 Andrés E. Martín Díaz
Andrés E. Martín Díaz James E. M. Lewis
James E. M. Lewis- Department of Chemistry, Imperial College London, Molecular Sciences Research Hub, London, United Kingdom
Metal-organic cages (MOCs) have emerged as a diverse class of molecular hosts with potential utility across a vast spectrum of applications. With advances in single-crystal X-ray diffraction and economic methods of computational structure optimisation, cavity sizes can be readily determined. In combination with a chemist’s intuition, educated guesses about the likelihood of particular guests being bound within these porous structures can be made. Whilst practically very useful, simple rules-of-thumb, such as Rebek’s 55% rule, fail to take into account structural flexibility inherent to MOCs that can allow hosts to significantly adapt their internal cavity. An often unappreciated facet of MOC structures is that, even though relatively rigid building blocks may be employed, conformational freedom can enable large structural changes. If it could be exploited, this flexibility might lead to behavior analogous to the induced-fit of substrates within the active sites of enzymes. To this end, in-roads have already been made to prepare MOCs incorporating ligands with large degrees of conformational freedom. Whilst this may make the constitution of MOCs harder to predict, it has the potential to lead to highly sophisticated and functional synthetic hosts.
Introduction
Metal-organic cages (MOCs) are self-assembled structures derived from carefully selected combinations of metal ions and ligands (Cook and Stang, 2015; Debata et al., 2019; Pilgrim and Champness, 2020). Porous MOCs of myriad shape and size are capable of binding guest molecules in their cavities (Rizzuto et al., 2019), leading to applications in catalysis (Yoshizawa et al., 2009; Sinha and Mukherjee, 2018), storage of reactive species (Galan and Ballester, 2016), molecular separations (Zhang et al., 2021), and drug delivery (Casini et al., 2017) amongst others. Guest-binding is a complex process involving contributions from enthalpic and entropic factors for both the encapsulation of the guest(s) and the liberation of solvent molecules (or displacement of extant guest molecules) (Metherell et al., 2018). The 55% rule originally established by Rebek (Mecozzi and Rebek, 1998) has been a good guiding principle when predicting molecular binding based on the packing coefficient of the guest inside the host’s cavity. This volume-based approach, however, assumes the cavity to be static and therefore does not take into consideration host flexibility, which can greatly affect cavity size and shape and is a relatively underexplored consideration. The ability to harness this structural flexibility could lead to enzyme-like conformational adaptability (Boehr et al., 2009; Feixas et al., 2014). Characterising host flexibility is therefore crucial to understanding its effects on any potential applications.
In this mini review we highlight examples from the literature that demonstrate the inherent structural flexibility of rigid metal-organic hosts, both in solution and the solid-state, and examine the limited number of examples wherein units with high degrees of conformational freedom have been purposefully incorporated into ligand frameworks. A focus is placed on assemblies that undergo conformational, rather than configurational, changes without requiring dissociation and rearrangement of components. Full-scale compositional changes will not be covered, although it is noted than for some examples the involvement of bond dissociation in the mechanism of observed adaptability cannot be absolutely excluded. Neither will assemblies with stimuli-responsive, switchable units be included (Han et al., 2013; Oldknow et al., 2018), which have been covered elsewhere (McConnell et al., 2015). This review is not intended to be comprehensive—selected examples from the literature have been chosen to demonstrate key principles.
Observation of Structural Flexibility in Solution
Various 1D and 2D NMR techniques are commonly used to characterise MOCs and to probe their guest-binding properties. Flexibility arising from conformational freedom, however, is likely to occur rapidly on the NMR timescale, resulting in a time-averaged structure. This makes it difficult to gain accurate information regarding the extent of conformational plasticity. Despite this, observations made during particular experiments can allow inference of structural flexibility, often aided by use in conjunction with SCXRD data.
In 2003 Shionoya and co-workers reported the synthesis of the helical sandwich-shaped assembly [Ag3(1)2]3+ as a racemic mixture of the M and P isomers (Figure 1A), demonstrated in solution through the formation of the Δ-TRISPHAT salt (Hiraoka et al., 2003). At 253 K (in 2:1 d6-acetone/CDCl3) two sets of equal intensity signals were observed for the two enantiomers, suggesting a lack of chiral induction from the anion. Upon increasing the temperature to 303 K coalescence to a single set of signals was observed, the result of rapid interconversion between the P and M enantiomers on the NMR timescale. In order to switch between the two enantiomers, the cage likely transitions through a high energy, achiral conformer with an increased distance between the core phenyl units of the ligands (< 1 Å). In an analogous complex, [Ag3(2)2]3+, interconversion between isomers occurred rapidly only at elevated temperatures, indicating a larger energy barrier to helical inversion.
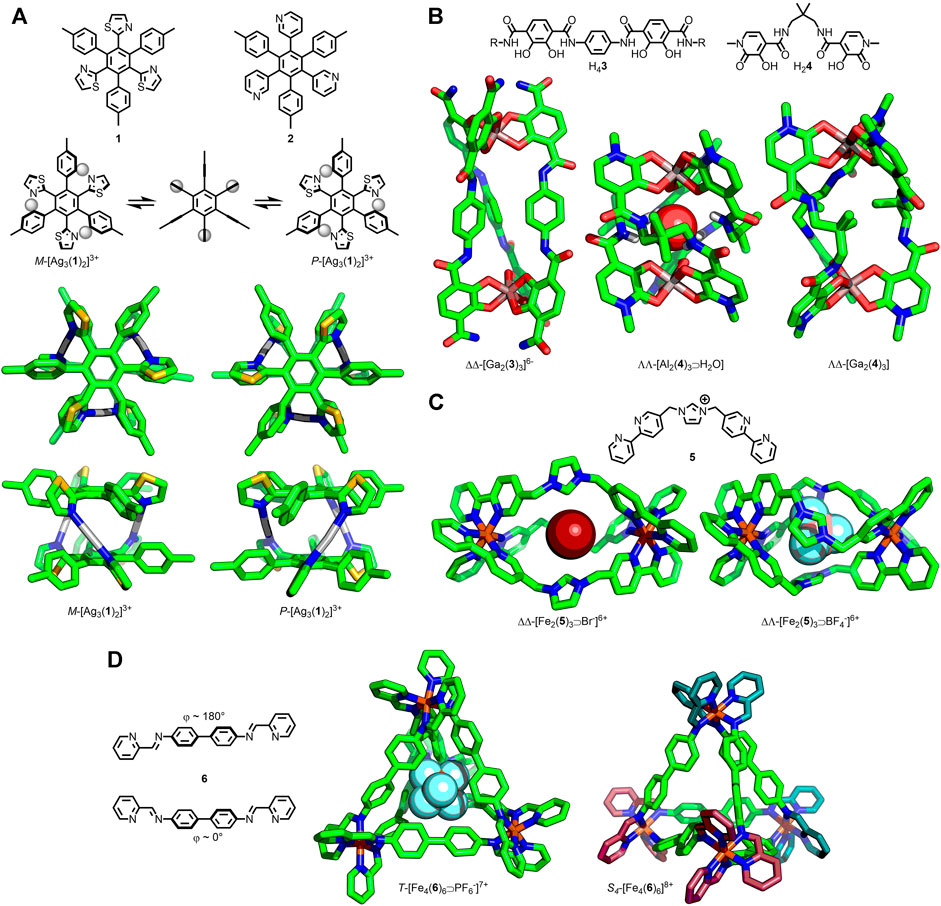
FIGURE 1. (A) Shionoya’s trinuclear P/M-[Ag3(1/2)2]3+ helicates capable of undergoing stereochemical inversion via an achiral intermediate. (B) Raymond’s [Ga2(3)3] helicate able to undergo helical inversion via a Bailar twist mechanism; SCXRD structures of [Al2(4)3⋑H2O] helicate and [Ga2(4)3] mesocate. (C) Wu’s triply-stranded [Fe2(5)3⋑X−]6+ assemblies with encapsulated anion-dependent formation of helicate (X− = Br−) or mesocate (X− = BF4−) structures. (D) Nitschke’s [Fe4(6)6]8+ tetrahedra, with encapsulation of a PF6− anion favouring the T-symmetric assembly, whilst the empty cage preferentially crystallised as the S4 structure with ∆ (cyan) and Λ (magenta) Fe(II) centers.
