational analysis, crystal simulations, DNA pairs and possible behavior. J. Biomol. Struct. Dynam. 32, 831–851. doi: 10.1080/07391102.2013.789402
Parker, B. R., and Van Everv, J. (1971). Quantum tunnelling in DNA. Chem. Phys. Lett. 8, 94–99. doi: 10.1016/0009-2614(71)80586-9
Parr, R. G., and Yang, W. (1989). Density-Functional Theory of Atoms and Molecules. Oxford: Oxford University Press.
Peng, C., Ayala, P. Y., Schlegel, H. B., and Frisch, M. J. (1996). Using redundant internal coordinates to optimize equilibrium geometries and transition states. J. Comput. Chem. 17, 49–56. doi: 10.1002/(SICI)1096-987X(19960115)17:1<49::AID-JCC5>3.0.CO;2-0
Poltev, V. I., Anisimov, V. M., Sanchez, C., Deriabina, A., Gonzalez, E., Garcia, D., et al. (2016). Analysis of the conformational features of Watson–Crick duplex fragments by molecular mechanics and quantum mechanics methods. Biophysics 61, 217–226. doi: 10.1134/S0006350916020160
Pous, J., Urpi, L., Subirana, J. A., Gouyette, C., Navaza, J., and Campos, J. L. (2008). Stabilization by extra-helical thymines of a DNA duplex with Hoogsteen base pairs. J. Am. Chem. Soc. 130, 6755–6760. doi: 10.1021/ja078022+
Pusuluk, O., Torun, G., and Deliduman, C. (2018). Quantum entanglement shared in hydrogen bonds and its usage as a resource in molecular recognition. Mod. Phys. Lett. 32:1850308. doi: 10.1142/S0217984918503086
Roßbach, S., and Ochsenfeld, Ch. (2017). Influence of coupling and embedding schemes on QM size convergence in QM/MM approaches for the example of a proton transfer in DNA. J. Chem. Theor. Comput. 13, 1102–1107. doi: 10.1021/acs.jctc.6b00727
Shekaari, A., and Jafari, M. (2019). Modeling the action of environment on proton tunneling in the adenine–thymine base pair. Prog. Biophys. Mol. Biol. doi: 10.1016/j.pbiomolbio.2019.07.002. [Epub ahead of print].
Smedarchina, Z., Siebrand, W., and Fernández-Ramos, A. (2018). Entanglement and co-tunneling of two equivalent protons in hydrogen bond pairs. J. Chem. Phys. 148, 102307–1–102307-15. doi: 10.1063/1.5000681
Sordo, J. A. (2001). On the use of the Boys–Bernardi function counterpoise procedure to correct barrier heights for basis set superposition error. J. Mol. Struct. 537, 245–251. doi: 10.1016/S0166-1280(00)00681-3
Sordo, J. A., Chin, S., and Sordo, T. L. (1988). On the counterpoise correction for the basis set superposition error in large systems. Theor. Chim. Acta. 74, 101–110. doi: 10.1007/BF00528320
Srivastava, R. (2019). The role of proton transfer on mutations. Front Chem. 7:536. doi: 10.3389/fchem.2019.00536
Szabat, M., and Kierzek, R. (2017). Parallel-stranded DNA and RNA duplexes: structural features and potential applications. FEBS J. 284, 3986–3998. doi: 10.1111/febs.14187
Tirado-Rives, J., and Jorgensen, W. L. (2008). Performance of B3LYP density functional methods for a large set of organic molecules. J. Chem. Theory Comput. 4, 297–306. doi: 10.1021/ct700248k
Turaeva, N., and Brown-Kennerly, V. (2015). Marcus model of spontaneous point mutation in DNA. Chem. Phys. 461, 106–110. doi: 10.1016/j.chemphys.2015.09.005
Watson, J. D., and Crick, F. H. C. (1953a). The structure of DNA. Cold Spring Harb. Symp. Quant. Biol. 18, 123–131. doi: 10.1101/SQB.1953.018.01.020
Watson, J. D., and Crick, F. H. C. (1953b). Molecular structure of nucleic acids: a structure for deoxyribose nucleic acid. Nature. 171, 737–738. doi: 10.1038/171737a0
Wigner, E. (1932). Über das Überschreiten von Potentialschwellen bei chemischen Reaktionen [Crossing of potential thresholds in chemical reactions]. Zeits Physik Chem. 19, 203–216. doi: 10.1515/zpch-1932-1920
Ye, M. Y., Zhu, R. T., Li, X., Zhou, X. S., Yin, Z. Z., Li, Q., et al. (2017). Adaptively recognizing parallel-stranded duplex structure for fluorescent DNA polarity analysis. Anal. Chem. 89, 8604–8608. doi: 10.1021/acs.analchem.7b02467
Zhou, H. (2016). Occurrence and Function of Hoogsteen Base Pairs in Nucleic Acids. PhD Thesis, Duke University. Available online at: https://dukespace.lib.duke.edu/dspace/handle/10161/12892
Keywords: single proton transfer, double proton transfer, conformer, reverse Löwdin base pair, Hoogsteen, reverse Hoogsteen, transition state, quantum-mechanical calculation
Citation: Brovarets' OO, Oliynyk TA and Hovorun DM (2019) Novel Tautomerisation Mechanisms of the Biologically Important Conformers of the Reverse Löwdin, Hoogsteen, and Reverse Hoogsteen G*·C* DNA Base Pairs via Proton Transfer: A Quantum-Mechanical Survey. Front. Chem. 7:597. doi: 10.3389/fchem.2019.00597
Received: 05 June 2019; Accepted: 12 August 2019;
Published: 18 September 2019.
Edited by:
Yong Wang, Ningbo University, ChinaReviewed by:
Santiago Tolosa Arroyo, University of Extremadura, SpainDingguo Xu, Sichuan University, China
Copyright © 2019 Brovarets', Oliynyk and Hovorun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dmytro M. Hovorun, ZC5tLmhvdm9ydW5AaW1iZy5vcmcudWE=
†ORCID: Ol'ha O. Brovarets' orcid.org/0000-0002-8929-293X
Timothy A. Oliynyk orcid.org/0000-0002-2276-033X
Dmytro M. Hovorun orcid.org/0000-0002-5579-5520