Raymond and co-workers investigated the stereochemical inversion of triply-stranded dinuclear helicates assembled from bis-catecholate ligands (3) and Ga(III) ions (Figure 1B) (Kerstin et al., 1996). From a comprehensive NMR study the authors were able to ascertain that the isomers were interconverting through an intramolecular helical inversion process (ΔΔ⇋ΛΛ), with independent chiral inversion occurring at each metal centre through a non-dissociative Bailar twist mechanism, via a mesocate intermediate. Subsequent work with related [M2(4)3] [M = Al(III) or Ga(III)] assemblies (Figue 1B) demonstrated an ability to controllably switch between the chiral helicate (M···M 7.13 Å) and larger, achiral mesocate (M···M 9.74 Å) using host-guest interactions (Xu et al., 1999). Despite crystallising as the mesocate, [Ga2(4)3] existed in solution as an equilibrium between the helicate and mesocate forms. The helicate was shown to be stabilised by an encapsulated solvent molecule (water or DMSO); increasing the water concentration served to shift the equilibrium further towards the helicate.
More recently Wu and co-workers (Cui et al., 2012) observed a similar change in the structure of a triply-stranded, dinuclear structure, assembled from bis-bidentate ligand 5 and Fe(II), with different anions bound in its cavity (Figure 1C). With Br− or NO3− encapsulated a helicate assembly was formed. In contrast, the tetrahedral anions BF4−, ClO4−, and SO42− resulted in mesocates, with the helicates being significantly longer (Fe···Fe ∼11.7 Å vs ∼10.6 Å). This switch between helicate and mesocate was reasoned to arise from conformational changes of the ligand in order to maximize favourable interactions with the encapsulated anion. In the case of the mesocate the ligands adopt a C-shaped conformation, whilst an S-conformer led to the helicate assembly.
A related concept has been studied by Nitschke and co-workers, wherein the relative stereochemistry of octahedral metal centres within a tetrahedral MOC could be affected by encapsulation of different anions, impacting the overall symmetry of the assembly (Clegg et al., 2013). Subcomponent self-assembly of 4,4′-diaminobiphenyl, 2-formylpyridine and Fe(II) with large triflimide counteranions resulted in a tetrahedral cage structure, [Fe4(6)6]8+ (Figure 1D). In the solid state, SCXRD data demonstrated that the cage possessed two metal centres of one stereochemistry and two of the opposite (i.e., ΔΔΛΛ), giving the tetrahedron an overall S4 symmetry [with two ligands adopting an S-shaped conformation (φ ≈ 180°), the remaining four a C-shape ( φ ≈ 0°)]. In solution, however, a mixture of S4-, T-, and C3-symmetric assemblies were observed in an approximately 5:3:2 ratio at equilibrium. Smaller anions were capable of binding within the cavity of the cage, and altering the equilibrium position of this diasteroeomeric mixture, with ClO4− and PF6− shifting the equilibrium in favor of the T-symmetric isomer (ΔΔΔΔ/ΛΛΛΛ; all ligands S-conformation). Remarkably NO3−, BF4−, Cl−, Br−, and I− encapsulation all resulted in quantitative conversion to the T diasteroisomer, as determined by solution-phase NMR analysis. In addition, SCXRD data demonstrated that the MOC framework was able to conformationally adapt to encapsulate the various anions; the cavity volume was found to expand from 69 Å3 with BF4− to 87 Å3 with PF6−.
In these studies solution phase data were able to demonstrate structural changes in the MOCs arising from alterations to the assembly’s symmetry. More subtle changes from, e.g., flexing and conformational adjustment of ligands, are not so readily detected in this manner. SCXRD, however, provides more precise structural information and was used effectively in combination with the solution-phase data in the previous examples to add quantitative spatial information. It is important to note that the solid-state data merely provides a snap-shot image of the assemblies, which can be affected by various packing interactions. Multiple sets of SCXRD data, however, can show differences in structural parameters, allowing inference of the extent of MOC flexibility.
Solid-State SCXRD Evidence for Structural Flexibility
Arguably one of the most studied MOCs in the literature is the hexanuclear octahedron ([Pd6(en)6(7)4]12+; Figure 2A) originally reported by the Fujita group (Fujita et al., 1995). For this assembly multiple sets of SCXRD data are available with different guests encapsulated within the host, allowing quantitative comparison of structural parameters in the solid state (Tashira et al., 2005; Nishioka et al., 2007; Kohyama et al., 2014; Takezawa et al., 2014; Cullen et al., 2019; Takezawa et al., 2020). These demonstrate that the M···M distance varies by less than 1% across numerous solid-state structures. Clearly, in this instance, significant distortion of the cage structure through host-guest interactions and crystal packing effects is not in evidence. For other MOCs, however, such effects have been observed.
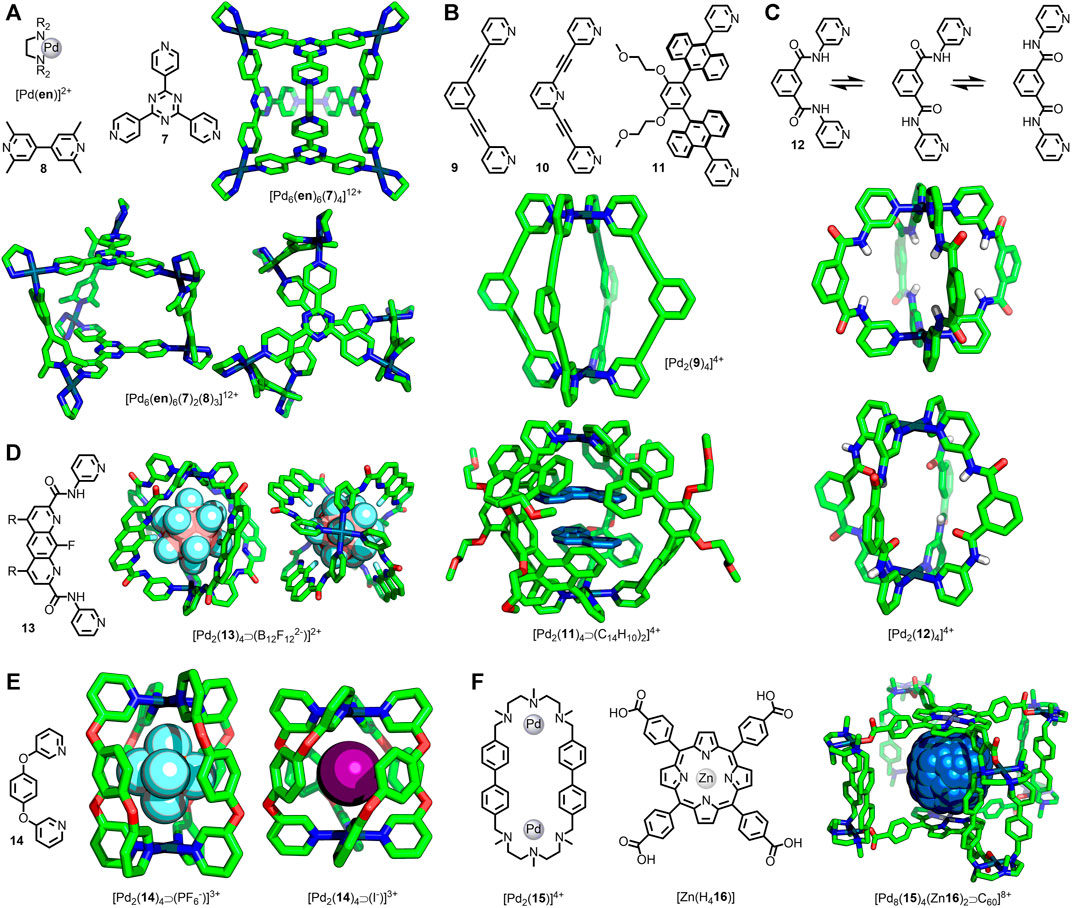
FIGURE 2. (A) Fujita’s octahedral cage, [Pd6(en)6(7)4]12+, and trigonal prismatic assembly [Pd6(en)6(8)3(7)2]12+ shown from the side and above. (B) Ditopic ligands 9, 10 and 11 used in the assembly of Pd2L4 cages. SCXRD structures of [Pd2(9)4]4+ and [Pd2(11)4⋑(C14H10)2]4+. (C) Puddephatt’s ditopic ligand 12 with three possible conformations accessed through rotation about the amide bonds; SCXRD structures of [Pd2(12)4]4+ with 12 exhibiting two different ligand conformations. (D) Gan’s [Pd2(13)4⋑(B12F122-)]2+ cage viewed from the side (left) and above (right). (E) McMorran and Steel’s [Pd2(14)4]4+ helicates with PF6−(left) and I−(right) anions bound. (F) Ribas’ porphyrin-based cage, [Pd8(15)4(Zn16)2]8+, with encapsulated C60 guest.
The heteroleptic trigonal prismatic cage [Pd6(en)6(7)2(8)3]12+ (Figure 2A), also reported by Fujita and co-workers (Yoshizawa et al., 2005a), was assembled from 7 and 2,2′-6,6′-tetramethyl-4,4′-bipyridine (8) with cis-PdII(en) metal nodes (Figure 1A). Due to the manner in which the ligands are arranged in this assembly, 7 and 8 are orthogonal in the most symmetric form. In this conformer the two 7 ligands would eclipse one another and the torsion angle between them would be 0°. As this angle increases, the 7 ligands will become more offset, and the angle between 7 and 8 will diverge from orthogonality. With extensive investigation of this cage’s host-guest properties having been undertaken, it was possible to examine multiple SCXRD structures of the assembly to analyse changes in structural parameters. From this it was seen that the average torsion angle varied between ∼19 and ∼39°, and the distance between triazines ranged from 9.9–10.6 Å (Yoshizawa et al., 2005a; Yoshizawa et al., 2005b; Ono et al., 2007; Ono et al., 2009; Yamauchi et al., 2010; Schmidt et al., 2016).
Torsional twisting of trigonal prismatic cages, leading to a dramatic change in cavity size, has been reported by others. Severin and co-workers prepared a hexanuclear cage from ligand 7 and diruthenium(II) clips. Upon binding two molecules of coronene in the cavity the torsion angle between the two 7 ligands decreased from ∼75–∼3°, increasing the distance between them from 3.4–11.1 Å, thereby expanding the cavity size from effectively zero to > 500 Å3 (Mirtschin et al., 2010). Likewise, Han and Jin have reported a trigonal prism assembled from 7 and diiridium(III) clips that collapsed (7···7 distance reduced from 6.9–3.3 Å) through increased torsion between the 7 ligands (∼16–∼30°) upon removal of a coronene guest molecule (Han and Jin, 2011).
Ditopic banana-shaped ligands (Han et al., 2014) are commonly combined with Pd(II) ions to form dinuclear, lantern-shaped architectures. These are often designed with the ligand framework fully conjugated; however, rotation about certain bonds is still feasible, potentially introducing some dynamic character to the resultant assemblies. In 2010 Hooley and co-workers reported the cage [Pd2(9)4]4+ (Figure 2B) and its SCXRD structure with an encapsulated OTf− anion (Liao et al., 2010). More recently, Lusby and co-workers have examined this cage for its catalytic potential (Spicer et al., 2020) and were able to obtain SCXRD data of the PF6− salt with pentacenedione bound in the cavity (August et al., 2016). Although relatively minor, there was a difference of 0.36 Å in the Pd···Pd distance between the cage SCXRD structures (11.85 and 12.21 Å), and the torsion angle between the core phenyl and terminal pyridyl rings were greater for the longer cage (12.5° on average compared to 5.1°).
The related [Pd2(10)4]4+ architecture, in which the central phenyl unit of the ligand is replaced with a pyridine, has been reported by Crowley and co-workers. They were able to obtain SCXRD structures of various salts of the cage (Lewis and Crowley, 2014), as well as a host-guest adduct with two cisplatin molecules (Lewis et al., 2012). Again, some variation in the Pd···Pd distance was observed (11.5–12.2 Å) with torsion angles between the core and coordinating pyridine rings ranging from 2.5–19.0°.
In a similar Pd2L4 system (Figure 2B) reported by Yoshizawa and co-workers (Kishi et al., 2011), with anthracene units linking the core phenyl ring and coordinating pyridines instead of alkynes (ligand 11), even more dramatic differences were observed with various host-guest adducts. For instance, in the 1:1 adduct formed between the cage and [2.2]-paracyclophane the Pd···Pd distance was found to be 13.8 Å. In contrast, with two molecules of corranulene encapsulated this was found to shrink to 12.4 Å, a reduction of 10% (Kishi et al., 2013).
Puddephatt and co-workers prepared ligand 12 with 3-pyridyl units attached to a meta-phenyl core through amide bonds. Rotation about these amide bonds ostensibly permitted three conformations of the ligand (Figure 2C). When combined with Pd(II) ions the expected [Pd2(12)4]4+ assembly was obtained, wherein adoption of different ligand conformations could alter the character of the cavity depending on whether the amide carbonyl or N-H units were directed internally. SCXRD structures of the cage (Figure 2C) with both F3CSO3− and F3CCO2− anions were obtained. In the case of the former both N-H units of each ligand were directed endohedrally, allowing the formation of hydrogen-bonds to partially encapsulated F3CSO3− anions. Conversely the F3CCO2− salt resulted in adoption of ligand conformations with one carbonyl directed exohedrally, the other endohedrally acting as a hydrogen-bond acceptor to encapsulated water molecules. This change from a higher to lower symmetry assembly was coincident with a lengthening of the Pd···Pd distance from 9.5–11.2 Å (Yue et al., 2003). Despite its lower symmetry in the solid state, the observation of a single set of signals for the pyridyl units by NMR demonstrated rapid conformational interconversion on the NMR timescale (Yue et al., 2004).
Likewise, Gan and co-workers were able to observe significant changes in structural parameters in a similarly designed system (Lin et al., 2018). [Pd2(13)4]4+ (Figure 2D) was assembled from ligand 13 and an appropriate Pd(II) source. SCXRD structures for this architecture were obtained with BF4−, NO3− (two polymorphs were reported), and B12F122− anions encapsulated within the internal cavity, with changes in the ligand conformation demonstrated through significant variation in the Pd···Pd distance (12.1–13.4 Å).
Unsurprisingly the introduction of saturated units within the ligand framework offers the potential for greater degrees of flexibility within assemblies. In 1998 McMorran and Steel reported the first example of a dipalladium(II) helical cage assembled from a ditopic ligand, namely 1,4-bis(3-pyridyloxy)benzene (14) (McMorran and Steel, 1998). In contrast to the previous examples, the ether linkage between the core and coordinating units engendered the ligands with more significant degrees of freedom. This work was recently extended to include a thorough examination of the anion-binding properties of this cage, including an extensive study in the solid state from SCXRD data (Steel and McMorran, 2019). This revealed a significant 16% reduction in the Pd···Pd distance upon exchanging a bound PF6− anion (8.84 Å) for an I− guest (7.44 Å) (Figure 2E), the result of a drastic change in the Pd-N···N-Pd torsion angle (average 41.5° for PF6− and 80.9° for I−). The associated reduced helical pitch (77–37 Å) reduced the effective cavity size from 92–41 Å3.
Molecular Dynamics Simulations of Metal-Organic Cage Flexibility
Computational modelling of supramolecular systems has become cheaper and more readily available in recent years, allowing combinations of experimental and theoretical data to be used to gain in-depth understanding of these artificial mimics of biological assemblies (Frederix et al., 2018). Given the previously mentioned constraints placed on information related to conformation dynamics that can be gleaned from solution and solid-state data, computational analysis offers a potential route to quantitatively examine flexibility of metal-organic assemblies. Perhaps most interestingly these techniques could be effectively used to aid in the design of such systems for specific applications (Young et al., 2020a).
Recently Lusby, Duarte and co-workers utilised molecular dynamics (MD) simulations to explore the conformational flexibility of the two dipalladium(II) cages assembled from 9 and 10 to explain observations related to their catalytic activity (Young et al., 2020b). In these simulations the Pd···Pd distance in the cages was found to vary between 11.3 and 12.9 Å for ligand 9, and 10.9 and 12.5 Å for ligand 10, with the torsion angle between the core ring and terminal pyridines ranging from 0–60°. Interestingly, for the [Pd2(10)4]4+ cage, conformations with significant twisting of the ligands were found to be of similar energy to the highly symmetric structure (conformer with Φ = 41° was only 2.6 kcal mol−1 higher in energy than the symmetric Φ = 0° conformer).
Ribas and co-workers reported the synthesis of a nanocage assembled from four dinuclear macrocycle Pd(II) complexes ([Pd2(15)]4+) and two tetracarboxylate Zn(II)-porphyrins ([Zn(H416)]) (García-Simón et al., 2014). These cages were observed to bind fullerene guests, and the SCXRD structures of the “empty” cage and C60 and C70 adducts were obtained (Figure 2F). For the empty cage the distance between Zn(II) ions was 14.1 Å, with a contraction seen for the C70 (13.8 Å) and C60 (13.1 Å) host-guest adducts. This was concomitant with a small decrease in the Pd···Pd distance within the macrocycles from 11.25–11.17 Å. Subsequent molecular dynamics simulations (2.5 μs) showed flexing of the cage to give Zn···Zn distances ranging between 11.3 and 15.8 Å. Binding of fullerene guests induced a compression of this distance, consistent with the SCXRD structures. To enable fullerene guests to enter the cage cavity a transient expansion of both the portal size, from ∼50 Å2 up to ∼90 Å2, and the Zn···Zn distance (∼15 Å) was observed, demonstrating a significant dynamic range for this host assembly (García-Simón et al., 2020).
Metal-Organic Assemblies Derived From Ligands With Large Amplitude Conformational Flexibility
The previous sections have sought to demonstrate that even MOCs assembled from what would normally be considered rigid ligands–including those with fully conjugated systems–can display a reasonable amount of flexibility. There is a paucity of examples in the literature, however, of purposeful introduction of units that display large-amplitude conformational freedom into metal-organic systems. A key facet of metallo-supramolecular self-assembly is predicting products of self-assembly based on constituent metal ion geometry and ligand conformation. The importance of ligand conformation has been amply demonstrated by the work of the Fujita group on PdnL2n cages assembled from ditopic ligands, in which the ligand angle determines the thermodynamically favoured value of n (Harris et al., 2013). When multiple ligand conformers are accessible, predicting/controlling self-assembly outcomes becomes more difficult. Adaptability, however, is a hallmark of natural self-assembled systems, and could offer an increased level of stimuli-responsiveness if incorporated into artificial systems seeking to mimic Nature’s success.
Ferrocene, first synthesised 70 years ago (Kealy and Pauson, 1951), is the most well-known of the organometallic sandwich complexes. The ability of the two cyclopentadiene ligands to rotate relative to each other has led to significant interest in ferrocene’s use as a “ball-bearing” component in molecular machines (Scottwell and Crowley, 2016). The ability of the ligands to undergo a full 360° rotation means that the dihedral angle between substituents of 1,1′-disubstituted ferrocenes can vary from 0 (syn)–180° (anti) (Figure 3A).
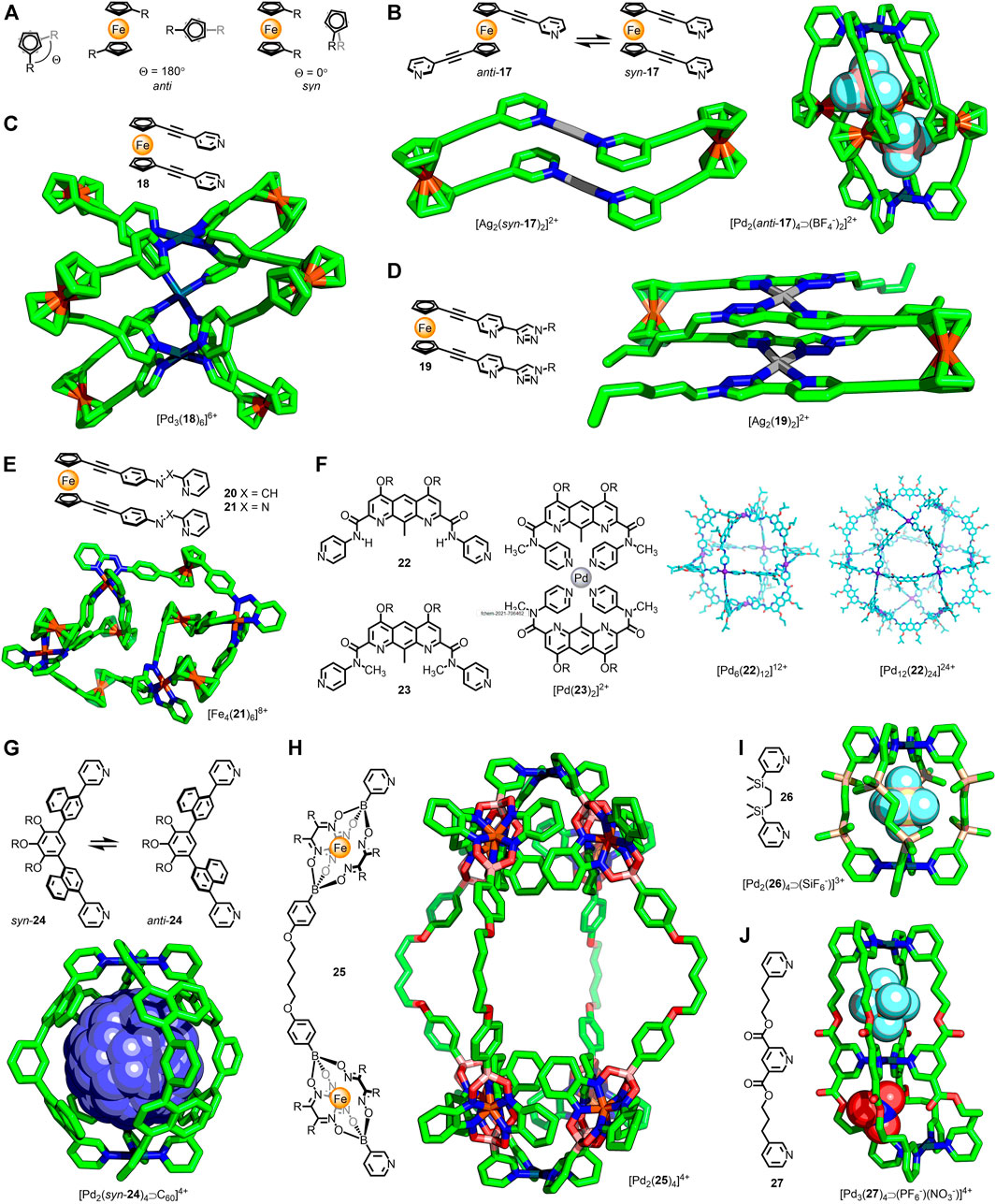
FIGURE 3. (A) The cyclopentadiene rings of ferrocene are able to undergo 360° rotation. The two extremes of conformation are the anti (Θ = 180°) and syn (Θ = 0°) conformers. (B) In the dinuclear metallacycle [Ag2(17)2]2+ ligand 17 is observed to adopt a syn conformation; in the quadruple-stranded [Pd2(17)4]4+ cage the anti conformer is observed. (C) Crowley’s trigonal prismatic [Pd3(18)6]6+ assembly with ligands in syn arrangements (Θ = 25–42°). (D) A dinuclear metallacycle, [Ag2(syn-18)2]2+ reported by Crowley. (E) Nitschke’s twisted parallelogram assembly, with ligand 21 in both syn and anti conformations. (F) Non-covalent interactions affect favoured ligand conformations: self-assembly with Pd(II) led to a Pd12L24 cuboctahedron with 22 (via a kinetic Pd6L12 cube; calculated structures shown), whilst ligand 23 gives the mononuclear [Pd(23)2]2+ complex. Adapted with permission from Liu et al. (2020). Copyright © 2020 Wiley-VCH GmbH. (G) Yoshizawa’s atropisomeric ditopic ligand that formed [Pd2(24)4]4+ assemblies; guest encapsulation biased the distribution of cage atropisomers. (H) Severin’s alkyl-linked metallo-ligand that formed the large (Pd···Pd ∼ 3 nm) [Pd2(25)4]4+ cage. (I) Alkyl-linked ligand 26 formed Pd2L4 cages capable of anion-encapsulation. (J) Jung’s trinuclear, double-cavity cage bound anions in each cavity, which adapt their shape to best match the encapsulated guest.
In 2003 Lindner and co-workers reported the self-assembly of 1,1′-bis(pyridin-3-ylethynyl)ferrocene (17) with Ag(I) and trans-Pd(II)Cl2 metal nodes (Lindner et al., 2003). In both instances dinuclear metallacycles were obtained (Figure 3B), with the ligands adopting a stacked conformation (Θ = 7°). Crowley and co-workers subsequently examined the self-assembly of this ligand with Pd(II) ions (Vasdev et al., 2019), observing formation of [Pd2L4]4+ cages (Figure 3B) with the ligands in anti conformations (Θ = 105–134°). SCXRD structures of the cage revealed that the Pd···Pd distance was affected by changes in the dihedral angle between the pyridyl arms (13.1 Å for one crystallographically independent molecule of [Pd2(17)4⋑(BF4−)2]2+ and Θave = 109°; 13.4 Å for [Pd2(17)4⋑(SbF6−)]3+ and Θave = 126°), demonstrating an ability for the cage to adapt its shape based on the ligand conformation.
Self-assembly of the isomeric 1,1′-bis(pyridin-4-ylethynyl)ferrocene ligand (18) with Pd(II) ions resulted in formation of a trigonal prismatic structure (Vasdev et al., 2021). SCXRD data of the BF4− salt revealed an eclipsed conformation adopted by the ferrocene units (Figure 3C), with a small variation in the dihedral angle between independent ligands (25–42°).
The self-assembly of twin-armed ferrocene ligands with bidentate coordinating groups has also been investigated. The formation and solid-state structures of dinuclear metallacycles (Figure 3D) assembled from bis(2-pyridyl-1,2,3-triazole) ligand 19 were reported by Crowley and co-workers (Findlay et al., 2018). For both the Ag(I) and Pd(II) metallacycles the ligands were eclipsed, with a slight difference in the dihedral angles observed between the two assemblies (0.9 and 16.2°, respectively). Nitschke and co-workers synthesised isostructural [Fe(II)4L6]8+ twisted parallelogram assemblies from imine and azo ligands 20/21 (Plajer et al., 2020). The SCXRD structure of one of these intriguing architectures (Figure 3E) revealed the co-existence of both syn (55–67°) and anti (118–121°) ligand conformations, demonstrating the ability of chemically identical ligands to occupy inequivalent environments through accessing different conformers. Attempts to oxidise the ferrocene units led to dis-assembly of the parallelogram, and formation of mononuclear [Fe(II)L]3+ complexes.
Singleton and co-workers synthesised dipyridyl ligands 22 and 23 (Figure 3F) capable of bond rotation about the amide units (Liu et al., 2020). For 22 the range of favourable conformations was restricted by intramolecular hydrogen bonds between the amide proton and diazaanthracene core. Initially self-assembly of this ligand with Pd(II) resulted in an M6L12 cube, with an average angle of 97° between the coordinating pyridyl groups in the calculated structure. This species was, however, revealed to be a kinetically trapped intermediate; prolonged heating of the sample led to rearrangement to form a larger M12L24 cuboctahedron, with the calculated average ligand angle increasing to 120°. In contrast, ligand 23, with methylated amide units preventing hydrogen bonds across the ligand framework, yielded a simple mononuclear ML2 complex (Figure 3F) as a result of increased conformational flexibility.
Recently Yoshizawa and co-workers investigated the dynamics of a Pd2L4 cage assembled from ditopic ligands with 1,4-naphthylene linkers (24) (Tsutsui et al., 2020). Hindered rotation of these units resulted in atropisomeric forms of the ligand. Although this did not inherently affect the relative orientations of the coordinating pyridyl groups, it did lead to an interconverting mixture of 42 virtually isoenergetic cage isomers. Guest encapsulation was found to bias the mixture towards individual isomers, with C60 inducing quantitative conversion to the all-syn assembly, as shown by SCXRD (Figure 3G). As such, the cage was able to undergo subtle structural modification in response to guest molecules in order to provide a favourable fit.
Alkyl spacers are inherently flexible and are therefore not usually used in MOC ligand design. Their conformational freedom, however, could allow for large amplitude adaptability through interactions with guest molecules. Severin and co-workers successfully synthesised Pd2L4 cages (Figure 3H) up to 3 nm in length using ditopic metallo-ligands with akyl spacer units (25), demonstrating the potential for the controlled incorporation of highly flexible alkyl units into metallo-assemblies (Jansze et al., 2016). Jung, Lee and co-workers obtained SCXRD structures for [Pd2L4⋑(X−)]3+ cages (Figure 3I), assembled from a dipyridyl ligand with a flexible disilylethane core (26), with a range of encapsulated anions (Lee et al., 2020). The flexibility of the alkyl linker enabled expansion of the capsule to suit the encapsulated anion (Pd···Pd = 8.49 and 9.58 Å for X− = BF4− and SiF6−, respectively). This concept was exploited in the double-cavity [Pd3(27)4⋑(X−)2]4+ architecture, with both homo and hetero-anion adducts formed (Sarada et al., 2020). In this instance each cavity was able to independently adopt an extended or crumpled shape, dependent on the anion encapsulated (Figure 3J).
An alternative method to introduce flexible, saturated units into MOCs is to use the Weak-Link Approach (WLA), developed by the Mirkin group (Giannesci et al., 2005). In this methodology kinetically stable metal-organic architectures assembled from flexible ligands are formed via ligand exchange from a rigid, thermodynamically-favourable precursor species. This enables access to flexible structures that may not be thermodynamic products and cannot be directly accessed from a combination of ligand and metal nodes. This was first demonstrated with ligand 28 which formed the metallacycle [Rh2(28)2]2+, with chelation to the metal centres between the phosphine and ether oxygen (Farrell et al., 1998). The hemilabile oxygen donors could be displaced through addition of competing ligands, such as CO, giving the expanded structure [Rh2(CO)6(28)2]2+ (Figure 4A). The Weak-Link Approach has since been shown to be suitable for the preparation of flexible MOCs (Ovchinnikov et al., 2002). The condensed cage [M3(29)2]3+ [M = Rh(I) or Ir(I)] could be opened through addition of CO and Cl− ligands, leading to the expanded structure, [M3(CO)3Cl3(29)2] (Figure 4B).
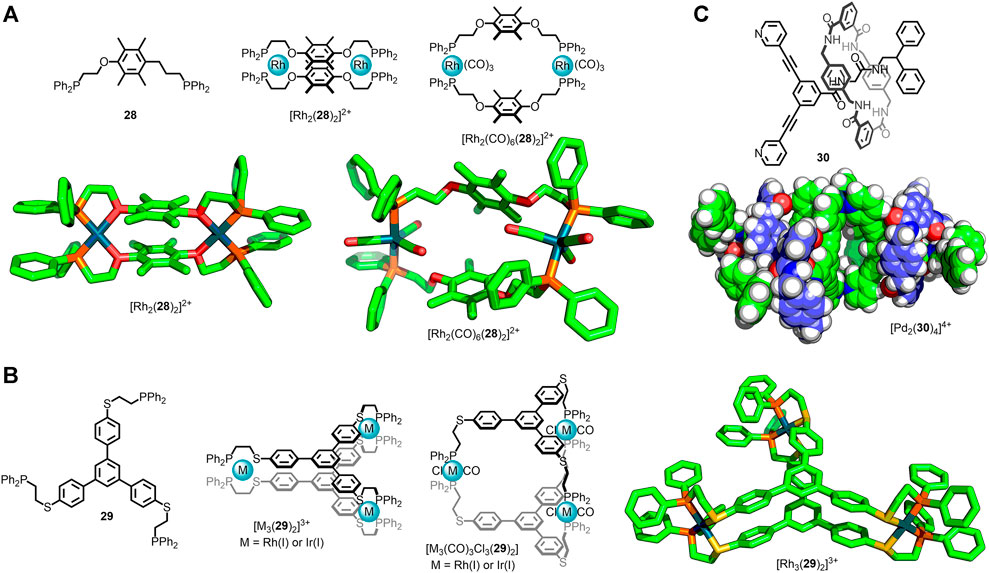
FIGURE 4. (A) Mirkin’s Weak-Link Approach in which a rigid metallacycle precursor, [Rh2(28)2]2+, is first formed under thermodynamic control. Subsequent displacement of the ether oxygen donors gave the flexible metallacycle [Rh2(CO)6(28)2]2+, with ligand 28 coordinated in a bis-monodentate fashion. (B) This principle has been applied to MOCs, with flexible cage [M3(CO)3Cl3(29)2] accessed via thermodynamic intermediate [M3(29)2]3+ [M = Rh(I) or Ir(I)]. (C) Lewis’ [2]rotaxane ligand, 30, that self-assembled with Pd(II) ions to form a Pd2L4 cage (molecular model) with dynamic interlocked macrocycle components (shown in blue).
Mechanically interlocked molecules have been widely investigated for their use as molecular machines due to the high degree of co-conformational freedom between their components afforded by the lack of covalent bonds (Heard and Goldup, 2020). Consequently there has been considerable interest in the development of mechanically interlocked ligands (Lewis et al., 2017) and the use of metal ions/nodes to periodically arrange them (Hoyas Pérez and Lewis, 2020; Wilson and Loeb, 2020). Lewis and co-workers recently reported the synthesis of [2]rotaxane ligand 30 (Figure 4C), wherein one of the stopper units preventing escape of the macrocycle component was based on dipyridyl ligand 9 (Wong et al., 2020). Self-assembly with Pd(II) ions led to the quadruple-stranded cage [Pd2(30)4]4+. NMR analysis of this structure revealed a single set of signals for the macrocycle, indicating rapid pirouetting on the NMR timescale. As such the large degree of co-conformational freedom inherent to rotaxane 30 was maintained in the metal-organic assembly, demonstrating the potential for mechanical bonds to be employed as flexible components within MOCs.
Conclusion and Future Outlooks
The complexity of MOCs that are routinely reported has increased dramatically since preliminary studies in the field over thirty years ago (Pullen et al., 2021). Heteroleptic (Bloch and Clever, 2017), mixed-metal (Li et al., 2015) and low symmetry (Lewis and Crowley, 2020) assemblies towards the development of more sophisticated host systems are becoming more commonplace. Coincident with this, easier access to and improvements within SCXRD and the advancement of computational power for theoretical investigations allow researchers to gain rapid and in-depth analysis of these systems. This has led to some remarkable applications for MOCs, particularly in the area of catalysis. Appreciation of the structural flexibility these systems are capable of, however, is largely overlooked, despite the parallels that are often drawn between MOCs and enzymes. Herein we have taken a brief look at some of the solution-phase, solid-state and computational data available to highlight how significant variations in the structural parameters of MOCs can be. If this flexibility could be harnessed it could open up the doors for developing more sophisticated artificial systems to mimic biological machinery. Steps towards this lofty goal have already been undertaken, with a handful of examples reported of MOCs derived from ligands containing moieties with significant conformational freedom. With continuing investigations into flexible metal-organic assemblies, elucidation of methodologies for their controlled self-assembly and manipulation will lead to increasingly sophisticated systems capable of displaying adaptive behaviour and allow their use in new and exciting applications.
Author Contributions
JL designed and supervised the writing of the review. Both authors contributed to writing and approved the final manuscript.
Funding
This work was supported by an Imperial College Research Fellowship (JL).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Professor Matthew Fuchter is thanked for useful discussions and access to resources.
References
August, D. P., Nichol, G. S., and Lusby, P. J. (2016). Maximizing Coordination Capsule-Guest Polar Interactions in Apolar Solvents Reveals Significant Binding. Angew. Chem. Int. Ed. 55, 15022–15026. doi:10.1002/anie.201608229
Bloch, W. M., and Clever, G. H. (2017). Integrative Self-Sorting of Coordination Cages Based on Naked Metal Ions. Chem. Commun. 53, 8506–8516. doi:10.1039/C7CC03379F
Boehr, D. D., Nussinov, R., and Wright, P. E. (2009). The Role of Dynamic Conformational Ensembles in Biomolecular Recognition. Nat. Chem. Biol. 5, 789–796. doi:10.1038/nchembio.232
Casini, A., Woods, B., and Wenzel, M. (2017). The Promise of Self-Assembled 3D Supramolecular Coordination Complexes for Biomedical Applications. Inorg. Chem. 56, 14715–14729. doi:10.1021/acs.inorgchem.7b02599
Clegg, J. K., Cremers, J., Hogben, A. J., Breiner, B., Smulders, M. M. J., Thoburn, J. D., et al. (2013). A Stimuli Responsive System of Self-Assembled Anion-Binding Fe4L68+cages. Chem. Sci. 4, 68–76. doi:10.1039/c2sc21486e
Cook, T. R., and Stang, P. J. (2015). Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination. Chem. Rev. 115, 7001–7045. doi:10.1021/cr5005666
Cui, F., Li, S., Jia, C., Mathieson, J. S., Cronin, L., Yang, X.-J., et al. (2012). Anion-Dependent Formation of Helicates versus Mesocates of Triple-Stranded M2L3 (M = Fe2+, Cu2+) Complexes. Inorg. Chem. 51, 179–187. doi:10.1021/ic201417y
Cullen, W., Takezawa, H., and Fujita, M. (2019). Demethylenation of Cyclopropanes via Photoinduced Guest‐to‐Host Electron Transfer in an M6L4 Cage. Angew. Chem. Int. Ed. 58, 9171–9173. doi:10.1002/anie.201904752
Debata, N. B., Tripathy, D., and Sahoo, H. S. (2019). Development of Coordination Driven Self-Assembled Discrete Spherical Ensembles. Coord. Chem. Rev. 387, 273–298. doi:10.1016/j.ccr.2019.02.016
Farrell, J. R., Mirkin, C. A., Guzei, I. A., Liable-Sands, L. M., and Rheingold, A. L. (1998). The Weak-Link Approach to the Synthesis of Inorganic Macrocycles. Angew. Chem. Int. Ed. 37, 465–467. doi:10.1002/(sici)1521-3773(19980302)37:4<465::aid-anie465>3.0.co;2-a
Feixas, F., Lindert, S., Sinko, W., and McCammon, J. A. (2014). Exploring the Role of Receptor Flexibility in Structure-Based Drug Discovery. Biophy. Chem. 186, 31–45. doi:10.1016/j.bpc.2013.10.007
Findlay, J. A., McAdam, C. J., Sutton, J. J., Preston, D., Gordon, K. C., and Crowley, J. D. (2018). Metallosupramolecular Architectures Formed with Ferrocene-Linked Bis-Bidentate Ligands: Synthesis, Structures, and Electrochemical Studies. Inorg. Chem. 57, 3602–3614. doi:10.1021/acs.inorgchem.7b02503
Frederix, P. W. J. M., Patmanidis, I., and Marrink, S. J. (2018). Molecular Simulations of Self-Assembling Bio-Inspired Supramolecular Systems and Their Connection to Experiments. Chem. Soc. Rev. 47, 3470–3489. doi:10.1039/c8cs00040a
Fujita, M., Oguro, D., Miyazawa, M., Oka, H., Yamaguchi, K., and Ogura, K. (1995). Self-assembly of Ten Molecules Into Nanometre-Sized Organic Host Frameworks. Nature 378, 469–471. doi:10.1038/378469a0
Galan, A., and Ballester, P. (2016). Stabilization of Reactive Species by Supramolecular Encapsulation. Chem. Soc. Rev. 45, 1720–1737. doi:10.1039/C5CS00861A
García-Simón, C., Colomban, C., Çetin, Y. A., Gimeno, A., Pujals, M., Ubasart, E., et al. (2020). Complete Dynamic Reconstruction of C60, C70, and (C59N)2 Encapsulation into an Adaptable Supramolecular Nanocapsule. J. Am. Chem. Soc. 142, 16051–16063. doi:10.1021/jacs.0c07591
García-Simón, C., Garcia-Borràs, M., Gómez, L., Parella, T., Osuna, S., Juanhuix, J., et al. (2014). Sponge-like Molecular Cage for Purification of Fullerenes. Nat. Commun. 5, 5557. doi:10.1038/ncomms6557
Giannesci, N. C., Masar, M. S., and Mirkin, C. A. (2005). Development of a Coordination Chemistry-Based Approach for Functional Supramolecular Structures. Acc. Chem. Res. 38, 825–837. doi:10.1021/ar980101q
Han, M., Engelhard, D. M., and Clever, G. H. (2014). Self-Assembled Coordination Cages Based on Banana-Shaped Ligands. Chem. Soc. Rev. 43, 1848–1860. doi:10.1039/C3CS60473J
Han, M., Michel, R., He, B., Chen, Y.-S., Stalke, D., John, M., et al. (2013). Light-Triggered Guest Uptake and Release by a Photochromic Coordination Cage. Angew. Chem. Int. Ed. 52, 1319–1323. doi:10.1002/anie.201207373
Han, Y.-F., and Jin, G.-X. (2011). Flexible Organometallic Cages: Efficient Formation by C–H Activation-Directed Muticomponent Assembly, Isomerization, and Host-Guest Properties. Chem. Asian J. 6, 1348–1352. doi:10.1002/asia.201100080
Harris, K., Fujita, D., and Fujita, M. (2013). Giant Hollow MnL2n Spherical Complexes: Structure, Functionalisation and Applications. Chem. Commun. 49, 6703–6712. doi:10.1039/C3CC43191F
Heard, A. W., and Goldup, S. M. (2020). Simplicity in the Design, Operation, and Applications of Mechanically Interlocked Molecular Machines. ACS Cent. Sci. 6, 117–128. doi:10.1021/acscentsci.9b01185
Hiraoka, S., Harano, K., Tanaka, T., Shiro, M., and Shionoya, M. (2003). Quantitative Formation of Sandwich-Shaped Trinuclear Silver(I) Complexes and Dynamic Nature of Their P⇌M Flip Motion in Solution. Angew. Chem. Int. Ed. 42, 5182–5185. doi:10.1002/anie.200351068
Hoyas Pérez, N., and Lewis, J. E. M. (2020). Synthetic Strategies towards Mechanically Interlocked Oligomers and Polymers. Org. Biomol. Chem. 18, 6757–6780. doi:10.1039/d0ob01583k
Jansze, S. M., Wise, M. D., Vologzhanina, A. V., Scopelliti, R., and Severin, K. (2016). PdII2L4-type Coordination Cages up to Three Nanometers in Size. Chem. Sci. 8, 1901–1908. doi:10.1039/c6sc04732g
Kealy, T. J., and Pauson, P. L. (1951). A New Type of Organo-Iron Compound. Nature 168, 1039–1040. doi:10.1038/1681039b0
Kerstin, B., Meyer, M., Powers, R. E., and Raymond, K. N. (1996). Dinuclear Catecholate Helicates: Their Inversion Mechanism. J. Am. Chem. Soc. 118, 7221–7222. doi:10.1021/ja9613522
Kishi, N., Li, Z., Sei, Y., Akita, M., Yoza, K., Siegel, J. S., et al. (2013). Wide-Ranging Host Capability of a PdII-Linked M2L4 Molecular Capsule With an Anthracene Shell. Chem. Eur. J. 19, 6313–6320. doi:10.1002/chem.201204010
Kishi, N., Li, Z., Yoza, K., Akita, M., and Yoshizawa, M. (2011). An M2L4 Molecular Capsule With an Anthracene Shell: Encapsulation of Large Guests up to 1 nm. J. Am. Chem. Soc. 133, 11438–11441. doi:10.1021/ja2037029
Kohyama, Y., Murase, T., and Fujita, M. (2014). Metal-Organic Proximity in a Synthetic Pocket. J. Am. Chem. Soc. 136, 2966–2969. doi:10.1021/ja412582k
Lee, J., Lim, S., Kim, D., Jung, O.-S., and Lee, Y.-A. (2020). Flexibility and Anion Exchange of [(X)@Pd2L4] Cages for Recognition of Size and Charge of Polyatomic Anions. Dalton Trans. 49, 15002–15008. doi:10.1039/d0dt03005h
Lewis, J. E. M., and Crowley, J. D. (2014). Exo- and Endo-Hedral Interactions of Counteranions with Tetracationic Pd2L4 Metallosupramolecular Architectures. Supramolecular Chem. 26, 173–181. doi:10.1080/10610278.2013.842644
Lewis, J. E. M., and Crowley, J. D. (2020). Metallo‐Supramolecular Self‐Assembly with Reduced‐Symmetry Ligands. ChemPlusChem 85, 815–827. doi:10.1002/cplu.202000153
Lewis, J. E. M., Galli, M., and Goldup, S. M. (2017). Properties and Emerging Applications of Mechanically Interlocked Ligands. Chem. Commun. 53, 298–312. doi:10.1039/C6CC07377H
Lewis, J. E. M., Gavey, E. L., Cameron, S. A., and Crowley, J. D. (2012). Stimuli-Responsive Pd2L4 metallosupramolecular Cages: Towards Targeted Cisplatin Drug Delivery. Chem. Sci. 3, 778–784. doi:10.1039/C2SC00899H
Li, H., Yao, Z.-J., Liu, D., and Jin, G.-X. (2015). Multi-component Coordination-Driven Self-Assembly toward Heterometallic Macrocycles and Cages. Coord. Chem. Rev. 293-294, 139–157. doi:10.1016/j.ccr.2014.08.014
Liao, P., Langloss, B. W., Johnson, A. M., Knudsen, E. R., Tham, F. S., Julian, R. R., et al. (2010). Two-component Control of Guest Binding in a Self-Assembled Cage Molecule. Chem. Commun. 46, 4932–4934. doi:10.1039/C0CC00234H
Lin, Q., Gao, L., Kauffmann, B., Zhang, J., Ma, C., Luo, D., et al. (2018). Helicity Adaptation Within a Quadruply Stranded Helicate by Encapsulation. Chem. Commun. 54, 13447–13450. doi:10.1039/c8cc07932c
Lindner, E., Zong, R., Eichele, K., Weisser, U., and Ströbele, M. (2003). Synthesis and Structure of Redox-Active Heterotetranuclear Molecular Polygons by Self-Assembly of Two Ferrocene-Bridged Bis(pyridines) With Two Transition Metals. Eur. J. Inorg. Chem. 2003, 705–712. doi:10.1002/ejic.200390097
Liu, C. L., Bobylev, E. O., Fu, Y., Poole, D. A., Robeyns, K., Fustin, C. A., et al. (2020). Balancing Ligand Flexibility versus Rigidity for the Stepwise Self‐Assembly of M12L24 via M6L12 Metal-Organic Cages. Chem. Eur. J. 26, 11960–11965. doi:10.110/chem.20200139910.1002/chem.202001399
McConnell, A. J., Wood, C. S., Neelakandan, P. P., and Nitschke, J. R. (2015). Stimuli-Responsive Metal-Ligand Assemblies. Chem. Rev. 115, 7729–7793. doi:10.1021/cr500632f
McMorran, D. A., and Steel, P. J. (1998). The First Coordinatively Saturated, Quadruply Stranded Helicate and its Encapsulation of a Hexafluorophosphate Anion, Angew. Chem. Int. Ed. Engl. 37. 3295–3297. doi:10.1002/(SICI)1521-3773(19981217)37:23<3295::AID-ANIE3295>3.0.CO;2-5
Mecozzi, S., and Rebek, J. (1998). The 55% Solution: A Formula for Molecular Recognition in the Liquid State. Chem. Eur. J. 4, 1016–1022. doi:10.1002/(SICI)1521-3765(19980615)4:6<1016::AID-CHEM1016>3.0.CO;2-B
Metherell, A. J., Cullen, W., Williams, N. H., and Ward, M. D. (2018). Binding of Hydrophobic Guests in a Coordination Cage Cavity Is Driven by Liberation of “High-Energy” Water. Chem. Eur. J. 24, 1554–1560. doi:10.1002/cem.201704163
Mirtschin, S., Slabon-Turski, A., Scopelliti, R., Velders, A. H., and Severin, K. (2010). A Coordination Cage with an Adaptable Cavity Size. J. Am. Chem. Soc. 132, 14004–14005. doi:10.1021/ja1063789
Nishioka, Y., Yamaguchi, T., Yoshizawa, M., and Fujita, M. (2007). Unusual [2+4] and [2+2] Cycloadditions of Arenes in the Confined Cavity of Self-Assembled Cages. J. Am. Chem. Soc. 129, 7000–7001. doi:10.1021/ja071591x
Oldknow, S., Martir, D. R., Pritchard, V. E., Blitz, M. A., Fishwick, C. W. G., Zysman-Colman, E., et al. (2018). Structure-Switching M3L2 Ir(iii) Coordination Cages With Photo-Isomerising Azo-Aromatic Linkers. Chem. Sci. 9, 8150–8159. doi:10.1039/C8SC03499K
Ono, K., Yoshizawa, M., Akita, M., Kato, T., Tsunobuchi, Y., Ohkoshi, S.-i., et al. (2009). Spin Crossover by Encapsulation. J. Am. Chem. Soc. 131, 2782–2783. doi:10.1021/ja8089894
Ono, K., Yoshizawa, M., Kato, T., Watanabe, K., and Fujita, M. (2007). Porphine Dimeric Assemblies in Organic-Pillared Coordination Cages. Angew. Chem. Int. Ed. 46, 1803–1806. doi:10.1002/anie.200604790
Ovchinnikov, M. V., Holliday, B. J., Mirkin, C. A., Zakharov, L. N., and Rheingold, A. L. (2002). Threefold Symmetric Trimetallic Macrocycles Formed via the Weak-Link Approach. Proc. Natl. Acad. Sci. 99, 4927–4931. doi:10.1073/pnas.072690599
Pilgrim, B. S., and Champness, N. R. (2020). Metal‐Organic Frameworks and Metal‐Organic Cages - A Perspective. ChemPlusChem 85, 1842–1856. doi:10.1002/cplu.202000408
Plajer, A. J., Rizzuto, F. J., von Krbek, L. K. S., Gisbert, Y., Martínez-Agramunt, V., and Nitschke, J. R. (2020). Oxidation Triggers Guest Dissociation During Reorganization of an FeII4L6 Twisted Parallelogram. Chem. Sci. 11, 10399–10404. doi:10.1039/d0sc04352d
Pullen, S., Tessarolo, J., and Clever, G. H. (2021). Increasing Structural and Functional Complexity in Self-Assembled Coordination Cages. Chem. Sci. 12, 7269–7293. doi:10.1039/D1SC01226F
Rizzuto, F. J., von Krbek, L. K. S., and Nitschke, J. R. (2019). Strategies for Binding Multiple Guests in Metal-Organic Cages. Nat. Rev. Chem. 3, 204–222. doi:10.1038/s41570-019-0085-3
Sarada, G., Kim, A., Kim, D., and Jung, O.-S. (2020). Diverse Anion Exchange of Pliable [X2@Pd3L4]4+ Double Cages: a Molecular Ruler for Recognition of Polyatomic Anions. Dalton Trans. 49, 6183–6190. doi:10.1039/d0dt01027h
Schmidt, B. M., Osuga, T., Sawada, T., Hoshino, M., and Fujita, M. (2016). Compressed Corannulene in a Molecular Cage. Angew. Chem. Int. Ed. 55, 1561–1564. doi:10.1002/anie201509963
Scottwell, S. Ø., and Crowley, J. D. (2016). Ferrocene-containing Non-interlocked Molecular Machines. Chem. Commun. 52, 2451–2464. doi:10.1039/c5cc09569g
Sinha, I., and Mukherjee, P. S. (2018). Chemical Transformations in Confined Space of Coordination Architectures. Inorg. Chem. 57, 4205–4221. doi:10.1021/acs.inorgchem.7b03067
Spicer, R. L., Stergiou, A. D., Young, T. A., Duarte, F., Symes, M. D., and Lusby, P. J. (2020). Host-Guest-Induced Electron Transfer Triggers Radical-Cation Catalysis. J. Am. Chem. Soc. 142, 2134–2139. doi:10.1021/jacs.9b11273
Steel, P. J., and McMorran, D. A. (2019). Selective Anion Recognition by a Dynamic Quadruple Helicate. Chem. Asian J. 14, 1098–1101. doi:10.1002/asia.201801262
Takezawa, H., Murase, T., Resnati, G., Metrangolo, P., and Fujita, M. (2014). Recognition of Polyfluorinated Compounds Through Self-Aggregation in a Cavity. J. Am. Chem. Soc. 136, 1786–1788. doi:10.1021/ja412893c
Takezawa, H., Tabuchi, R., Sunohara, H., and Fujita, M. (2020). Confinement of Water-Soluble Cationic Substrates in a Cationic Molecular Cage by Capping the Portals with Tripodal Anions. J. Am. Chem. Soc. 142, 17919–17922. doi:10.1021/jacs.0c08835
Tashiro, S., Tominaga, M., Kawano, M., Therrien, B., Ozeki, T., and Fujita, M. (2005). Sequence-Selective Recognition of Peptides within the Single Binding Pocket of a Self-Assembled Coordination Cage. J. Am. Chem. Soc. 127, 4546–4547. doi:10.1021/ja044782y
Tsutsui, T., Catti, L., Yoza, K., and Yoshizawa, M. (2020). An Atropisomeric M2L4 Cage Mixture Displaying Guest-Induced Convergence and Strong Guest Emission in Water. Chem. Sci. 11, 8145–8150. doi:10.1039/d0sc03223a
Vasdev, R. A. S., Findlay, J. A., Garden, A. L., and Crowley, J. D. (2019). Redox Active [Pd2L4]4+ Cages Constructed From Rotationally Flexible 1,1′-disubstituted Ferrocene Ligands. Chem. Commun. 55, 7506–7509. doi:10.1039/c9cc03321a
Vasdev, R. A. S., Findlay, J. A., Turner, D. R., and Crowley, J. D. (2021). Self‐Assembly of a Redox Active, Metallosupramolecular [Pd3L6]6+ Complex Using a Rotationally Flexible Ferrocene Ligand. Chem. Asian J. 16, 39–43. doi:10.1002/asia.202001277
Wilson, B. H., and Loeb, S. J. (2020). Integrating the Mechanical Bond Into Metal-Organic Frameworks. Chem 6, 1604–1612. doi:10.1016/j.chempr.2020.06.016
Wong, K. K. G., Hoyas Pérez, N., White, A. J. P., and Lewis, J. E. M. (2020). Self-assembly of a Porous Metallo-[5]rotaxane. Chem. Commun. 56, 10453–10456. doi:10.1039/D0CC04780E
Xu, J., Parac, T. N., and Raymond, K. N. (1999). Meso Myths: What Drives Assembly of Helical versus meso-[M2L3] Clusters? Angew. Chem. Int. Ed. 38:2, 2878–2882. doi:10.1002/(SICI)1521-3773(19991004)38:19<2878::aid-anie2878>3.0.co;2-w
Yamauchi, Y., Yoshizawa, M., Akita, M., and Fujita, M. (2010). Engineering Double to Quintuple Stacks of a Polarized Aromatic in Confined Cavities. J. Am. Chem. Soc. 132, 960–966. doi:10.1021/ja904063r
Yoshizawa, M., Klosterman, J. K., and Fujita, M. (2009). Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts. Angew. Chem. Int. Ed. 48, 3418–3438. doi:10.1002/anie.200805340
Yoshizawa, M., Nakagawa, J., Kumazawa, K., Nagao, M., Kawano, M., Ozeki, T., et al. (2005a). Discrete Stacking of Large Aromatic Molecules within Organic-Pillared Coordination Cages. Angew. Chem. Int. Ed. 44, 1810–1813. doi:10.1002/anie.200462171
Yoshizawa, M., Ono, K., Kumazawa, K., Kato, T., and Fujita, M. (2005b). Metal−Metal d−d Interaction through the Discrete Stacking of Mononuclear M(II) Complexes (M = Pt, Pd, and Cu) Within an Organic-Pillared Coordination Cage. J. Am. Chem. Soc. 127, 10800–10801. doi:10.1021/ja053009f
Young, T. A., Gheorghe, R., and Duarte, F. (2020a). Cgbind: A Python Module and Web App for Automated Metallocage Construction and Host-Guest Characterization. J. Chem. Inf. Model. 60, 3546–3557. doi:10.1021/acs.jcim.0c00519
Young, T. A., Martí-Centelles, V., Wang, J., Lusby, P. J., and Duarte, F. (2020b). Rationalizing the Activity of an “Artificial Diels-Alderase”: Establishing Efficient and Accurate Protocols for Calculating Supramolecular Catalysis. J. Am. Chem. Soc. 142, 1300–1310. doi:10.1021/jacs.9b10302
Yue, N. L. S., Eisler, D. J., Jennings, M. C., and Puddephatt, R. J. (2004). Macrocyclic and Lantern Complexes of Palladium(II) With Bis(amidopyridine) Ligands: Synthesis, Structure, and Host−Guest Chemistry. Inorg. Chem. 43, 7671–7681. doi:10.1021/ic048893+
Yue, N., Qin, Z., Jennings, M. C., Eisler, D. J., and Puddephatt, R. J. (2003). Host Complexes that Adapt to Their Guests: Amphitopic Receptors. Inorg. Chem. Commun. 6, 1269–1271. doi:10.1016/S1387-7003(03)00246-6
Keywords: self-assembly, cages, flexibility, host-guest, metallosupramolecular
Citation: Martín Díaz AE and Lewis JEM (2021) Structural Flexibility in Metal-Organic Cages. Front. Chem. 9:706462. doi: 10.3389/fchem.2021.706462
Received: 07 May 2021; Accepted: 02 June 2021;
Published: 17 June 2021.
Edited by:
Chao Deng, Soochow University, ChinaReviewed by:
Narayanan Selvapalam, Kalasalingam University, IndiaRichard J. Hooley, University of California, Riverside, United States
Copyright © 2021 Martín Díaz and Lewis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James E. M. Lewis, amFtZXMubGV3aXNAaW1wZXJpYWwuYWMudWs=